

Abstract
Background
Reactions between nitric oxide (NO), nitrite (NO2-), and unsaturated fatty acids give rise to electrophilic nitro-fatty acids (NO2-FAs), such as nitro-oleic acid (OA-NO2) and nitro-linoleic acid (LNO2). Endogenous electrophilic fatty acids (EFAs) mediate anti-inflammatory responses by modulating metabolic and inflammatory signal transduction reactions. Hence, there is considerable interest in employing NO2-FAs and other EFAs for the prevention and treatment of inflammatory disorders. Thus, we sought to determine whether OA-NO2, an exemplary nitro-fatty acid, has the capacity to inhibit cutaneous inflammation.
Methods
We evaluated the effect of OA-NO2 on allergic contact dermatitis (ACD) using an established model of contact hypersensitivity (CHS) in C57Bl/6 mice utilizing 2,4-dinitrofluorobenzene (DNFB) as the hapten.
Results
We found that subcutaneous (SC) OA-NO2 injections administered 18 h prior to sensitization and elicitation suppresses ACD in both preventative and therapeutic models. In vivo SC OA-NO2 significantly inhibits pathways that lead to inflammatory cell infiltration and the production of inflammatory cytokines in the skin. Moreover, OA-NO2 is capable of enhancing regulatory T cell activity. Thus, OA-NO2 treatment results in anti-inflammatory effects capable of inhibiting ACD by inducing immunosuppressive responses.
Conclusion
Overall, these results support the development of OA-NO2 as a promising therapeutic for ACD and provides new insights into the role of electrophilic fatty acids in the control of cutaneous immune responses potentially relevant to a broad range of allergic and inflammatory skin diseases.
Keywords: Allergic contact dermatitis, contact hypersensitivity, electrophilic fatty acids, nitro-oleic acid, T regulatory cells
Introduction
Reactions between nitric oxide (NO), nitrite (NO2-), and unsaturated fatty acids give rise to electrophilic nitro-fatty acids (NO2-FAs) such as nitro-oleic acid (OA-NO2) and nitro-linoleic acid (LNO2). These fatty acid nitration products and their metabolites are found endogenously in plants, insects, and mammals (1). NO2-FAs have been shown to mediate anti-inflammatory responses both in vitro and in vivo. For example, OA-NO2 and LNO2 inhibit NF-κB signal transduction by blocking p65 DNA binding, thereby suppressing multiple pro-inflammatory cytokines. Similarly, in LPS-stimulated macrophages, OA-NO2 suppresses the secretion of IL-6, TNF-α, and MCP-1 in an NF-κB-dependent manner (2). In vivo, OA-NO2 has been shown to prevent vascular NF-κB activation and subsequent TLR4 surface recruitment following LPS stimulation (3). Additionally, NO2-FAs stimulate anti-inflammatory, antioxidant, and cytoprotective gene expression by acting as partial agonist of peroxisome proliferator-activated receptor-γ (PPAR-γ) and activating heme oxygenase-1 (HMOX-1), nuclear factor E2-related factor 2 (Nrf2), and the heat shock response pathways (4-7). The beneficial health effects and therapeutic potential of NO2-FAs have been demonstrated in several in vivo murine models of inflammatory disease. In this regard, administration of OA-NO2 attenuated endotoxemia, renal ischemia and reperfusion (I/R) injury, angiotensin II-induced hypertension, and atherosclerosis (8-11). Thus, there is considerable interest in employing NO2-FAs and other electrophilic fatty acids (EFA) for the prevention and treatment of inflammatory disorders.
Allergic contact dermatitis (ACD) is a delayed-type hypersensitivity response and one of the most common cutaneous inflammatory diseases, affecting as many as 15-20% of the population worldwide. ACD is an antigen-specific T cell mediated cutaneous inflammatory disease typically triggered through hapten sensitization and elicitation. Hapten exposure results in the secretion of a variety of inflammatory mediators in skin in vivo, and activates NF-κB pathways in human and mouse cutaneous dendritic cells (12-14). Based on this rationale, we evaluated potential anti-inflammatory effects of NO2-FAs in a murine model of ACD.
Materials and Methods
Mice
Female Balb/c mice were purchased from Jackson Laboratories (Bar Harbor, Maine) and used between the ages of 6 and 12 weeks. Male and Female Foxp3tm3(DTR/GFP)Ayr (FoxP3DTR) mice on the C56Bl/6 background (15), developed and granted by Alexander Rudensky and a kind gift by Dario Vignali, were bred and housed in the University of Pittsburgh animal facility. Mice were housed under specific-pathogen-free conditions and treated according to the University of Pittsburgh's institutional animal care guidelines and according to the NIH guide for the care and use of laboratory animals.
Chemicals and reagents
10-OA-NO2 and NO2-13C18OA was synthesized and purified as previously described (16, 17). The concentration of 10-OA-NO2 was calculated gravimetrically or spectrophotometrically using the following extinction coefficient in phosphate buffer, λ268 8.22.
Contact hypersensitivity response (CHS)
0.2 mg/mse (0.1%) of OA-NO2 was dissolved in DMSO and Balb/c or FoxP3DTR mice were injected at each of two subcutaneous (SC) sites (100μl/injection) 18 h prior to sensitization with 1.0% 2,4-dinitrofluorobenzene (DNFB) in acetone and olive oil, 4:1 (v:v) (Sigma Aldrich; St. Louis, MO) on the shaved abdomen, unless otherwise indicated. Elicitation of the CHS response was performed 5 d later by administering SC injections of 0.2 mg/mse OA-NO2 (100μl/injection) 18 h prior to DNFB application on dorsal surface of the right ear. CHS responses were assessed as an increase in ear thickness, and data is expressed as the percentage of ear thickness increase using the formula: [(thickness of challenged ear – thickness of control ear)/(thickness of control ear)] × 100.
Real-time qRT-PCR
Total RNA was extracted from ears utilizing TRI-reagent (Molecular Research Center; Cincinnati, OH) and reverse transcribed using the QuantiTect Reverse Transcription Kit (Qiagen; Hilden, Germany) according to manufacturers' instructions. Quantitative real-time PCR was performed utilizing the Taqman Gene Expression Master Mix (Life Technologies, Carlsbad, CA) according to manufacturer's instructions with IDT PrimeTime qPCR assays (IDT; Coralville, IA) specific for ActB (endogenous control), IL-1β, IL-6, IL-10, and HMOX-1. Reactions were run in triplicates and analyzed on a StepOne Plus sequence detection system (Applied Biosystems). Expression levels were normalized based on the 2-ΔΔCt method.
Cutaneous microscopy
To assess histology and cellular infiltration, cross-sections of mouse ears were prepared and stained as previously described (18). Briefly, frozen cross-sections were embedded in Tissue-Tek OCT (Miles Laboratories; Elkhart, IN) and snap frozen in pre-chilled methyl-butane (Sigma Aldrich). Cryostat sections (8 μm) were mounted onto slides pre-treated with Vectabond (Vector Laboratories; Burlingame, CA), fixed in 96% EtOH, and used for H&E staining. Images were acquired using an Olympus Provis AX-70 microscope system (Olympus) with FluoView 500 software.
Regulatory T cell (Treg) ablation
To ablate Treg cells, FoxP3DTR mice were intraperitoneally (i.p.) injected with 50 μg/kg DT (Sigma Aldrich) on day 1 (day 1 is determined according to Fig 1A) and then every other day until day 9. In these experiments control mice were FoxP3DTR littermates not treated with DT.
Figure 1. OA-NO2 suppresses cutaneous ear swelling in a model of ACD.
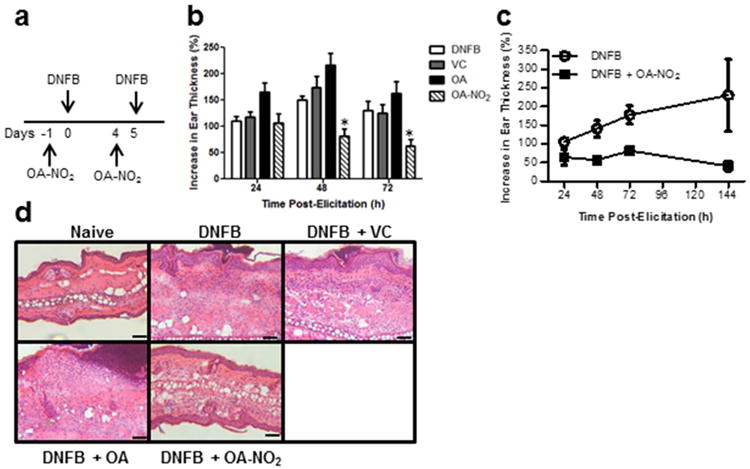
(a) Balb/c mice were sensitized and CHS was elicited with DNFB ± contralateral SC injections of OA-NO2, OA (negative control), or VC (vehicle control), according to diagram. The CHS response was analyzed at time points indicated following elicitation. (b) Bars represent the increase in ear thickness mean ± SEM of 4-5 mice per group. One representative of four individual experiments. *indicates a significant difference compared to the DNFB treated group at each time point, p < 0.05. (c) Ear swelling over an extended time course. Each point represents the mean ± SEM of 4-5 mice per group. (d) 72 h following elicitation ears were excised and cross-sections were stained with H&E. Scale bar = 50 μm.
Statistical analysis
Results from multiple different groups were compared using a two-way analysis of variance (ANOVA) followed by the Bonferroni post-hoc test to compare groups over time. A one-way ANOVA was utilized followed by Newman-Keuls multiple comparison post-hoc test if only one time point was assessed. Comparison of two means was performed by a 2-tailed Student's T-test. A p value of < 0.05 was considered statistically significant.
Results
Bioavailability of OA-NO2
To assess the capacity of OA-NO2 to access the systemic circulation following SC injection, we determined OA-NO2 serum levels over time after treatment. Initially, titrations of OA-NO2 were performed from 0.01-1%, and 0.1% OA-NO2 was selected as the highest dose that did not induce a local irritant effect (data not shown). 0.1% OA-NO2 (18:1) was injected SC (two injection sites, 100 μl each) and it and its major metabolites (16:1 and 14:1) were quantified in the serum 3, 6, and 18 h following SC injections. OA-NO2 was present in the circulation and active based on the metabolites detected (Fig S1). Importantly, physiologically-relevant levels of OA-NO2 (3) were detected as early as 3 h following injection of 0.1% OA-NO2, and levels were sustained within the physiological range for at least 18 h following treatment (Fig S1). Overall, these data confirm the bioavailability of SC-administered OA-NO2 for at least 18 h following treatment.
Induction of cutaneous inflammatory immune responses are inhibited by OA-NO2
To determine whether OA-NO2 can inhibit a cutaneous inflammatory response, we established CHS as a murine model of ACD in Balb/c mice utilizing the hapten DNFB. The effect of OA-NO2 on CHS induction was determined by injecting mice SC at a distal site with OA-NO2 18 h prior to abdominal sensitization with DNFB, then again 5 d later and 18 h prior to DNFB elicitation of the CHS response in the ear (Fig 1a). We chose to treat mice with OA-NO2 18 h prior to DNFB sensitization and elicitation based on our pharmacokinetic studies that indicated the presence of physiologically-relevant levels of OA-NO2 within 18 h enabling sufficient time to initiate anti-inflammatory responses prior to hapten exposure. CHS, assessed by increases in ear thickness, was significantly decreased in OA-NO2 treated mice at 48 and 72 h following elicitation, compared to DNFB treatment alone (Fig 1b). Moreover, this response was sustained for at least 144 h post-elicitation (Fig 1c). Native OA control did not suppress CHS nor did vehicle control (VC) (Fig 1b). Histological analysis demonstrated an absence of epidermal micro-abscesses and a decrease in epidermal/dermal thickness and cellular infiltrate in mice treated with OA-NO2 compared to those administered DNFB alone, or OA or VC controls (Fig 1d), consistent with the inhibitory effect observed by ear thickness measurements (Fig 1b and c). These results demonstrate the anti-inflammatory electrophilic effects of OA-NO2 in the cutaneous microenvironment.
OA-NO2 suppresses inflammation induced cutaneous angiogenesis
There is substantial evidence demonstrating the interdependence of angiogenesis and inflammation. In this regard, one of the hallmarks of ACD is increased vascularization and inhibition of vascularization can block CHS (19). In vivo, OA-NO2 prevents vascular NF-κB activation (3). Thus, we evaluated the cutaneous expression of the endothelial cell marker CD31 following induction of CHS to determine the effect of SC OA-NO2 treatment on cutaneous angiogenesis. A significant increase in vascular CD31 expression was observed in the DNFB treatment group 72 h following elicitation, compared to control mice (Fig 2a and b). In the presence of OA-NO2, CD31 expression was markedly and significantly decreased 72 and 144 h post-elicitation, compared to DNFB alone. Moreover, with OA-NO2 treatment vessels that were present appeared to be collapsed compared to open vessels observed following DNFB alone. Our results demonstrate that OA-NO2 inhibits vascularization in the setting of ACD, which potentially limits homing of inflammatory cells into the cutaneous microenvironment.
Figure 2. CHS-dependent dermal vascularization is inhibited by OA-NO2.
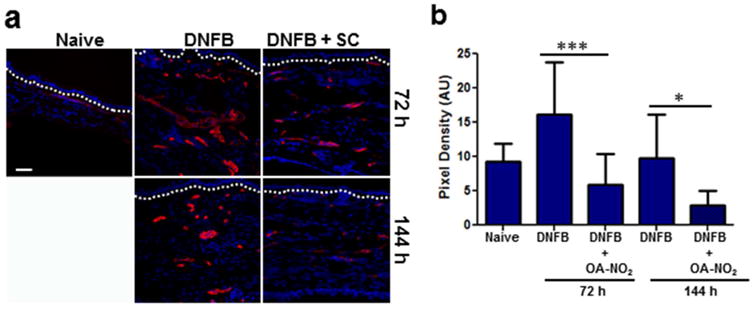
DNFB CHS responses were initiated following treatment with SC OA-NO2. (a) 72 h and 144 h following elicitation ears were excised and cross-sections were immunofluorescently labeled with CD31-specific antibodies (red) in addition to DAPI nuclear counterstain (blue). Dashed line indicates epidermal-dermal junction. Scale bar = 50 μm. (b) CD31 expression was quantitated by assessing the red pixel density. Bars represent the mean density of 10 independent high powered fields ± SD. Asterisks indicate a significant difference compared to DNFB alone at each time point. *** = p < 0.001 and * = p < 0.05.
Molecular phenotype of skin following subcutaneous OA-NO2 injection
To evaluate the molecular effects of OA-NO2 in the cutaneous microenvironment, mice were treated SC with OA-NO2 prior to DNFB treatments, and skin was evaluated for changes in gene expression 12 h after elicitation. There was a significant decrease in mRNA expression levels for the inflammatory factor IL-1β in the OA-NO2 treated group, compared to those receiving DNFB alone or DNFB + OA control treatment (Fig 3a). Furthermore, IL-1β transcripts were suppressed in the skin for at least 72 h (data not shown). IL-6 levels were not decreased by 12 h after elicitation in the presence of OA-NO2, compared to DNFB alone. Though, IL-6 was significantly increased in the native OA control group compared to both the DNFB and OA-NO2 treatment groups (Fig 3b, left panel). Importantly, 72 h following elicitation, compared to DNFB alone, OA-NO2 significantly decreased IL-6 expression (Fig 3b, right panel). Thus, OA-NO2 suppresses pro-inflammatory cytokines at both 12 h and 72 h following elicitation.
Figure 3. OA-NO2 initiates an anti-inflammatory response with cytoprotective responses.
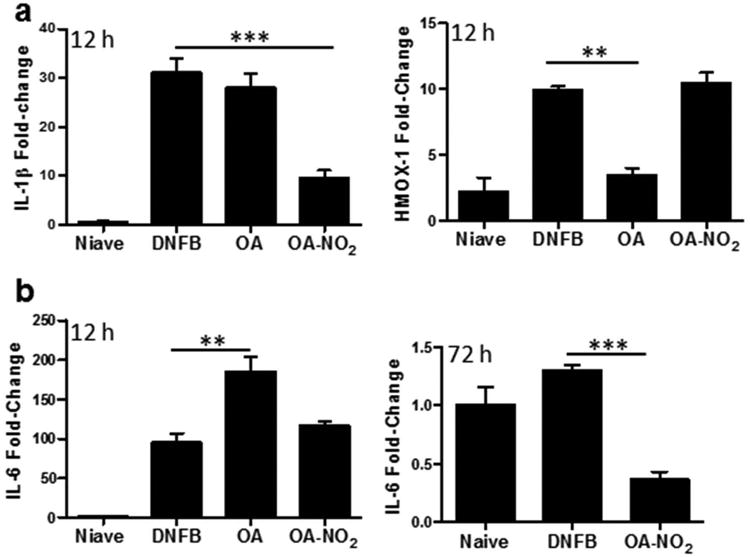
Graphs demonstrate the relative fold change in mRNA expression of (a) IL-1β and HMOX-1 at 12 h following elicitation and (b) IL-6 at 12 h (left panel) and 72 h (right panel). Fold-change was determined using the relative qRT-PCR 2-ΔΔCt method. The bar indicates the mean of three mice for each treatment group. One representative of two independent experiments. Asterisks indicate a significant difference compared to DNFB treatment alone, as indicated, **=p < 0.01, ***= p < 0.001.
EFAs inherently initiate antioxidant and cytoprotective gene expression by activating the Nrf2/Keap1/HMOX-1 pathway, which is the prototypical signaling pathway activated by electrophilic compounds (7, 20). Thus, we assessed HMOX-1 expression in the skin following SC injections of OA-NO2. We observed a significant increase in HMOX-1 12 h after elicitation in the DNFB alone CHS group, which was maintained in the OA-NO2 treatment group, but significantly suppressed by native OA (Fig 3a, right panel).
OA-NO2 enhances regulatory cell functions
In addition to various cutaneous regulatory pathways, Tregs secreting IL-10 play a major role in the resolution of CHS (21). Thus, we next determined whether OA-NO2 could potentiate Treg responses in order to resolve inflammation. We first examined the ability of OA-NO2 to induce IL-10, a regulatory cytokine secreted by CD4+CD25+Foxp3+ regulatory T cells. IL-10 expression was not affected by OA-NO2 in the first 24 h following elicitation. However, by 72 h after elicitation IL-10 transcripts were significantly increased in OA-NO2 treated skin (Fig 4a). Subsequently, we examined the presence of Foxp3+ cells in the skin. 72 h after elicitation Foxp3+ Tregs accumulate in the skin regardless of OA-NO2 treatment (Fig 4b and c). While there was not a significant increase in Tregs at 72 h, there was a significant increase in IL-10 transcript expression, suggesting that OA-NO2 promoted Treg activation. By 144 h following elicitation there was a significant increase in dermal Foxp3+ regulatory T cells in OA-NO2 treated animals compared to those with DNFB treatments alone (Fig 4b and c). In line with this finding there was also a significant increase in cutaneous Foxp3 transcript expression (Fig 4d).
Figure 4. OA-NO2 enhances immune regulatory responses.
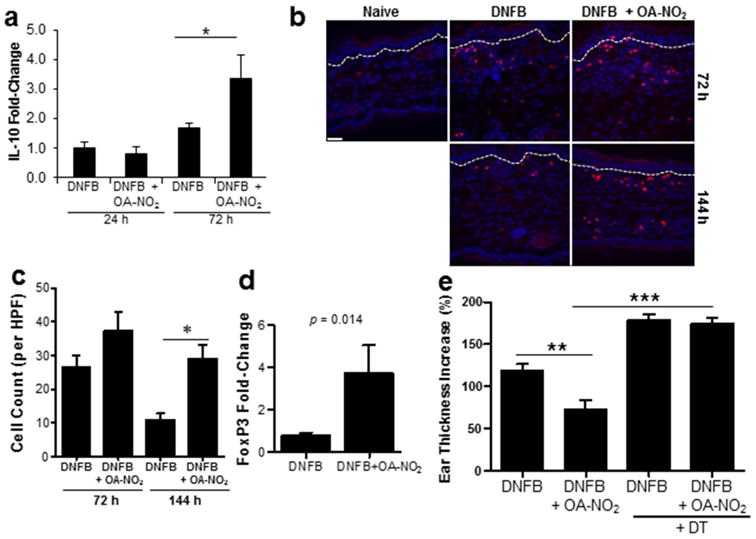
CHS was induced with DNFB ± OA-NO2. (a) Bars represent the mean relative fold-change in IL-10 mRNA expression ± SEM of 3 mice per group, 24 and 72 h following elicitation. (b) 72 and 144 h following elicitation ears were immunofluorescently labeled with FoxP3-specific antibodies (red) in addition to DAPI nuclear counterstain (blue). Dashed line indicates epidermal-dermal junction. Scale bar = 50 μM. (c) FoxP3+ cells were quantitated. Bars represent the mean ± SEM of 3 mice, 7-10 independent high powered fields per mouse were averaged. (d) FoxP3 transcripts were assessed 144 h following elicitation. Bar indicates the mean of each treatment group ± SEM of 3-5 mice. Significance value is indicated. (e) CHS was induced in FoxP3DTR mice ± OA-NO2. Bars represent the mean increase in ear thickness at 48 h ± SEM of 4-5 mice. One representative of two individual experiments for each study. Asterisk indicates a significant difference between the indicated treatment groups, *= p < 0.05, **= p < 0.01, and ***= p < 0.001.
To more specifically evaluate whether enhanced Treg activity contributes to OA-NO2 induced CHS inhibition, we evaluated OA-NO2 effects in Treg depleted animals. Tregs were depleted by treating FoxP3DTR mice with 50 μg/kg of DT starting 1 day following sensitization (day 1) and continuing every other day until 4 days following elicitation (day 9) (15). FoxP3DTR littermates not treated with DT were used as control mice. In FoxP3DTR mice OA-NO2 significantly suppressed the CHS response by 48 h post-elicitation, as determined by a decrease in ear thickness (Fig 4e). However, when Treg cells were depleted by treatment with DT, OA-NO2 had a minimal effect compared to DNFB treatment alone (Fig 4e). Taken together, these results indicate that OA-NO2 supports Treg accumulation and function in reducing CHS.
OA-NO2 treatment resolves established CHS responses
Given the observed efficacy of OA-NO2 in the prevention of CHS, and underlying mechanisms suggestive of a potent anti-inflammatory effect, we sought to ascertain the effectiveness of OA-NO2 in the treatment of established CHS. For this, we first determined if OA-NO2 is critical during sensitization and/or elicitation. Mice were sensitized and elicited with DNFB and treated with OA-NO2 only during either the sensitization phase (SO), elicitation phase (EO), or during sensitization and elicitation (SE). There was a significant decrease in the ear swelling in the EO and SE treatment groups compared to the SO treatment group (Fig 5a), indicating that OA-NO2 is essential only during the elicitation phase, and suggests that OA-NO2 could be effective therapeutically. To address this directly, we sought to determine whether OA-NO2 has the capacity to suppress an established CHS response. Mice were sensitized (day 0) and elicited (day 5) with DNFB. 48 h following elicitation (day 7) mice were treated with OA-NO2, vehicle control, or left untreated. Ear thickness was measured 24 h after elicitation (day 6) and again at 48 h (day 7, treatment day 0), 72 h (day 8, post-treatment day 1), and 144 h (day 11, post-treatment day 4) after elicitation. Within 24 h following OA-NO2 injection there was a significant decrease in ear swelling in the OA-NO2 treated group compared to untreated animals and vehicle controls (Fig 5b). Over the subsequent 3 days, significant ear swelling persisted in vehicle control and untreated animals, while ear swelling was reduced to near baseline in OA-NO2 treated animals. Taken together, these results demonstrate that OA-NO2 has the capacity to therapeutically suppress established CHS.
Figure 5. OA-NO2 treatment suppresses established CHS.
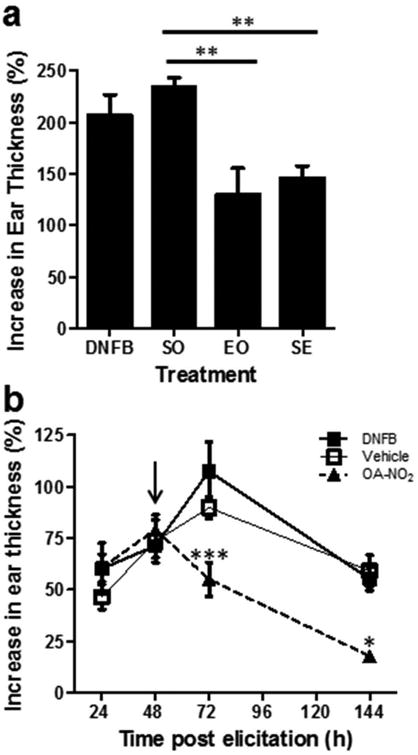
(a) CHS was induced with DNFB ± OA-NO2 administered during sensitization only (SO), elicitation only (EO), or during sensitization and elicitation (SE). Bars represent the mean increase in ear thickness ± SEM of 4-5 mice per group at 144 hr post-elicitation. Asterisks indicate a significant difference between indicated groups, ** = p < 0.01. (b) CHS was induced in mice with 0.5% DNFB. 48 h following elicitation (arrow), groups of mice received OA-NO2, vehicle control, or were untreated. Each data point represents the mean increase in ear thickness ± SEM of 3-5 mice. Asterisk indicates a significant difference compared to DNFB treatment group, * = p < .05 and *** = p < 0.001. Both data sets are one representative of three independent experiments each.
Discussion
The therapeutic potential of NO2-FAs has been demonstrated in several in vivo murine models of inflammatory disease. Herein, we reveal that a distant SC injection of an exemplary NO2-FA, OA-NO2, can prevent and treat CHS in mice. NO2-FAs appear to affect multiple immune regulatory mechanisms in the skin. First, OA-NO2 reduced IL-1β and IL-6, essential pro-inflammatory cytokines for the differentiation of Th17 cells involved in ACD (22). This reduced cytokine expression is consistent with the capacity of EFAs to inhibit inflammation through the blockade of NF-κB translocation and downstream expression of IL-1β and IL-6 (3, 8). Specifically, OA-NO2 and LNO2 inhibit NF-κB signal transduction at various signaling stages by disrupting the upstream assembly of adaptor protein complexes (TRAF6/IKKβ/IκBα) in lipid rafts and blocking IκBα phosphorylation and p65 DNA binding, thereby suppressing multiple pro-inflammatory cytokines (2, 3, 23). Second, in addition to these cytokine effects, our results demonstrate that OA-NO2 inhibits vascularization in the setting of ACD. There is substantial evidence demonstrating the interdependence of angiogenesis and inflammation. In this regard, one hallmark of ACD is increased vascularization and inhibition of vascularization can block CHS (19). By limiting vascularization OA-NO2 can limit the homing of inflammatory cells into the cutaneous microenvironment. Third, our studies show that OA-NO2 affects the accumulation of Tregs in the skin. Tregs secreting IL-10 play a major role in the resolution of CHS (21). The regulatory mechanisms engaged by Tregs are involved in both suppression of effector T cell responses in the LNs, and in restraining peripheral inflammation in the skin following ACD induction (24-28). Consistent with studies by Lehtimaki et al, we find that Tregs gradually accumulate in the dermis of DNFB challenged mice (27), presumably as a compensatory mechanism facilitating inflammatory resolution. Importantly, we show that OA-NO2 enhances this recruitment of Tregs into inflamed dermis and potentiates the IL-10 response.
EFAs have been shown to inherently initiate antioxidant and cytoprotective gene expression by activating the Nrf2/Keap1/HMOX-1 pathway, which is the prototypical signaling pathway activated by electrophilic compounds (7, 20). HMOX-1 is induced in response to cellular stress and diverse oxidative stimuli. HMOX-1 contributes to the reduction of oxidative stress and the attenuation of inflammatory responses. We assessed HMOX-1 expression in the skin following SC injections of OA-NO2. In the context of the enhanced pro-inflammatory DNFB CHS cytokine response, we observed a significant increase in HMOX-1, in both OA-NO2 treated and untreated groups. In vivo, exposure to sensitizers can activate Nrf2 signaling by increasing the levels of reactive oxygen species, other radical species, and lipid oxidation products, a response paralleled by extents of EFA generation (29). Despite the similarity between these responses, several characteristics differentiate the signaling induced by sensitizers and EFAs. Sensitizers, their metabolites and byproducts of the inflammatory responses are typically harder electrophiles and thus more reactive not only with cysteine but also more readily alkylate lysine, histidine, and DNA bases (30). Hard electrophile reactions with lysine and histidine are long lived compared to thiol reactions and are accompanied by both DNA base alkylation and more indiscriminate cysteine reactions. This is an important consideration, as DNA damage furthers inflammation and cell death. As a consequence, the activation of Nrf2 and downstream HMOX-1 expression by sensitizers is accompanied by widespread lysine, histidine, and cysteine adduction that can promote cell toxicity, increased inflammation, and cell death (31). In contrast, NO2-FA are soft electrophiles that preferentially target Keap1 over other competing cellular thiols, minimally modify histidine, and do not react with lysines (32). This mechanism of action allows for HMOX-1 activation and the attenuation rather than exacerbation of inflammatory responses that would be induced by indiscriminate hard electrophile-mediated alkylation reactions. In addition, EFAs react reversibly with thiols, a characteristic not shared by reactive oxygen species and harder electrophiles, resulting in reduced residency time on target proteins and lower potential for toxicity (30, 33). Thus, EFAs will inhibit CHS by mechanisms not shared by sensitizers, inhibiting inflammation by limiting NF-κB-dependent proinflammatory mediator expression and promoting Nrf2-dependent NF-κB downregulation (2, 34). Therefore, while we demonstrate that DNFB induces the HMOX-1 expression to a similar extent as OA-NO2, it is important to consider that the environment and underlying conditions resulting from EFAs actions are mechanistically distinct and are documented to account for the protective actions of OA-NO2.
In addition to reversing Nrf2/HMOX-1 pathway activation, natural OA potentiated the ACD inflammatory response, characterized by intense epidermal infiltration, thickness, and microabscess formation. There are conflicting reports regarding the role of OA in inflammation. For instance, studies have suggested that OA has an anti-inflammatory role by, in part, inhibiting T-cell activation and proliferation (35). Contrary to these findings, studies utilizing OA-induced lung injury and peritoneal exudate models suggest that OA enhances inflammatory responses by inducing neutrophil activation and migration to sites of inflammation (36-38). In this regard, neutrophils in skin are highly inflammatory and are required for the development of CHS (39). Thus, in an ACD model the observed enhancement of the inflammatory response in the skin by natural OA may be a result of attraction and activation of neutrophils, distinct from the observed electrophilic OA-NO2 suppression of cutaneous infiltration and inflammation.
Importantly, the levels of OA-NO2 detected in the serum in our studies were physiologically-relevant in the context of instigating systemic signaling responses. For instance, Villacorta et al. (3) demonstrated in a murine model of vascular inflammation that 25 nM (8 ng/ml) serum concentrations of OA-NO2 were sufficient to inhibit NF-κB translocation and DNA binding. In parallel, we observed systemic serum concentrations ranging from 25-60 nM (8-20 ng/ml). These levels of OA-NO2 were sufficient to significantly decrease the expression of the NF-κB dependent cytokines IL-β and IL-6 in the context of CHS. These murine-based observations have a significant potential to be translated into clinical trials, as the pharmacokinetics of OA-NO2 have been extensively characterized in multiple animal models in the course of IND-enabling studies of both intravenous and oral formulations of OA-NO2 for administration to humans. FDA-approved Phase 1a and 1b investigations of both intravenous and oral OA-NO2 show bioavailability and affirm drug safety, with these human trials exceeding the 25-50 nM serum concentrations that were achieved in the present murine model studies.
In aggregate, the data presented here provides experimental evidence that OA-NO2 delivered SC is capable of preventing and treating CHS. In the setting of CHS, OA-NO2 demonstrates multiple effects on cutaneous immune function. It inhibits the production of inflammatory cytokines and reduces vascularization. Importantly, OA-NO2 favors the accumulation of functional Tregs in inflamed skin over time, contributing to the resolution of the inflammatory response. Overall, these results support the development of OA-NO2 as a promising therapeutic for ACD and provides new insights into the role of EFAs in the control of cutaneous immune responses potentially relevant to a broad range of allergic and inflammatory skin diseases.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number R01AR066548 (to A.R.M.). Additionally, this work was supported by P50 CA121973 (to L.D.F.) and R01 AR068249 (to L.D.F.). Tissue sample imaging was performed in the Center for Biological Imaging, supported by NIH Grant U54 RR022241. The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- NO2-FA
nitro-fatty acid
- OA
oleic acid
- OA-NO2
nitro-oleic acid
- LNO2
nitro-linoleic acid
- HMOX-1
heme oxygenase-1
- PPAR-γ
peroxisome proliferator-activated receptor-γ
- Nrf2
nuclear factor E2-related factor 2
- EFA
electrophilic fatty acids
- ACD
Allergic contact dermatitis
- SC
subcutaneous
- DNFB
2,4-dinitrofluorobenzene
- CHS
contact hypersensitivity
- Treg
regulatory T
Footnotes
Conflict of Interest: BAF and FJS acknowledge financial interest in Complexa, Inc. LDF has a financial interest in SkinJect and Brainstage. The other authors state no conflict of interest.
Author Contributions: ARM, CDC, MEK, JDP, and SRS all contributed to the acquisition and analysis of data. ARM, FJS, BAF, and LDF contributed substantially to the conception and design of the manuscript and interpretation of data. ARM drafted the article and CDC, MEK, JDP, SRS, FJS, BAF, and LDF had input on article revision and final approval.
References
- 1.Delmastro-Greenwood M, Freeman BA, Wendell SG. Redox-Dependent Anti-Inflammatory Signaling Actions of Unsaturated Fatty Acids. Annual Rev Physiol. 2014;76:79–105. doi: 10.1146/annurev-physiol-021113-170341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cui T, Schopfer FJ, Zhang J, Chen K, Ichikawa T, Baker PR, et al. Nitrated fatty acids: Endogenous anti-inflammatory signaling mediators. J Biol Chem. 2006;281(47):35686–35698. doi: 10.1074/jbc.M603357200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Villacorta L, Chang L, Salvatore SR, Ichikawa T, Zhang J, Petrovic-Djergovic D, et al. Electrophilic nitro-fatty acids inhibit vascular inflammation by disrupting LPS-dependent TLR4 signalling in lipid rafts. Cardiovas Res. 2013;98(1):116–124. doi: 10.1093/cvr/cvt002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferreira AM, Minarrieta L, Lamas Bervejillo M, Rubbo H. Nitro-fatty acids as novel electrophilic ligands for peroxisome proliferator-activated receptors. Free Rad Biol Med. 2012;53(9):1654–1663. doi: 10.1016/j.freeradbiomed.2012.08.572. [DOI] [PubMed] [Google Scholar]
- 5.Tsujita T, Li L, Nakajima H, Iwamoto N, Nakajima-Takagi Y, Ohashi K, et al. Nitro-fatty acids and cyclopentenone prostaglandins share strategies to activate the Keap1-Nrf2 system: a study using green fluorescent protein transgenic zebrafish. Genes Cells. 2011;16(1):46–57. doi: 10.1111/j.1365-2443.2010.01466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Freeman BA, Baker PR, Schopfer FJ, Woodcock SR, Napolitano A, d'Ischia M. Nitro-fatty acid formation and signaling. J Biol Chem. 2008;283(23):15515–15519. doi: 10.1074/jbc.R800004200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kansanen E, Jyrkkanen HK, Volger OL, Leinonen H, Kivela AM, Hakkinen SK, et al. Nrf2-dependent and -independent responses to nitro-fatty acids in human endothelial cells: identification of heat shock response as the major pathway activated by nitro-oleic acid. J Biol Chem. 2009;284(48):33233–33241. doi: 10.1074/jbc.M109.064873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu H, Jia Z, Soodvilai S, Guan G, Wang MH, Dong Z, et al. Nitro-oleic acid protects the mouse kidney from ischemia and reperfusion injury. Am J Physiol Renal Physiol. 2008;295(4):F942–949. doi: 10.1152/ajprenal.90236.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang H, Liu H, Jia Z, Olsen C, Litwin S, Guan G, et al. Nitro-oleic acid protects against endotoxin-induced endotoxemia and multiorgan injury in mice. Am J Physiol Renal Physiol. 2010;298(3):F754–762. doi: 10.1152/ajprenal.00439.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang J, Villacorta L, Chang L, Fan Z, Hamblin M, Zhu T, et al. Nitro-oleic acid inhibits angiotensin II-induced hypertension. Circ Res. 2010;107(4):540–548. doi: 10.1161/CIRCRESAHA.110.218404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rudolph TK, Rudolph V, Edreira MM, Cole MP, Bonacci G, Schopfer FJ, et al. Nitro-fatty acids reduce atherosclerosis in apolipoprotein E-deficient mice. Arterioscl Throm Vas. 2010;30(5):938–945. doi: 10.1161/ATVBAHA.109.201582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cruz MT, Goncalo M, Figueiredo A, Carvalho AP, Duarte CB, Lopes MC. Contact sensitizer nickel sulfate activates the transcription factors NF-kB and AP-1 and increases the expression of nitric oxide synthase in a skin dendritic cell line. Exp Dermatol. 2004;13(1):18–26. doi: 10.1111/j.0906-6705.2004.00105.x. [DOI] [PubMed] [Google Scholar]
- 13.Martin SF, Esser PR, Weber FC, Jakob T, Freudenberg MA, Schmidt M, et al. Mechanisms of chemical-induced innate immunity in allergic contact dermatitis. Allergy. 2011;66(9):1152–1163. doi: 10.1111/j.1398-9995.2011.02652.x. [DOI] [PubMed] [Google Scholar]
- 14.Cruz MT, Duarte CB, Goncalo M, Figueiredo A, Carvalho AP, Lopes MC. Differential activation of nuclear factor kappa B subunits in a skin dendritic cell line in response to the strong sensitizer 2,4-dinitrofluorobenzene. Arch Dermatol Res. 2002;294(9):419–425. doi: 10.1007/s00403-002-0342-y. [DOI] [PubMed] [Google Scholar]
- 15.Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8(2):191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- 16.Woodcock SR, Marwitz AJ, Bruno P, Branchaud BP. Synthesis of nitrolipids. All four possible diastereomers of nitrooleic acids: (E)- and (Z)-, 9- and 10-nitro-octadec-9-enoic acids. Org Lett. 2006;8(18):3931–3934. doi: 10.1021/ol0613463. [DOI] [PubMed] [Google Scholar]
- 17.Baker PR, Lin Y, Schopfer FJ, Woodcock SR, Groeger AL, Batthyany C, et al. Fatty acid transduction of nitric oxide signaling: multiple nitrated unsaturated fatty acid derivatives exist in human blood and urine and serve as endogenous peroxisome proliferator-activated receptor ligands. J Biol Chem. 2005;280(51):42464–42475. doi: 10.1074/jbc.M504212200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mathers AR, Janelsins BM, Rubin JP, Tkacheva OA, Shufesky WJ, Watkins SC, et al. Differential capability of human cutaneous dendritic cell subsets to initiate Th17 responses. J Immunol. 2009;182(2):921–933. doi: 10.4049/jimmunol.182.2.921. [DOI] [PubMed] [Google Scholar]
- 19.Watanabe H, Mamelak AJ, Wang B, Howell BG, Freed I, Esche C, et al. Anti-vascular endothelial growth factor receptor-2 (Flk-1/KDR) antibody suppresses contact hypersensitivity. Exp Dermatol. 2004;13(11):671–681. doi: 10.1111/j.0906-6705.2004.00240.x. [DOI] [PubMed] [Google Scholar]
- 20.Kansanen E, Bonacci G, Schopfer FJ, Kuosmanen SM, Tong KI, Leinonen H, et al. Electrophilic Nitro-fatty Acids Activate NRF2 by a KEAP1 Cysteine 151-independent Mechanism. J Biol Chem. 2011;286(16):14019–14027. doi: 10.1074/jbc.M110.190710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ring S, Schäfer SC, Mahnke K, Lehr HA, Enk AH. CD4+CD25+ regulatory T cells suppress contact hypersensitivity reactions by blocking influx of effector T cells into inflamed tissue. Eur J Immunol. 2006;36(11):2981–2992. doi: 10.1002/eji.200636207. [DOI] [PubMed] [Google Scholar]
- 22.Peiser M. Role of Th17 cells in skin inflammation of allergic contact dermatitis. Clin Dev Immunol. 2013;2013:261037. doi: 10.1155/2013/261037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rudolph V, Rudolph TK, Schopfer FJ, Bonacci G, Woodcock SR, Cole MP, et al. Endogenous generation and protective effects of nitro-fatty acids in a murine model of focal cardiac ischaemia and reperfusion. Cardiovas Res. 2010;85(1):155–166. doi: 10.1093/cvr/cvp275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chow Z, Mueller SN, Deane JA, Hickey MJ. Dermal regulatory T cells display distinct migratory behavior that is modulated during adaptive and innate inflammation. J Immunol. 2013;191(6):3049–3056. doi: 10.4049/jimmunol.1203205. [DOI] [PubMed] [Google Scholar]
- 25.Christensen AD, Skov S, Kvist PH, Haase C. Depletion of regulatory T cells in a hapten-induced inflammation model results in prolonged and increased inflammation driven by T cells. Clin Exp Immunol. 2014;179:485–499. doi: 10.1111/cei.12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gorbachev AV, Fairchild RL. Induction and regulation of T-cell priming for contact hypersensitivity. Crit Rev Immunol. 2001;21(5):451–472. [PubMed] [Google Scholar]
- 27.Lehtimaki S, Savinko T, Lahl K, Sparwasser T, Wolff H, Lauerma A, et al. The Temporal and Spatial Dynamics of Foxp3+ Treg Cell-Mediated Suppression during Contact Hypersensitivity Responses in a Murine Model. J Invest Dermatol. 2012;132(12):2744–2751. doi: 10.1038/jid.2012.212. [DOI] [PubMed] [Google Scholar]
- 28.Schwarz A, Navid F, Sparwasser T, Clausen BE, Schwarz T. In vivo reprogramming of UV radiation–induced regulatory T-cell migration to inhibit the elicitation of contact hypersensitivity. J Allergy Clin Immunol. 2011;128(4):826–833. doi: 10.1016/j.jaci.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 29.Migdal C, Botton J, El Ali Z, Azoury ME, Guldemann J, Giménez-Arnau E, et al. Reactivity of Chemical Sensitizers Toward Amino Acids In Cellulo Plays a Role in the Activation of the Nrf2-ARE Pathway in Human Monocyte Dendritic Cells and the THP-1 Cell Line. Toxicol Sci. 2013;133(2):259–274. doi: 10.1093/toxsci/kft075. [DOI] [PubMed] [Google Scholar]
- 30.Schopfer FJ, Cipollina C, Freeman BA. Formation and Signaling Actions of Electrophilic Lipids. Chem Rev. 2011;111(10):5997–6021. doi: 10.1021/cr200131e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Codreanu SG, Ullery JC, Zhu J, Tallman KA, Beavers WN, Porter NA, et al. Alkylation Damage by Lipid Electrophiles Targets Functional Protein Systems. Mol Cell Proteo. 2014;13(3):849–859. doi: 10.1074/mcp.M113.032953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Batthyany C, Schopfer FJ, Baker PRS, Durán R, Baker LMS, Huang Y, et al. Reversible Post-translational Modification of Proteins by Nitrated Fatty Acids in Vivo. J Biol Chem. 2006;281(29):20450–20463. doi: 10.1074/jbc.M602814200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De L, Saleh S, Liebler DC. Reversibility of Covalent Electrophile-Protein Adducts and Chemical Toxicity. Chem Res Ttoxicol. 2008;21(12):2361–2369. doi: 10.1021/tx800248x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Groeger AL, Cipollina C, Cole MP, Woodcock SR, Bonacci G, Rudolph TK, et al. Cyclooxygenase-2 generates anti-inflammatory mediators from omega-3 fatty acids. Nat Chem Biol. 2010;6(6):433–441. doi: 10.1038/nchembio.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carrillo C, Cavia Mdel M, Alonso-Torre S. Role of oleic acid in immune system; mechanism of action; a review. Nutr Hosp. 2012;27(4):978–990. doi: 10.3305/nh.2012.27.4.5783. [DOI] [PubMed] [Google Scholar]
- 36.Watts FL, Oliver BL, Johnson GM, Thrall RS. Superoxide production by rat neutrophils in the oleic acid model of lung injury. Free Radic Biol Med. 1990;9(4):327–332. doi: 10.1016/0891-5849(90)90007-6. [DOI] [PubMed] [Google Scholar]
- 37.Goncalves-de-Albuquerque CF, Burth P, Silva AR, de Moraes IM, de Jesus Oliveira FM, Santelli RE, et al. Oleic acid inhibits lung Na/K-ATPase in mice and induces injury with lipid body formation in leukocytes and eicosanoid production. J Inflamm. 2013;10(1):34. doi: 10.1186/1476-9255-10-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rodrigues HG, Vinolo MA, Magdalon J, Fujiwara H, Cavalcanti DM, Farsky SH, et al. Dietary free oleic and linoleic acid enhances neutrophil function and modulates the inflammatory response in rats. Lipids. 2010;45(9):809–819. doi: 10.1007/s11745-010-3461-9. [DOI] [PubMed] [Google Scholar]
- 39.Weber FC, Németh T, Csepregi JZ, Dudeck A, Roers A, Ozsvári B, et al. Neutrophils are required for both the sensitization and elicitation phase of contact hypersensitivity. J Exp Med. 2015;212(1):15–22. doi: 10.1084/jem.20130062. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.