

Abstract
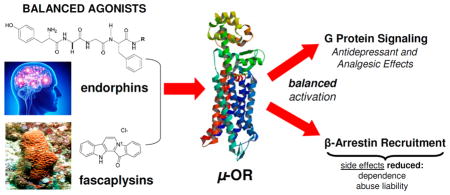
Opioid therapeutics are excellent analgesics, whose utility is compromised by dependence. Morphine (1) and its clinically relevant derivatives such as OxyContin (2), Vicodin (3), and Dilaudid (4) are “biased” agonists at the μ opioid receptor (OR), wherein they engage G protein signaling but poorly engage β-arrestin and the endocytic machinery. In contrast, endorphins, the endogenous peptide agonists for ORs, are potent analgesics, show reduced liability for tolerance and dependence, and engage both G protein and β-arrestin pathways as “balanced” agonists. We set out to determine if marine-derived alkaloids could serve as novel OR agonist chemotypes with a signaling profile distinct from morphine and more similar to the endorphins. Screening of 96 sponge-derived extracts followed by LC-MS-based purification to pinpoint the active compounds and subsequent evaluation of a mini library of related alkaloids identified two structural classes that modulate the ORs. These included the following: aaptamine (10), 9-demethyl aaptamine (11), demethyl (oxy)–aaptamine (12) with activity at the δ-OR (EC50: 5.1, 4.1, 2.3 μM, respectively) and fascaplysin (17), and 10-bromo fascaplysin (18) with activity at the μ-OR (EC50: 6.3, 4.2 μM respectively). An in vivo evaluation of 10 using δ-KO mice indicated its previously reported antidepressant-like effects are dependent on the δ-OR. Importantly, 17 functioned as a balanced agonist promoting both G protein signaling and β-arrestin recruitment along with receptor endocytosis similar to the endorphins. Collectively these results demonstrate the burgeoning potential for marine natural products to serve as novel lead compounds for therapeutic targets in neuroscience research.
Keywords: opioid, endorphins, signaling profile, biased agonism, balanced agonism, G protein-coupled receptor (GPCR), tolerance, dependence, aaptamine, fascaplysin
Graphical Abstract
INTRODUCTION
Opioids are exceptionally effective analgesics and the mainstay for the treatment of severe pain in many settings both acute and chronic.1,2 The first opioid (the alkaloid morphine, 1, Figure 1) has been used for centuries for pain relief and its euphoric properties, long before its target of action, the mu opioid receptor (μ-OR), and related delta (δ-OR) and kappa (κ-OR) opioid receptors were identified.3 Since its discovery in 1804 from extracts of the opium plant Papaver somniferum, a number of different small molecules have emerged that share the mechanism of action (MOA) of morphine in terms of how they target the μ-OR.4 Selected examples along with more detailed history are presented in Figure S1 (Supporting Information) with discovery dates in parentheses. Conversely, the endogenous ligands for the opioid receptors, collectively referred to as endorphins, are neuropeptides, which are structurally unrelated to morphine.2 Because the endorphins are rapidly hydrolyzed, do not easily cross the blood brain barrier, and were discovered many years after morphine, historically most therapeutic development of opioids has focused on compounds that resemble morphine rather than the endorphins.5,6 However, the utility of morphine and other clinically important therapeutic derivatives including oxycodone (2, OxyContin), hydrocodone (3, Vicodin), and hydromorphone (4, Dilaudid) is compromised by the development of tolerance to the analgesic effects of the drug,7 which, in turn, leads to dose escalation and a growing epidemic of increased liability for dependence.8 Consequently, there remains a great resistance among clinicians and the general population to use opioids for pain management because of perceived risk of addiction. These fears are not unwarranted. It is estimated that 2.5 million people begin abusing opioid painkillers each year and the number of users of both heroin and prescription opioids has doubled in the past 10 years.9 Some have become dependent as a consequence of recreational use of prescription painkillers. Nevertheless, many abusers of prescription painkillers are among the 40 million people suffering from chronic pain, and have developed dependence after using the opioids for legitimate purposes. Both of these trends highlight the need to develop new opioid therapeutics with a reduced abuse liability. Intriguingly, the endorphins show excellent analgesic activity but with reduced liability for tolerance and dependence compared to morphine providing a clue as to how to approach this goal.10,11
Figure 1.

Possible signaling profiles at the μ-opioid receptor (μ-OR): biased versus balanced agonism and reported side effects.
Recently, new information has emerged regarding key signaling differences between μ-ORs activated by classic morphine-like opioid agonists and those activated by the endorphin chemotype such as Met-enkephalin (5) and DAMGO(6). Specifically, it was discovered that morphine (1) and its derivatives are “biased” opioid agonists that potently activate one effector downstream of the receptor, the G protein, while only poorly engaging β-arrestins and the endocytic machinery, an important second effector of G protein-coupled receptor (GPCR) signaling (see Figure 1). More specifically, endorphin-occupied receptors are much better substrates for GPCR kinases (GRKs) than are morphine-occupied receptors.12,13 Consequently, morphine-occupied receptors engage weakly with arrestins14 and are poorly endocytosed12,15 unless GRKs12 and/or arrestins15 are overexpressed. Importantly, increases in GRK/arrestin expression, not receptor expression, are required to achieve an Emax for arrestin recruitment14 and endocytosis.12,15 Consequently, morphine and its derivatives are not partial agonists in the classical sense whereby maximal effect is dependent on receptor reserve. Instead morphine and its derivatives are biased agonists for arrestin recruitment, signaling poorly to the arrestin effector regardless of receptor number. Therefore, any mathematical model used to quantify bias would benefit from incorporating such innate partial agonism that cannot be overcome with increased receptor expression into the calculation.
This signaling bias toward G protein is shared by all the opioid therapeutics derived from the scaffold of morphine (1), and it is distinct from the “balanced” engagement of G protein and arrestin effectors that is promoted by the endorphins (Figure 1). Interestingly, the small-molecule agonists methadone (S6), fentanyl (S8), and etonitazine (S9) (shown in Figure S1), which are structurally distinct from morphine, all have a signaling profile somewhere between that of the morphinan and endorphin chemotype. Furthermore, these compounds all show a reduced liability to produce tolerance compared to morphine in rodent models when administered at equi-analgesic doses.16–24 On the other hand, buprenorphine (S5) has a signaling bias for G protein even greater than that of morphine and produces increased tolerance.24
While there are now many structurally distinct G protein biased agonists for the μ-OR (such as PZM21 (S14), TRV130 (S13), herkinorin (S12), and the morphinan chemotype shown in Figure S1), there is a scarcity of reported balanced opioid agonist chemotypes with a signaling profile similar to the endorphins. Among the existing opioids, only methadone (S6) approaches a signaling profile similar to the endogenous ligands. However, methadone’s variable and unpredictable pharmacokinetics and pharmacodynamics in the human population make it an unfavorable first-line analgesic. Therefore, to more thoroughly examine the hypothesis that a balanced opioid agonist could ameliorate the side effects of tolerance and dependence associated with long-term opioid use, there is a need to identify additional balanced opioid agonists with a signaling profile comparable to the endorphins. The question was where to search for such small molecules.
The fact that marine natural products show a chemical diversity often strikingly different from their terrestrial-derived counterparts suggest that novel scaffolds could be obtained from these sources.25 In terms of structural classes, marine natural products represent a burgeoning source of distinct lead compounds in neurobiological research.26,27 Several have now emerged as therapeutics or are in clinical development for the treatment of neurological disorders as shown in Figure 2.28 Their structures include ziconotide (7, syn. Prialt)29 and tetrodotoxin (8),30,31 used for the treatment of severe pain, and bryostatin-1 (9), which is being investigated for Alzheimer’s disease (AD).32 Over a dozen chemotypes have displayed neuroactive efficacy in vivo, and nearly all have at least one or more ascribed MOA.25,28–35 The first compounds to show in vivo antidepressant activity were bryostatin-1,36 5,6-dibromo-N, N-dimethyltryptamine and aaptamine. However, a MOA for the latter two compounds has not been clearly defined.37 To date, greater than 50 different chemotypes of marine-derived compounds have been reported as either neuroactive chemical probes or in vitro therapeutic leads for peripheral and central nervous system (CNS) indications, particularly for pain, AD, or as neuroprotective agents.26,27,38–40 Most were initially investigated based on their (a) fortuitous discovery of biological activities of interest,25–27 (b) unique chemical scaffold that fit well-defined pharmacophore models,41 or (c) environmental health and food safety issues.42 Surprisingly, there are few reports of neuroactive marine-derived lead compounds discovered using target-based high-throughput screening (HTS) to identify novel chemical probes or affect targets important in disease or treatment.43,44 One reason cited for this was that traditional methods for profiling natural products for therapeutic lead discovery were not amenable to HTS platforms.45 This has since been addressed by industry and academic research groups that have shown that using purified natural product libraries can streamline lead compound discovery using HTS methods.46–50
Figure 2.

Selected examples of neuroactive marine natural products approved by the FDA as therapeutics (7) or in various stages of clinical development (8–9). Images of selected organisms above were reproduced with permission from Jeanette and Scott Johnson (C. magus and D. liturosus) and SFBay: 2K, California Academy of Sciences (B. neritina).
RESULTS AND DISCUSSION
To examine whether novel opioid agonists with distinct scaffolds could be identified from marine sources, we profiled 96 marine sponge-derived extracts and a purified mini library of marine sponge-derived alkaloids using activity based HTS.46 Our HTS utilized HEK293T cells stably expressing the human μ-OR and δ-OR.51 Our approach, shown in Scheme 1, generated purified libraries of marine natural products compatible with cell and target based HTS programs. The process involved four steps, which included the following: (a) generating a marine-sponge derived crude extract library, which was screened for μ-OR and δ-OR agonist and antagonist activity in cells expressing each of these two targets, (b) processing bioactive extracts using LC-MS based purification to provide (c) a library of purified marine natural products from each active extract, which were rescreened for μ-OR and δ-OR agonist and antagonist activity, in order to (d) identify the lead compound(s) using dereplication and or structure elucidation strategies.
Scheme 1.

Approach To Prepare Peak Libraries of Purified Marine Natural Products for Target-Based High-Throughput Screening (HTS) To Identify Lead Compounds Demethyl (oxy)–Aaptamine (11) and 9-Demethyl–Aaptamine (12) with μ-Opioid Receptor (μ-OR) and δ-Opioid Receptor (δ-OR) Agonist Activity
Our first lead was a crude methanolic extract from an Indonesian sponge Aaptos aaptos (coll. no. 92553) that exhibited both μ-OR and δ-OR receptor agonist activity (not shown). Follow up evaluation of the LC-MS based purification library pinpointed two LC-MS m/z ion peaks (213 m/z, and 215 m/z) shown in Figure 3a. These peaks corresponded to measurable δ-OR agonist activity (blue bars – A8, B4) on par with the standard D-Pen(2), D-Pen(5)]-enkephalin (DPDPE, S16) (A1, 100 nM) and greater than the crude extract (92553 FM, A2,) in Figure 3b, but unfortunately, only very weak partial agonism at the μ-OR. Nevertheless, we quickly dereplicated these two LC-MS peaks as demethyl (oxy)–aaptamine (11) and 9-demethyl–aaptamine (12) shown in Figure 4 using taxonomy, and mass spectrometry m/z ion and 1H NMR data that matched literature values.52,53
Figure 3.

(a) LC-MS library f ractions of parent 96-well plate with annotations including m/z ions of demethyl (oxy)–aaptamine (11) and 9-demethyl–aaptamine (12), (b) corresponding wells A3-C5 evaluated in the delta opioid receptor (δ-OR) assay. Wells A1 and A2 contained positive controls D-Pen(2), D-Pen(5)]-enkephalin (DPDPE) and the crude extract (92553 FM). LC-MS fractions corresponding to wells C6-D12 (not shown) were inactive. Structures are shown in Figure 4.
Figure 4.

Structures of marine-derived alkaloid heterocycles (10–20) and mitragynine (21) evaluated in Table 1 for δ-opioid receptor (δ-OR) and μ-opioid receptor (μ-OR) agonist activity. Selection criterion based on chemotypes containing a secondary or tertiary nitrogen atom (bold) separated by 3–4 bonds from an adjacent tertiary amine or quaternary nitrogen (gray).
Aaptamine (10) is reported to have a significant effect on the immobility time of mice in the forced swim test (FST) mediated by an unknown target.37 The FST is commonly used to assess antidepressant like activity in mice. Several structurally distinct opioid receptor agonists have been shown to reduce immobility time on the FST.54–58 In addition mice lacking δ-ORs display increased depression like behavior on this test.59 We speculated that aaptamine, like demethyl (oxy)–aaptamine (11) and 9-demethyl–aaptamine (12) identified in our HTS, would exhibit δ-OR agonist activity based on its structural homology to 11–12, which it did as shown in Table 1. We further speculated that the antidepressant activity of aaptamine could be mediated through its agonist activity at the δ-OR.
Table 1.
Agonist Activity against the δ-OR and μ-OR
| compound | EC50 (μM)a | EC50 (μM)a |
|---|---|---|
|
| ||
| δ-OR | μ-OR | |
| morphine (1) | 0.426 ±0.1 | 0.0068 ±0.03 |
| DAMGO (6) | ND | 0.002 ± 0.002 |
| DPDPE (S16) | 0.007 ± 0.002 | ND |
| aaptamine (10) | 5.1 ± 0.2 | 10.1 ± 1.4 |
| 9-demethyl aaptamine (11) | 4.1 ± 0.1 | 6.03 ± 1.2 |
| demethyl (oxy) –aaptamine (12) | 2.3 ± 0.1 | 4.1 ± 1.2 |
| makaluvamine C (13) | NA | NA |
| makaluvamine H (14) | NA | NA |
| makaluvamine D (15) | NA | NA |
| makaluvamine J (16) | NA | NA |
| fascaplysin (17) | NA | 6.3 ± 0.2 |
| 10 bromofascaplysin (18) | NA | 4.2 ± 3.7 |
| plakinidine A (19) | NA | NA |
| plakinidine B (20) | NA | NA |
| mitragynine (21) | ND | 9.9 ± 0.2 |
Opioid agonist activity of 1, 6, and 10–21 was measured using the calcium mobilization assay in HEK-293 cells individually expressing the delta or mu opioid receptor (δ-OR, μ-OR). ND, not determined. NA (not active) ≤ 25.0 μM. Compounds 10–20 were inactive against κ-OR at ≤25.0 μM.
To examine this hypothesis, we assessed the reduction in time spent immobile in the FST with administration of aaptamine (10) in both wild type (WT) mice and δ-OR knockout mice (δ-KO)60 (see Methods). Similar to a previous report,37 aaptamine (40 mg/kg, ip) showed antidepressant-like activity in WT mice as shown in Figure 5 (**p = 0.003, WT saline vs WT 10, each circle represents an individual animal). In Figure 5a, the WT mice treated with vehicle (saline, black open bar) were immobilized on average for ~30 s while those exposed to aaptamine (gray solid bar) were immobilized on average for ~10 s, with one-third of the mice swimming the entire duration of the test, indicative of an antidepressant-like effect. This dose of aaptamine had no effect on general locomotion in WT mice (Figure 5b compare saline, open black bars to 10, solid gray bars, p = 0.76)), indicating a specific effect of aaptamine on forced swim. Consistent with previous reports,59 the δ-KO mice showed increased depression-like behavior on this forced swim test when exposed to saline (Figure 5a compare WT saline, black open bars, to δ-KO treated with saline, red open bars, #p = 0.013). More importantly, aaptamine had no effect on the δ-KO mice in the forced swim test (Figure 5a compare saline red open bars to 10 red solid bars, p = 0.20). Aaptamine likewise had no effect on general locomotion in δ-KO mice (Figure 5b, compare open and closed red bars), although δ-KO mice had reduced locomotion compared to WT mice as previously reported59,60 (Figure 5b compare WT saline, black open bar to δ-KO saline, red open bar, **p = 0.01). Taken together, this data suggested the antidepressant like activity of aaptamine is mediated, at least in part, by activity at the δ-OR. Future experiments will determine the structure activity relationship (SAR) requirements of this class for the δ-OR, and whether it modulates other targets to influence anxiety. More importantly, the discovery of aaptamine as a δ-OR agonist provided incentive to continue to mine the marine environment for other opioid ligands in our search for a balanced μ-OR agonist.
Figure 5.

Effects of aaptamine (10) in wild-type and δ-OR knock-out (δ-KO) mice. Each individual animal is represented by a circle, and the average is shown by the histogram. (a) Wild type (WT) mice (black and gray bars), and their littermates, mice with a disruption of the δ-OR (δ-KO, red bars), were injected with saline or 10 (40 mgs/kg ip) and then placed in a water bucket. Swimming was video recorded and time immobile scored. Aaptamine showed antidepressant-like activity in WT but not δ-KO mice (**p = 0.003). The δ-KO mice showed greater antidepressant-like activity than WT mice (# p = 0.013). (b) Following the swim test, mice were placed in a locomotor chamber and distance traveled was recorded automatically for 30 min. Aaptamine had no effect on general locomotion. δ-KO showed decreased locomotion compared to WT mice (**p = 0.01).
From here it seemed logical to evaluate compounds 10–12 alongside a mini library of marine-derived alkaloid heterocycles to gain insight into their SAR at the opioid receptor targets. This was based on a growing number of recent reports of structurally distinct marine-derived alkaloids that have shown potential as lead compounds by serving as GPCR ligands (including the opioid receptor agonists).41,61,62 We began by using compounds drawn from the UCSC marine natural products repository shown in Figure 4. We selected the following chemotypes with either a secondary or tertiary amine nitrogen atom (in bold) separated by either three or four bonds to an adjacent tertiary amine or quaternary nitrogen atom (in gray). This pattern has been observed in other selective μ-OR and δ-OR agonist chemotypes such as fentany1,63 etonitazine,64 and mitragynine.65 The structures we further evaluated included the aaptamines (10–12) the makaluvamines (13–16),66 fascaplysins (17,18)67 and the plakinidines (19,20).68 Compounds 10–20 were screened for their agonist and antagonist activity at the δ-OR, μ-OR, and κ-OR using as standard controls the peptide agonists DPDPE, DAMGO (6), hydrolysis resistant forms of the endorphins, and dynorphin.69 These results are summarized in Table 1. None of the compounds were more potent agonists than the standards (DPDPE, δ-OR: EC50 = 0.007 ± 0.002 μM or 6, μ-OR: EC50 = 0.001±0.002 μM). However, as expected the aaptamines (10–12) displayed low micromolar dual δ-OR and μ-OR agonist activity (10, δ-OR: EC50 = 5.1 ± 0.2 μM, μ-OR: EC50 = 10.1 ± 1.4 μM, 11, δ-OR: EC50 = 4.1 ± 0.1 μM, μ-OR: EC50 = 6.03 ± 1.2 μM, 12, δ-OR: EC50 = 2.3 ± 0.1 μM, μ-OR: EC50 = 4.1 ± 1.2 μM). While 10–12 were full agonists at the δ-OR, they were only weak partial agonists at the μ-OR (Emax < 10% at 50 μM) and showed no κ-OR activity (not shown). The other tricyclic chemotypes (13–16) were not active in any of these assays up to 50.0 μM. Conversely, two of the pentacyclic chemotypes, fascaplysin (17) and 10-bromo-fascaplysin (18) displayed selective low micromolar potency and full efficacy for the μ-OR as shown in Tables 1, 2 and Figure 6 (17, EC50 = 6.3 ± 0.2 μM, 18, EC50 = 4.2 ± 3.7 μM, Emax 100%) with no significant agonist or antagonist activity at δ-OR or κ-OR activity up to 50.0 μM (data not shown). Compounds 19,20 were not active in any of μ-OR, δ-OR, κ-OR assays up to 50.0 μM.
Table 2.
G Protein Signaling
| Gi | ||
|---|---|---|
|
| ||
| compound | EC50 | Emax(%) |
| DAMGO (6) | 1.844e-009 | 100 |
| morphine (1) | 1.670e-008 | 99.43 |
| fascaplysin (17) | 3.843e-006 | 111.4 |
| mitragynine (21) | 4.919e-006 | 107.5 |
| methadone (S6) | 8.538e-009 | 100.9 |
Figure 6.

Effects of μ-opioid receptor (μ-OR) agonists on inhibition of cAMP accumulation in HEK 293T cells. The EC50 and Emax values are shown in Table 2. Data are normalized to DAMGO and are mean ± SEM of normalized results (n = 3 measurements).
We found the identification of fascaplysin (17) as a μ-OR agonist intriguing, as it is structurally distinct from the major classes of the opioid chemotypes shown in Figure S1. We therefore proposed that fascaplysin might engage the μ-OR differently than the other opioid chemotypes and, thereby, signal with a different bias. Indeed, fascaplysin not only promoted G protein signaling as a full agonist (Figure 6, Table 2) but also promoted endocytosis of the μ-OR as shown in Figure 7. As previously reported13,70 the endorphin chemotype DAMGO (6, Figure 7b) and methadone (Figure 7c) also promoted endocytosis of the μ-OR, as evidenced by a redistribution of labeled receptors from the cell surface to intracellular compartments. Conversely morphine (Figure 7d) did not promote significant endocytosis, as evidenced by the receptors remaining on the plasma membrane as in the untreated control (no treatment, Figure 7a). In contrast, although fascaplysin (EC50 = 6.3 μM) is significantly less potent than morphine (EC50 = 0.0068 μM) for G protein signaling at the μ-OR as shown in Table 1, 2 and Figure 6, it promoted substantial endocytosis as shown in Figure 7e. Fascaplysin (17) shares structural homology with another natural product μ-OR agonist, mitragynine (21), as shown in Figure 8. Surprisingly, we found that mitragynine functions as a G protein biased agonist, as it did not promote receptor endocytosis as shown in Figure 7f, consistent with recent reports.71,72 We then examined endocytosis in a second non visual, biotin protection assay as previously described.73 In accordance with the results of the visual assay (Figure 7a–f), the biotin protection assay showed that DAMGO (6) and methadone promoted significant endocytosis (Figure 9). This was revealed by the intense signal (produced by the large protected pool of endocytosed receptors) compared to the no treatment (NT) control. In contrast, morphine induced no significant endocytosis beyond that in the cells that were not treated. Also, in agreement with results in Figure 7, fascaplysin promoted substantial endocytosis of the receptor while mitragynine did not do so.
Figure 7.

Assessment of μ-opioid receptor (μ-OR) endocytosis by immunofluorescence. (a–f) Fluorescent endocytosis assay. Cells stably expressing μ-OR were incubated with antibody to the extracellular epitope then treated with (a) water - no treatment (NT), (b) DAMGO (1 μM), (c) methadone (10 μM), (d) morphine (10 μM), (e) fascaplysin (10 μM), or (f) mitragynine (10 μM), for 30 min, fixed, permeabilized, and stained for receptor. DAMGO, methadone, and fascaplysin promoted μ-OR endocytosis while morphine and mitragynine did not.
Figure 8.

3D overlay of fascaplysin (17) (top) and mitragynine (21) (bottom) highlighting structural similarity.
Figure 9.

Assessment of μ-opioid receptor (μ-OR) endocytosis using the biotin protection assay. Cells stably expressing μ-OR were incubated with thio-cleavable biotin to label surface proteins then treated with compound (10 μM each) as listed, or left untreated (NT). Remaining surface receptors were stripped of biotin with reducing agent, and endocytosed receptors were immunoprecipitated and those that were protected from reducing agent visualized with streptavidin overlay. DAMGO, methadone, and fascaplysin promoted μ-OR endocytosis while morphine and mitragynine did not.
To better quantify the varying bias of these compounds for G protein and β-arrestin effectors, we assessed β-arrestin-2 recruitment to the μ-OR using a dynamic BRET-based assay shown in Figure 10, Table 3. Fascaplysin was toxic at concentrations higher than 50 μM, so a full dose–response curve was not achieved. Consequently, we could not determine whether fascaplysin is a full agonist at this effector. Nevertheless, the efficacy of fascaplysin at the β-arrestin-2 effector was significantly greater than that of morphine (Figure 10, Table 3, Emax morphine 22%, Emax fascaplysin 78%), In short, fascaplysin by engaging both G protein and β-arrestin-2 effectors at a ratio comparable to DAMGO (6) functions as a new balanced μ-OR agonist similar to the endorphins and methadone and distinct from the traditional morphinan opioid chemotypes.
Figure 10.

Effects of μ-opioid receptor (μ-OR) agonists on β-arrestin-2 recruitment in HEK 293T cells. The EC50 and Emax values are shown in Table 3. Data are normalized to DAMGO and are mean ± SEM of normalized results (n = 3 measurements).
Table 3.
β-Arrestin-2 Recruitment
| β-arrestin-2 | ||
|---|---|---|
|
| ||
| compound | EC50 | Emax (%) |
| DAMGO (6) | 1.731e-007 | 100 |
| morphine (1) | 4.190e-007 | 22.98 |
| fascaplysin (17) | 1.360e-005 | 76.80* |
| mitragynine (21) | 0.0003581 | 54.09 |
| methadone (S6) | 1.871e-006 | 92.62 |
Emax shown is that achieved before compound became toxic
A number of natural and synthetic analogues of fascaplysin (17)67,74–77 and mitragynine (21)65,71,72 have been reported. Specifically, there are analogs of mitragynine from the medicinal plant Mitragyna speciosa that show 100-fold greater potency at the μ-OR versus morphine with significant analgesic activity and potentially reduced side effect liability in animal models.65,71,72 Extracts of M. speciosa (known as Kratom) have long been utilized in S. East Asia for the treatment of pain and for withdrawal symptoms of morphine (see ref 71 and references therein). The recent classification of Kratom as schedule I by the U.S. Drug Enforcement Agency (DEA) reflects that the active compound(s) derived from this extract are potent opioid agonists on par with morphine. Kratom use worldwide has grown considerably in recent years, while definitive evidence for its therapeutic efficacy or addiction potential in humans is conflicting and warrants further investigaton. (see ref 71,72 and references therein). Moving forward it will be helpful to assess the SAR of the fascaplysin analogues and other chemotypes with regards to their μ-OR signaling profile with the goal of identifying additional balanced agonists with greater potency than fascaplysin. Future work will focus on initiating new collaborations to screen other novel μ-OR agonists to (a) determine if they engage G protein and the arrestins comparable to the endorphins and (b) more fully explore whether balanced opioid agonists retain analgesic potency that demonstrate reduced liability for tolerance and dependence similar to the endogenous ligands of the endorphin chemotype.
Several conclusions can be drawn from our pilot study screening marine sponge-derived extracts and our mini library of marine-derived alkaloids. First, crude extract and purified marine natural product libraries compatible with neuroactive target-based high-throughput screening can accelerate the discovery of lead compounds and their MOA. This approach allowed us to rapidly identify the aaptamines (10–12) as a new structural class of δ-OR agonists with a previously undisclosed MOA. We believe this MOA for aaptamine could be responsible, at least in part, for its reported in vivo antidepressant-like effects37 as other δ-OR agonists have shown anxiolytic and antidepressant activity in vivo.78–84 Second, using marine-derived alkaloids as seed compounds for GPCR SAR analysis can lead to the discovery of new chemotypes with unreported neuroactive MOA, and altered signaling bias. Using this process we discovered the fascaplysins (17–18) to be a novel class of μ-OR agonists. Here we report that fascaplysin functions as a balanced agonist that activates G protein, recruits β-arrestin, and promotes endocytosis, similar to the endogenous agonists (the endorphins) and distinct from the traditional opioid chemotypes.
There is mounting evidence that signaling bias can be leveraged to alter the side effect profiles of GPCR therapeutics, and opioids in particular. For example, opioid agonists that are G protein biased, including the clinical candidate TRV130 (S13), show excellent analgesic potency with reduced side effects of constipation and respiratory depression in animal models85 and healthy volunteers.86,87 However, buprenorphine, a clinically important drug with an extreme G protein bias on par with TRV130 produces significant respiratory depression and constipation both in rodent models and humans, suggesting that G protein bias per se is not sufficient to reduce these side effects.88 Instead, the high intrinsic efficacy of TRV130 for G protein signaling compared to morphine and buprenorphine and the consequent lower receptor occupancy necessary for pain control with TRV-130 could explain its reduced side effects. In fact, there is clear evidence that at least some of the effects of opioids, on respiration are mediated by activity from the G protein.89 Another G protein biased agonist, PZM21(S14)90 also shows reduced respiratory depression and a reduced ability to produce conditioned place preference (CPP), an indicator of reward. These results are consistent with the finding that constipation and respiratory depression in response to opioids are reduced in mice that lack β-arrestin-2.91 (see Figure 1). However, they conflict with the observation that disruption of β-arrestin-2 actually increases the rewarding properties of opioids,92 suggesting that the reduced CPP to PZM21 is due to another property of this compound rather than its bias away from β-arrestin-2. One likely explanation is the potent antagonist activity of PZM21 at the κ-OR, which has been shown to have significant influences on reward processing (for review, see ref 93).
To date, there remains conflicting evidence for the role of signaling bias in the development of tolerance and dependence. On one hand, balanced agonists that both signal to G protein and recruit β-arrestin along with driving endocytosis, such as methadone, fentanyl, and the endorphins have reduced liability for producing tolerance and dependence when administered at equi-analgesic doses.16–24 In addition, morphine produces reduced tolerance, reduced dependence, and reduced “addiction” while also showing enhanced analgesia and reward in mice who express a mutant μ-OR that engages both G protein and arrestins when occupied by morphine.94,95 These results suggest that balanced signaling can reduce the side effects of tolerance and dependence without compromising analgesic efficacy (see Figure 1). On the other hand, mice that lack β-arrestin-2 show enhanced analgesia96 and reward92 as well as reduced tolerance to morphine (but still show dependence),97 suggesting that preventing engagement with arrestin can reduce tolerance. Taken together, these results seem to contradict one another. Consequently, an important goal in this field should be to resolve this conundrum. Several experiments have provided important clues as to how to proceed. For example, tolerance to opioids that normally engage arrestins (e.g., methadone) is not reduced in the β-arrestin-2 knock out mice.22 This suggests that receptors occupied by morphine may be interacting with arrestins in a distinct way, and that receptors occupied by other balanced agonists (like the endorphins or fascaplysin) would not do so. Indeed, there is evidence that this is the case. For example, morphine promotes receptor desensitization (one suggested candidate for acute tolerance) via phosphorylation of protein kinase C (PKC), whereas endorphins do not promote (PKC) phosphorylation (for review, see ref 98).
When considering the role of arrestins in the side effect profile of opioid drugs, it is also important to keep in mind that arrestins serve at least three important roles in modulating signaling from GPCRs: (1) they “arrest” G protein signaling by uncoupling the receptor from G protein; (2) they promote receptor-mediated signaling to diverse kinase cascade molecules including ERK, Akt, and Src, by scaffolding the proteins close to the receptor; and (3) they serve as a linker between the receptor and the endocytic machinery (see Figure S2, Supporting Information). Hence, G protein biased ligands that poorly recruit arrestins could be deficient in three distinct ways: (a) desensitization of G protein signaling, (b) interaction with intracellular kinase cascades, and (c) endocytosis, recycling, and resenstitization of receptors and receptor-mediated signaling. To date, there is no method that selectively disrupts only one of these three arrestin-mediated events. For example, genetic deletion of β-arrestin-2 prevents arrestin-mediated signaling (to ERK for example), but it also eliminates its modulatory role in titrating G protein signaling. Loss of either (or both) of these signal transduction roles could be responsible for the changes in effect/side effect profile of opioid drugs in β-arrestin-2 knock out mice. By extension, since the degree of bias of individual ligands is often measured by assessing recruitment of arrestin (as in Figure 10 here), or monitoring endocytosis (as in Figure 7 and 9 here), whether distinct G protein biased ligands are equally deficient at all three signaling roles of arrestin remains unclear.
In summary, one of the most effective ways to examine the role of signaling bias in the side effects of tolerance and dependence is to add new chemotypes to the short list of balanced μ-OR small molecule agonists and compare them to the many biased agonist chemotypes with regard to effect/side-effect profiles. That being said, every compound has its own intrinsic set of pharmacological properties, and no two ligands differ only in signaling bias. Therefore, it is important to use caution when drawing conclusions as to the role of signaling bias. Furthermore, although changing the signaling profile of an opioid agonist can alter the rate at which tolerance and dependence develop, all the opioids, even the endorphins, produce tolerance and dependence with enough exposure (for review, see ref 99). It may not be possible to alleviate all the side effects of opioids with a single approach. G protein fully biased agonists may provide pain relief for acute indications with reduced risk of constipation and respiratory suppression. Balanced agonists (similar to the endorphin chemotype) may provide pain relief for chronic conditions with reduced liability for dependence. Ultimately identifying the correct signaling profile to reduce the development of tolerance to opioids could potentially solve all these issues, as reducing dose escalation would lower the risks of both fatal respiratory events and dependence.
In conclusion, we have identified two new marine-sponge-derived natural product agonists at the opioid receptors. The first, aaptamine (10) is a δ-OR agonist on a scaffold distinct from other known δ-OR ligands. While aaptamine has previously been shown to have antidepressant like activity, here we demonstrate that activity is mediated at least in part by the δ-OR. The second, fascaplysin (17) is a selective μ–OR agonist. Most importantly, we have demonstrated that fascaplysin is a balanced agonist, adding a new chemotype to the short list of compounds with a signaling profile that resembles endorphins. Future work will interrogate the SAR of this structural class, with the hopes of improving potency without altering its signaling profile.
METHODS
General Experimental Procedures
LC-MS based purification was performed using a Waters purification system running Empower 2 software that utilized the following: (a) 717 Autosampler, (b) 510 HPLC pumps, (c) 996 PDA detector. The elution was split between a (1) Sedex model 55 evaporative light scattering detector (ELSD), (2) an Applied Biosystems Mariner electrospray ionization time-of-flight (ESI-TOF) mass spectrometer, and (3) a sample collection tube. Sample collection was performed using a Gilson 215 liquid handler controlled with Gilson Unipoint LC software as reported in detail previously.46 NMR experiments were run on a Varian Unity 500 spectrometer (500 and 125 MHz for 1H and 13C respectively). High accuracy mass spectrometry measurements were obtained using the Applied Biosystems Mariner ESI-TOF mass spectrometer.
Biological Material, Collection, and Identification
Specimens used for the 96-well plate crude extract library were prepared from extracts provided using the UCSC marine natural products repository. These extracts were derived from sponges obtained during a 1992 expedition in Waigeo, Indonesia. The active crude extract from sponge Aaptos aaptos (coll. no. 92553, FM) was taxonomically identified as A. aaptos by Dr. Christina Diaz (1992). Additional raw material of A. aaptos was obtained from a 2011 collection of Aaptos aaptos (coll. no. 11308) from Waigeo, Indonesia.
Scaled Up Isolation of Aaptamine for In Vivo Evaluation
Using a standard solvent partitioning scheme reported elsewhere,66 aaptamine (10) was obtained from the butanol extract (coded WB) of coll. no. 11308. The WB extract was further purified using repeated reverse phase HPLC (Phenomenex Luna 250 × 10 mm with a 4.6 μm column) using gradient conditions of 10% → 100% CH3CN:H2O with 0.1% formic acid (45 min.) using a λmax = 254 nm, sensitivity = 2.0 AU and a flow rate of 2.0 mL/min. A series of injections ([4.0 mg/100 μL] × 30) of the WB extract were made which liberated approximately 41.2 mg of 10 which was used to conduct the Porsolt swim test and locomoter tests using 36 mice reported in Figure 5. Purity of 10 was confirmed from 1H NMR (Figure S3 in Supporting Information) and mass spectral data (not shown) that matched literature values.52,53
Extraction and 96-Well Plate Crude Extract Library Preparation for HTS Bioassay
Sponge extracts evaluated in the 96-well plate crude extract library (shown in Scheme 1, step a) were prepared using a traditional solvent partition scheme to provide four sample codes, and a more detailed description is provided elsewhere.66 The process involved generating a butanol soluble extract (coded WB), hexanes soluble extract (coded FH), methanol soluble extract (coded FM), and dichloromethane soluble extract (coded FD). Approximately 1.0 mg of 24 selected sponge specimens from a 1992 expedition (which included their WB, FH, FM, and FD extracts) were prepared for evaluation into a 96-deep-well plate extract library and dissolved in methanol using 1000 μL. A 50.0 μL portion of DMSO was added to each well containing extract. The 96-well plate was then counter balanced with a duplicate blank 96-well plate containing 1000 μL of methanol and evaporated using a Savant AES2010 speed vac concentrator (room temperature) for 24 h. The remaining solution was devoid of methanol and contained a concentrated soluble extract solution of 1.0 mg/50 μL (20 mg/mL) in DMSO. This 96-well plate was shipped on dry ice (to prevent spillage and or chemical degradation) to UCSF for initial bioassay evaluation.
LC-MS-Based Purification To Generate a Library of Purified Marine Natural Products for HTS Bioassay
Approximately 15.0 mg/100 μL of the bioactive lead extract (coded 92553 FM) was processed using LCMS based purification into a 96-deep-well plate to generate a library of purified marine natural products as shown in Scheme 1, steps b and c. Automated separation (reported elsewhere46) was performed on a Luna 4.6 μm, C18(2) 100 Å 10 × 250 mm column (Phenomenex, Inc.) in conjunction with a guard column using a larger 10.0 × 10.0 mm C18 (ODS) cartridge (holder part number: AJ0-7220, cartridge part number: AJ0-7221). This involved a gradient elution that consisted of 10:90 to 20:80 (acetonitrile:water, with 0.1% formic acid) over 30 min with a flow rate of 2.0 mL/min which eluted 2.0 mL fractions into each of 48 wells. The library was split into a second 96-well plate for reference using a 12-channel pipet withdrawing 1.0 mL/well fraction to create an exact copy and counter balance for centrifugal drying. A 25 μL aliquot of DMSO was then added to 48 of the original 96-well plate fractions and the solution was evaporated using a Savant AES2010 SpeedVac. After 24 h, the plates were removed, and the original purified natural product library well fractions remained suspended in a solution of approximately ~0.15 mg/25 ul (~1–3 mg/mL) of DMSO. These values are based on assumed weights of an average of 0.15mg/well estimated from the 15.0 mg/100 μL injection divided into the two library plates (containing fractionated material of 7.5 mg each total) that were equally fractionated into 48 wells. The library of purified marine natural products was then transported on dry ice (to prevent spillage and or chemical degradation) to UCSF for follow up bioassay to pinpoint the lead compound(s) responsible for the activity. Once received it was stored at 4 °C and later evaluated using the δ-OR and μ-OR HTS assays.
G-Protein Signaling Assay
The 96 marine-sponge-derived extracts were assayed at a concentration of 10 μg/mL suspended in DMSO. The LC-MS-based natural product library of the bioactive methanol crude extract (coded 92553 FM) shown in Scheme 1, step c, and Figure 3, involved testing all LC-MS based library well fractions (wells A3-D12) at a concentration of approximately 20.0 μg/mL. The standard controls DPDPE (well A1) and the crude extract 92553 FM (well A2) were evaluated in the δ-OR assay at 100 nM and 20.0 μg/mL, respectively, in DMSO. Sponge-derived extracts, LCMS library fractions, and compound-induced intracellular calcium release in HEK-293 cells was detected using cells stably expressing the human δ and μ opioid receptors (δ-OR) and (μ-OR) cDNAS and a chimeric Gi/q G protein to couple receptor activity to release of intracellular calcium stores as previously reported.51 Results are expressed as the mean (±SEM) relative fluorescence units (RFU, calculated as agonist-induced maximum Ca2+ peak/cell number × 1000) and as (mean ± SEM for n = 3) percentage of baseline response (calculated as the ratio of the agonist-induced RFU to that of untreated cells). Activation of δ-OR and μ-OR targets was detected using real-time calcium detection in a FLEX-3 apparatus. Full dose–response curves were generated for the purified compounds in Table 1. In all cases, ligand was added, and the calcium response was measured for 5 min to detect agonist activity. Then control ligand (DAMGO for μ-OR cell line and DPDPE for the δ-OR cell line) was added, and once again calcium release measured to detect whether the compounds had antagonist activity.
cAMP Assays
To measure μ opioid receptor (μ-OR) Gαi-mediated cAMP inhibition, HEK 293T (ATCC CRL-11268) cells were cotransfected with human μ-OR along with a luciferase-based cAMP biosensor (GloSensor, Promega) and assays performed similar to those previously described.90 After at least 16 h, transfected cells were plated into polylysine-coated 384-well white clear-bottom cell culture plates with DMEM+ 1%dialyzed FBS at a density of 15 000–20 000 cells per 40 μL per well and incubated at 37 °C with 5% CO2 overnight. The next day, drug solutions were prepared in fresh assay buffer (20 mM HEPES, 1× HBSS, pH 7.4, 0.1% bovine serum album) at 3× drug concentration. Plates were decanted and received 20 μL per well of assay buffer (20mM HEPES, 1× HBSS, pH 7.4) followed by addition of 10 μL of drug solution (3 wells per condition) for 15 min in the dark at room temperature. To stimulate endogenous cAMP via β adrenergic-Gs activation, 10 μL luciferin (4 mM final concentration) supplemented with isoproterenol (400 nM final concentration) were added per well. Cells were again incubated in the dark at room temperature for 15 min, and luminescence intensity was quantified using a Wallac TriLux microbeta (PerkinElmer) luminescence counter. Data were normalized to DAMGO-induced cAMP inhibition and analyzed using nonlinear regression in GraphPad Prism5.0.
β-Arrestin-2 Recruitment Assays
To measure μ-OR-mediated β-arrestin recruitment by BRET, HEK 293T cells were cotransfected in a 1:15 ratio with human μ-OR containing C-terminal renilla luciferase (Rluc8) and venus-tagged N-terminal β-arrestin-2, respectively. After at least 16 h, transfected cells were plated in polylysine-coated 96-well white clear bottom cell culture plates in DMEM + 1% dialyzed FBS at a density of 500 000–750 000 cells per 200 μL per well and incubated overnight. The next day, media was decanted and cells were washed twice with 100 μL of drug buffer. Then, 60 μL of the RLuc substrate, coelenterazine H (Promega, 5 μM final concentration) was added per well. After 5 min, 30 μL of drug (3 wells per condition) was added per well and incubated for 5 min in the dark, and plates were read for both luminescence at 485 nm and fluorescent eYFP emission at 530 nm for 1s per well using a Mithras LB940 microplate reader. The ratio of eYFP/RLuc was calculated per well and the net BRET ratio was calculated by subtracting the eYFP/RLuc per well from the eYFP/RLuc ratio without venus–arrestin present. Data were normalized to DAMGO-induced stimulation and analyzed using nonlinear regression in GraphPad Prism 5.0.
Fluorescent Endocytosis Assay
HEK293 cells stably expressing N-terminally FLAG-tagged μ-OR were plated to coverslips and allowed to recover overnight. To examine endocytosis, live cells were incubated with an antibody to the extracellular epitope tag to label-only receptors on the cell surface. Cells were then left untreated (Figure 7a) or treated with selective μ-OR agonists (Figure 7b–f) for 30 min. Following endocytosis, cells were fixed, permeabilized, and stained with a fluorescently tagged secondary antibody to examine the distribution of the receptors that started on the cell surface. Endocytosis is visualized as a redistribution of receptors from the plasma membrane (Figure 7a, no treatment) to intracellular compartments, as only receptors that were on the cell surface and had access to antibody are revealed in intracellular compartments.
Biotin Protection Endocytosis Assay
HEK293 cells stably expressing N-terminal FLAG-tagged μ-OR were grown to 100% confluency in 10 cm poly D-lysine-coated plates and subjected to the biotin protection assay protocol as described previously.73 Briefly, cells were treated with 3 μg/mL disulfide-cleavable biotin (Pierce) for 30 min at 4 °C. Cells were then washed in PBS and placed in prewarmed media for 15 min before treatment with ligand (or no treatment) for 30 min with 10 μm of DAMGO, methadone, morphine, fascaplysin, ormitragynine. Concurrent with ligand treatment, 100% and strip plates remained at 4 °C. After ligand treatment, plates were washed in PBS, and the remaining cell-surface-biotinylated receptors were stripped in 50 mm glutathione, 0.3 m NaCl, 75 mm NaOH, 1% FBS at 4 °C for 30 min. Cells were quenched with Tris-HCl buffer (pH 7.4) and then lysed in immunoprecipitation buffer (IPB), 0.1% Triton X-100, 150 mm NaCl, 25 mm KCl, 10 mm Tris-HCl (pH 7.4), with protease inhibitors (Roche Applied Science). Cleared lysates were immunoprecipitated with anti-FLAG M2 antibodies for 1 h at 4 °C and incubated for 1 h with protein G-Sepharose (Invitrogen). Samples were washed four times in 1 mL of IPB and twice in 1 mL of 10 mm Tris-HCl (pH 7.4) and deglycosylated by incubation with peptide: N-glycosidase F (New England Biolabs) for 1 h at 37 °C, resolved by SDS-PAGE, and visualized with streptavidin overlay (Vectastain ABC immunoperoxidase reagent, Vector Laboratories).
Forced Swim and Locomoter Tests
Animals
Male WT or δ-KO C57BL/6 mice (9 per group) between 8 and 10 weeks old were housed in a temperature controlled colony room (21 °C) on a 12-h reversed light/dark cycle (lights off at 10 a.m.). All experiments were performed during the dark (active) cycle. Food and water were available ad libitum. Experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (NIH) and were approved by the UCSF Institutional Animal Care and Use Committee.
Forced Swim Test
Mice were allowed to habituate to the room and lights for 2 h before the start of the experiment and then were placed in a swimming task. Cylinders measured 30 cm in diameter and 36 cm high, water depth ranged from 22 to 24 cm, and temperature was 36.0 ± 0.2 °C. Mice were placed into the center of the bucket and allowed to swim freely for 6 min before they were removed and towel dried. Sessions were video recorded and mice scored for seconds immobile from these videos.
Locomotor Activity
After towel drying, mice were placed in a locomotor exploration task. Clear plexiglass cubes measuring 19.5 cm in each direction were inserted in the corner of standard 43 cm locomotor chambers (Med Associates, St Albans, VT). Mice were placed into the cube, and locomotor activity was automatically digitally recorded for 30 min.
3D Overlay of 17 and 21
Molecular graphics and analyses were performed with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311).100
Spectral Data of Isolated and Pure Compounds
Aaptamine (10) 1H NMR (D6-DMSO, 500 MHz) δ 7.84 (d, J = 7.0 Hz, 1H), 7.38 (d, J = 7.0, 1H), 7.11 (s, 1H), 6.86 (d, J = 7.0, 1H), 6.48 (d, J = 7.0, 1H), 3.97 (s, 3H, OCH3), 3.80 (s, 3H, OCH3). ESI-TOF-HRMS 229.0977 m/z [M + H]+ (calcd for C13H13N2O2, 229.0856). 9-demethyl aaptamine (11). 1H NMR (D6-DMSO, 500 MHz) δ 7.69 (d, J = 6.5 Hz, 1H), 7.15 (d, J = 7.2, 1H), 7.04 (s, 1H), 6.76 (d, J = 7.2, 1H), 6.24 (d, J = 6.5, 1H), 4.01 (s, 3H, OCH3). ESI-TOF-HRMS 215.0956 m/z [M + H]+ (calcd for C12H11N2O2, 215.0921). 9-demethyl(oxy) aaptamine (12). 1HNMR (D6-DMSO, 500 MHz) 9.17 (d, J = 4.4 Hz, 1H), δ 9.12 (d, J = 5.5 Hz, 1H), 8.25 (d, J = 5.5 Hz, 1H), 7.78 (d, J = 4.4 Hz, 1H), 7.20 (s, 1H), 3.94 (s, 3H, OCH3). ESI-TOF-HRMS 213.0768 m/z [M + H]+ (calcd for C12H8N2O2, 213.0729). The marine sponge-derived alkaloids including the makaluvamines (13–16), the fascaplysins (17,18), and the plakinidines (19,20) were drawn from the UCSC marine natural products pure compound repository where their purity (≥95% HPLC) and spectral data (NMR) have been reported previously.66–68
Supplementary Material
Acknowledgments
We thank Carsten K. Nielsen and Selena Bartlett for assistance in screening compounds (10–20). T.A.J. thanks David Barnes for inspiring philosophical discussions related to the power of the endorphins. This work was supported by grants from the NIH, R01DA019958 (J.L.W.), R01DA015232 (J.L.W.), R21DA031574 (J.L.W.), R01CA47135 (P.C.), NSF MS Instrument Grant UCSC CHE-147922 (P.C.), and the Fletcher Jones Fund of Dominican University of California (T.A.J.).
Footnotes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschemneuro.6b00167.
Selected examples of major chemotypes that function as potent agonists for μ-OR, δ-OR, and κ-OR are shown in Figure S1; signaling profile of a classic balanced agonist at the mu-opioid receptor (μ-OR) emphasizing three key roles of β-arrestin; 1H NMR spectra for the scaled up sample of aaptamine (10) for in vivo evaluation is shown Figure S3 (PDF)
References
- 1.McQuay H. Opioids in pain management. Lancet. 1999;353:2229–2232. doi: 10.1016/S0140-6736(99)03528-X. [DOI] [PubMed] [Google Scholar]
- 2.Akil H, Watson SJ, Young E, Lewis ME, Khachaturian H, Walker JM. Endogenous opioids: biology and function. Annu Rev Neurosci. 1984;7:223–255. doi: 10.1146/annurev.ne.07.030184.001255. [DOI] [PubMed] [Google Scholar]
- 3.Dhawan BN, Cesselin F, Raghubir T, Reisine PB, Bradley PS, Portoghese PS, Hamon M. International Union of Pharmacology. XII Classification of opioid receptors. Pharmacol Rev. 1996;48:567–592. [PubMed] [Google Scholar]
- 4.Trescot AM, Datta S, Lee M, Hansen H. Opioid pharmacology. Pain Physician. 2008;11:S133. [PubMed] [Google Scholar]
- 5.Davis TP, Abbruscato TJ, Egleton RD. Peptides at the blood brain barrier: Knowing me knowing you. Peptides. 2015;72:50–56. doi: 10.1016/j.peptides.2015.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Corbett AD, Henderson G, McKnight AT, Paterson SJ. 75 years of opioid research: the exciting but vain quest for the Holy Grail. Br J Pharmacol. 2006;147(Suppl 1):S153–S162. doi: 10.1038/sj.bjp.0706435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ballantyne JC, Shin NS. Efficacy of opioids for chronic pain: a review of the evidence. Clin J Pain. 2008;24:469–478. doi: 10.1097/AJP.0b013e31816b2f26. [DOI] [PubMed] [Google Scholar]
- 8.Manchikanti L, Helm S, 2nd, Fellows B, Janata JW, Pampati V, Grider JS, Boswell MV. Opioid epidemic in the United States. Pain Physician. 2012;15:ES9. [PubMed] [Google Scholar]
- 9. [accessed April 1, 2016];America’s Addiction to Opioids: Heroin and Prescription Drug Abuse. https://www.drugabuse.gov/about-nida/legislative-activities/testimony-to-congress/2016/americas-addiction-to-opioids-heroinprescription-drug-abuse.
- 10.Stevens CW, Yaksh TL. Time course characteristics of tolerance development to continuously infused antinociceptive agents in rat spinal cord. J Pharmacol Exp Ther. 1989;251:216–223. [PubMed] [Google Scholar]
- 11.Whistler JL. Examining the role of mu opioid receptor endocytosis in the beneficial and side-effects of prolonged opioid use: From a symposium on new concepts in mu-opioid pharmacology. Drug Alcohol Depend. 2012;121:189–204. doi: 10.1016/j.drugalcdep.2011.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang J, Ferguson SS, Barak LS, Bodduluri SR, Laporte SA, Law PY, Caron MG. Role for G protein-coupled receptor kinase in agonist-specific regulation of mu-opioid receptor responsiveness. Proc Natl Acad Sci U S A. 1998;95:7157–7162. doi: 10.1073/pnas.95.12.7157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whistler JL, Chuang HH, Chu P, Jan LY, von Zastrow M. Functional dissociation of mu opioid receptor signaling and endocytosis: implications for the biology of opiate tolerance and addiction. Neuron. 1999;23:737–746. doi: 10.1016/s0896-6273(01)80032-5. [DOI] [PubMed] [Google Scholar]
- 14.Winpenny D, Clark M, Cawkill D. Biased ligand quantification in drug discovery: from theory to high throughput screening to identify new biased μ opioid receptor agonists. Br J Pharmacol. 2016;173:1393–1403. doi: 10.1111/bph.13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whistler JL, von Zastrow M. Morphine-activated opioid receptors elude desensitization by beta-arrestin. Proc Natl Acad Sci U S A. 1998;95:9914–9919. doi: 10.1073/pnas.95.17.9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duttaroy A, Yoburn BC. The effect of intrinsic efficacy on opioid tolerance. Anesthesiology. 1995;82:1226–1236. doi: 10.1097/00000542-199505000-00018. [DOI] [PubMed] [Google Scholar]
- 17.Enquist J, Ferwerda M, Milan-Lobo L, Whistler JL. Chronic methadone treatment shows a better cost/benefit ratio than chronic morphine in mice. J Pharmacol Exp Ther. 2012;340:386–392. doi: 10.1124/jpet.111.187583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grecksch G, Bartzsch K, Widera A, Becker A, Hollt V, Koch T. Development of tolerance and sensitization to different opioid agonists in rats. Psychopharmacology (Berl) 2006;186:177–184. doi: 10.1007/s00213-006-0365-8. [DOI] [PubMed] [Google Scholar]
- 19.Kumar P, Sunkaraneni S, Sirohi S, Dighe SV, Walker EA, Yoburn BC. Hydromorphone efficacy and treatment protocol impact on tolerance and mu-opioid receptor regulation. Eur J Pharmacol. 2008;597:39–45. doi: 10.1016/j.ejphar.2008.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Madia PA, Dighe SV, Sirohi S, Walker EA, Yoburn BC. Dosing protocol and analgesic efficacy determine opioid tolerance in the mouse. Psychopharmacology (Berl) 2009;207:413–422. doi: 10.1007/s00213-009-1673-6. [DOI] [PubMed] [Google Scholar]
- 21.Pawar M, Kumar P, Sunkaraneni S, Sirohi S, Walker EA, Yoburn BC. Opioid agonist efficacy predicts the magnitude of tolerance and the regulation of μ-opioid receptors and dynamin-2. Eur J Pharmacol. 2007;563:92–101. doi: 10.1016/j.ejphar.2007.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raehal KM, Bohn LM. The role of beta-arrestin2 in the severity of antinociceptive tolerance and physical dependence induced by different opioid pain therapeutics. Neuropharmacology. 2011;60:58–65. doi: 10.1016/j.neuropharm.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sirohi S, Dighe SV, Walker EA, Yoburn BC. The analgesic efficacy of fentanyl: relationship to tolerance and muopioid receptor regulation. Pharmacol, Biochem Behav. 2008;91:115–120. doi: 10.1016/j.pbb.2008.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walker EA, Young AM. Differential tolerance to antinociceptive effects of mu opioids during repeated treatment with etonitazene, morphine, or buprenorphine in rats. Psychopharmacology (Berl) 2001;154:131–142. doi: 10.1007/s002130000620. [DOI] [PubMed] [Google Scholar]
- 25.Villa FA, Gerwick L. Marine natural product drug discovery: Leads for treatment of inflammation, cancer, infections, and neurological disorders. Immunopharmacol Immunotoxicol. 2010;32:228–237. doi: 10.3109/08923970903296136. [DOI] [PubMed] [Google Scholar]
- 26.Kochanowska-Karamyan AJ, Hamann MT. Marine Indole Alkaloids: potential new drug leads for the control of depression and anxiety. Chem Rev. 2010;110:4489–4497. doi: 10.1021/cr900211p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakai R, Swanson GT. Recent progress in neuroactive marine natural products. Nat Prod Rep. 2014;31:273–309. doi: 10.1039/c3np70083f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Butler MS, Robertson ABA, Cooper MA. Natural product and natural product derived drugs in clinical trials. Nat Prod Rep. 2014;31:1612–1661. doi: 10.1039/c4np00064a. [DOI] [PubMed] [Google Scholar]
- 29.Molinski TF, Dalisay DS, Lievens SL, Saludes JP. Drug development from marine natural products. Nat Rev Drug Discovery. 2009;8:69–85. doi: 10.1038/nrd2487. [DOI] [PubMed] [Google Scholar]
- 30.Hagen NA, du Souich P, Lapointe B, Ong-Lam M, Dubuc B, Walde D, Love R, Ngoc AH. Tetrodotoxin for moderate to severe cancer pain: a randomized, double blind, parallel design multicenter study. J Pain Symptom Manage. 2008;35:420–429. doi: 10.1016/j.jpainsymman.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 31. [accessed Jan 26, 2016];Study to determine if tetrodotoxin (TTX) is an effective treatment of pain resulting from chemotherapy treatment (TTX-CINP-201) https://clinicaltrials.gov/ct2/show/NCT01655823.
- 32. [accessed Sep 14, 2016];Study assessing bryostatin in treatment of moderately severe to severe Alzheimer’s disease. https://clinicaltrials.gov/ct2/show/NCT02431468.
- 33.Davies LP, Baird-Lambert J, Hall JG. 1-Methylisoguanosine: interaction with central adenosine receptors and lack of antagonism of its in vivo effects by a benzodiazepine antagonist. Neuropharmacology. 1987;26:493–497. doi: 10.1016/0028-3908(87)90033-5. [DOI] [PubMed] [Google Scholar]
- 34.Wen Z, Chao C, Wu M, Sheu J. A neuroprotective sulfone of marine origin and the in vivo anti-inflammatory activity of an analogue. Eur J Med Chem. 2010;45:5998–6004. doi: 10.1016/j.ejmech.2010.09.067. [DOI] [PubMed] [Google Scholar]
- 35.Xiao A, Chen W, Xu B, Liu R, Turlova E, Barszczyk A, Sun CLF, Liu L, Deurloo M, Wang G, Feng Z, Sun H. Marine compound xyloketal B reduces neonatal hypoxic-ischemic brain injury. Mar Drugs. 2015;13:29–47. doi: 10.3390/md13010029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun M, Alkon DL. Dual effects of bryostatin-1 on spatial memory and depression. Eur J Pharmacol. 2005;512:43–51. doi: 10.1016/j.ejphar.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 37.Diers JA, Ivey KD, El-Alfy A, Shaikh J, Wang J, Kochanowska AJ, Stoker JF, Hamann MT, Matsumoto RR. Identification of antidepressant drug leads through the evaluation of marine natural products with neuropsychiatric pharmacophores. Pharmacol, Biochem Behav. 2008;89:46–53. doi: 10.1016/j.pbb.2007.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brogan JT, Stoops SL, Crews BC, Marnett LJ, Lindsley CW. Total synthesis of (+)-7- bromotrypargine and unnatural analogues: biological evaluation uncovers activity at CNS targets of therapeutic relevance. ACS Chem Neurosci. 2011;2:633–639. doi: 10.1021/cn200075n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choi D, Choi H. Natural products from marine organisms with neuroprotective activity in the experimental models of Alzheimer’s disease, Parkinson’s disease and ischemic brain stroke: their molecular targets and action mechanisms. Arch Pharmacal Res. 2015;38:139–170. doi: 10.1007/s12272-014-0503-5. [DOI] [PubMed] [Google Scholar]
- 40.Leiros M, Alonso E, Rateb ME, Houssen WE, Ebel R, Jaspars M, Alfonso A, Botana LM. Bromoalkaloids protect primary cortical neurons from induced oxidative stress. ACS Chem Neurosci. 2015;6:331–338. doi: 10.1021/cn500258c. [DOI] [PubMed] [Google Scholar]
- 41.Brogan JT, Stoops SL, Lindsley CW. Total synthesis and biological evaluation of phidianidines A and B uncovers unique pharmacological profiles at CNS targets. ACS Chem Neurosci. 2012;3:658–664. doi: 10.1021/cn300064r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stewart I, Mcleod C. The laboratory mouse in routine food safety testing for marine algal biotoxins and harmful algal bloom toxin research: Past, present and future. J AOAC Int. 2014;97:356–372. doi: 10.5740/jaoacint.sgestewart. [DOI] [PubMed] [Google Scholar]
- 43.Balansa W, Islam R, Fontaine F, Piggott AM, Zhang H, Webb TI, Gilbert DF, Lynch JW, Capon RJ. Ircinialactams: Subunit-selective glycine receptor modulators from Australian sponges of the family Irciniidae. Bioorg Med Chem. 2010;18:2912–2919. doi: 10.1016/j.bmc.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 44.Williams P, Sorribas A, Liang Z. New methods to explore marine resources for Alzheimer’s therapeutics. Curr Alzheimer Res. 2010;7:210–213. doi: 10.2174/156720510791050812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koehn FE, Carter GT. The evolving role of natural products in drug discovery. Nat Rev Drug Discovery. 2005;4:206–220. doi: 10.1038/nrd1657. [DOI] [PubMed] [Google Scholar]
- 46.Johnson TA, Sohn J, Inman WI, Estee SA, Loveridge SL, Vervoort HC, Tenney K, Liu J, Ang KK, Ratnam J, Bray WM, Gassner NC, Shen YY, Lokey RS, McKerrow JH, Boundy-Mills K, Nukanto A, Kanti A, Julistiono H, Kardono LBS, Bjeldanes LF, Crews P. Natural Product Libraries to Accelerate the High Throughput Discovery of Therapeutic Leads. J Nat Prod. 2011;74:2545–2555. doi: 10.1021/np200673b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Camp D, Davis RA, Campitelli M, Ebdon J, Quinn RJ. Drug-like Properties: Guiding Principles for the Design of Natural Product Libraries. J Nat Prod. 2012;75:72–81. doi: 10.1021/np200687v. [DOI] [PubMed] [Google Scholar]
- 48.Schulze CJ, Bray WM, Woerhmann MH, Stuart J, Lokey RS, Linington RG. Function-First” lead discovery: mode of action profiling of natural product libraries using image-based screening. Chem Biol. 2013;20:285–295. doi: 10.1016/j.chembiol.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Q, Grkovic T, Font J, Bonham S, Pouwer RH, Bailey CG, Moran AM, Ryan RM, Rasko JEJ, Jormakka M, Quinn RJ, Holst J. Monoterpene glycoside ESK246 from pittosporum targets LAT3 amino acid transport and prostate cancer cell growth. ACS Chem Biol. 2014;9:1369–1376. doi: 10.1021/cb500120x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Butler MS, Fontaine F, Cooper MA. Natural Product Libraries: Assembly, Maintenance, and Screening. Planta Med. 2014;80:1161–1170. doi: 10.1055/s-0033-1360109. [DOI] [PubMed] [Google Scholar]
- 51.Waldhoer M, Fong J, Jones RM, Lunzer MM, Sharma SK, Kostenis E, Portoghese PS, Whistler JL. A heterodimer-selective agonist shows in vivo relevance of G protein-coupled receptor dimers. Proc Natl Acad Sci U S A. 2005;102:9050–9055. doi: 10.1073/pnas.0501112102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nakamura HKJ, Kobayashi J, Ohizumi Y, Hirata Y. Aaptamines Novel Benzo [de] [1,6] naphthyridines from the Okinawan Marine Sponge Aaptos aaptos. J Chem Soc, Perkin Trans. 1987;1:173–176. [Google Scholar]
- 53.Calcul L, Longeon A, Al Mourabit A, Guyot M, Bourguet-Kondracki ML. Novel alkaloids of the aaptamine class from an Indonesian marine sponge of the genus. Tetrahedron. 2003;59:6539–6544. [Google Scholar]
- 54.Fujii H, Takahashi T, Nagase H. Non-peptidic δ opioid receptor agonists and antagonists (2000 – 2012) Expert Opin Ther Pat. 2013;23:1181–1208. doi: 10.1517/13543776.2013.804066. [DOI] [PubMed] [Google Scholar]
- 55.Jutkiewicz EM. The antidepressant-like effects of delta-opioid receptor agonists. Mol Interventions. 2006;6:162–169. doi: 10.1124/mi.6.3.7. [DOI] [PubMed] [Google Scholar]
- 56.Saitoh A, Yamada M. Antidepressant-like effects of δ 0pioid receptor agonists in animal models. Curr Neuropharmacol. 2012;10:231–238. doi: 10.2174/157015912803217314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pradhan AA, Befort K, Nozaki C, Gavériaux-Ruff C, Kieffer BL. The delta opioid receptor: an evolving target for the treatment of brain disorders. Trends Pharmacol Sci. 2011;32:581–590. doi: 10.1016/j.tips.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Broom DC, Jutkiewicz EM, Rice KC, Traynor JR, Woods JH. Behavioral effects of delta-opioid receptor agonists: potential antidepressants? Jpn J Pharmacol. 2002;90:1–6. doi: 10.1254/jjp.90.1. [DOI] [PubMed] [Google Scholar]
- 59.Filliol D, Ghozland S, Chluba J, Martin M, Matthes HW, Simonin F, Befort K, Gavériaux-Ruff C, Dierich A, LeMeur M, Valverde O, Maldonado R, Kieffer BL. Mice deficient for delta- and mu-opioid receptors exhibit opposing alterations of emotional responses. Nat Genet. 2000;25:195–200. doi: 10.1038/76061. [DOI] [PubMed] [Google Scholar]
- 60.van Rijn RM, Whistler JL. The delta(1) opioid receptor is a heterodimer that opposes the actions of the delta(2) receptor on alcohol intake. Biol Psychiatry. 2009;66:777. doi: 10.1016/j.biopsych.2009.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gross H, Goeger DE, Hills P, Mooberry SL, Ballantine DL, Murray TF, Valeriote FA, Gerwick WH. Lophocladines, bioactive alkaloids from the red alga Lophocladia sp. J Nat Prod. 2006;69:640–644. doi: 10.1021/np050519e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Carroll AR, Kaiser SM, Davis RA, Moni RW, Hooper JN, Quinn RJ. A bastadin with potent and selective delta-opioid receptor binding affinity from the Australian sponge Ianthella flabelliformis. J Nat Prod. 2010;73:1173–1176. doi: 10.1021/np100010z. [DOI] [PubMed] [Google Scholar]
- 63.Stanley TH. The history and development of the fentanyl series. J Pain Symptom Manage. 1992;7(3):S3–S7. doi: 10.1016/0885-3924(92)90047-l. [DOI] [PubMed] [Google Scholar]
- 64.Carroll FI, Coleman MC. Etonitazene. An improved synthesis. J Med Chem. 1975;18(3):318–20. doi: 10.1021/jm00237a024. [DOI] [PubMed] [Google Scholar]
- 65.Takayama H, Ishikawa H, Kurihara M, Kitajima M, Aimi N, Ponglux D, Koyama F, Matsumoto K, Moriyama T, Yamamoto LT, Watanabe K, Murayama T, Horie S. Studies on the synthesis and opioid agonistic activities of mitragynine-related indole alkaloids: discovery of opioid agonists structurally different from other opioid ligands. J Med Chem. 2002;45:1949–1956. doi: 10.1021/jm010576e. [DOI] [PubMed] [Google Scholar]
- 66.Johnson TA, Morgan M, Aratow N, Estee SA, Sashidhara KV, Loveridge S, Segraves NL, Crews P. Assessing pressurized liquid extraction for the high throughput extraction of marine- sponge derived natural products. J Nat Prod. 2010;73:359–364. doi: 10.1021/np900565a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Segraves NL, Lopez S, Johnson TA, Said SA, Fu X, Schmitz FJ, Pietraszkiewicz H, Valeriote FA, Crews P. Structures and cytotoxicities of fascaplysin and related alkaloids from two marine phyla - Fascaplysinopsis sponges and Didemnum tunicates. Tetrahedron Lett. 2003;44:3471–75. [Google Scholar]
- 68.Inman WD, O’Neill-Johnson M, Crews P. Novel marine sponge alkaloids. Plakinidine-A and Plakinidine-B, Anthelmintic active alkaloids from a Plakortis sponge. J Am Chem Soc. 1990;112:1–4. [Google Scholar]
- 69.Chavkin C, James IF, Goldstein A. Dynorphin is a specific endogenous ligand of the kappa- opioid receptor. Science. 1982;215:413–415. doi: 10.1126/science.6120570. [DOI] [PubMed] [Google Scholar]
- 70.Koch T, Widera A, Bartzsch K, Schulz S, Brandenburg LO, Wundrack N, Beyer A, Grecksch G, Höllt V. Receptor endocytosis counteracts the development of opioid tolerance. Mol Pharmacol. 2005;67:280–287. doi: 10.1124/mol.104.004994. [DOI] [PubMed] [Google Scholar]
- 71.Kruegel AC, Gassaway MM, Kapoor A, Váradi A, Majumdar S, Filizola M, Javitch JA, Sames D. Synthetic and Receptor Signaling Explorations of the Mitragyna Alkaloids: Mitragynine as an Atypical Molecular Framework for Opioid Receptor Modulators. J Am Chem Soc. 2016;138:6754. doi: 10.1021/jacs.6b00360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Váradi A, Marrone GF, Palmer TC, Narayan A, Szabó MR, Le Rouzic V, Grinnell SG, Subrath JJ, Warner E, Kalra S, Hunkele A, Pagirsky J, Eans SO, Medina JM, Xu J, Pan YX, Borics A, Pasternak GW, McLaughlin JP, Majumdar S. Mitragynine/Corynantheidine pseudoindoxyls as opioid analgesics with mu agonism and delta antagonism, which do not recruit β-Arrestin-2. J Med Chem. 2016;59:8381. doi: 10.1021/acs.jmedchem.6b00748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Finn AK, Whistler JL. Endocytosis of the mu opioid receptor reduces tolerance and a cellular hallmark of opiate withdrawal. Neuron. 2001;32:829–839. doi: 10.1016/s0896-6273(01)00517-7. [DOI] [PubMed] [Google Scholar]
- 74.Manda S, Sharma S, Wani A, Joshi P, Kumar V, Guru SK, Bharate S, Bhushan S, Vishwakarma RA, Kumar A, Bharate SB. Discovery of a marine-derived bis- indole alkaloid fascaplysin, as a new class of potent P-glycoprotein inducer and establishment of its structure activity relationship. Eur J Med Chem. 2016;107:1–11. doi: 10.1016/j.ejmech.2015.10.049. [DOI] [PubMed] [Google Scholar]
- 75.Bharate SB, Manda S, Mupparapu N, Battini N, Vishwakarma RA. Chemistry and Biology of Fascaplysin, a Potent Marine-Derived CDK-4 Inhibitor. Mini-Rev Med Chem. 2012;12:650–664. doi: 10.2174/138955712800626719. [DOI] [PubMed] [Google Scholar]
- 76.Aubry C, Wilson AJ, Jenkins PR, Mahale S, Chaudhuri B, Marechal JD, Sutcliffe MJ. Design, synthesis and biological activity of new CDK4-specific inhibitors, based on fascaplysin. Org Biomol Chem. 2006;4:787–801. doi: 10.1039/b518019h. [DOI] [PubMed] [Google Scholar]
- 77.Segraves NL, Robinson SJ, Garcia D, Said SA, Fu X, Schmitz FJ, Pietraszkiewicz H, Valeriote FA, Crews P. Comparison of fascaplysin and related alkaloids: A study of structures, cytotoxicities, and sources. J Nat Prod. 2004;67:783–792. doi: 10.1021/np049935+. [DOI] [PubMed] [Google Scholar]
- 78.Hudzik TJ, Maciag C, Smith MA, Caccese R, Pietras MR, Bui KH, Coupal M, Adam L, Payza K, Griffin A, Smagin G, Song D, Swedberg MD, Brown W. Preclinical pharmacology of AZD2327: a highly selective agonist of the δ-opioid receptor. J Pharmacol Exp Ther. 2011;338:195–204. doi: 10.1124/jpet.111.179432. [DOI] [PubMed] [Google Scholar]
- 79.Vergura R, Balboni G, Spagnolo B, Gavioli E, Lambert DG, McDonald J, Trapella C, Lazarus LH, Regoli D, Guerrini R, Salvadori S, Calo G. Anxiolytic- and antidepressant-like activities of H-Dmt-Tic-NH-CH(CH2-COOH)-Bid (UFP-512), a novel selective delta opioid receptor agonist. Peptides. 2008;29:93–103. doi: 10.1016/j.peptides.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 80.Perrine SA, Hoshaw BA, Unterwald EM. Delta opioid receptor ligands modulate anxiety- like behaviors in the rat. Br J Pharmacol. 2006;147:864–872. doi: 10.1038/sj.bjp.0706686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Saitoh A, Kimura Y, Suzuki T, Kawai K, Nagase H, Kamei J. Antidepressant-like effects of δ opioid receptor agonists in animal models. J Pharmacol Sci. 2004;95:374–378. doi: 10.1254/jphs.fpj04014x. [DOI] [PubMed] [Google Scholar]
- 82.Saitoh A, Sugiyama A, Yamada M, Inagaki M, Oka J, Nagase H, Yamada M. The novel δ opioid receptor agonist KNT-127 produces distinct anxiolytic-like effects in rats without producing the adverse effects associated with benzodiazepines. Neuropharmacology. 2013;67:485–493. doi: 10.1016/j.neuropharm.2012.11.025. [DOI] [PubMed] [Google Scholar]
- 83.van Rijn RM, Brissett DI, Whistler JL. Dual efficacy of delta opioid receptor-selective ligands for ethanol drinking and anxiety. J Pharmacol Exp Ther. 2010;335:133–139. doi: 10.1124/jpet.110.170969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Richards EM, Mathews DC, Luckenbaugh DA, Ionescu DF, Machado-Vieira R, Niciu MJ, Duncan WC, Nolan NM, Franco-Chaves JA, Hudzik T, Maciag C, Li S, Cross A, Smith MA, Zarate CA. Psychopharmacology (Berl) 2016;233(233):1119–30. doi: 10.1007/s00213-015-4195-4. A randomized, placebo-controlled pilot trial of the delta opioid receptor agonist AZD2327 in anxious depression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, Chen XT, Pitis PM, Gotchev D, Yuan C, Koblish M, Lark MW, Violin JD. A G protein-biased ligand at the μ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther. 2013;344:708–717. doi: 10.1124/jpet.112.201616. [DOI] [PubMed] [Google Scholar]
- 86.Soergel DG, Subach RA, Burnham N, Lark MW, James IE, Sadler BM, Skobieranda F, Violin JD, Webster LR. Biased agonism of the μ-opioid receptor by TRV130 increases analgesia and reduces on-target adverse effects versus morphine: A randomized, double-blind, placebo- controlled, crossover study in healthy volunteers. Pain. 2014;155:1829–1835. doi: 10.1016/j.pain.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 87.Viscusi ER, Webster L, Kuss M, Daniels S, Bolognese JA, Zuckerman S, Soergel DG, Subach RA, Cook E, Skobieranda F. A randomized, phase 2 study investigating TRV130, a biased ligand of the μ-opioid receptor, for the intravenous treatment of acute pain. Pain. 2016;157:264–272. doi: 10.1097/j.pain.0000000000000363. [DOI] [PubMed] [Google Scholar]
- 88.Kuo A, Wyse BD, Meutermans W, Smith MT. In vivo profiling of seven common opioids for antinociception, constipation and respiratory depression: no two opioids have the same profile. Br J Pharmacol. 2015;172:532–548. doi: 10.1111/bph.12696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Levitt ES, Abdala AP, Paton JF, Bissonnette JM, Williams JT. μ opioid receptor activation hyperpolarizes respiratory-controlling Kölliker-Fuse neurons and suppresses post-inspiratory drive. J Physiol. 2015;593:4453–4469. doi: 10.1113/JP270822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G, Levit A, Kling RC, Bernat V, Hübner H, Huang X, Sassano MF, Giguère PM, Löber S, Duan D, Scherrer G, Kobilka BK, Gmeiner P, Roth BL, Shoichet BK. Nature. 2016;537:185. doi: 10.1038/nature19112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Raehal KM, Walker JK, Bohn LM. Morphine side effects in beta-arrestin 2 knockout mice. J Pharmacol Exp Ther. 2005;314:1195–1201. doi: 10.1124/jpet.105.087254. [DOI] [PubMed] [Google Scholar]
- 92.Bohn LM, Gainetdinov RR, Sotnikova TD, Medvedev IO, Lefkowitz RJ, Dykstra LA, Caron MG. Enhanced rewarding properties of morphine, but not cocaine, in beta(arrestin)2 knock-out mice. J Neurosci. 2003;23:10265–10273. doi: 10.1523/JNEUROSCI.23-32-10265.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lalanne L, Ayranci G, Kieffer BL, Lutz PE. The kappa opioid receptor: from addiction to depression, and back. Front Psychiatry. 2014;5:1–17. doi: 10.3389/fpsyt.2014.00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kim JA, Bartlett S, He L, Nielsen CK, Chang AM, Kharazia V, Waldhoer M, Ou CJ, Taylor S, Ferwerda M, Cado D, Whistler JL. Morphine-induced receptor endocytosis in a novel knockin mouse reduces tolerance and dependence. Curr Biol. 2008;18:129–135. doi: 10.1016/j.cub.2007.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Berger AC, Whistler JL. Morphine-induced mu opioid receptor trafficking enhances reward yet prevents compulsive drug use. EMBO Mol Med. 2011;3:385–397. doi: 10.1002/emmm.201100144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science. 1999;286:2495–2498. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- 97.Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000;408:720–723. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- 98.Kelly E, Bailey CP, Henderson G. Agonist-selective mechanisms of GPCR desensitization. Br J Pharmacol. 2008;153(Suppl 1):S379–S388. doi: 10.1038/sj.bjp.0707604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Raehal KM, Schmid CL, Groer CE, Bohn LM. Functional selectivity at the μ-opioid receptor: implications for understanding opioid analgesia and tolerance. Pharmacol Rev. 2011;63:1001–1019. doi: 10.1124/pr.111.004598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.