

Abstract
Ammonium is one of the most important nitrogen sources for bacteria, fungi, and plants, but it is toxic to animals. The ammonium transport proteins (methylamine permeases/ammonium transporters/rhesus) are present in all domains of life; however, functional studies with members of this family have yielded controversial results with respect to the chemical identity (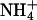 or NH3) of the transported species. We have solved the structure of wild-type AmtB from Escherichia coli in two crystal forms at 1.8- and 2.1-Å resolution, respectively. Substrate transport occurs through a narrow mainly hydrophobic pore located at the center of each monomer of the trimeric AmtB. At the periplasmic entry, a binding site for
or NH3) of the transported species. We have solved the structure of wild-type AmtB from Escherichia coli in two crystal forms at 1.8- and 2.1-Å resolution, respectively. Substrate transport occurs through a narrow mainly hydrophobic pore located at the center of each monomer of the trimeric AmtB. At the periplasmic entry, a binding site for 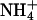 is observed. Two phenylalanine side chains (F107 and F215) block access into the pore from the periplasmic side. Further into the pore, the side chains of two highly conserved histidine residues (H168 and H318) bridged by a H-bond lie adjacent, with their edges pointing into the cavity. These histidine residues may facilitate the deprotonation of an ammonium ion entering the pore. Adiabatic free energy calculations support the hypothesis that an electrostatic barrier between H168 and H318 hinders the permeation of cations but not that of the uncharged NH3. The structural data and energetic considerations strongly indicate that the methylamine permeases/ammonium transporters/rhesus proteins are ammonia gas channels. Interestingly, at the cytoplasmic exit of the pore, two different conformational states are observed that might be related to the inactivation mechanism by its regulatory partner.
is observed. Two phenylalanine side chains (F107 and F215) block access into the pore from the periplasmic side. Further into the pore, the side chains of two highly conserved histidine residues (H168 and H318) bridged by a H-bond lie adjacent, with their edges pointing into the cavity. These histidine residues may facilitate the deprotonation of an ammonium ion entering the pore. Adiabatic free energy calculations support the hypothesis that an electrostatic barrier between H168 and H318 hinders the permeation of cations but not that of the uncharged NH3. The structural data and energetic considerations strongly indicate that the methylamine permeases/ammonium transporters/rhesus proteins are ammonia gas channels. Interestingly, at the cytoplasmic exit of the pore, two different conformational states are observed that might be related to the inactivation mechanism by its regulatory partner.
Keywords: conformational change, x-ray structure
Ammonium is an excellent nitrogen source for many bacteria, fungi, and plants, but it can be cytotoxic, especially for animal cells at high concentration. Its transport across cellular membranes is thus of high biological relevance. Ammonia is a weak base (pKa, 9.25) and exists predominantly as the ammonium ion (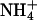 ) in biological fluids. At high ammonium concentration in the environment (typically >1 mM), passive membrane permeation of the uncharged species is usually sufficient to promote cell growth in the case of enteric bacteria and fungi. However, at lower concentration, when diffusion becomes limiting for nitrogen uptake, “ammonium” (
) in biological fluids. At high ammonium concentration in the environment (typically >1 mM), passive membrane permeation of the uncharged species is usually sufficient to promote cell growth in the case of enteric bacteria and fungi. However, at lower concentration, when diffusion becomes limiting for nitrogen uptake, “ammonium” (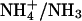 ) transport systems are needed (1). The molecular identity of these transporters has been identified recently in many different organisms (2).
) transport systems are needed (1). The molecular identity of these transporters has been identified recently in many different organisms (2).
The ammonium transport mechanism in different species has remained controversial. Energy-dependent uptake of ammonium under limiting nitrogen supply has been described for bacteria (1) and fungi (3, 4). Genetic studies identified genes of ammonium transporters (Amts) and methylamine permeases (Meps) and permitted investigation of their activity and regulation (3, 5). The most widely accepted view is that they are secondary transporters mediating the uptake of the ammonium cation (6–8). This conclusion was mainly based on the apparent accumulation of the substrate analog [14C] methylammonium in whole cells in an Amt/Mep-dependent manner. This view has been challenged (9–12) by accumulating evidence showing that the Amt/Mep proteins are channels that simply increase the rate of equilibration of ammonia across the cytoplasmic membrane.
The first sequenced Amt and Mep genes were from Arabidopsis thaliana and Saccharomyces cerevisiae (13, 14). Many additional members have subsequently been identified in plants, yeast, and bacteria, indicating the presence of an extended Amt family comprising the Amt and Mep genes (7, 15–18). Another important discovery, based on sequence analysis, was that the human rhesus (Rh) blood group proteins appear related to the Mep/Amt family (19). Functional complementation experiments later demonstrated that human Rh proteins can rescue the growth of a yeast mutant deficient in ammonium uptake (20), suggesting they could function as Amts (21). The Rh family (22) comprises the erythroid blood group antigens and nonerythroid homologues that are expressed in the kidney, liver, skin, ovary, testes, and CNS (23, 24). Although most evidence suggests that Rh proteins are Amts, they also have been proposed to be gas channels for CO2 (9–12). Supporting evidence is that at elevated CO2 concentration (25, 26), the expression level of RH1 in Chlamydomonas reinhardtii has increased.
Presently >200 genes belonging to the Mep/Amt/Rh family are known. The corresponding proteins are 400–450 aa in length and are predicted to have 10–12 transmembrane (TM) helices with a C-terminal cytoplasmic extension (13–15). Here we report on the structure determination of the AmtB protein from Escherichia coli in two crystal forms in the presence and absence of substrate at 2.1- and 1.8-Å resolution, respectively, and on the resulting mechanistic implications.
Methods
Protein Expression and Purification. The E. coli AmtB gene was cloned into the pET22b (Novagen) vector by using NdeI/VspI and XhoI restriction sites, and 12 aa were placed before the C-terminal hexahistidine tag to facilitate affinity purification. AmtB was overexpessed in E. coli strain C43(DE3), as described (27). Cells were harvested, lysed by French press, and sonicated in a buffer containing 25 mM sodium phosphate (pH 7.8), 500 mM NaCl, 10% glycerol, and 1 mM PMSF. The membrane fraction was solubilized with 40 mM dodecyl-β-maltoside (Anatrace, Maumee, OH), and its supernatant was loaded onto a Ni2+-affinity column (HisTrap, Pharmacia). The column was step-washed with 20 and 40 mM imidazole containing 6 mM N,N-dimethyldodecylamine-N-oxide (LDAO). Then, the AmtB protein was eluted with buffer containing 300 mM imidazole and 4 mM LDAO. Further purification was carried out by size exclusion chromatography (Superdex 200 16/60) with a buffer containing 10 mM Tris·HCl (pH 7.6), 100 mM NaCl, 10% glycerol, and 4 mM LDAO. The purified AmtB was concentrated with Centricon 100-K (Millipore) to 20 mg/ml. For the production of selenomethionine-labeled protein, the same expression and purification procedure was followed except for including seleno-l-methionine (obtained from Sigma) in the medium. The incorporation of selenomethionine was confirmed by MALDI-MS.
Crystallization. Crystals of AmtB (native or selenomethionine-labeled) were grown at 20°C in sitting drops by mixing 3 μl of the concentrated protein solution with reservoir solution [100 mM sodium acetate buffer, pH 4.6/200 mM (NH4)2SO4/20–25% polyethylene glycol 550]. As an additive for improving crystal quality, we used Deoxy-BigChap (Hampton Research, Riverside, CA) at a final concentration of 1.4 mM. Two different crystal forms (R3 and P63) were obtained, both diffracting to high resolution. The P63 form was initially observed with the selenomethionine-labeled protein but could subsequently also be produced with the native protein when substituting ammonium sulfate (200 mM) for magnesium sulfate (200 mM).
Structure Determination. For data collection, crystals were mounted in cryoloops and flash-cooled by fast transfer into a nitrogen gas stream with a temperature of 100 K. All data sets were collected as consecutive series of 0.5° rotation images on a marCCD detector at the protein beamline X06SA at the Swiss Light Source in Villigen, Switzerland (Table 1). The data sets were processed with xds (28). Anomalous data for the selenomethionine derivative crystals were collected at the peak wavelength of 0.9794 Å derived from an x-ray fluorescence spectrum. Using the anomalous differences of the peak data set, we could locate 10 of 11 expected selenium sites with shelxd (29). These 10 selenium sites were refined with sharp (30), followed by density modification with solomon (31). An initial model of the protein backbone was automatically built with arp/warp (32), and the side chains were manually added by using the interactive graphics program moloc (33). The initial model from the P63 crystal form was then used as a search model for molecular replacement with amore (34) into the R3 crystal form. All models were refined by iterative rounds of model building with moloc and maximum-likelihood refinement with refmac (35) (see Table 1). Figures were prepared with pymol (36).
Table 1. Data collection and refinement statistics.
| SeMet-AmtB (Am2SO4) | Native-1 (Am2SO4) | Native-2 (MgSO4) | |
|---|---|---|---|
| Data collection | |||
| Space group | P63 | R3 | P63 |
| Cell dimensions a and c, Å | 109.3, 84.8 | 116.6, 130.5 | 111.9, 86.5 |
| Beamline | X06SA at SLS | X06SA at SLS | X06SA at SLS |
| Wavelength, Å | 0.9794 | 0.9797 | 0.9797 |
| Resolution,* Å | 40-2.6 (2.7 - 2.6) | 60-2.1 (2.15 - 2.1) | 60-1.8 (1.85 - 1.8) |
| Completeness,* % | 99.9 (99.8) | 99.7 (99.9) | 95.6 (98.5) |
| Rsym,* % | 8.1 (69.8) | 7.3 (70.7) | 5.4 (69.3) |
| I/σ* | 13.5 (2.7) | 13.6 (2.5) | 13.2 (2.0) |
| Reflections (total/unique) | 203,925/34,832 | 222,042/38,583 | 209,103/54,526 |
| Refinement statistics | |||
| Rcryst, % | 15.9 | 18.1 | |
| Rfree, % | 18.6 | 20.8 | |
| Nonglycine residues in Ramachandran plot, % | |||
| Most favorable regions | 93 | 93 | |
| Generously allowed regions | 7 | 7 | |
| Protein/sulfate/acetate/water atoms in a.u. | 2661/15/4/93 | 2633/10/4/103 | |
| rms deviation bond lengths, Å | 0.012 | 0.013 | |
| rms deviation bond angles, ° | 1.2 | 1.1 |
Rsym = Σhkl Σi |/(hkl;i) - 〈/(hkl)〉|/Σhkl Σi/(hkl;i). Rcryst; Rfree = Σhkl|Fobs(hkl) - Fcalc(hkl)|/Σhkl Fobs(hkl), whereby the summation runs over the working set and test set reflections, respectively. SLS, Swiss Light Source.
Values in parentheses are for the outmost resolution shell.
Transport Rate Estimates. One estimate for the nitrogen uptake per bacterial cell per second was based on total nitrogen consumption per hour (11 mmol N for 1 g of E. coli ≈ 3·1012 cells) (37). Furthermore, we have assumed that most of this nitrogen uses the Amt under nitrogen-limiting conditions. The second estimate uses the known limiting external ammonia concentration for which an amtB–strain can normally grow (≈100 nM) (9). Together with the membrane permeability coefficient for ammonia (38), this yields a lower estimate for the uptake that must be provided by the transporter under limiting conditions.
To arrive at rates per single transporter, we have assumed there may be between 10 and 1,000 active transporters per μm2 of membrane area. Despite uncertainties in some of the numbers used, both estimates lead to rates in the range of 101 to 104 ammonium molecules transported per second per transporter.
The details of the adiabatic free energy calculations are available in Supporting Text, which is published as supporting information on the PNAS web site.
Structure
Native AmtB and selenomethionine-labeled protein have been crystallized under nearly identical conditions in space groups R3 and P63, respectively (see Methods). The P63 crystals could also be obtained with nonlabeled protein in the absence of ammonium sulfate. The structure was solved by using single wavelength anomalous dispersion data from the P63 SeMet crystals (Table 1). Both crystal forms can be described as being built from stacked layers of disk-shaped crystallographic trimers (Fig. 1). One single but different protein–protein contact between trimers of adjacent layers fully determines each crystal packing. One protein subunit comprises 11 TM helices (TM1–TM11), with the N terminus located on the periplasmic side of the membrane and the C terminus on the cytoplasmic side. The two refined structures are largely identical, and the only functionally relevant structural difference is observed at the N terminus of TM10 (see below). The refined models comprise 366 (R3 form) and 362 (P63 form) of the 406 residues with the missing residues occurring in four disordered peptide segments located at the two termini and in two cytoplasmic loops.
Fig. 1.

Overall structure of the AmtB trimer. (a) Surface view of the AmtB trimer from the extracellular side. Two monomers of an AmtB trimer are drawn in salmon and slate; one monomer is shown as a ribbon. (b) Side view of the molecular surface of the AmtB trimer.
The fold of AmtB (Figs. 1 and 2) is not related to any of the known membrane protein structures. It can be characterized as consisting of two contiguous α-helical bundles, TM1–TM5 and TM6–TM10, and a long C-terminal helix (TM11). The two five-helix bundles are related by an approximately twofold axis in the plane of the membrane (Fig. 2b). AmtB shows an antiparallel architecture (pseudotwofold perpendicular to membrane plane) like aquaporin (39), the TM subunit of BtuCD (40), and the ClC chloride channel (41), but unlike AcrB (42), LacY (43), and GlpT (44), which show a parallel architecture. In contrast to the aquaporins, where the twofold relatedness was recognized in the sequence and where the equivalent TM helices superpose very well (45), the pseudosymmetry is poorer in the case of AmtB, both in sequence and structure.
Fig. 2.

Topological representation of the AmtB monomer. (a) Side view from the membrane. (b) Top view from the extracellular side.
The 11 helices TM1–TM11 have very different lengths ranging from 15 (TM5 and TM8) to 33 (TM11) residues (Fig. 3). The longest helix, TM11, is tilted ≈45° with respect to the membrane plane and lies across the membrane-exposed side of the monomer. For the other 10 helices, a tilt of <20° is observed. TM1 is kinked at P26, which is observed in the cis-peptide conformation. TM2 has an almost 90° kink at G63, and its two-turn periplasmic extension (TM2b) runs nearly parallel to the membrane plane. At the TM2a–TM2b junction, there is a highly conserved tripeptide sequence F(Y)G-Y(F) (Fig. 3). In both crystal forms, all five periplasmic loops, which range from 8 to 28 residues in length, are very well ordered, whereas two of the five cytoplasmic loops and the cytoplasmic C terminus appear highly disordered.
Fig. 3.

Sequence alignment of selected members of the Mep/Amt/Rh family. Secondary structure according to dspp (28, 52) and sequence numbering (AmtB of E. coli) are indicated. Highly conserved residues are in red. Amino acid sequences are as follows: AmtB, E. coli (P37905); AmtA, Dictyostelium discoideum (BAB39709); Amt, Schizosaccharomyces pombe (NP_593983); Amt, Chlamydomonas reinhardtii (AAL38652); AtAMT1, Arabidopsis thaliana (P54144); and RhAG, Homo sapiens (Q02094). The alignment was made with multalin (53) and adjusted manually based on the structural information
The presence of a tightly packed clover-shaped trimer (Fig. 1) in the crystals is consistent with the finding that detergent-solubilized, purified AmtB is a very stable trimer (46). The extensive, predominantly hydrophobic subunit interface involves mainly residues of the TM helices but also some that are located on connecting loops. This lateral pseudotwofold helical interface involves TM1–TM3 from one subunit and TM6–TM8 from the adjacent one. On the periplasmic side of the threefold axis, hydrophobic side chains from the three-symmetry-related TM1 helices form a tight trimer contact for ≈20 Å into the membrane.
Pathway of Ammonium Translocation. One major interest regarding the structure of AmtB is the identification of the ammonium translocation pathway. A pathway along the threefold axis could readily be ruled out in view of the observed tight packing. Although there is no open channel, closer inspection of the structure strongly indicated that translocation occurs through a pore located at the center of each monomer (Fig. 4b), right between the pseudosymmetry-related helical domains. Depressions on the periplasmic and cytoplasmic surfaces lead into this central pore. On its periplasmic side, it is blocked by the π -face of F215. The opposite face of F215 is partly stacked against the phenyl ring of F107, which is located at the bottom of the deep periplasmic depression (Fig. 4a). Many of the most conserved residues within the Mep/Amt/Rh family cluster around this narrow, predominantly hydrophobic pore (Figs. 3 and 4c).
Fig. 4.

Ammonium-binding site and hydrophobic pore. (a) Stereoview from the extracellular side of the putative substrate-binding site at the extracellular pore entry. The 2 Fo–Fc electron density map of AmtB is contoured at 1.0 σ in slate; the Fo–Fc electron density map is contoured at 3.0 σ in yellow. (b) A cut through the AmtB monomer surface within the plane of the membrane. The dark elongated feature in the center indicates the location and shape of the conduction pore. The pore is seen to be blocked by F215 and F104 on its extracellular side, and H168 and H318 are seen to be adjacent in its central part. (c) Stereoview of the AmtB pore-lining residues in the R3 structure. Highly conserved residues (Fig. 3) are shown in salmon, less conserved ones in green. A final omit difference electron density (Fo–Fc) contoured at 3.0 σ is shown in the pore region in red. (d) Representation as in c for the P63 structure (crystal grown in the absence of ammonium sulfate). Note the different structure at the cytoplasmic exit at V314 and the different peaks in the omit difference density.
Residues from helices TM1, TM3, and TM5 and their pseudosymmetry mates TM6, TM8, and TM10 form the entries and walls of the blocked pore (Figs. 3 and 4c). Very prominently, the side chains of two histidine residues (H168 and H318), both highly conserved, lie adjacent, with their imidazole ring edges facing the cavity such that a hydrogen bond can be formed between their δ nitrogens. Assuming both imidazole rings to be neutral in this hydrophobic environment requires them to be in different tautomeric states, such that their ε nitrogens would offer one acceptor and one donor function to molecules diffusing through the pore. The two histidines, H168 and H318, are located at the N termini of helices TM5 and TM10, respectively, and are related by the pseudotwofold symmetry. They are preceded by two highly conserved aspartates, D160 and D310 (Fig. 3). D310 is disordered in the P63 structure. In the R3 structure, both acidic side chains are involved in multiple H-bonding interactions with main chain N-H groups (residues 163–165 and 314–316, respectively) and appear to have an important structural role in fixing the backbone of the somewhat irregular helical structure preceding H168 and H318, respectively. The conserved D160 has been considered a potential candidate for a periplasmic ammonium-binding site (15), which is clearly ruled out. Segments 160–168 and 310–318 have similar backbone structures in the R3 structure, with the consequence that in both cases a main-chain carbonyl (A162 and C312, respectively) points into the pore and has no intramolecular hydrogen-bonding partners. Although that of C312 is located at the open cytoplasmic end of the pore and appears solvent-accessible, that of A162 is buried and packed against the edge of the pore-blocking aromatic ring of F107. It is tempting to speculate that this carbonyl oxygen could serve as a transient H-bond acceptor for translocating substrate molecules in a transient open state of the periplasmic pore entry.
In the P63 crystal form, segment 311–315 has quite a different structure corresponding to a 310 helical turn at the N terminus of TM10. In particular, the V314 side chain faces the pore exit and occupies the position of water 111 of the R3 structure. As a result, the cytoplasmic pore exit is more “closed” and purely hydrophobic in the P63 structure, but more “open” and more polar in the R3 structure. An interesting question is whether both local structures are functionally relevant. There is a possibility that the more closed structure observed in the P63 form represents a nonconducting state, which may be induced by binding of the regulatory GlnK protein. In E. coli, the GlnK protein has been shown to bind to membranes in an AmtB-dependent manner and to be a negative regulator of the transport activity of AmtB (46).
The residues lining the pore and its entry sites (Fig. 4c) are predominantly hydrophobic and can be grouped into pairs related by the pseudotwofold symmetry [TM1/TM6: M23/L208, I28/W212, F31/F215, Y32/N216; TM3/TM8: (F107/S263), I110/I266, L114/V270; TM5/TM10: V167/V317, and H168/H318]. The pore structure with one end blocked does not obey the striking degree of pseudosymmetry, as it has been observed for the aquaporin channels (45). Examination of the conservation of the pore residues across representatives from the Mep/Amt/Rh family (Fig. 3) shows that those at the closed extracellular end of the pore (F107, L208, W212, F215, and N216) are much better conserved than their quasitwofold related mates at the open cytoplasmic side (S263, M23, I28, F31, and Y32). Two other strictly conserved residues across all Mep/Amt/Rh family members are T234 and T273. Although not directly lining the pore, they are strategically located to orient and position the side chains of the conserved residues around the periplasmic end of the pore (Fig. 4c). This strongly suggests that the well conserved precisely packed structure at the closed periplasmic side of the pore is functionally relevant.
Transport Mechanism and Selectivity. All functional studies on the Mep/Amt/Rh proteins have been carried out by using wild-type and mutant whole cells or with heterologous expression systems. Because it is difficult to measure ammonium directly, its transport has usually been inferred from its inhibitory effect on methylamine uptake (1, 4, 7) or through measurement of ion currents in oocytes expressing selected transporter genes (47–50). The interpretation of these experimental results is not straightforward, and this may be one reason why the literature is still rather controversial with respect to the nature of the transported species as well as to the type of energy coupling. The majority of these data indicate that the Mep/Amt/Rh proteins mediate the transport of the ammonium ion along the electrochemical gradient.
Many studies report apparent ammonium affinities of these proteins, and they are typically 1–20 μM for the high-affinity and in the low millimolar range for the low-affinity transporters. For the discussion of possible transport mechanisms, it is essential to know the approximate rate of transport through a single channel. Using two different types of experimental data (see Methods), we estimate values in the range of 101 to 104 substrate molecules per second per channel. Such a rate is far below that of open channels operating close to the diffusion limit (108 to 109 ions or molecules s–1) but rather typical for transporters and carriers that usually undergo larger conformational changes. Another important question concerns specificity. There is no compelling reason that these transporters should discriminate strongly between NH3 and 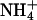 , although their different physical characteristics may naturally lead to discrimination. However, one would expect discrimination against the physiologically important cations Na+ and K+, both present at relatively high concentrations compared to ammonium (Fig. 5).
, although their different physical characteristics may naturally lead to discrimination. However, one would expect discrimination against the physiologically important cations Na+ and K+, both present at relatively high concentrations compared to ammonium (Fig. 5).
Fig. 5.

Conformational change at the cytoplasmic exit between R3 and P63 crystal structures. The pore is viewed from the cytoplasmic side. (a) R3 structure (“open” exit conformation). (b)P63 structure (“closed” exit conformation).
We have identified a possible substrate-binding site that accounts for the observed saturation of the transport rate. The site is located at the bottom of the periplasmic depression at a strategic position to control entry into the pore (Fig. 4). The corresponding difference electron density peak is observed in both crystal forms, and it is also present in the crystal grown in the presence of MgSO4 instead of (NH4)2SO4. The findings suggest that in these crystals the site is occupied by ammonium and/or water.
Crystallographically, it is impossible at this resolution to discriminate bound 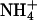 from bound water. That the site is occupied by water in the absence of ammonium does not imply it is not specific for ammonium but merely reflects the high water concentration (55 M). The binding cavity is surrounded by the aromatic side chains of F103, F107, and W148, which are at 3.8–4.0 Å from the density peak. The hydroxyl group of S219 and one surface water molecule are within hydrogen bonding distance. No significant competitive inhibition of the Mep/Amt/Rh transporters by alkali cations has been observed (7, 47), suggesting specificity for ammonium. Adiabatic free energy calculations imply that Na+ is disfavored compared with ammonium at this site (Fig. 6, which is published as supporting information on the PNAS web site), although entropic contributions that are not considered in this calculation could reduce the observed difference in relative binding energies.
from bound water. That the site is occupied by water in the absence of ammonium does not imply it is not specific for ammonium but merely reflects the high water concentration (55 M). The binding cavity is surrounded by the aromatic side chains of F103, F107, and W148, which are at 3.8–4.0 Å from the density peak. The hydroxyl group of S219 and one surface water molecule are within hydrogen bonding distance. No significant competitive inhibition of the Mep/Amt/Rh transporters by alkali cations has been observed (7, 47), suggesting specificity for ammonium. Adiabatic free energy calculations imply that Na+ is disfavored compared with ammonium at this site (Fig. 6, which is published as supporting information on the PNAS web site), although entropic contributions that are not considered in this calculation could reduce the observed difference in relative binding energies.
The relatively slow translocation through the pore may have different causes. First, if there is no stable open state of the pore, transient structural fluctuations at the blocked pore entry might limit the rate. Second, entry into the predominantly hydrophobic pore may be energetically unfavorable. The adiabatic free energy profile (Fig. 6) shows an electrostatic barrier (≈10 kcal/mol for 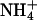 ) at the level of the two histidine residues (around Z =–6 Å), suggesting that diffusion of charged substrates is hindered. The barrier height appears surprisingly low in view of the large dehydration cost for an ammonium ion (≈80 kcal/mol). Despite this, the calculations support the view that ammonia is very likely to be the preferred conducted species.
) at the level of the two histidine residues (around Z =–6 Å), suggesting that diffusion of charged substrates is hindered. The barrier height appears surprisingly low in view of the large dehydration cost for an ammonium ion (≈80 kcal/mol). Despite this, the calculations support the view that ammonia is very likely to be the preferred conducted species.
We are intrigued by the unusual lateral H-bonded association of the two conserved histidine side chains in the pore (Fig. 4b). Whether these histidines serve as proton acceptors for entering ammonium ions or whether deprotonation/reprotonation occurs at the water-accessible pore entries remains open at this point. The neutral (His-HisH) system (HisH meaning that the Nε carries a proton) may have an elevated pKa because the protonated (HisH-HisH)+ system has a symmetric hydrogen bond (between the two δ nitrogens) and thus permits increased charge delocalization. However, such an ammonium deprotonation mechanism would require that the proton can subsequently diffuse back to the aqueous phase.
Conclusion
It appears plausible that the identified periplasmic entry site is specific for ammonium and contributes to discrimination against water and monovalent metal cations. The estimated slow transport rate may be due to slow conformational changes needed for transient pore opening at the periplasmic constriction. The pore structure and energetic considerations suggest that AmtB facilitates the conduction of the uncharged species ammonia and strongly hinders the passage of ions. AmtB may thus be considered a slowly conducting channel rather than a transporter. The structure determination of AmtB provides a powerful basis for carrying out further mechanistic investigations of Mep/Amt/Rh family members.
Note. During the reviewing process of this manuscript, the structure of the E. coli AmtB protein was reported by another group (51). Experimentally, there are the following notable differences between the two independent structure determinations: (i) we crystallized the wild-type AmtB protein, whereas Khademi et al. (51) crystallized the mutant protein, which carries three single mutations (F68S, S126P, and K255L); (ii) we obtained two crystal forms (R3 and P63), whereas Khademi et al. got one crystal form (P63) (the cell constants of the two P63 crystal forms differ by >10%) (iii) different detergents, lauryldimethylamine oxide (this work), or β-octyl-glucoside (51) were used for crystallization; and (iv) different crystallization conditions, particularly pH 4.6 (this work) or 6.5 (51), were used. The structural comparison reveals that our P63 structure is very similar to that of Khademi et al. (51), except that all cytoplasmic loops are ordered in the latter. Despite different crystal-packing contacts, both P63 structures (this work and ref. 51) show the same narrow and more hydrophobic cytoplasmic pore exit as compared with the R3 structure. The constriction at the pore entry by the side chains of F107 and F215 is identical in all of the structures (this work and ref. 51). We have emphasized that transient opening of this obstruction may limit the conduction rate and Khademi et al. (51) similarly argue that these side chains must move dynamically during any conduction event. In the highest-resolution structure of Khademi et al. (PDB ID code 1U7G), a β-octyl glucoside molecule is bound to the cytoplasmic side with its alkyl chain reaching up to the pore entry adjacent to V314. It is unclear how far the experimental differences (pH, detergent, and crystal packing) influence the different conformational order seen in the cytoplasmic loops. Another important point is that our final difference maps (Fig. 4 c and d) reveal that the small peaks in the central part of the channel assigned to be ammonia binding sites by Khademi et al. are similarly present in the absence of ammonia in our P63 structure. We think that the pore will be partly hydrated for energetic reasons, and that the structural rigidity of the pore structure may influence the average distribution of these water molecules. One of the three mutations in the structure of Khademi et al. (51), F68S, lies adjacent to the periplasmic ammonium-binding site. We note that the carboxamide group of Q104, a conserved residue in the Amt family, points into this binding site in our structure (Fig. 4a), whereas this group is reoriented toward the mutated S68 side chain in the structure of Khademi et al. In the wild-type protein, the bulky F68 side chain does not allow this glutamine side chain conformation. In the Q104 conformation that we observe, the methyl group of the methylammonium bound in the 1U7C structure of Khademi et al. (51) structures would be unfavorably close to the Q104 carboxamide.
Supplementary Material
Acknowledgments
Data were collected at beamline X06SA of the Swiss Light Source (Paul Scherrer Institut, Villigen, Switzerland), and we thank Clemens Schulze and Takashi Tomizaki for excellent help. This work was supported by the Swiss National Science Foundation, within the framework of the National Center of Competence in Research Structural Biology program. S.B. is a fellow of the Human Frontier Science Program.
Author contributions: L.Z. and X.-D.L. designed research; L.Z., S.B., and X-D.L. performed research; L.Z., D.K., S.B., F.K.W., and X.-D.L. analyzed data; and L.Z., S.B., F.K.W., and X.-D.L. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: Amt, ammonium transporter; Mep, methylamine permease; Rh, rhesus; TM, transmembrane.
Data deposition: The atomic coordinates and structure factore have been deposited in the Protein Data Bank, www.pdb.org [PDB ID codes 1XQE (R3) and 1XQF (P63)].
References
- 1.Kleiner, D. (1985) FEMS Microbiol. Rev. 32, 87–100. [Google Scholar]
- 2.Ludewig, U., von Wiren, N., Rentsch, D. & Frommer, W. B. (2001) Genome Biol. 2, REVIEWS1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dubois, E. & Grenson, M. (1979) Mol. Gen. Genet. 175, 67–76. [DOI] [PubMed] [Google Scholar]
- 4.Hackette, S. L., Skye, G. E., Burton, C. & Segel, I. H. (1970) J. Biol. Chem. 245, 4241–4250. [PubMed] [Google Scholar]
- 5.Jayakumar, A., Hwang, S. J., Fabiny, J. M., Chinault, A. C. & Barnes, E. M., Jr. (1989) J. Bacteriol. 171, 996–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kleiner, D. (1993) in Alkali Cation Transport Systems in Prokaryotes, ed. Bakker, E. P. (CRC, Boca Raton, FL), pp. 379–395.
- 7.Marini, A. M., Soussi-Boudekou, S., Vissers, S. & Andre, B. (1997) Mol. Cell. Biol. 17, 4282–4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.von Wiren, N., Gazzarrini, S., Gojon, A. & Frommer, W. B. (2000) Curr. Opin. Plant Biol. 3, 254–261. [PubMed] [Google Scholar]
- 9.Soupene, E., He, L., Yan, D. & Kustu, S. (1998) Proc. Natl. Acad. Sci. USA 95, 7030–7034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soupene, E., Ramirez, R. M. & Kustu, S. (2001) Mol. Cell. Biol. 21, 5733–5741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soupene, E., Chu, T., Corbin, R. W., Hunt, D. F. & Kustu, S. (2002) J. Bacteriol. 184, 3396–3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Soupene, E., Lee, H. & Kustu, S. (2002) Proc. Natl. Acad. Sci. USA 99, 3926–3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marini, A. M., Vissers, S., Urrestarazu, A. & Andre, B. (1994) EMBO J. 13, 3456–3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ninnemann, O., Jauniaux, J. C. & Frommer, W. B. (1994) EMBO J. 13, 3464–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomas, G. H., Mullins, J. G. & Merrick, M. (2000) Mol. Microbiol. 37, 331–344. [DOI] [PubMed] [Google Scholar]
- 16.Gazzarrini, S., Lejay, L., Gojon, A., Ninnemann, O., Frommer, W. B. & von Wiren, N. (1999) Plant Cell 11, 937–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meier-Wagner, J., Nolden, L., Jakoby, M., Siewe, R., Kramer, R. & Burkovski, A. (2001) Microbiology 147, 135–143. [DOI] [PubMed] [Google Scholar]
- 18.Monahan, B. J., Fraser, J. A., Hynes, M. J. & Davis, M. A. (2002) Eukaryot. Cell 1, 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marini, A. M., Urrestarazu, A., Beauwens, R. & Andre, B. (1997) Trends Biochem. Sci. 22, 460–461. [DOI] [PubMed] [Google Scholar]
- 20.Marini, A. M., Matassi, G., Raynal, V., Andre, B., Cartron, J. P. & Cherif-Zahar, B. (2000) Nat. Genet. 26, 341–344. [DOI] [PubMed] [Google Scholar]
- 21.Heitman, J. & Agre, P. (2000) Nat. Genet. 26, 258–259. [DOI] [PubMed] [Google Scholar]
- 22.Huang, C. H. & Liu, P. Z. (2001) Blood Cells Mol. Dis. 27, 90–101. [DOI] [PubMed] [Google Scholar]
- 23.Liu, Z., Peng, J., Mo, R., Hui, C. & Huang, C. H. (2001) J. Biol. Chem. 276, 1424–1433. [DOI] [PubMed] [Google Scholar]
- 24.Liu, Z., Chen, Y., Mo, R., Hui, C., Cheng, J. F., Mohandas, N. & Huang, C. H. (2000) J. Biol. Chem. 275, 25641–25651. [DOI] [PubMed] [Google Scholar]
- 25.Soupene, E., King, N., Feild, E., Liu, P., Niyogi, K. K., Huang, C. H. & Kustu, S. (2002) Proc. Natl. Acad. Sci. USA 99, 7769–7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soupene, E., Inwood, W. & Kustu, S. (2004) Proc. Natl. Acad. Sci. USA 101, 7787–7792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blakey, D., Leech, A., Thomas, G. H., Coutts, G., Findlay, K. & Merrick, M. (2002) Biochem. J. 364, 527–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kabsch, W. (1993) J. Appl. Crystallogr. 26, 795–800. [Google Scholar]
- 29.Schneider, T. R. & Sheldrick, G. M. (2002) Acta Crystallogr. D 58, 1772–1779. [DOI] [PubMed] [Google Scholar]
- 30.De La Fortelle, E. & Bricogne, G. (1997) in Methods in Enzymology, eds. Carter, C. W. & Sweet, R. M. (Academic, New York), pp. 472–494. [DOI] [PubMed]
- 31.Abrahams, J. P. (1996) Acta Crystallogr. D 52, 30–42. [DOI] [PubMed] [Google Scholar]
- 32.Lamzin, V. S., Perrakis, A. & Wilson, K. S. (2001) in Crystallography of Biological Macromolecules, eds. Rossmann, M. G. & Arnold, E. (Kluwer, Dordrecht, The Netherlands), pp. 720–722.
- 33.Gerber, P. R. & Muller, K. (1995) J. Comput. Aided Mol. Des. 9, 251–268. [DOI] [PubMed] [Google Scholar]
- 34.Navaza, J. (1994) Acta Crystallogr. A 50, 157–163. [Google Scholar]
- 35.Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997) Acta Crystallogr. D 53, 240–255. [DOI] [PubMed] [Google Scholar]
- 36.DeLano, W. L. (2002) The Pymol Molecular Graphics System (DeLano Scientific, San Carlos, CA).
- 37.Reitzer, L. (2003) Annu. Rev. Microbiol. 57, 155–176. [DOI] [PubMed] [Google Scholar]
- 38.Kleiner, D. (1985) FEBS Lett. 187, 237–239. [DOI] [PubMed] [Google Scholar]
- 39.Murata, K., Mitsuoka, K., Hirai, T., Walz, T., Agre, P., Heymann, J. B., Engel, A. & Fujiyoshi, Y. (2000) Nature 407, 599–605. [DOI] [PubMed] [Google Scholar]
- 40.Locher, K. P., Lee, A. T. & Rees, D. C. (2002) Science 296, 1091–1098. [DOI] [PubMed] [Google Scholar]
- 41.Dutzler, R., Campbell, E. B., Cadene, M., Chait, B. T. & MacKinnon, R. (2002) Nature 415, 287–294. [DOI] [PubMed] [Google Scholar]
- 42.Murakami, S., Nakashima, R., Yamashita, E. & Yamaguchi, A. (2002) Nature 419, 587–593. [DOI] [PubMed] [Google Scholar]
- 43.Abramson, J., Smirnova, I., Kasho, V., Verner, G., Kaback, H. R. & Iwata, S. (2003) Science 301, 610–615. [DOI] [PubMed] [Google Scholar]
- 44.Huang, Y., Lemieux, M. J., Song, J., Auer, M. & Wang, D. N. (2003) Science 301, 616–620. [DOI] [PubMed] [Google Scholar]
- 45.Stroud, R. M., Savage, D., Miercke, L. J., Lee, J. K., Khademi, S. & Harries, W. (2003) FEBS Lett. 555, 79–84. [DOI] [PubMed] [Google Scholar]
- 46.Coutts, G., Thomas, G., Blakey, D. & Merrick, M. (2002) EMBO J. 21, 536–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bakouh, N., Benjelloun, F., Hulin, P., Brouillard, F., Edelman, A., Cherif-Zahar, B. & Planelles, G. (2004) J. Biol. Chem. 279, 15975–15983. [DOI] [PubMed] [Google Scholar]
- 48.Ludewig, U., von, W. N. & Frommer, W. B. (2002) J. Biol. Chem. 277, 13548–13555. [DOI] [PubMed] [Google Scholar]
- 49.Ludewig, U., Wilken, S., Wu, B., Jost, W., Obrdlik, P., El, B. M., Marini, A. M., Andre, B., Hamacher, T., Boles, E., et al. (2003) J. Biol. Chem. 278, 45603–45610. [DOI] [PubMed] [Google Scholar]
- 50.Westhoff, C. M., Ferreri-Jacobia, M., Mak, D. O. & Foskett, J. K. (2002) J. Biol. Chem. 277, 12499–12502. [DOI] [PubMed] [Google Scholar]
- 51.Khademi, S., O'Connell, J., III, Remis, J., Robles-Colmenares, Y., Miercke, L. J. & Stroud, R. M. (2004) Science 305, 1587–1594. [DOI] [PubMed] [Google Scholar]
- 52.Kabsch, W. & Sander, C. (1983) Biopolymers 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- 53.Corpet, F. (1988) Nucleic Acids Res. 16, 10881–10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}