

ABSTRACT
As the blockade of inhibitory surface-molecules such as CTLA-4 on T cells has led to recent advances in antitumor immune therapy, there is great interest in identifying novel mechanisms of action of CD8+ T cells to evoke effective cytotoxic antitumor responses. Using in vitro and in vivo models, we investigated the molecular pathways underlying the CTLA-4-mediated differentiation of IL-17-producing CD8+ T cells (Tc17 cells) that strongly impairs cytotoxicity. Our studies demonstrate that Tc17 cells lacking CTLA-4 signaling have limited production of STAT3-target gene products such as IL-17, IL-21, IL-23R and RORγt. Upon re-stimulation with IL-12, these cells display fast downregulation of Tc17 hallmarks and acquire Tc1 characteristics such as IFNγ and TNF-α co-expression, which is known to correlate with tumor control. Indeed, upon adoptive transfer, these cells were highly efficient in the antigen-specific rejection of established OVA-expressing B16 melanoma in vivo. Mechanistically, in primary and re-stimulated Tc17 cells, STAT3 binding to the IL-17 promoter was strongly augmented by CTLA-4, associated with less binding of STAT5 and reduced relative activation of STAT1 which is known to block STAT3 activity. Inhibiting CTLA-4-induced STAT3 activity reverses enhancement of signature Tc17 gene products, rendering Tc17 cells susceptible to conversion to Tc1-like cells with enhanced cytotoxic potential. Thus, CTLA-4 critically shapes the characteristics of Tc17 cells by regulating relative STAT3 activation, which provides new perspectives to enhance cytotoxicity of antitumor responses.
KEYWORDS: CTL, IFNγ, IL-17, plasticity, STATs, T cell differentiation
Introduction
Differentiated CD8+ T cells play a crucial role in host protection, as they are the main effector cells that eliminate infected and malignant cells in an antigen (Ag)-specific manner. Encounters with endogenous antigens on antigen-presenting cells (APCs), either from intracellular pathogens or tumor neoantigens, orchestrate naive CD8+ T cell activation and differentiation into effector cells, mainly into cytotoxic T lymphocytes (CTLs). CTLs directly kill their target cells by releasing the cytotoxic molecules granzyme B and perforin into the immunological synapse.1,2 CTLs that express IL-2, IFNγ, and/or TNF-α, called Tc1 cells, are generated upon stimulation in a pro-inflammatory milieu containing IL-12/IFNγ. These cells can overcome immunosuppressive tumor escape mechanisms in tumor-bearing mice.3 In mice and humans, Tc1 cells co-expressing two or three of their signature cytokines are correlated with reduced bacterial load and tumor burden.4-6 Nevertheless, depending on the activation conditions, a cytokine milieu containing transforming growth factor β (TGF-β) and IL-67,8 drives CD8+ T cells to differentiate into IL-17-producing Tc17 cells, which have only limited cytotoxic activity.9
In addition to TCR and co-stimulation-mediated signals, a third signal provided by cytokine receptor engagement with its respective cytokine, strongly determines the outcome of T cell differentiation as well as the stability of the initial phenotype. Cytokine engagement with its receptor induces the phosphorylation of STAT molecules, which translocate into the nucleus and bind to the promoters of their target genes.10 For Tc17 cells, IL-6/IL-6R mediates the activation of pSTAT3, which binds to the promoters of Tc17 signature genes, including the transcription factor RORγt and the cytokine IL-17. STAT3 also induces the transcription of IL-21 and IL-23R, further increasing pSTAT3 expression and ultimately leading to stabilization of the Tc17 phenotype.9,11-13 T cells lacking STAT3 do not express the Tc17 hallmarks IL-17 and RORγt.14 The cytokines IL-2 and IFNγ, which strongly induce STAT5 and STAT1, are known to prevent the differentiation of IL-17-producing T cells. In the nucleus, STAT3 and STAT5 compete for binding to the IL-17 promoter, leading to gene activation or silencing, respectively.14,15 As STAT1 and STAT3 inhibit each other, relatively higher expression of STAT3 than STAT1 is required for the optimal activation of the transcriptional machinery for Tc17-related genes.16,17
Tc17 cells have been shown to be tumor-promoting cells, but they can also contribute to tumor rejection.18-20 The first sight controversy about the role of Tc17 cells in tumor biology might occur because Tc17 cells easily convert into Tc1-like cells in a pro-inflammatory environment, which rescues their killing capacity while maintaining some of their Tc17 characteristics, such as cell longevity.21,22 Indeed, some studies have shown that adoptive transfer of tumor-specific Tc17 cells into tumor-bearing mice induces potent antitumor activity in an IFNγ-dependent manner. Furthermore, adoptively transferred Tc17 cells were plastically converted into Tc1-like cells, improving the long-term cure rates in mice.20,21,23,24 Thus, the factors that determine Tc17 plasticity or stability remain of major interest to improve antitumor therapies.
The first target with reported effectiveness in immune checkpoint therapy25,26 was CTLA-4 (CD152), a glycoprotein expressed on the surface of activated T cells.27-28 On CD8+ T cells, it is expressed at particularly high levels and for long period of time.29 Blockade of CTLA-4 in patients having advanced melanoma is effective as a single agent for antitumor therapy30-32 and is even better in combination with agents that block other inhibitory molecules.33,34 Focusing on the major effector cells that eliminate tumors, CTLA-4-deficient CD8+ T cells exhibited increased production of IFNγ and granzyme B.29,35 In a Tc17 milieu, CTLA-4 supports Tc17 differentiation and limits the lineage plasticity of the Tc17 phenotype by inhibiting the expression of central factors of the Tc1 program, namely, IFNγ and granzyme B.36 This indicates that CTLA-4 affects the Tc1 and Tc17 lineage programs. Surprisingly, restoration of the downregulated factors, such as RORγt and IRF4 which are key to Tc17 cells, did not rescue the Tc17 program in CTLA-4-deficient cells.36 Therefore, the CTLA-4-mediated mechanism of Tc17 differentiation and plasticity is not understood so far.
In the present study, we report that CTLA-4 regulates Tc17 differentiation and stability by controlling STAT3 binding to the IL-17 promoter. Using ChIP assays, the DNA binding capacities of STAT3 and STAT5 were assessed in Tc1 and Tc17 conditions; there was more binding of STAT3 than STAT5 to the IL-17 promoter following CTLA-4 signaling. Consequently, blockade of STAT3 activation hampered the effect of CTLA-4 on Tc17 differentiation. Looking at the signaling network, the results demonstrate that two distinct mechanisms are mediated by CTLA-4. In the presence of a Tc17 cytokine milieu, CTLA-4 enhances STAT3 activation, whereas Tc17 cells exposed to a Tc1 environment (IL-2 and IL-12) exhibits restricted STAT1 activation. Indeed, in tumor-bearing mice, CTLA-4−/− Tc17 cells exhibit plasticity and differentiate into Tc1-like cells that were highly efficient at controlling tumor growth. These studies extend the knowledge of how CTLA-4 controls CD8+ T cell differentiation and plasticity, namely by altering the third signal of T cell activation.
Results
STAT3 activity is enhanced by CTLA-4 in Tc17 cells
In our previous studies, overexpression of RORγt and IRF4 in CTLA-4−/− Tc17 cells did not rescue IL-17 production, indicating that neither RORγt nor IRF4 alone is responsible for the CTLA-4-mediated effect on IL-17 production.36 To investigate the molecular mechanism of CTLA-4-mediated effects on Tc17 differentiation (Fig. 1A, left and right panels),36 we used an in vitro differentiation model that excluded extrinsic signals from B7 ligands on APCs by crosslinking cells with immobilized anti-CD3, anti-CD28 and anti-CTLA-4 antibodies (Ab) (Fig. 1B).37,38 As expected, CD8+ T cells that were crosslinked with anti-CD3, anti-CD28 and anti-CTLA-4 (+agon. (agonistic) αCTLA-4) displayed a 4-fold increase in the frequency of IL-17-producing cells (23.4%) compared with cells that were engaged with anti-CD3, anti-CD28 and isotype control antibody (αCD3) (4.8%) (Fig. 1B). Although the increase in CD28 concentration enhanced the frequency of IL-17-producing cells, the cells that were treated additionally with agonistic αCTLA-4 still had a significantly increased frequency of IL-17 producers (αCD3 32.6% vs. +agon. αCTLA-4 43.3%) (Fig. 1B). In the absence of CD28 signals, CTLA-4 signaling still resulted in a 3-fold increase in the frequency of IL-17 producers on day 3 (Fig. 1C, left and right panels). All further experiments in this study were performed with similar concentrations of immobilized anti-CD3 and anti-CTLA-4 (+agon. αCTLA-4) or Isotype (αCD3) (as in Fig. 1C). Similar expression of the activation-induced surface molecules CD44, CD25 and CD69 between the conditions excluded the possibility of differences in activation (Fig. 1D).
Figure 1.
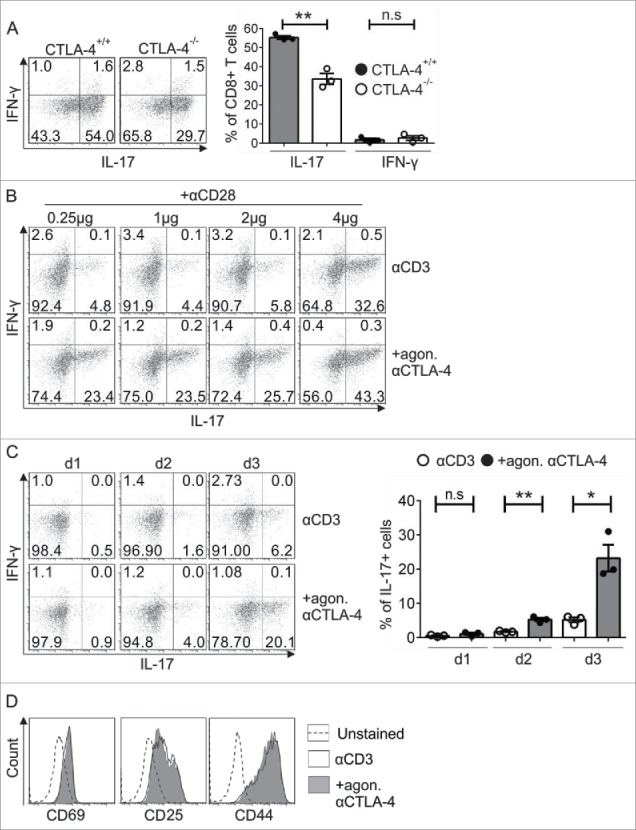
Analysis of the exclusive role of CTLA-4 in Tc17 differentiation. (A) Naive CD8+ T cells from CTLA-4+/+ and CTLA-4−/− OT.1 mice were activated with the specific antigen OVA257–264 in the presence of APCs under Tc17 conditions. IL-17 and IFNγ expression in these cells was analyzed by flow cytometry for 72 h after primary stimulation (left). Cumulative staining results are shown on the right. The data are representative of three independent experiments. (B) CD8+ T cells from C57BL/6JRj mice were stimulated under Tc17 conditions by crosslinking the cells with plate-bound immobilized anti-CD3 (3 μg/mL) and anti-CD28 (0.25–4 µg/mL) in the presence (+agon. αCTLA-4) or absence (αCD3) of immobilized anti-CTLA-4 (10 μg/mL). Three days after the primary stimulation, IL-17 expression in these cells was analyzed by flow cytometry. The data are from one representative experiment. (C) IL-17 and IFNγ expression in CD3-stimulated (3 μg/mL) cells in the presence or absence of CTLA-4 crosslinking (10 μg/mL) was analyzed by flow cytometry every day until day 3. Cumulative staining results are shown on the right. The data are representative of three independent experiments. (D) CD8+ T cells from C57BL/6JRj mice were cultured as in (C) and analyzed for the surface expression of CD69, CD25 and CD44 on day 3 by flow cytometry. The data are from a single experiment that is representative of three independent experiments. The error bars denote ± SEM. **p < 0.01, *p < 0.05, n.s.: not significant, unpaired t-test.
To identify target molecules of CTLA-4 signal transduction, we analyzed the lysates of CTLA-4+/+ and CTLA-4−/− Tc17 cells (that had been stimulated for 3 d) using a peptide array (PepChip) with different kinase consensus substrates spotted in triplicate. Analysis of the kinomic profile revealed substantial differences in the phosphorylation of kinase substrates in the presence and absence of the CTLA-4 signal. Interestingly, increased phosphorylation of STAT3 and decreased phosphorylation of STAT1 consensus substrates were observed in lysates of CTLA-4+/+ Tc17 cells, whereas the phosphorylation of the STAT5 consensus substrate was similar under both conditions (Fig. 2A).
Figure 2.
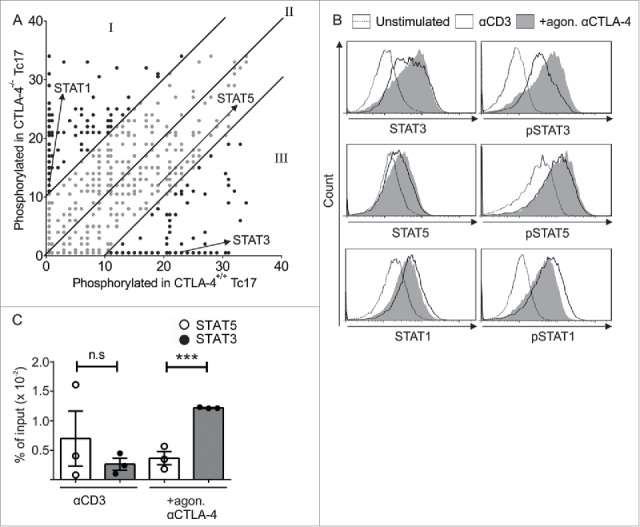
CTLA-4-regulated STAT phosphorylation determines Tc17 differentiation. (A) Dot plot representing the phosphorylation status of the kinase-specific peptide substrates spotted on the PepChip array. Different kinase activities in the lysates from the CTLA-4+/+ and CTLA-4−/− Tc17 cells are shown using a ranking method; each spot represents the extent of phosphorylation of a specific peptide substrate. Using the ranking method, a bisymmetric distribution of peptides is generated, in which phosphorylation was either significantly increased or decreased by CTLA-4 signaling. Peptide substrates that were phosphorylated in the absence of CTLA-4 signaling are reflected by an equivalent peptide with altered or unaltered phosphorylation in response to CTLA-4 signaling. The ranks of the differentially phosphorylated peptides of interest (STAT3, STAT5 and STAT1) are marked by arrows. Spots representing peptides with significantly decreased (I), increased (III) or unaltered (II) phosphorylation as a result of CTLA-4 signaling are shown. (B) Tc17 cells were stimulated with anti-CD3 in the presence or absence of additional CTLA-4 crosslinking for 3 d, harvested, and analyzed for the expression of total and phosphorylated STAT3, STAT1 and STAT5. The data are from a single experiment that is representative of two independent experiments. (C) ChIP analysis of Tc17 cells that were stimulated with CD3 in the presence or absence of additional CTLA-4 crosslinking for 3 d. Tc17 cells were stimulated with IL-6+IL-23 or IL-2 for 30 min, and protein–DNA complexes were crosslinked with formaldehyde and immunoprecipitated with anti-STAT3 or anti-STAT5. The bound DNA was purified and amplified by quantitative PCR with primers designed for the IL-17a promoter site. The results are presented relative to the input DNA. The data are representative of three independent experiments. The error bars denote ± SEM. ***p < 0.001, n.s.: not significant, unpaired t-test.
To support the PepChip results, the altered phosphorylation levels of the target proteins were analyzed by flow cytometry (Fig. 2B). As STAT3 binds to the IL-17a promoter to initiate its transcription, and also competes with STAT5 binding which interferes with IL-17 transcription,14,15 we evaluated the binding of both STAT proteins using chromatin immunoprecipitation (ChIP) assays followed by quantitative PCR (qPCR). STAT3 binding to the IL-17 promoter was diminished, whereas STAT5 binding was elevated in cells that lacked CTLA-4 engagement. In contrast, STAT3 binding to the IL-17 promoter was increased 3-fold compared with STAT5 in the cells that were crosslinked with CTLA-4 Ab (Fig. 2C).
Next, we determined the impact of CTLA-4-induced STAT3 activity on Tc17 differentiation by inactivating STAT3 with S31–201 a chemical probe inhibitor, which selectively blocks STAT3 phosphorylation, dimerization, DNA binding and STAT3-dependent transcription. Flow cytometric analysis of the CTLA-4 targets revealed that the 2-fold increase in the expression of the Tc17 signature transcription factor RORγt (+agon. αCTLA-4 DMSO) was significantly reduced in cells crosslinked with CTLA-4 upon STAT3 inactivation (+agon. αCTLA-4 S31–201), similar to the expression observed for IL-17 (Figs. 3A, B, left and right panels). In addition, CTLA-4 mediated a significant increase in IL-23R expression, which was reduced by more than half upon STAT3 inactivation (Fig. 3C).
Figure 3.
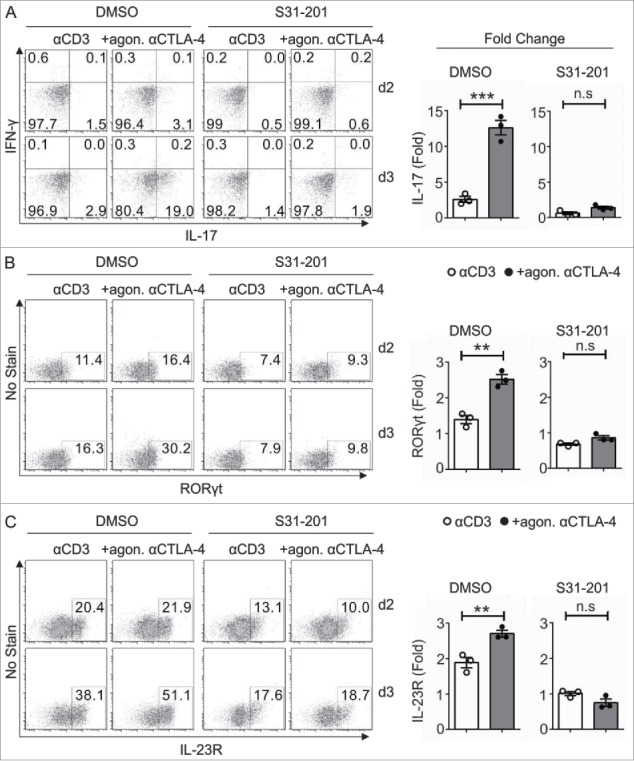
Impact of STAT3 on CTLA-4-mediated Tc17 differentiation. CD8+ T cells from C57BL/6JRj mice primed under Tc17 conditions with anti-CD3 in the presence or absence of additional CTLA-4 crosslinking were untreated (DMSO) or treated with the STAT3 inhibitor (S31–201) 24 h after stimulation. The expression levels of the Tc17 signature molecules RORγt (A) IL-17, IFNγ (B) and IL-23R (C) were analyzed by flow cytometry at the indicated time points (left), and the d3 results are shown as the fold increase compared with the d2 control cells (right). The data are from a single experiment that is representative of three independent experiments. The error bars denote ± SEM. ***p < 0.001, **p < 0.01, n.s.: not significant, unpaired t-test.
Taken together, these results suggest that CTLA-4 promotes STAT3 activation and binding to the IL-17 promoter region and thus enhances IL-17 expression in Tc17 cells. Consequently, the data also show that CTLA-4 induces the expression of many proteins encoded by STAT3-dependent target genes, including IL-23R, which is essential for maintaining the Tc17 phenotype (Fig. 3C).
CTLA-4 restricts the cytotoxic function of Tc17 cells
Considering the above-reported induction of STAT3 activity by CTLA-4, we hypothesized that CTLA-4-deficient Tc17 cells which cannot efficiently upregulate STAT3 activity, strongly support tumor rejection in vivo. For this purpose, adoptively transferred CTLA-4+/+ and CTLA-4−/− Tc17 cells were analyzed for their capacity to control the progression of pre-established melanoma by B16 OVA257–264-expressing melanoma cells in a mouse model. Recipient tumor-bearing mice on a Ly5.1 background were used to differentiate adoptively transferred congenic OT.1 CD45.2 CD8+ T cells. Tumor progression was measured for up to 6 d following adoptive T cell transfer (Fig. 4A). In PBS-treated tumor-bearing mice tumor outgrowth was progressing dramatically. In mice receiving adoptively transferred T cells, tumor growth measurements clearly showed that CTLA-4−/− Tc17 cells significantly restricted tumor progression, whereas CTLA-4+/+ Tc17 cells were not able to control the tumor growth (d2 to d6, Fig. 4B and C). In fact, CTLA-4−/− Tc17 cells displayed a 2-fold higher efficiency in controlling tumor progression than did CTLA-4+/+ Tc17 cells. Additionally, the in vivo re-stimulated Tc17 cells showed enhanced expression of Tc1-like characteristics; for example, a 4-fold higher frequency of IFNγ/TNF-α double producers was observed in CTLA-4−/− Tc17 cells compared with CTLA-4+/+ Tc17 cells (Fig. 4D). These kind of double producers are well known to control tumor progression in mice and men.5,6,39,40 Collectively, these results show that CTLA-4 deficiency in vivo or absence of CTLA-4 signals in vitro enhances the functional and transcriptional plasticity of Tc17 cells and thus profoundly augments their antitumor activity.
Figure 4.
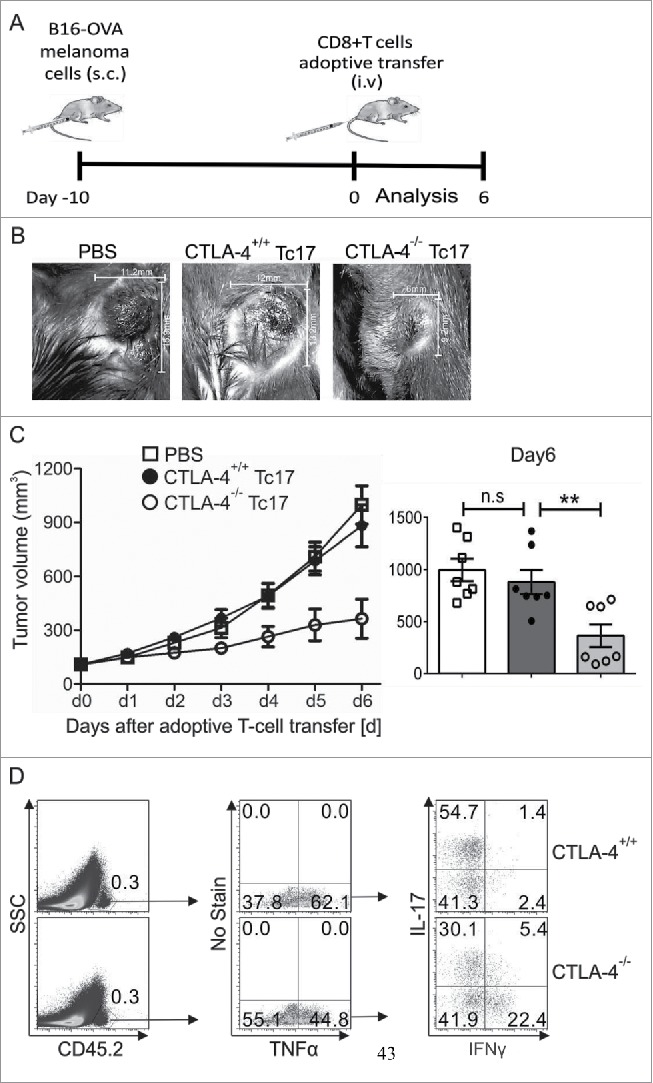
CTLA-4 regulates the cytotoxic activity of Tc17 cells. (A) Schematic of the tumor experiment. Recipient Ly5.1 mice were s.c. injected with B16-OVA melanoma cells. Approximately 10 d later, when a visible tumor was present, CTLA-4+/+ and CTLA-4−/− OT.1 CD8+ T cells that had been stimulated under Tc17 conditions for 3 d were adoptively transferred into the recipient mice through intravenous (i.v.) injection, and tumor growth was measured for the next 6 d. (B) Pictorial representation of tumor size in the recipient mice on day 6 after adoptive transfer with PBS or CTLA-4+/+ or CTLA-4−/− OT.1 Tc17 cells. (C) Tumor growth in the mice receiving PBS or CTLA-4+/+ or CTLA-4−/− OT.1 Tc17 cells was measured on a daily basis until day 6. Results represent ± SEM of seven mice per group from three independent experiments. Cumulative bar graphs of tumor volume in the recipient mice on day 6 are shown on the right. Results represent ± SEM of seven mice per group from three independent experiments. (D) Adoptively transferred CD45.2+ cells were surface stained ex vivo in the splenocytes of the tumor-bearing mice 6 d after the transfer of CTLA-4+/+ or CTLA-4−/− OT.1 Tc17 cells and were analyzed for TNF-α, IL-17 and IFNγ production by flow cytometry. The data are from one representative experiment. The error bars denote ± SEM. **p < 0.01, n.s.: not significant, Mann–Whitney U-test.
CTLA-4-mediated effects on STATs stabilize Tc17 differentiation
Next, we hypothesized that CTLA-4 signaling involving STATs might be responsible for the resistance of Tc17 cells to convert to Tc1-like cells. Initially, downstream targets of the Tc1 lineage were analyzed to confirm the Tc1 conversion, as it is well established that inhibiting CTLA-4 enhances Tc1 differentiation.29,35 First, flow cytometric analysis showed that the CTLA-4-crosslinked cells more effectively retained their Tc17 profile, even under Tc1-skewing conditions, compared with the activated control cells (Fig. 5A). Additionally, a 2-fold decrease in the mRNA levels of the Tc17-supporting factors HIF-1α and IRF4 was observed in the activated control cells (αCD3) (Fig. 5B). The mRNA expression of the main Tc1-supporting transcription factor Eomes, but not that of T-bet, was elevated in the re-activated control cells that did not receive a CTLA-4 signal. Flow cytometric analysis extended these mRNA findings to the respective proteins (Fig. 5C). In addition, similar results were observed with other Tc1-supporting molecules, such as IFNγ (Fig. 5B). Together, these results indicate that Eomes is a major downstream target of CTLA-4 signaling in Tc17 cell plasticity.
Figure 5.

Impact of CTLA-4 on Tc17 stability and plasticity. (A) Tc17 cells that had been stimulated for 3 d (as in Fig. 1C) were washed twice with medium and stimulated again with fresh plate-bound immobilized anti-CD3 in the presence or absence of additional crosslinking with anti-CTLA-4 coupled with IL-12 and IL-2 cytokines. Twenty-four hours later, IL-17 and IFNγ expression in these cells was determined using flow cytometry (left). Cumulative staining results are shown on the right. The data are representative of four independent experiments. (B) Tc17 cells were harvested on day 3 after primary stimulation (considered as 0 h before re-stimulation), re-activated as described above, and harvested at the indicated time points. The harvested cells were lysed, RNA was extracted, and RT was used to synthesize cDNA. The expression levels of the indicated genes relative to the housekeeping gene, HPRT, are shown as the mean ± SEM of duplicates from a single experiment that is representative of three experiments. (C) Tc17 cells were stimulated as in (B) and analyzed for the expression of Eomes and T-bet by flow cytometry at the indicated time points. The data are from a single experiment that is representative of 2 independent experiments. The error bars denote ± SEM. ***p < 0.001, **p < 0.01, *p < 0.05, n.s.: not significant, unpaired t-test.
In accordance with the recognized importance of STAT3 in CTLA-4-mediated Tc17 differentiation, we next evaluated the role of STAT3 in controlling CTLA-4-mediated Tc17 plasticity. Although STAT3 and STAT5 phosphorylation remained the same after re-stimulation with Tc1-inducing cytokines in the presence or absence of CTLA-4 crosslinking (Fig. 6A), ChIP followed by qPCR showed a 3-fold increase in STAT5 binding to the IL-17 promoter in cells that were not crosslinked with CTLA-4 Ab. In contrast, a 2-fold increase in STAT3 binding to the IL-17 promoter was identified in the cells that were crosslinked with CTLA-4 Ab (Fig. 6B). Studies have shown that the relative protein abundance of STAT1 versus STAT3 may reciprocally influence the activity of the other protein.16,17 Therefore, we measured STAT1 phosphorylation. Interestingly, a significant increase in STAT1 phosphorylation was observed in the cells that were not exposed to CTLA-4 crosslinking (Fig. 6A). The normalized pSTAT3 and pSTAT1 levels were used to compare the pSTAT3/pSTAT1 ratio. Tc17 cells exposed to CTLA-4 crosslinking displayed a 2-fold increase in the pSTAT3/pSTAT1 ratio compared with control cells (Fig. 6C).
Figure 6.

CTLA-4-regulated STAT phosphorylation determines Tc17 plasticity. (A) Tc17 cells were re-activated in a Tc1 environment for 24 h (as described in Fig. 5A), harvested, and analyzed for the expression of the total and phosphorylated forms of STAT3, STAT1 and STAT5. The data are from a single experiment that is representative of two independent experiments. (B) ChIP analysis of Tc17 cells that were re-stimulated with Tc1-inducing cytokines for 24 h. The re-activated Tc17 cells were stimulated with IL-6+IL-23 or IL-2 for 30 min, and the protein–DNA interactions were crosslinked with formaldehyde and immunoprecipitated with anti-STAT3 or anti-STAT5. The bound DNA was purified and amplified by quantitative PCR with primers designed for the IL-17a promoter site. The results are presented relative to the input DNA. The data are representative of three independent experiments. (C) STAT3 and STAT1 phosphorylation levels in the re-stimulated cells were normalized to those in unstimulated cells, and the pSTAT3/pSTAT1 ratio was compared among cells that were crosslinked with anti-CD3 in the presence or absence of CTLA-4. The data are representative of three independent experiments. (D) Tc17 cells were harvested on day 3 after primary stimulation (considered 0 h before re-stimulation), re-stimulated as described in (A), and harvested at the indicated time points. The harvested cells were lysed, RNA was extracted, and RT was used to synthesize cDNA. The expression levels of the indicated genes relative to the housekeeping gene HPRT are shown as the mean ± SEM of duplicates from a single experiment that is representative of three experiments. (E, F) Tc17 cells were stimulated as in (D) and analyzed for the expression of (E) RORγt and (F) IL-23R by flow cytometry at the indicated time points. The data are representative of three independent experiments. (G) 3-d-cultured Tc17 cells in the presence (S31–201) or absence (DMSO) of STAT3 inhibitor were re-stimulated as in (A) and analyzed for the expression of degranulation-associated surface molecule CD107a on CD8+ T cell. The data are from a single experiment that is representative of three independent experiments. The error bars denote ± SEM. ***p < 0.001, **p < 0.01, *p < 0.05, n.s.: not significant, unpaired t-test.
As CTLA-4 has a potential binding site for STATs, direct binding to the phosphorylated YVKM motif of CTLA-4 was analyzed. Therefore, we performed a peptide pulldown experiment with 18O/16O labeling of proteins in lysates from anti-CD3/CD28 stimulated Tc17 cells. As baits, phosphorylated and non-phosphorylated peptides comprising the YVKM sequence were used. The previously shown phosphorylation specific binding of p85 subunit of PI3K to the CTLA-4 peptide,41 was identified with a 18O/16O (phosphorylated/non-phosphorylated peptide) ratio of > 100, indicating the accuracy of the 18O labeling method to track proteins binding to the phosphorylated CTLA-4. Analysis of the peptide bound proteins to CTLA-4-bait revealed that neither STAT1 nor STAT3 directly associate with the phosphorylated cytoplasmic tail of CTLA-4.
The increased mRNA expression of the Tc17-supporting factors IL-17, RORc, IL-21 and IL-23R in the qPCR analysis (Fig. 6D) correlated well with the relative increase in the amount of pSTAT3 in cells that were crosslinked with CTLA-4 Ab (Fig. 6C). In support of the qPCR data, we also detected increased RORγt and IL-23R expression by flow cytometry in the cells that underwent CTLA-4 crosslinking (Figs. 6E and F). Considering the previous reported observation that inactivation of STAT3 enhances the cytotoxicity of CD8+ T cells,42,43 we next evaluated the role of STAT3 in regulating the cytotoxic potential of Tc17 cells which get a CTLA-4 signal. For this purpose, expression of a degranulation marker CD107a was analyzed which is well known to determine cytolytic activity of CD8+ T cells.44 Flow cytometric analysis showed that the diminished expression of CD107a in CTLA-4-crosslinked Tc17 cells restimulated under Tc1 environment was significantly enhanced upon STAT3 inactivation (Fig. 6G). Collectively, we report for the first time that CTLA-4 mediates an increase in the relative amount of pSTAT3 which helps to stabilize Tc17 differentiation and hamper cytotoxic activity of Tc17 cells.
Discussion
This is the first study to report that CTLA-4 regulates the third signal for CD8+ T cell activation, namely, cytokine receptor signaling, by affecting STAT molecules. In the current study, we found that CTLA-4 stabilizes Tc17 differentiation and increases the resistance of Tc17 cells to plasticity by increasing the relative amount of pSTAT3 compared with pSTAT1 and pSTAT5 (which are known to curb Tc17 differentiation). Ultimately, STAT3-activated target molecules, such as RORγt, IL-17, IL-21 and IL-23R, were strongly expressed.
Our data show that CTLA-4 signaling in CD8+ T cells specifically enhances the expression of IL-17 and Tc17-related molecules in a STAT3-dependent manner irrespective of IL-1β synergizing effect in Tc17 differentiation (Fig. S1) in combination with IL-6, IL-23 and TGF-β. Consequently, STAT3 inactivation reversed the stimulatory effect of CTLA-4 on Tc17 differentiation. Our data from CTLA-4-deficient T cells agree with the phenotype of STAT3-deficient T cells, which exhibit defective RORc, IL-21 and IL-17 expression.45-47 Previously, it was assumed that ectopic overexpression of RORγt or IRF4, which are essential for Tc17 cells, would rescue the diminished IL-17 expression in CTLA-4-incompetent cells. However, this hypothesis was not confirmed,36 indicating that the effects of CTLA-4 on these downstream transcription factors do not govern the increase in Tc17 differentiation. Our data show that STAT3 manipulation alone reverses the effects of CTLA-4, likely by inhibiting a signaling knot upstream of RORγt, IL-23R, IL-17 and other molecules. Even though our 18O-labeling analysis reveal that CTLA-4 does not directly associate with STAT1, a close interaction is still conceivable, as upon T cell activation CTLA-4, IFNGR and STAT1 are expressed in a polarized manner in the immunological synapse.48 Thus, CTLA-4 could negatively regulate the IFN-induced STAT1 phosphorylation by phosphatases such as SHP2 recruited to its cytoplasmic tail.49,50
Tc17 cells are known to be phenotypically unstable.21,22,51 Here, we show that CTLA-4 plays a central role in controlling Tc17 stability in vitro and in vivo (Figs. 5A and 4D). In vivo, CTLA-4−/− Tc17 cells displayed a significant ability to control tumor progression (Figs. 4B and C). Indeed, adoptively transferred CTLA-4−/− Tc17 cells gave rise to an increased number of IFNγ/TNF-α co-producers (Fig. 4D), which are known to correlate with tumor rejection.24 In addition absence of CTLA-4 signal also displayed a significantly enhanced CD107a expression (Fig. 6G) correlating well with enhanced cytotoxic activity against tumors.44 It is likely that this mechanism occurs in response to anti-CTLA-4 therapy (Ipilimumab) in melanoma patients.5,6 In addition, strategies that enhance the plasticity of Tc17 cells in tumor-bearing subjects and the development of Tc1-like cytokine patterns have been shown to result in stronger antitumor immunity due to increased cell persistence and cytotoxicity.20,21 Efforts to inhibit Tc17 cell activity are likely to result in a valuable strategy for treating cancer;52 indeed, CTLA-4 blockade is well known to augment the antitumor activity of T cells in patients with advanced melanoma.32 Unwanted IL-17 production by continuous CTLA-4 signaling may be the major contributor to the pathological effects of Tc17 cells, not only in the tumor environment but also in autoimmunity.53-55
The CTLA-4-crosslinked cells exhibited prolonged expression of the Tc17 hallmarks and diminished upregulation of the Tc1 markers implicating that CTLA-4 promotes Tc17 differentiation and inhibits Tc1-like factors. According to our data, this mechanism involves regulating both STAT1 and STAT3, whereas STAT5 is only indirectly involved. Although STAT5 activation was not affected by CTLA-4, more STAT5 bound to the IL-17 promoter in cells lacking CTLA-4 signal, indicating that it was sufficient to outcompete the binding of low amounts of relative STAT3 (pSTAT3/pSTAT1 ratio) to the IL-17 promoter (Figs. 6B and C). CTLA-4-mediated induction of the Tc17 program by default is unlikely, as absence of T-bet and Eomes would have been an obligatory prerequisite,56 but CTLA-4 only downregulates Eomes (Figs. 5B and C). However, STAT1-knockout CD4+ T cells have been shown to strongly upregulate IL-17 production independent of T-bet and Eomes manipulation.17,57 Therefore, the CTLA-4-mediated downregulation of STAT1 is likely an intrinsic inducer of IL-17 expression. This assumption is supported as even in the absence of Tc17 stimulatory conditions CTLA-4 strongly downregulates STAT1 concomitantly with IL-17 upregulation (Figs. 6A and D). Indeed, individuals with inactivating mutations of STAT1 have a severely impaired capacity (often lethal in children) to mount immune responses against the intracellular pathogens like mycobacteria and viruses.58 Thus, for antitumor responses CTLA-4-mediated STAT1 inhibition is wanted to be prevented, especially as STAT1-induced expression of Eomes59 is needed for granzyme B upregulation and cytotoxicity.1
STAT3 is known to stabilize Tc17 phenotype.9 Relatively more STAT3 binds to the IL-17 promoter than STAT5, regardless of whether the CTLA-4-induced Tc17 cells are re-stimulated under Tc1 conditions (Fig. 6B). However, similar levels of STAT3 and STAT5 phosphorylation were detected. The initial discrepancy is explained by the fact that STAT molecules regulate each other. STAT3 activation is enhanced in STAT1-null cells, and conversely, STAT1 activation is enhanced in STAT3-null cells.16,60-63 Although our studies did not show a difference in STAT3 phosphorylation during re-stimulation in a Tc1 environment, STAT1 phosphorylation was significantly altered. As STAT3 and STAT1 have different functions and even oppose each other, their ratio determines the Tc17 cell phenotype.17 Therefore, the higher pSTAT3/pSTAT1 ratio in the CTLA-4-crosslinked Tc17 cells, which were re-stimulated in a Tc1 environment, explains the CTLA-4-mediated stability of Tc17 differentiation. The increased mRNA expression of the STAT3 target molecules IL-17, IL-23R, IL-21 and RORc in these cells further strengthens the role of the relative amount of pSTAT3 in stabilizing Tc17 differentiation. STATs can also have indirect effects on one another; for instance, STATs regulate SOCS3, which in turn impedes Tc17 cell differentiation.64 However, SOCS3 mRNA expression was upregulated by no more than 1.5-fold in the absence of CTLA-4 signals (data not shown). The effect of CTLA-4-STAT3 axis may also be mediated by RORγt expression, but RORγt overexpression on its own did not reverse suppressed IL-17 production.36
In addition to the reported regulation of Tc17 plasticity by STATs and its impact on tumor biology, these results also show that the surface molecule CTLA-4 regulates the third signal for T cell differentiation, namely, cytokine receptor signaling, which is an important feature in the decision-making process that determines the type of T cell differentiation. Although we focused on Tc17 cells, this relationship between CTLA-4 and STAT1 or STAT3 is likely to impact other STAT1- or STAT3-producing lineages, such as Tc1 cells, T-helper subpopulations and Treg cells, and is likely to influence various immune settings, making this pathway acutely relevant in the context of immune checkpoint therapy and cytokine-based drug design.
Materials and methods
Mice and cell line
All animal experiments were performed under license approved from Landesverwaltungsamt Sachsen-Anhalt in Halle. C57BL/6JRj mice were obtained from Janvier laboratories, Ly5.1, CTLA-4+/+ and CTLA-4−/− OT.1 (OVA specific TCRtg) mice have been described previously.36 All mice were bred under pathogen-free conditions following institutional guidance, at the central animal facility of University clinic of Magdeburg (Germany). Sex- and age-matched mice were used for all experiments. All mice have been backcrossed for more than 15 generations to the C57BL/6JRj strain. Efforts were put to minimize stress and suffering of the animals used for in vivo experiments. Animals used for the experiments were killed by cervical dislocation. The OVA-transfected B16 tumor cell line (B16-OVA) was described previously.65 Cells were maintained in RPMI 1640, supplemented with 10% heat-inactivated FCS, 25 mM HEPES, 1 mM Sodium pyruvate (Sigma Aldrich), 50 µM 2-ME and 1 mg/mL G418 (Carl Roth).
Antibodies and reagents
The following fluorescently labeled Ab against murine Ags were used: anti-TCR Vα2 (B20.1), anti-CD90.2, anti-CD45.2 (104), anti-CD25, anti-CD69, anti-TNFα, anti-IFNγ (XMG1.2), (all from BD Biosciences), anti-Eomes, anti-RORγt, anti-pSTAT5 (all from eBioscience), anti-CD8α (53–6.7), anti-IL-17 (Tc11–18H10), anti-T-bet, anti-IL-23R, anti-CD44 and anti-CD62L (all from Biolegend), anti-STAT1, anti-pSTAT1, anti-STAT3, anti-pSTAT3 and anti-STAT5 (all from Cell signaling). Control antibody donkey anti-rabbit IgG was purchased from Biolegend. STAT3 inhibitor S31–201 was purchased from Merck Millipore.
T-cell differentiation
Spleens, inguinal, axillary and mesenteric lymph nodes of CTLA-4+/+ and CTLA-4−/− OT.1 mice were collected to obtain naive CD8+ T-cells (CD8+ CD62Lhigh). Naive CD8+ T-cells were isolated to a purity of ≥ 98% by magnetic beads separation using AutoMACSpro (Miltenyi Biotec). Comparable levels of CD44, CD69 and CD62L expression were routinely found using flow cytometery. For Ag-specific activation CD8+ T cells were stimulated with 0.5 μg/mL of LPS-free SIINFEKL (OVA257–264) peptide (Invivogen) and CD90-depleted splenocytes from C57BL/6JRj mice at a ratio of 4:1. For Ab-specific agonistic stimulation, CD8+ T-cells isolated from spleen, inguinal, axillary and mesenteric lymph nodes of C57BL/6JRj mice were used. Cells were stimulated with plate bound immobilized anti-CD3 (3 μg/mL), anti-CD28 (Biolegend) (0.25–4 µg/mL) and anti-CTLA-4 (4F10) (10 μg/mL) or Isotype (BD Biosciences) Ab. All cells were cultured in serum free x-vivo 15 medium (Lonza). For Tc17 differentiation the cells were conditioned with 2 ng/mL TGF-β, 10 ng/mL IL-6 (R&D systems), 25 ng/mL IL-23, 5 ng/mL IL-1β (Biolegend) and 10 μg/mL anti-IFNγ XMG.1.2 (DRFZ, Berlin). For Tc1 differentiation, the cells were primed with 5 ng/mL IL-12 and 1 ng/mL IL-2 (Biolegend). To determine Tc17 plasticity, primary stimulated Tc17 cells were re-stimulated with fresh plate bound immobilized anti-CD3 (Biolegend) in the presence or absence of anti-CTLA-4 and were conditioned with 5 ng/mL IL-12 and 1 ng/mL IL-2.
Flow cytometry, surface and intracellular staining
The cells were harvested and stained with the indicated surface markers in PBS/0.2% BSA. Prior to intracellular cytokine analysis, the cells were re-stimulated with PMA, ionomycin and Brefeldin A for 4 h. Intracellular staining was performed after the cells were fixed with 2% paraformaldehyde (Merck) in PBS for 20 min on ice and permeabilized in 0.5% saponin (Sigma-Aldrich) in PBS/BSA. The transcription factors were measured by fixing the cells with 4% formaldehyde (Carl Roth) in PBS for 10 min at 37°C, followed by permeabilization in ice-cold 90% methanol (Carl Roth) for 30 min. For detection of STATs, Tc17 cells were treated with IFNγ (STAT1) or IL-2 (STAT5) or IL-6+IL-23 (STAT3) or IL-1β (STAT3, Fig. S1) for the last 10 min. Variations in FACS analyses were corrected by normalizing the measurements and considering the MFI of unstimulated cells as the basal level. All cytometric analyses were performed using a FACS-Canto II™ (BD Biosciences) and FlowJo™ software (FlowJo LLC).
Adoptive T cell transfer and melanoma model
CD45.1 (Ly5.1) C57BL/6 mice received a subcutaneous (s.c.) injection into the right flank with 2×105 B16-OVA melanoma cells in PBS. Approximately 10 d after the tumor cell injection, mice that had developed a substantial tumor (∼100 mm3) received an i.v. injection with either PBS or in vitro generated CD45.2-expressing CTLA-4+/+ or CTLA-4−/− OT.1 Tc17 cells. Tumor growth was then monitored on a daily basis. Mice with large tumors were humanely killed. Adoptively transferred CD8+ CD45.2+ cells were analyzed by flow cytometry as described above.
Kinome array analysis
For kinome array, CTLA-4+/+ and CTLA-4−/− Tc17 cells that had been cultured for 3 d were washed twice with PBS and lysed in complete lysis buffer. The protein concentration in the cell lysate was determined using a BCA assay. A 10-µL activation mix containing 50% glycerol, 50 mM MgCl2, 50 mM MnCl2, 0.25 mg/mL PEG 8000, 0.25 mg/mL bovine serum albumin and 2,000 µCi/mL [γ-33P]ATP was added to 90 µL of cell lysate to ascertain kinase activity. The peptide arrays, which contain 1,024 different kinase pseudo-substrates in triplicate (Pepscan), were incubated with the activation mix and the cell lysate for 2 h in a humidified chamber at 37°C. Subsequently, the arrays were washed with each of the following solutions: PBS containing 1% Triton X-100, 1% SDS in demineralized and distilled water. The slides were air-dried and exposed to a phosphoimaging screen for 72 h.
Data acquisition and analysis of PepChip array
The data on the phosphoimaging screen were acquired using a Phosphoimager (GE Healthcare Lifesciences) and quantified using ScanAlyze software (Leland Stanford Junior University). Subsequently, the data were exported to a spreadsheet program (Microsoft Excel 2010; Microsoft Co.). The spot densities were corrected for the individual backgrounds to diminish interarray variance. The variation between arrays and individual experiments was reduced by normalizing the data to the 99th percentile of the intensity of each array. The averaged spots were included in dissimilarity measurements using a ranking method to identify peptides with either significantly increased or decreased phosphorylation.
Pulldown experiments and mass spectrometric analysis
The peptides CSPLT TGV (p)YVKMPPTEPESEKQFQPYFIPIN with the indicated tyrosine either phosphorylated or non-phosphorylated were used in the pulldown experiments to profile the phosphorylation-dependent interaction partners. The serine in the sequence was introduced instead of cysteine in the original sequence to allow for cysteine-mediated covalent coupling to the beads. Thirty million CTLA4−/− CD8+ T cells were lysed and the soluble fraction of the lysate was incubated with the peptide beads before tryptic on-bead digest. The digest was performed either in 18O or 16O water with swapped labels in the two replicate experiments regarding the phosphorylated or unphosphorylated peptide bait. LC-MS analysis was subsequently performed on an Orbitrap LTQ XL machine and data was analyzed by Mascot Distiller.
Chromatin immunoprecipitation (ChIP)
Tc17 cells were stimulated with either the STAT3-inducing cytokines IL-6 and IL-23 or the STAT5-inducing cytokine IL-2 for 30 min, and the protein–DNA complexes were crosslinked with 1% formaldehyde. The cells were then lysed by sonication, and the protein–DNA complexes were immunoprecipitated with magnetic μMACS™ Protein G MicroBeads (Miltenyi Biotech) coated with either an anti-STAT3 antibody or an anti-STAT5 antibody (Cell Signaling Technology). The magnetic immune complexes were passed through a separation column placed in the magnetic field of a MACS Separator (Miltenyi Biotech). The labeled complexes were retained on the column, and the other proteins were efficiently washed away. The immunoprecipitated protein–DNA complex was eluted from the column using elution buffer, and the protein–DNA crosslinks were reversed at 65°C overnight. The DNA was then purified from the sample, eluted and analyzed by quantitative PCR with custom designed primers (Table S2) using a CFX96™ Real-Time PCR detection system (Bio-Rad). The Ct value for each sample was normalized to the corresponding input value.
Real-time RT-PCR
RNA was isolated from re-stimulated Tc17 cells at the indicated time points using the NucleoSpin RNA/Protein Isolation Kit (Macherey-Nagel) and was reverse transcribed using the Applied Biosystems Reverse Transcription Kit. The cDNAs were stored at –20°C. Gene expression was analyzed using Fermentas Maxima™ (Thermo Scientific) SYBR Green qPCR Master Mix on a CFX96™ Real-Time PCR detection system (Bio-Rad). HPRT was used as a control. Primer pairs for quantitative real-time PCR were purchased from TIB MOLBIOL (primer sequences are shown in Table S1).
Statistics
Data were analyzed by Microsoft Excel 2010 (Microsoft Co.) and Prism 6 (Graphpad software Inc.). Data are presented as ± SEM. p-values are computed by unpaired Student's t-test or Mann–Whitney U-tests. ***p < 0.001, **p < 0.01, *p < 0.05, n.s: not significant.
Supplementary Material
Disclosure of potential conflicts of interest
The authors declare no financial or commercial conflict of interest.
Acknowledgement
We thank Dr E. Krause (FMP Berlin) for help with the MS experiments.
Funding
The study was supported by Sonderforschungsbereich (SFB) 854 TP 14 (MCBW) and DFG Br1860/11.
References
- 1.Pearce EL, Mullen AC, Martins GA, Krawczyk CM, Hutchins AS, Zediak VP, Banica M, DiCioccio CB, Gross DA, Mao CA et al.. Control of effector CD8+ T cell function by the transcription factor eomesodermin. Science 2003; 302:1041-43; PMID:14605368; http://dx.doi.org/ 10.1126/science.1090148 [DOI] [PubMed] [Google Scholar]
- 2.Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, Mullen AC, Gasink CR, Kaech SM, Miller JD et al.. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol 2005; 6:1236-44; PMID:16273099; http://dx.doi.org/ 10.1038/ni1268 [DOI] [PubMed] [Google Scholar]
- 3.Nguyen HH, Kim T, Song SY, Park S, Cho HH, Jung S, Ahn J, Kim H, Lee J, Kim H et al.. Naive CD8(+) T cell derived tumor-specific cytotoxic effectors as a potential remedy for overcoming TGF-beta immunosuppression in the tumor microenvironment. Sci Rep 2016; 6:28208; PMID:27306834; http://dx.doi.org/ 10.1038/srep28208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pedicord VA, Montalvo W, Leiner IM, Allison JP. Single dose of anti-CTLA-4 enhances CD8+ T-cell memory formation, function, and maintenance. Proc Natl Acad Sci U S A 2011; 108:266-71; PMID:21173239; http://dx.doi.org/ 10.1073/pnas.1016791108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yuan J, Adamow M, Ginsberg BA, Rasalan TS, Ritter E, Gallardo HF, Xu Y, Pogoriler E, Terzulli SL, Kuk D et al.. Integrated NY-ESO-1 antibody and CD8+ T-cell responses correlate with clinical benefit in advanced melanoma patients treated with ipilimumab. Proc Natl Acad Sci U S A 2011; 108:16723-28; PMID:21933959; http://dx.doi.org/ 10.1073/pnas.1110814108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yuan J, Gnjatic S, Li H, Powel S, Gallardo HF, Ritter E, Ku GY, Jungbluth AA, Segal NH, Rasalan TS et al.. CTLA-4 blockade enhances polyfunctional NY-ESO-1 specific T cell responses in metastatic melanoma patients with clinical benefit. Proc Natl Acad Sci U S A 2008; 105:20410-15; PMID:19074257; http://dx.doi.org/ 10.1073/pnas.0810114105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 2006; 24:179-89; PMID:16473830; http://dx.doi.org/ 10.1016/j.immuni.2006.01.001 [DOI] [PubMed] [Google Scholar]
- 8.Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-β induces development of the T H17 lineage. Nature 2006; 441:231-34; PMID:16648837; http://dx.doi.org/ 10.1038/nature04754 [DOI] [PubMed] [Google Scholar]
- 9.Huber M, Heink S, Grothe H, Guralnik A, Reinhard K, Elflein K, Hünig T, Mittrücker H, Brüstle A, Kamradt T et al.. A Th17-like developmental process leads to CD8(+) Tc17 cells with reduced cytotoxic activity. Eur J Immunol 2009; 39:1716-25; PMID:19544308; http://dx.doi.org/ 10.1002/eji.200939412 [DOI] [PubMed] [Google Scholar]
- 10.Delgoffe GM, Vignali DAA. STAT heterodimers in immunity: A mixed message or a unique signal? JAKSTAT 2013; 2:e23060; PMID:24058793; http://dx.doi.org/ 10.4161/jkst.23060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol 2007; 8:967-74; PMID:17581537; http://dx.doi.org/ 10.1038/ni1488 [DOI] [PubMed] [Google Scholar]
- 12.Korn T, Bettelli E, Gao W, Awasthi A, Jäger A, Strom TB, Oukka M, Kuchroo VK. IL-21 initiates an alternative pathway to induce proinflammatory T H17 cells. Nature 2007; 448:484-87; PMID:17581588; http://dx.doi.org/ 10.1038/nature05970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM et al.. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature 2007; 448:480-83; PMID:17581589; http://dx.doi.org/ 10.1038/nature05969 [DOI] [PubMed] [Google Scholar]
- 14.Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan F, Siegel R, Hennighausen L et al.. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity 2007; 26:371-81; PMID:17363300; http://dx.doi.org/ 10.1016/j.immuni.2007.02.009 [DOI] [PubMed] [Google Scholar]
- 15.Yang X, Ghoreschi K, Steward-Tharp SM, Rodriguez-Canales J, Zhu J, Grainger JR, Hirahara K, Sun H, Wei L, Vahedi G et al.. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol 2011; 12:247-54; PMID:21278738; http://dx.doi.org/ 10.1038/ni.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dimberg LY, Dimberg A, Ivarsson K, Fryknäs M, Rickardson L, Tobin G, Ekman S, Larsson R, Gullberg U, Nilsson K et al.. Stat1 activation attenuates IL-6 induced Stat3 activity but does not alter apoptosis sensitivity in multiple myeloma. BMC Cancer 2012; 12:318; PMID:22838736; http://dx.doi.org/ 10.1186/1471-2407-12-318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peters A, Fowler KD, Chalmin F, Merkler D, Kuchroo VK, Pot C. IL-27 induces Th17 differentiation in the absence of STAT1 signaling. J Immunol 2015; 195:4144-53; PMID:26408664; http://dx.doi.org/ 10.4049/jimmunol.1302246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nam J, Terabe M, Kang M, Chae H, Voong N, Yang Y, Laurence A, Michalowska A, Mamura M, Lonning S et al.. Transforming growth factor beta subverts the immune system into directly promoting tumor growth through interleukin-17. Cancer Res 2008; 68:3915-23; PMID:18483277; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-0206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kryczek I, Wei S, Zou L, Altuwaijri S, Szeliga W, Kolls J, Chang A, Zou W. Cutting edge: Th17 and regulatory T cell dynamics and the regulation by IL-2 in the tumor microenvironment. J Immunol 2007; 178:6730-33; PMID:17513719; http://doi.org/ 10.4049/jimmunol.178.11.6730 [DOI] [PubMed] [Google Scholar]
- 20.Hinrichs CS, Kaiser A, Paulos CM, Cassard L, Sanchez-Perez L, Heemskerk B, Wrzesinski C, Borman ZA, Muranski P, Restifo NP. Type 17 CD8+ T cells display enhanced antitumor immunity. Blood 2009; 114:596-99; PMID:19471017; http://dx.doi.org/ 10.1182/blood-2009-02-203935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bowers JS, Nelson MH, Kundimi S, Bailey SR, Huff LW, Schwartz KM, Cole DJ, Rubinstein MP, Paulos CM. Dendritic cells in irradiated Mice trigger the functional plasticity and antitumor activity of adoptively transferred Tc17 cells via IL12 signaling. Clin Cancer Res 2015; 21:2546-57; PMID:25904754; http://dx.doi.org/ 10.1158/1078-0432.CCR-14-2294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yen HR, Harris TJ, Wada S, Grosso JF, Getnet D, Goldberg MV, Liang KL, Bruno TC, Pyle KJ, Chan SL et al.. Tc17 CD8 T cells: functional plasticity and subset diversity. J Immunol 2009; 183:7161-68; PMID:19917680; http://dx.doi.org/ 10.4049/jimmunol.0900368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu Y, Cho H, Wang D, Kaosaard K, Anasetti C, Celis E, Yu X. Adoptive transfer of Tc1 or Tc17 cells elicits antitumor immunity against established melanoma through distinct mechanisms. J Immunol 2013; 190:1873-81; PMID:23315072; http://dx.doi.org/ 10.4049/jimmunol.1201989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garcia-Hernandez Maria de la Luz, Hamada H, Reome JB, Misra SK, Tighe MP, Dutton RW. Adoptive transfer of tumor-specific Tc17 effector T cells controls the growth of B16 melanoma in mice. J Immunol 2010; 184:4215-27; PMID:20237297; http://dx.doi.org/ 10.4049/jimmunol.0902995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12:252-64; PMID:22437870; http://dx.doi.org/ 10.1038/nrc3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol 2015; 33:1974-82; PMID:25605845; http://dx.doi.org/ 10.1200/JCO.2014.59.4358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, Baker J, Jeffery LE, Kaur S, Briggs Z et al.. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 2011; 332:600-03; PMID:21474713; http://dx.doi.org/ 10.1126/science.1202947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev 2009; 229:12-26; PMID:19426212; http://dx.doi.org/ 10.1111/j.1600-065X.2009.00770.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pandiyan P, Hegel JK, Krueger M, Quandt D, Brunner-Weinzierl MC. High IFN-γ production of individual CD8 T lymphocytes is controlled by CD152 (CTLA-4). J Immunol 2007; 178:2132-40; PMID:17277117; http://doi.org/ 10.4049/jimmunol.178.4.2132 [DOI] [PubMed] [Google Scholar]
- 30.Egen JG, Kuhns MS, Allison JP. CTLA-4: new insights into its biological function and use in tumor immunotherapy. Nat Immunol 2002; 3:611-18; PMID:12087419; http://dx.doi.org/ 10.1038/ni0702-611 [DOI] [PubMed] [Google Scholar]
- 31.Sandin LC, Eriksson F, Ellmark P, Loskog AS, Totterman TH, Mangsbo SM. Local CTLA4 blockade effectively restrains experimental pancreatic adenocarcinoma growth in vivo. Oncoimmunology 2014; 3:e27614; PMID:24701377; http://dx.doi.org/ 10.4161/onci.27614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC et al.. Improved survival with ipilimumab in patients with metastatic melanoma. New Engl J Med 2010; 363:711-23; PMID:20525992; http://dx.doi.org/ 10.1056/NEJMoa1003466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS et al.. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 2015; 372:2006-17; PMID:25891304; http://dx.doi.org/ 10.1056/NEJMoa1414428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dai M, Yip YY, Hellstrom I, Hellstrom KE. Curing mice with large tumors by locally delivering combinations of immunomodulatory antibodies. Clin Cancer Res 2015; 21:1127-38; PMID:25142145; http://dx.doi.org/ 10.1158/1078-0432.CCR-14-1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hegel JK, Knieke K, Kolar P, Reiner SL, Brunner-Weinzierl MC. CD152 (CTLA-4) regulates effect or functions of CD8+ T lymphocytes by repressing Eomesodermin. Eur J Immunol 2009; 39:883-93; PMID:19224637; http://dx.doi.org/ 10.1002/eji.200838770 [DOI] [PubMed] [Google Scholar]
- 36.Pick J, Arra A, Lingel H, Hegel JK, Huber M, Nishanth G, Jorch G, Fischer K, Schlüter D, Tedford K et al.. CTLA-4 (CD152) enhances the Tc17 differentiation program. Eur J Immunol 2014; 44:2139-52; PMID:24723371; http://dx.doi.org/ 10.1002/eji.201343497 [DOI] [PubMed] [Google Scholar]
- 37.Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med 1995; 182:459-65; PMID:7543139; http://dx.doi.org/ 10.1084/jem.182.2.459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakaseko C, Miyatake S, Iida T, Hara S, Abe R, Ohno H, Saito Y, Saito T. Cytotoxic T lymphocyte antigen 4 (CTLA-4) engagement delivers an inhibitory signal through the membrane-proximal region in the absence of the tyrosine motif in the cytoplasmic tail. J Exp Med 1999; 190:765-74; PMID:10499915; http://doi.org/ 10.1084/jem.190.6.765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wimmers F, Aarntzen EHJG, Duiveman-deBoer T, Figdor CG, Jacobs JFM, Tel J, Vries IJM de. Long-lasting multifunctional CD8+ T cell responses in end-stage melanoma patients can be induced by dendritic cell vaccination. Oncoimmunology 2016; 5:e1067745; PMID:26942087; http://dx.doi.org/ 10.1080/2162402X.2015.1067745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seder RA, Darrah PA, Roederer M. T-cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol 2008; 8:247-58; PMID:18323851; http://dx.doi.org/ 10.1038/nri2274 [DOI] [PubMed] [Google Scholar]
- 41.Schneider H, Prasad KV, Shoelson SE, Rudd CE. CTLA-4 binding to the lipid kinase phosphatidylinositol 3-kinase in T cells. J Exp Med 1995; 181:351-55; PMID:7807015; http://dx.doi.org/ 10.1084/jem.181.1.351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Herrmann A, Priceman SJ, Swiderski P, Kujawski M, Xin H, Cherryholmes GA, Zhang W, Zhang C, Lahtz C, Kowolik C et al.. CTLA4 aptamer delivers STAT3 siRNA to tumor-associated and malignant T cells. J Clin Invest 2014; 124:2977-87; PMID:24892807; http://dx.doi.org/ 10.1172/JCI73174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kujawski M, Zhang C, Herrmann A, Reckamp K, Scuto A, Jensen M, Deng J, Forman S, Figlin R, Yu H. Targeting STAT3 in adoptively transferred T cells promotes their in vivo expansion and antitumor effects. Cancer Res 2010; 70:9599-610; PMID:21118964; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rubio V, Stuge TB, Singh N, Betts MR, Weber JS, Roederer M, Lee PP. Ex vivo identification, isolation and analysis of tumor-cytolytic T cells. Nat Med 2003; 9:1377-82; PMID:14528297; http://dx.doi.org/ 10.1038/nm942 [DOI] [PubMed] [Google Scholar]
- 45.Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, Dong C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem 2007; 282:9358-63; PMID:17277312; http://dx.doi.org/ 10.1074/jbc.C600321200 [DOI] [PubMed] [Google Scholar]
- 46.Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, Ma L, Shah B, Panopoulos AD, Schluns KS et al.. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity 2008; 28:29-39; PMID:18164222; http://dx.doi.org/ 10.1016/j.immuni.2007.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wei L, Laurence A, Elias KM, O'shea JJ. IL-21 is produced by Th17 cells and drives IL-17 production in a STAT3-dependent manner. J Biol Chem 2007; 282:34605-10; PMID:17884812; http://dx.doi.org/ 10.1074/jbc.M705100200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maldonado RA, Soriano MA, Perdomo LC, Sigrist K, Irvine DJ, Decker T, Glimcher LH. Control of T helper cell differentiation through cytokine receptor inclusion in the immunological synapse. J Exp Med 2009; 206:877-92; PMID:19349465; http://dx.doi.org/ 10.1084/jem.20082900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.You M, Yu DH, Feng GS. Shp-2 tyrosine phosphatase functions as a negative regulator of the interferon-stimulated Jak/STAT pathway. Mol Cell Biol 1999; 19:2416-24; PMID:10022928; http://doi.org/ 10.1128/MCB.19.3.2416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baron M, Davignon JL. Inhibition of IFN-γ-induced STAT1 tyrosine phosphorylation by human CMV is mediated by SHP2. J Immunol 2008; 181:5530-36; PMID:18832710; http://doi.org/ 10.4049/jimmunol.181.8.5530 [DOI] [PubMed] [Google Scholar]
- 51.Tajima M, Wakita D, Satoh T, Kitamura H, Nishimura T. IL-17/IFN-γ double producing CD8+ T (Tc17/IFN-γ) cells: a novel cytotoxic T-cell subset converted from Tc17 cells by IL-12. Inter Immunol 2011; 23:751-59; PMID:22039016; http://dx.doi.org/ 10.1093/intimm/dxr086 [DOI] [PubMed] [Google Scholar]
- 52.Zhuang Y, Peng L, Zhao Y, Shi Y, Mao X, Chen W, Pang KC, Liu X, Liu T, Zhang J et al.. CD8(+) T cells that produce interleukin-17 regulate myeloid-derived suppressor cells and are associated with survival time of patients with gastric cancer. Gastroenterology 2012; 143:951-62.e8; PMID:22710190; http://dx.doi.org/ 10.1053/j.gastro.2012.06.010 [DOI] [PubMed] [Google Scholar]
- 53.Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, Fugger L. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol 2008; 172:146-55; PMID:18156204; http://dx.doi.org/ 10.2353/ajpath.2008.070690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hu Y, Ma D, Shan N, Zhu Y, Liu X, Zhang L, Yu S, Ji C, Hou M. Increased number of Tc17 and correlation with Th17 cells in patients with immune thrombocytopenia. PLoS One 2011; 6:e26522; PMID:22039505; http://dx.doi.org/ 10.1371/journal.pone.0026522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ciric B, El-behi M, Cabrera R, Zhang GX, Rostami A. IL-23 drives pathogenic IL-17-producing CD8+ T cells. J Immunol 2009; 182:5296-305; PMID:19380776; http://dx.doi.org/ 10.4049/jimmunol.0900036 [DOI] [PubMed] [Google Scholar]
- 56.Intlekofer AM, Banerjee A, Takemoto N, Gordon SM, Dejong CS, Shin H, Hunter CA, Wherry EJ, Lindsten T, Reiner SL. Anomalous type 17 response to viral infection by CD8+ T cells lacking T-bet and eomesodermin. Science 2008; 321:408-11; PMID:18635804; http://dx.doi.org/ 10.1126/science.1159806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Villarino AV, Gallo E, Abbas AK. STAT1-activating cytokines limit Th17 responses through both T-bet-dependent and -independent mechanisms. J Immunol 2010; 185:6461-71; PMID:20974984; http://dx.doi.org/ 10.4049/jimmunol.1001343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi C, Zaid Al-Mohsen I, Al-Jumaah S, Yang K, Chapgier A, Eidenschenk C, Eid P et al.. Impaired response to interferon-α/β and lethal viral disease in human STAT1 deficiency. Nat Genet 2003; 33:388-91; PMID:12590259; http://dx.doi.org/ 10.1038/ng1097 [DOI] [PubMed] [Google Scholar]
- 59.Martinet V, Tonon S, Torres D, Azouz A, Nguyen M, Kohler A, Flamand V, Mao C, Klein WH, Leo O et al.. Type I interferons regulate eomesodermin expression and the development of unconventional memory CD8(+) T cells. Nat Commun 2015; 6:7089; PMID:25953241; http://dx.doi.org/ 10.1038/ncomms8089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Costa-Pereira AP, Tininini S, Strobl B, Alonzi T, Schlaak JF, Is'harc H, Gesualdo I, Newman SJ, Kerr IM, Poli V. Mutational switch of an IL-6 response to an interferon-γ-like response. Proc Natl Acad Sci U S A 2002; 99:8043-47; PMID:12060750; http://dx.doi.org/ 10.1073/pnas.122236099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Avalle L, Pensa S, Regis G, Novelli F, Poli V. STAT1 and STAT3 in tumorigenesis: A matter of balance. JAKSTAT 2012; 1:65-72; PMID:24058752; http://dx.doi.org/ 10.4161/jkst.20045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qing Y, Stark GR. Alternative activation of STAT1 and STAT3 in response to interferon-γ. J Biol Chem 2004; 279:41679-85; PMID:15284232; http://dx.doi.org/ 10.1074/jbc.M406413200 [DOI] [PubMed] [Google Scholar]
- 63.Ho HH, Ivashkiv LB. Role of STAT3 in type I interferon responses. Negative regulation of STAT1-dependent inflammatory gene activation. J Biol Chem 2006; 281:14111-18; PMID:16571725; http://dx.doi.org/ 10.1074/jbc.M511797200 [DOI] [PubMed] [Google Scholar]
- 64.Chen Z, Laurence A, Kanno Y, Pacher-Zavisin M, Zhu BM, Tato C, Yoshimura A, Hennighausen L, O'shea JJ. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc Natl Acad Sci U S A 2006; 103:8137-42; PMID:16698929; http://dx.doi.org/ 10.1073/pnas.0600666103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Klages K, Mayer CT, Lahl K, Loddenkemper C, Teng MWL, Ngiow SF, Smyth MJ, Hamann A, Huehn J, Sparwasser T. Selective depletion of Foxp3+ regulatory T cells improves effective therapeutic vaccination against established melanoma. Cancer Res 2010; 70:7788-99; PMID:20924102; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-1736 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.