

ABSTRACT
Purpose: Patients with regionally advanced melanoma were treated with neoadjuvant ipilimumab in a previously reported study (PLOS One 2014). Gene expression profiles of tumors of treated patients were investigated for their association with immunotherapeutic benefit. Methods: Patients were treated with ipilimumab (10 mg/kg intravenously every 3 weeks × 2 doses) before and after surgery. Tumor specimens were obtained at baseline and at definitive surgery (weeks 6–8). Gene expression profiling was performed on the tumor biopsies of 27 patients. The primary endpoint was mRNA expression profiling using U133A 2.0 Affymetrix gene chips. Significance analysis of microarrays was performed to test the association of each gene with outcome. Pathway analysis was performed using Ingenuity Pathway Analysis software. The Benjamini and Hochberg method was used to adjust for multiple testing in the pathway analysis. Results: Pathway analysis identified biologically relevant pathways enriched with genes that are significantly associated with clinical outcome at baseline in relation to relapse-free survival (RFS) and disease non-progression (as assessed preoperatively at week 6) as well as early on-treatment (RFS and overall survival). The molecules and pathways that achieved differential expression of highest statistical significance were notably immune related. Association of the gene signature with clinical outcome overlapped between baseline and on-treatment specimens and across clinical endpoints tested. Conclusion: Gene expression profiling identified a signature reflecting an immune active and proinflammatory tumor microenvironment that derived clinical benefit from neoadjuvant ipilimumab at baseline and early on-treatment. These findings warrant further investigation in relation to ipilimumab and other immunotherapeutics.
KEYWORDS: Cytotoxic T lymphocyte antigen-4, gene expression profiling, ipilimumab, melanoma, neoadjuvant
Introduction
Cytotoxic T lymphocyte antigen-4 (CTLA4) blockade with ipilimumab has demonstrated significant and durable clinical activity in the management of patients with metastatic melanoma. The MDX-1020, phase III study that lead to the FDA approval of ipilimumab at 3 mg/kg for advanced inoperable melanoma demonstrated significant overall survival (OS) prolongation in favor of ipilimumab as compared to the Gp100 peptide vaccine in previously treated patients.1 Later, the MDX-024 phase III trial showed that ipilimumab at 10 mg/kg plus dacarbazine has significant survival benefit over dacarbazine alone as first-line treatment in metastatic melanoma.2 More recently, a pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in metastatic melanoma showed a plateau in OS that began at approximately 3 y with follow-up to 10 y and demonstrated greater durability of survival relative to historical data.3
The current paradigm of adjuvant immunotherapy in melanoma involves the indiscriminate treatment of all patients clinically considered at high risk for melanoma recurrence and mortality. Among the 70,000 new cases of invasive melanoma diagnosed annually, about 30,000 will be surgically operable melanoma that are at intermediate to high risk for recurrence and death and for which adjuvant therapy is indicated.4 High-dose IFNα-2b (HDI) has established but limited efficacy as an adjuvant therapy and is associated with significant toxicity and high cost.5 Ipilimumab offers a potentially superior adjuvant therapy, with differing patterns of toxicity.6 In the Eastern Cooperative Oncology Group (ECOG) adjuvant trial E1684 testing HDI vs. observation, the 5-year relapse-free survival (RFS) and OS rates were 37% vs. 26% and 46% vs. 37%, respectively.7 However, we have no means to predict which patients will be among the roughly one-third who will benefit, and who will be among the two-thirds who could be spared from the adverse events and cost due to lack of predicted efficacy. Recently, EORTC 18071 trial reported significant reduction in the risk of relapse among patients with AJCC Stage III melanoma treated with adjuvant ipilimumab at 10 mg/kg, HR 0.75, 95% CI (0.64–0.90); p = 0.0013.8 The rate of grade 3/4 immune-related adverse events was 42% and drug-related grade 5 events occurred in 1.1%. In addition, E1609 trial is a randomized phase III trial that is testing adjuvant therapy with ipilimumab at 10 mg/kg and 3 mg/kg versus the standard HDI regimen in patients with surgically resectable Stages IIIB/C and M1a/b melanoma. This study completed subject accrual in August 2014, but the clinical outcome results are currently pending. Therefore, there is a need to identify baseline and/or early on-treatment predictive biomarker(s) capable of classifying patients according to the degree of benefit they will receive from treatment with ipilimumab, IFNα or the newer generation of immunotherapeutics.
We recently published the results of a clinical trial testing neoadjuvant ipilimumab in patients with regionally advanced but operable melanoma.9 Here, we investigated the tumor microenvironment of patients treated with neoadjuvant ipilimumab by conducting RNA microarray studies on tumor specimens obtained before and after neoadjuvant therapy. We hypothesized that an immune-related gene expression signature will be significantly associated with clinical benefit after neoadjuvant ipilimumab. We identified genes that were significantly differentially expressed in association with clinical benefit from neoadjuvant ipilimumab at baseline and early on-treatment. These genes were invariably immune related.
Results
Table 1 summarizes patient demographics and baseline disease characteristics of the 27 patients included in the gene expression profiling analysis.
Table 1.
Patient demographics and baseline disease characteristics (N = 27 patients).
| Variable | No. of patients (%) |
|---|---|
| Age, years; median (range) | 53 (40–87) |
| Cutaneous primary | 23 (85) |
| Mucosal primary | 3 (11) |
| Unknown primary | 1 (4) |
| Gender | |
| Female | 9 (33) |
| Male | 18 (67) |
| Performance status (ECOG) | |
| 0 | 19 (70) |
| 1 | 8 (30) |
| Recurrent disease after prior surgery | 21 (78) |
| Prior adjuvant HDI | 10 (37) |
| Presence of in-transit metastases | 15 (55) |
| Estimated risk stage | |
| IIIB | 3 (11) |
| IIIC | 24 (89) |
| Tumor mutational status | |
| BRAFV600 | 9 (33) |
| NRASQ61 | 7 (26) |
| NRAS R73 | 1 (4) |
| Unknown | 3 (11) |
HDI: high dose interferon-α; ECOG: Eastern Cooperative Oncology Group.
We selected the top molecules that were significantly differentially expressed at baseline by non-progression (NP), RFS and OS at a p-value of 0.05 and used these genes to conduct pathway analysis. Pathway analysis identified biologically relevant pathways enriched with genes that were significantly associated with clinical outcome at baseline in relation to RFS and NP as well as early on-treatment in relation to RFS and OS. These pathways and the top associated molecules were notably immune related and highly statistically significant. Associations with clinical outcome overlapped between baseline and on-treatment specimens as well as across the clinical endpoints tested. Table 2 summarizes the top canonical pathways identified at baseline (PRE) and on-treatment (POST) and their association with RFS, NP and OS.
Table 2.
Top canonical pathways identified at baseline (PRE) and on-treatment (POST) and their association with relapse free survival (RFS), disease non-progression (NP) and overall survival (OS).
| Pathways | PRE/RFS (Adjusted P) | PRE/NP (Adj. p) | POST/RFS (Adj. p) | POST/OS (Adj. p) |
|---|---|---|---|---|
| Antigen presentation | 0.0002 | 4.22 × 10−05 | 0.004 | 3.08 × 10−05 |
| Cytotoxic T lymphocyte-mediated apoptosis | 0.0004 | 3.92 × 10−07 | 0.004 | 0.0009 |
| T helper cell differentiation | 0.001 | 0.0005 | 0.05 | 0.023 |
| B cell development | 6.98 × 10−12 | 1.14 × 10−13 | 2.26 × 10−06 | 4.26 × 10−07 |
| iCOS-iCOSL signaling in T helper | 0.006 | 4.23 × 10−07 | 0.11 | 0.11 |
| OX40 signaling | 0.007 | 1.29 × 10−05 | 0.005 | 0.0002 |
| CD28 signaling in T helper cells | 0.04 | 8.3 × 10−05 | 0.04 | 0.15 |
| IL-4 signaling | 0.02 | 0.0008 | 0.06 | 0.002 |
| PKCθ signaling in T lymphocytes | 0.04 | 7.97 × 10−05 | 0.14 | 0.04 |
| Nur77 signaling in T lymphocytes | 0.03 | 0.0001 | 0.03 | 0.008 |
| SLE signaling | 1.52 × 10−05 | 4.27 × 10−06 | ||
| Allograft rejection signaling | 0.0003 | 4.27 × 10−06 | 0.004 | 0.0006 |
| Autoimmune thyroid signaling | 0.004 | 4.51 × 10−05 | 0.021 | 0.003 |
The genes that belonged to the top pathways were selected (22 genes, 49 probe sets). These genes are defined in Table 3. Time to event analysis was conducted utilizing the 49 probe sets corresponding to the 22 genes. Supervised Principle Components (SPC) were applied to the top genes (probe sets) that are associated with progression-free survival (PFS). The plots demonstrated the ability to separate the survival curves by dichotomized value of the first SPC. Log rank tests were used to compare the two groups of patients with different genomic profiles in each plot. For the baseline expression prediction of RFS, HR = 2.70, 95% CI for HR estimate (1.1, 6.8). Log rank test p-value = 0.029. For week 6 expression prediction of RFS, HR = 2.71, 95% CI for HR estimate (1.04, 7.04). Log rank test p-value = 0.034. These data are shown in Figs. 1 and 2.
Table 3.
The genes that overlapped among the top pathways significantly associated with clinical outcome (49 probe sets/22 genes).
| Gene ID | Gene description |
|---|---|
| HLA-DMA | Major histocompatibility complex, class II, DM alpha |
| HLA-DOA | Major histocompatibility complex, class II, DO alpha |
| CD79B | CD79b molecule, immunoglobulin-associated beta |
| IGH | Immunoglobulin heavy locus |
| IGKC | Immunoglobulin kappa constant |
| IGLC1 | Immunoglobulin lambda constant 1 (Mcg marker) |
| HLA-DQA1 | Major histocompatibility complex, class II, DQ alpha 1 |
| IGHM | Immunoglobulin heavy constant mu |
| CD79A | CD79a molecule, immunoglobulin-associated alpha |
| IGHD | Immunoglobulin heavy constant delta |
| CD3G | CD3g molecule, gamma (CD3-TCR complex) |
| CD3D | CD3d molecule, delta (CD3-TCR complex) |
| HLA-DPA1 | Major histocompatibility complex, class II, DP alpha 1 |
| GZMB | Granzyme B (granzyme 2, cytotoxic T-lymphocyte-associated serine esterase 1) |
| LAT | Linker for activation of T cells |
| VAV1 | vav 1 guanine nucleotide exchange factor |
| INPP5D | Inositol polyphosphate-5-phosphatase, 145 kDa |
| IL2RB | Interleukin 2 receptor, β |
| IGHG1 | Immunoglobulin heavy constant gamma 1 (G1m marker) |
| CIITA | Class II, major histocompatibility complex, transactivator |
| IL21R | Interleukin 21 receptor |
| STAT1 | Signal transducer and activator of transcription 1, 91 kDa |
Figure 1.
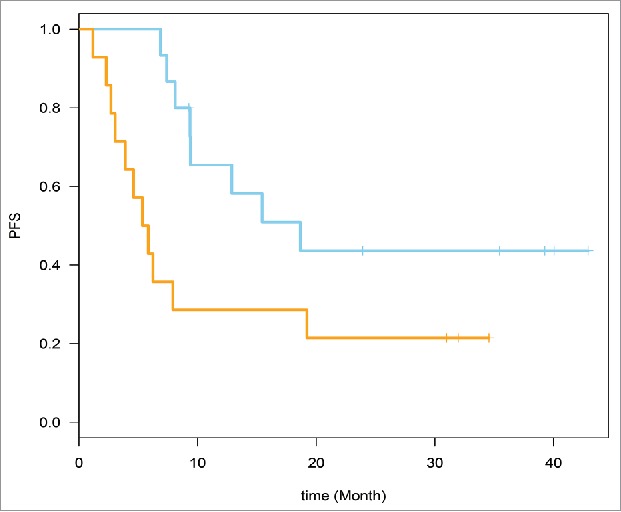
Time to event analysis was conducted. Supervised Principle Components (SPC) were applied to the top genes (probe sets) that are associated with relapse-free survival (RFS) (49 probe sets from the 22 genes). Baseline expression prediction of RFS is shown here. RFS, HR = 2.70, 95% CI for HR estimate (1.1, 6.8). Log rank test p-value = 0.029.
Figure 2.
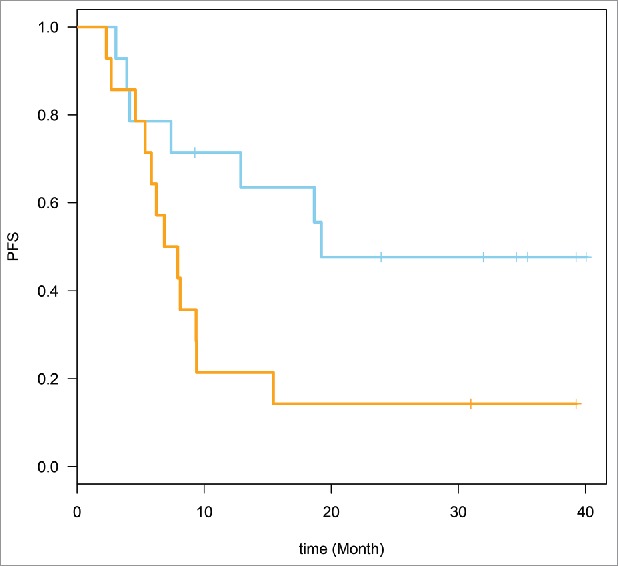
Week 6 expression prediction of relapse-free survival (RFS). HR = 2.71, 95% CI for HR estimate (1.04, 7.04). Log rank test p-value = 0.034.
We evaluated correlation with clinical outcome at 6–8 weeks (disease progression, PD vs. non-PD) as assessed clinically and radiologically preoperatively. Some probe sets were not significantly associated with PD vs. non-PD endpoint. For the PD vs. non-PD heatmap, we used only the probe sets that are significantly differentially expressed between the two groups. We ended up using 27 probe sets from 19 genes. Therefore, we generated a PD vs. non-PD heatmap using only the probe sets that are significantly differentially expressed between the two groups (27 probe sets from 19 genes). Hierarchical clustering (unsupervised) was performed and the PD group was found to have a lower expression of these genes. We observed that only one PD and one non-PD subjects clustered within the wrong group. Upon further investigation, we discovered that wrongly clustering non-PD subject actually had stable disease (SD) on the pre-operative imaging. However, the patient actually relapsed approximately 3 mo after surgery (4.5 mo after the initiation of neoadjuvant ipilimumab). On the other hand, the wrongly clustering PD subject had PD by preoperative PET-CT but was rendered disease free surgically and continues to be disease free on latest follow-up (over 4 y later). These results are shown in Fig. 3.
Figure 3.
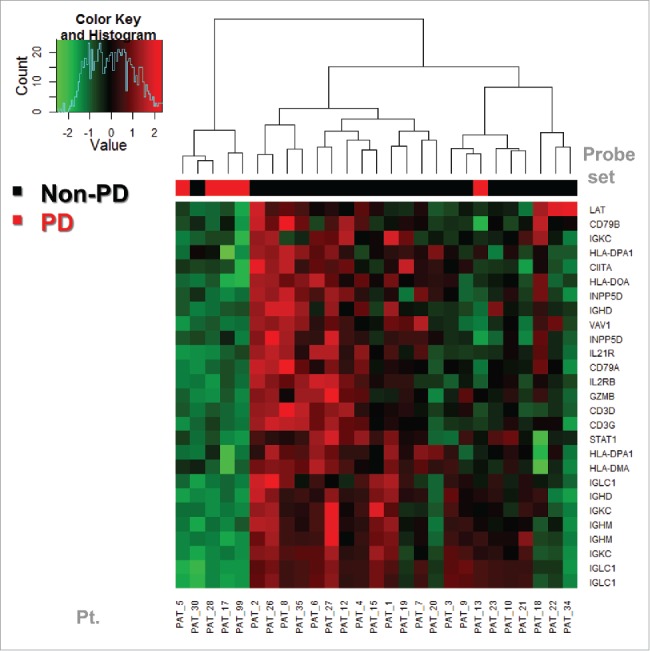
Baseline expression prediction of PD (disease progression as assessed at week 6 by PET-CT preoperatively) vs. non-PD. We evaluated correlation with PD vs. non-PD, using only the probe sets that are significantly differentially expressed between the two groups (27 probe sets from 19 genes). Hierarchical clustering was performed generating a heat map of low expression vs. high expression that correlated well with the PD vs. non-PD status of the patients. The PD group was found to have a lower expression of these genes. One PD and one non-PD subjects were in the wrong group, although by RFS they appeared to cluster within similar prognosis groups. These findings supported the investigation of relapse-free survival (RFS) as a more relevant clinical endpoint as shown in Fig. 4.
We noticed a sub-group within the non-PD group that has a genomic profile between the PD group and the other non-PD subgroup. Kaplan–Meier (KM) plots were applied to evaluate PFS of the three groups and showed that this subgroup (in gray) has slightly worse RFS than the others (in black) and both are superior to the low expression (red) group. These results are shown in Fig. 4.
Figure 4.
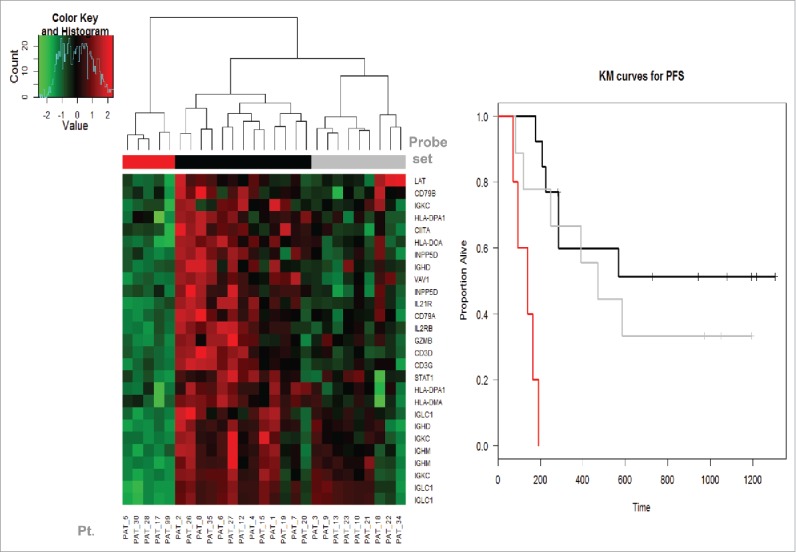
The Kaplan–Meier (KM) plot of RFS by the expression levels of PD (disease progression as assessed at week 6 by PET-CT preoperatively) and non-PD groups. We noticed a sub-group within the high expression (non-PD group) group that had a genomic expression profile in-between the very low expression (PD group) and the other high expression (non-PD subgroup). KM curve showed that this subgroup (in gray) has slightly worse RFS than the other high expression subgroup (in black). Both had significantly lower risk of relapse than the low expression (PD) group (in red).
Discussion
The recent advances in the field of immune checkpoint modulation and the unprecedented clinical activity in advanced melanoma opened the doors for novel agents and combinations that can potently overcome tumor tolerogenic mechanisms in multiple malignancies.10 The clinical management of advanced melanoma with anti-CTLA4 and anti-PD1 monoclonal antibodies has propelled the field into an era of long-term survival and potential cures. However, not all patients benefit from this form of therapy, and there is a need for predictive biomarkers that may allow treatment to be focused on those patients that have the capacity to respond while saving others from the unwanted toxicities and cost. CTLA4 is a member of the CD28:B7 immunoglobulin superfamily where low levels of expression are normally seen at the surface of regulatory T-cells (Tregs) and naive effector T-cells under normal conditions. CTLA-4 expression at the plasma membrane increases after stimulation of naive T cells through the binding of the MHC/antigen complex to T cell receptor and competes with CD28 for B7, leading to the downregulation of T cell receptor signaling.11 CTLA-4 signaling blockade through ipilimumab prolongs T-cell activation and restores T-cell proliferation, leading to enhanced antitumor T cell immunity.12,13 Clinical testing of ipilimumab in two pivotal phase III trials yielded significant results, leading to global regulatory approvals for the treatment of metastatic melanoma.1,2 Long-term survival benefit from ipilimumab was demonstrated in an analysis of 1861 melanoma patients treated in clinical trials where 21% were still alive at 3 y. Importantly, a survival plateau was observed with a maximum follow-up of about 10 y.6 Ipilimumab was approved in the US as adjuvant therapy based on the results of the EORTC 18071 trial that randomized stage III melanoma patients to receive ipilimumab at 10 mg/kg or placebo following complete surgical resection. After a median follow-up of 2.7 y, the study met its primary endpoint where 46.5% and 34.8% (p = 0.0013) of patients were relapse free in the ipilimumab and placebo arms, respectively.8 The US Intergroup E1609 trial is currently comparing standard HDI versus ipilimumab (at either 3 mg/kg or 10 mg/kg) in patients with surgically resected stage IIIB, IIIC, M1a and M1b melanoma and concluded accrual in August 2014.14
In the current study, we tested ipilimumab at 10 mg/kg in the neoadjuvant setting. We were able to collect tumor tissue at baseline and 6–8 weeks after the initiation of induction immunotherapy allowing for the RNA microarray studies at both time points. Our goal was to test the hypothesis that an immune-related gene expression profile will be significantly associated with clinical benefit using baseline and early on-treatment tumor specimens. Our analysis identified 22 genes that were invariably immune related and were significantly associated with clinical benefit. These genes were characteristic of a proinflammatory gene expression profile consisting of chemokines and other immune-related genes associated with a Type I immune active tumor microenvironment, antigen presentation and cytotoxic and helper T cell activity. Recent rapid advances in cancer genomics and proteomics highlight the importance of the proinflammatory gene expression profile as a potential predictor of immunotherapeutic benefit. Chemokine expression in melanoma metastases was reported to be important for CD8+ T-cell recruitment into the tumor microenvironment (TME). In an RNA microarray study of metastatic melanoma, Harlin et al. observed a major segregation of tumor samples based on the differential expression of T-cell-associated genes.15 More importantly, the expression of defined chemokine genes was significantly associated with the presence of tumor-infiltrating lymphocytes. Using protein array and/or quantitative reverse transcription-PCR, they confirmed that T-cell-infiltrated tumors preferentially expressed a subset of six chemokines (CCL2, CCL3, CCL4, CCL5, CXCL9 and CXCL10). Further, they demonstrated the upregulation of corresponding chemokine receptors on human CD8+ T-cells. These chemokines were shown to promote CD8+ T cell migration in vitro where migration of CD8+ T cells was inhibited through chemokine blockade with specific antibodies. These data suggest that specific chemokines are critical for T cell migration in melanoma metastases which could be essential for promoting cell-mediated antitumor immunity. Further, a proinflammatory gene expression signature was associated with survival following GSK MAGE-A3 protein vaccine in a phase II trial of immunization with recombinant MAGE-A3 protein using two different immune stimulants as an adjuvant (AS15 and AS02B). This study was conducted by GSK Biologicals in 72 patients with MAGE-A3-positive unresectable stage III or stage IV M1a metastatic melanoma. Microarray gene expression profiling was performed on baseline biopsies.16 A proinflammatory signature was found to be associated with a significant improvement in median OS; 16.2 mo in signature (–ve) versus 28 mo in signature (+ve) patient population. Quantitative reverse transcription PCR was later used to confirm the high expression of immune-related genes in patients with improved clinical outcome.16 These findings suggest that a subset of patients have pro-inflammatory infiltrates of T cells and chemokines in their tumors, making them more susceptible to immunotherapeutic interventions, a tumor vaccine in this case. Further, a phase II study in surgically resected stage IB or II non-small cell lung cancer evaluated the MAGE-A3 recombinant protein in 182 patients with MAGE-A3-positive tumors. Here again, increased expression of immune-related genes was associated with improved clinical activity after MAGE-A3 vaccination and these genes overlapped with the gene signature identified in the melanoma study.17 A Type I immune active tumor microenvironment may be required for therapeutic benefit from immunotherapy and could have a predictive value. A similar pro-inflammatory gene expression profile on pre-treatment biopsies may be associated with clinical response to high-dose interleukin-2 (IL2). Data presented at the 2009 ASCO Meeting reported a similar pro-inflammatory tumor microenvironment to be potentially predictive of IL-2 clinical responses.18 In patients with metastatic melanoma treated with ipilimumab, a prior study reported that a pro-inflammatory/immune-reactive tumor microenvironment is associated with clinical activity. In that phase II study that enrolled 45 patients, Affymetrix gene expression profiling was conducted on tumor biopsies obtained from before and 3 weeks after the start of treatment.19,20 High-baseline (pretreatment) expression of immune-related genes was significantly associated with clinical benefit from ipilimumab. The genes selected included CD8A, CD2, CD247, CD27, CD38, CD3 (T cell surface markers), CD40, FAS and TNFRSF9 (members of the TNF receptor family), CXCL9, CXCL10, CXCL11, CCL4 and CCL5 (chemokines), IL10RA, IL12RB2, IL15RA, IL21R, CXCR6 and CCR5 (immune receptors), cytotoxic factors, including perforin 1 and various granzymes, in addition to various types of immunoglobulin genes, T cell receptors and MHC molecules consistent with our findings in the neoadjuvant high-risk setting. The top functional pathways included inflammatory response, immune-cell trafficking, proliferation, and activation. 22 unique genes were selected in the baseline expression samples that had at least 2.5-fold difference in expression between the clinical benefit groups and most were associated with immune function. The selected list included genes encoding for Th1 chemokines (CCL4, CCL5, CXCL9, CXCL10 and CXCL11), CD8+ T cell surface markers (CD8A,GZMB, PRF1), HLA-DQA1 and other immune-related genes including IDO1, NKG7, CD38 and IGL. Investigating post-treatment samples, it was interesting to observe increased expression of genes involved in immune response, decreased expression of genes involved in cell proliferation and melanoma-specific antigens and genes. Further, there was evidence of increased expression of IFNγ-inducible genes and Th1-associated markers after ipilimumab, suggesting that ipilimumab may induce proinflammatory changes within the tumor microenvironment that mediate antitumor activity.
Together with our findings, these data point toward pro-inflammatory T cell immunity within the tumor microenvironment as a likely predictor of immunotherapeutic benefit in patients with advanced melanoma. They support the testing of the therapeutic predictive value of the pro-inflammatory tumor microenvironment reflected in the gene expression profile.
Conclusion
Gene expression profiling identified immune-related genes whose expression levels are significantly associated with clinical benefit from neoadjuvant ipilimumab at baseline and early on-treatment. These findings warrant further investigation in relation to ipilimumab therapeutic benefit and perhaps other immunotherapeutics.
Patients and methods
Patients
Patients with regionally advanced melanoma were treated with neoadjuvant ipilimumab in a previously reported study.9 Ipilimumab was planned to be given at 10 mg/kg intravenously every 3 weeks for up to two doses before definitive surgery and two doses after recovery from surgery. Tumor specimens were obtained at baseline and at definitive surgery (week 6–8). Among 35 patients enrolled in the original study, one was excluded from this project due to lack of adequate consent. In addition, seven patients did not have the adequate tumor tissue or RNA quality needed for the RNA microarray studies. The Institutional Review Board of the University of Pittsburgh approved the study and the written informed consent that was obtained from all patients participating in the study.
Laboratory methods
Gene expression profiling was performed on the tumor biopsies of 27 patients. Microdissection of FFPE tumor specimens was performed manually using an inverted microscope (Nikon Eclipse TE200) as needed to obtain a minimum of 90% tumor cells for RNA purification. Dissection involved scraping cells from unstained sections of 5 micron thickness on slides aligned in register with serially cut hematoxylin and eosin stained specimens including tumor domains demarcated by a surgical pathologist (U.R.). RNA purification was performed using the Qiagen miRNeasy FFPE Kit and protocol (Qiagen, Valencia, CA) with isolated RNA suspended in nuclease-free water. Inclusion in subsequent in vitro amplification (IVT) assays was determined both by spectrophotometric absorption ratio [260/280 > 1.8 (NanoDrop, Wilmington, DE)] and RIN values (RNA Integrity Index) determined via microchip electrophoretic analysis (Agilent Bioanalyzer 2100, Agilent Technologies, Santa Clara, CA). We have previously established that RIN values ranging from 5.0 to 8.0 in RNA from FFPE specimens can undergo successful in vitro transcription and amplification using a multiplex primer approach. Amplification was performed using the NuGen whole transcription method comprising the Ovation FFPE WTA assay (NuGEN, San Carlos, CA) employing random and 3′ primers to eliminate amplification bias beginning with 100 ng total RNA. Confirmation of cDNA diversity was obtained using the Bioanalyzer 2100 to generate an electrophoretogram for each amplification reaction regarding sample yield, integrity and size diversity compared to a laboratory human RNA standard and a Universal Human Reference RNA (Stratagene, La Jolla, CA). 5 µg of purified cDNA were incubated with fragmentation buffer (NuGEN, San Carlos, CA) at 37°C for 30 min, then 95°C for 2 min. All cDNA samples underwent hybridization on Affymetrix GeneChip HG U133A 2.0 arrays which contain overlapping probe sets for transcripts comprehensively representing the functionally characterized human genome. Briefly, fragmented cDNAs were mixed in a hybridization cocktail with water to a final volume of 220 μL. 130 µL of hybridization cocktail is hybridized on each array at 45°C for 18 h. The arrays were then washed and stained with streptavidin-phycoerythrin in a GeneChip Fluidics Station 450 (Affymetrix) and scanned using a GeneChip Scanner 3000 (Affymetrix). Quality control (QC) parameters and expression intensity data were derived from the MAS 5.0 (Microarray Suite) and RMA algorithms (Robust Multi-array Average) of the Expression Console software (version 1.2.0.20; Affymetrix). Comparisons of global and individual gene expression data were performed using median normalized and log2 values transferred to Partek Genomics Suite v6.5 (Partek Inc., St Louis, MI). Statistical significance was performed at a false discovery rate (FDR) of less than 5% (q value) to control for Type 1 errors arising from multiple tests.
Statistical methods
Significance analysis of microarrays (SAM) was performed to determine the differential expression of each gene correcting for FDR; FDR controlled at 5% (q value <0 .05). Pathway analysis was performed using Ingenuity Pathway Analysis software. The Benjamini and Hochberg method was used to adjust for multiple testing in the pathway analysis. The clinical endpoints evaluated were disease NP as assessed clinically and radiologically at 6 weeks prior to definitive surgery, RFS and OS.
Disclosure of potential conflicts of interest
AT received contracted research support from Bristol-Myers Squibb.
Funding
This study was supported by NIH award P50CA121973 and by a grant from Bristol-Myers Squibb. UPCI shared resources that are supported in part by NIH/NCI award P30CA047904 were used for this project. Its content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute.
References
- 1.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC et al.. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711-23; PMID:20525992; http://dx.doi.org/ 10.1056/NEJMoa1003466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robert C, Thomas L, Bondarenko I, O'Day S, Weber J, Garbe C, Lebbe C, Baurain JF, Testori A, Grob JJ et al.. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011; 364:2517-26; PMID:21639810; http://dx.doi.org/ 10.1056/NEJMoa1104621 [DOI] [PubMed] [Google Scholar]
- 3.Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, Patt D, Chen TT, Berman DM, Wolchok JD. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 2015; 33:1889-94; PMID:25667295; http://dx.doi.org/ 10.1200/JCO.2014.56.2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR, Buzaid AC, Cochran AJ, Coit DG, Ding S et al.. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol 2009; 27:6199-206; PMID:19917835; http://dx.doi.org/ 10.1200/JCO.2009.23.4799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tarhini AA, Kirkwood JM. How much of a good thing? What duration for interferon alfa-2b adjuvant therapy? J Clin Oncol 2012; 30:3773-6; PMID:23008298; http://dx.doi.org/ 10.1200/JCO.2012.44.9975 [DOI] [PubMed] [Google Scholar]
- 6.Schadendorf DHF, Robert C et al.. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in metastatic or locally advanced, unresectable melanoma. European Cancer Congress 2013. (ECCO-ESMO-ESTRO), 2013 [Google Scholar]
- 7.Kirkwood JM, Strawderman MH, Ernstoff MS, Smith TJ, Borden EC, Blum RH. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. J Clin Oncol 1996; 14:7-17; PMID:8558223 [DOI] [PubMed] [Google Scholar]
- 8.Eggermont AM, Chiarion-Sileni V, Grob JJ, Dummer R, Wolchok JD, Schmidt H, Hamid O, Robert C, Ascierto PA, Richards JM et al.. Adjuvant ipilimumab vs. placebo after complete resection of high-risk stage III melanoma (EORTC 18071): a randomised, double-blind, phase 3 trial. Lancet Oncol 2015; 16:522-30; PMID:25840693; http://dx.doi.org/ 10.1016/S1470-2045(15)70122-1 [DOI] [PubMed] [Google Scholar]
- 9.Tarhini AA, Edington H, Butterfield LH, Lin Y, Shuai Y, Tawbi H, Sander C, Yin Y, Holtzman M, Johnson J et al.. Immune monitoring of the circulation and the tumor microenvironment in patients with regionally advanced melanoma receiving neoadjuvant ipilimumab. PloS One 2014; 9:e87705; PMID:24498358; http://dx.doi.org/ 10.1371/journal.pone.0087705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tarhini AA. Immunotherapy of melanoma. Curr Mol Pharmacol 2015; PMID:26177647 [DOI] [PubMed] [Google Scholar]
- 11.Linsley PS, Bradshaw J, Greene J, Peach R, Bennett KL, Mittler RS. Intracellular trafficking of CTLA-4 and focal localization towards sites of TCR engagement. Immunity 1996; 4:535-43; PMID:8673700; http://dx.doi.org/ 10.1016/S1074-7613(00)80480-X [DOI] [PubMed] [Google Scholar]
- 12.Peggs KS, Quezada SA, Korman AJ, Allison JP. Principles and use of anti-CTLA4 antibody in human cancer immunotherapy. Curr Opin Immunol 2006; 18:206-13; PMID:16464564; http://dx.doi.org/ 10.1016/j.coi.2006.01.011 [DOI] [PubMed] [Google Scholar]
- 13.Robert C, Ghiringhelli F. What is the role of cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma? Oncologist 2009; 14:848-61; PMID:19648604; http://dx.doi.org/ 10.1634/theoncologist.2009-0028 [DOI] [PubMed] [Google Scholar]
- 14.Clinical Trials .gov [Internet]. Bethesda (MD): national Library of Medicine (US) 2000 Feb 29 - Identifier: NCT01274338, Ipilimumab or High-Dose Interferon Alfa-2b in Treating Patients With High-Risk Stage III-IV Melanoma That Has Been Removed by Surgery. 2011 January 8 [cited 2016 March 24]; [about 7 screens] p https://clinicaltrials.gov/ct2/show/NCT01274338 [Google Scholar]
- 15.Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, McKee M, Gajewski TF. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res 2009; 69:3077-85; PMID:19293190; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-2281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Louahed J, Gruselle O, Gaulis S, Coche T, Eggermont AM, Kruit W, Dreno B, Chiarion SV, Lehmann F, Brichard VG. Expression of defined genes identified by pre-treatment tumor profiling: association with clinical responses to the GSK MAGE-A3 immunotherapeutic in metastatic melanoma patients. J Clin Oncol. 2008; 26(suppl): 9045. [Google Scholar]
- 17.Vansteenkiste JF, Zielinski M, Dahabreh IJ, Linder A, Lehmann F, Gruselle O, Therasse P, Louahed J, Brichard VG. Association of gene expression signature and clinical efficacy of MAGE-A3 antigen-specific cancer immunotherapeutic (ASCI) as adjuvant therapy in resected stage IB/II non-small cell lung cancer (NSCLC). J Clin Oncol 2008; 26(suppl): 7501 [Google Scholar]
- 18.Sullivan RJ, Hoshida Y, Brunet J, Tahan S, Aldridge J, Kwabi C, Gardiner E, McDermot D, Golub T, Atkins MA. A single center experience with high-dose IL-2 treatment for patients with advanced melanoma and pilot investigation of a novel gene expression signature as a predictor of response. J Clin Oncol 2009; 27:15S: 9003 [Google Scholar]
- 19.Ji RR, Chasalow SD, Wang L, Hamid O, Schmidt H, Cogswell J, Alaparthy S, Berman D, Jure-Kunkel M, Siemers NO et al.. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother 2011; 61:1019-31; PMID:22146893; http://dx.doi.org/140.1007/s00262-011-1172-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamid O, Schmidt H, Nissan A, Ridolfi L, Aamdal S, Hansson J, Guida M, Hyams DM, Gomez H, Bastholt L et al.. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J Transl Med 2011; 9:204; PMID:22123319; http://dx.doi.org/ 10.1186/1479-5876-9-204 [DOI] [PMC free article] [PubMed] [Google Scholar]