

Abstract
Over the years, significant progress has been made in reducing metabolic instability due to cytochrome P450-mediated oxidation. High throughput metabolic stability screening has enabled advancement of compounds with little to no oxidative metabolism. Furthermore, high lipophilicity and low aqueous solubility of presently pursued chemotypes reduces the probability of renal excretion. As such, these low microsomal turnover compounds are often substrates for non CYP-mediated metabolism. UGTs, esterases and aldehyde oxidase are major enzymes involved in catalyzing such metabolism. Hepatocytes provide an excellent tool to identify such pathways including elucidation of major metabolites. To predict human PK parameters for P450-mediated metabolism, in vitro–in vivo extrapolation using hepatic microsomes, hepatocytes, and intestinal microsomes has been actively investigated. However, such methods have not been sufficiently evaluated for non-P450 enzymes. In addition to the involvement of liver, extra-hepatic enzymes (intestine, kidney, lung) are also likely to contribute to these pathways. While there has been considerable progress in predicting metabolic pathways and clearance primarily mediated by the liver, progress in characterizing extra-hepatic metabolism and prediction of clearance has been slow. Well-characterized in vitro systems or in vivo animal models to assess drug-drug interaction potential and inter-subject variability due to polymorphism are not available. Here we focus on the utility of appropriate in vitro studies to characterize non CYP-mediated metabolism; understand the enzymes involved followed by pharmacokinetic studies in the appropriately characterized surrogate species. The review will highlight progress made in establishing in vitro-in vivo correlation; predicting human clearance and avoid costly clinical failures when non-CYP mediated metabolic pathways are predominant.
INTRODUCTION
Optimizing ADME (absorption, distribution, metabolism, excretion) properties of novel chemical entities has become routine in drug discovery and resulted in dramatic reduction in attrition due to poor pharmacokinetics. There is an increasing trend in medicinal chemistry strategy to reduce the lipophilicity of new chemical entities, which consequently leads to reduction in cytochrome P450 (CYP) mediated metabolism. High throughput metabolic stability screening assays have been successful in eliminating compounds with high metabolic turnover in liver microsomes, hence compounds with little to no oxidative metabolism advance further in lead optimization. As such, these low microsomal turnover compounds are often substrates for NADPH-independent oxidation, conjugation and hydrolysis. Uridine diphosphoglucuronosyl transferases (UGTs), esterases and aldehyde oxidase (AO) are major enzymes involved in catalyzing such metabolism. These non CYP enzymes then become major metabolic routes and clearance pathways in animals and humans and a better understanding of these enzymes is required for drug development.
The non CYP enzymes pose significant challenges with respect to their characterization, availability of in vitro and in vivo models for prediction of human clearance and susceptibility for induction, inhibition and polymorphism. Additional challenges arise due to their extra-hepatic expression, lack of relevant probe substrates and inhibitors and species differences. This review will highlight the utility of appropriate in vitro studies to characterize non CYP-mediated metabolism; understand the enzymes involved followed by pharmacokinetic studies in the appropriately characterized surrogate species. It will also detail progress made in establishing in vitro-in vivo correlation; predicting human clearance and avoid costly clinical failures when non-CYP mediated metabolic pathways are predominant.
ALDEHYDE OXIDASE
Aldehyde oxidase (AO) is a molybdenum cofactor (MoCo)-containing drug-metabolizing enzyme localized in the cytosolic subcellular fraction. AO is active as a homodimer, with each monomer composed of two identical 150 kDa subunits divided into three conserved domains: a 20 kDa amino-terminal iron-sulfur (Fe-S) domain, a 40 kDa flavin adenine dinucleotide (FAD) binding domain, and an 85 kDa carboxy-terminal domain containing the MoCo and active site where substrate binds (1, 2). Aldehyde oxidase catalyzes oxidation of some aldehydes to the corresponding carboxylic acid, but is also involved in oxidation of carbon atoms adjacent to nitrogens within the heteroaromatic ring systems in drug molecules by a nucleophilic mechanism (3, 4). In addition, AO has been demonstrated to play a role in the reductive ring-opening metabolic pathways for zonisamide and ziprasidone (5, 6), and more recently has been reported to play a novel role in the amide hydrolysis of GDC-0834, a Bruton’s Tyrosine Kinase (BTK) inhibitor (7). Examples of AO substrates are presented in Fig. 1. While liver is the primary site of expression of the human AO enzyme (AOX1), several extrahepatic tissues, including kidney, small and large intestine, lung, and adrenal gland, express AOX1, which suggests an endogenous role for AO beyond xenobiotic metabolism (8, 9). Recently, the crystal structure of human AOX1 has been published (10), which will be of significant value to the metabolism community as it relates to structural modelling and predicting binding requirements for AO-mediated metabolism.
Fig. 1.

Structures of known AO substrates
Due to a number of early clinical failures of drug candidates that are substrates of AO, the pharmaceutical industry has been the victim of a potential negative impact when AO-mediated metabolism is not identified. Examples of early termination of several clinical programs due to unanticipated rapid first-pass metabolism and thus, poor systemic exposure following oral dose, have been published (11–15). In addition, toxicological findings due to AO-mediated metabolism of SGX523 to an insoluble metabolite have also been reported (16). These clinical failures could have been avoided if the AO metabolism mechanism was identified early during lead optimization. The intent of this section of the review will be to highlight the most important features of AO-mediated metabolism, phenotyping strategies, inter-subject variability, and challenges toward the prediction of human clearance.
Reaction Phenotyping
Mechanism
The mechanism of oxidation by AO is distinct from other drug metabolizing enzymes, and may be leveraged for early phenotyping. For example, cytochrome (CYP) P450 enzymes generally oxidize carbons with high electron density, whereas AO prefers to oxidize carbons with low electron density and operates by a nucleophilic mechanism, with water as the source of oxygen. This mechanistic difference can be used as an advantage, particularly in an in vitro system where multiple oxidative enzymes such as CYP, flavin monooxygenase (FMO), and AO are present (e.g. S9 fraction or hepatocytes). For example, when incubations are conducted in the presence of isotopically labelled water (H218O) (50/50 mixture as one option), a characteristic isotope pattern is observed for an AO-derived metabolite, with a +16 and +18 mass shift of approximately equal intensity. Conversely, for a substrate of CYP, this isotopic pattern is not observed due to molecular oxygen being the source of oxygen in the metabolite. However, even if this characteristic +16/+18 isotopic pattern is observed, it does not rule out the possible role for xanthine oxidase (XO), as this enzyme is closely related to AO, and operates by a similar mechanism (17).
Cytosol and S9 Fraction
Another methodology for differentiating AO from CYP (or FMO) in an in vitro system, is by cofactor manipulation. For example, metabolite profiles following addition of NADPH as the necessary cofactor for CYP and FMO could be compared to profiles from incubations devoid of this cofactor. If oxidative metabolites (or metabolic clearance) are observed in S9 fractions devoid of NADPH, then the enzyme(s) involved can be narrowed down to AO or XO. Since both AO and XO are expressed in the cytosolic and S9 fractions, differentiation between these related enzymes would have to be achieved by co-incubation of specific inhibitors (e.g. raloxifene, menadione or hydralazine for AO and allopurinol for XO).
Hepatocytes
Human hepatocytes are the optimal in vitro metabolism system, as they contain the full complement of drug-metabolizing enzymes. This benefit however, can also be a challenge when attempting to dissect the enzyme(s) involved in the metabolism of a drug candidate. A reported approach for estimating the contribution of AO compared to other routes of metabolism (fraction metabolized by AO, or fm,AO) in hepatocytes is the use of the AO-selective inhibitor hydralazine (30 minute pre-incubation at 25 μM) (18). Employment of raloxifene or menadione as inhibitors of AO would be inappropriate in human hepatocytes due to their lack of inhibition specificity (19, 20). Alternatively, 1-aminobenzotriazole (ABT) may be used to effectively knock down CYP-mediated metabolism without inhibiting AO metabolism, or other non-CYP pathways such as UGT (30 minute pre-incubation at 1 mM).
Challenges in Prediction of Human Clearance
Prediction of clearance for AO substrates has been problematic, as reports demonstrate a bias towards under-prediction when using in vitro liver cytosol, S9 fraction, and hepatocytes (21–23). One issue is the substantial difference in AO activity in donor-matched liver cytosol and S9 fraction depending on where and how these fractions are obtained (24). Thus, when conducting in vitro studies using these matrices, numerous lots of cytosol and/or S9 fractions should be tested with a range of AO substrates in order to find lots with the highest activity.
Variability and Polymorphism of Human AOX1
Highly variable AO activity across human donors is yet another complexity for prediction of clearance. For example, methotrexate 7-hydroxylase activity in liver cytosol varied 48-fold (25), and an 18-fold range of intrinsic clearance was observed for N-[(2-dimethylamino)ethyl] acridine-4-carboxamide (DACA) when testing 13 donor livers (26). More recently, following incubation in liver cytosol from 20 donors, three probe substrates for AO (carbazeran, zoniporide, and phthalazine) were shown to have highly variable activity (17 to 90-fold) (27). Hutzler and colleagues expanded this research by determining AO activity using O6-benzylguanine in cryopreserved human hepatocytes prepared from 75 individual donors (28). Results from this study showed that activity varied by at least 17-fold, with the lowest activity not measurable by substrate depletion (≤5.4 mL/min/kg) and the highest activity translating to ~90 mL/min/kg. No significant trends were observed when comparing ethnic background, gender, age, or body mass index (28). Among the factors proposed to contribute to variability of AO activity are molybdenum deficiency, potential for de-dimerization of the enzyme, and single nucleotide polymorphisms (SNPs) for AOX1. In addition, diet may play a role in the observed variability (29, 30). The translation of in vitro findings to in vivo clearance appears to be substrate-dependent. For example, the clearance of DACA across 28 individuals ranged from 5.2 to 35.8 mL/min/kg (about 7-fold) (31). Similarly, oral exposures (Cmax and AUC) reported for FK3453 (12) and BIBX1382 (13) were notably variable, with ranges spanning ~23-fold, and 27-fold, respectively. However, a well-controlled study evaluating the variability of exposure for an AO substrate that is not confounded by low oral bioavailability still needs to be performed. To address the extreme in vitro variability, the AO activity of individual donors should be determined prior to pooling, such that custom-made pools of cytosol, S9 fraction, or cryopreserved hepatocytes composed of high activity donors can be prepared to minimize under-predictions of metabolic clearance (32).
Additionally complicating the prediction of metabolic clearance by AO is its extra-hepatic activity. Expression of AOX1 has been reported in kidney, pancreas, intestine, and prostate, with high expression in the zona reticularis of the adrenal gland (8). More recently, AO activity has also been determined in human skin (33). All of this data suggests that only accounting for metabolism of AO substrates using liver tissue may lead to under-prediction of clearance. It is likely that the hepatic and extra-hepatic activity of AO, once quantitatively determined, may be used to develop physiologically based pharmacokinetic (PBPK) models for the purpose of estimating pharmacokinetic profiles of AO substrate drug candidates.
Species Differences
Profound species differences in expression and activity of AO complicates early interpretation of pharmacokinetic behaviour of drug candidates (34). AO expression differences are particularly an issue when coupled with the common drug discovery paradigm where pharmacokinetic (PK) studies are first conducted in the rat and dog, two species which generally have minimal to no AO activity (35). This common in vivo discovery approach, in addition to early in vitro metabolic stability screening in HLMs (a subcellular fraction devoid of AO) has resulted in drug discovery teams overlooking this pathway altogether. An additional risk associated with species differences is a disproportionate or unique human metabolite for an AO substrate. An example of an adverse clinical finding was due to an AO-derived insoluble metabolite of SGX523 crystallizing in the kidney, causing acute renal toxicity (16). Pre-clinically, SGX523 demonstrated a comparable metabolite profile in liver microsomes from rat, dog, monkey and human, which was used to rationalize conducting regulatory toxicology studies in rats and dogs. Despite an apparently favorable safety profile in those species, doses >80 mg administered to human subjects resulted in acute renal failure. Retrospectively, a major oxidative metabolite was observed when profiling human plasma that was not observed from in vitro studies using liver microsomes. Additional metabolite profiling studies in human liver S9 fraction revealed a predominant NADPH-independent, AO-derived metabolite (M11, 2-quinolinone-SGX523). In addition, while M11 was not formed in rat and dog liver S9, it was generated in monkey liver S9. This example highlights the importance of comprehensive metabolite profiling studies using human in vitro systems that contain the full complement of drug-metabolizing enzymes.
Recent literature suggests that rhesus monkey and guinea pig represent the best in vivo surrogate for studies with AO substrates (36). BIBX1382 was a clinical candidate being evaluated as an inhibitor of epidermal growth factor receptor (EGFR) in the treatment of cancer. Following the observation of extremely low oral exposure in humans, it was discovered that the pharmacokinetic properties of BIBX1382 in rhesus monkey aligned with humans (13). When considering that the cynomolgus and rhesus monkey AOX1 enzymes are 96% homologous to human AOX1, and 99% homologous to each other, it is possible that cynomolgus monkey, which is a preferred primate for pharmacokinetic studies may also represent a suitable surrogate species for human AO. In that context, cynomolgus monkey was found to reproduce the low oral exposure observed with BIBX1382 (37). The AO-mediated metabolism of several other AO substrates (e.g. SGX523, zaleplon, SB-277011 and RS-8359) have been reported to be comparable between cynomolgus monkey and human (37). Since rhesus and cynomolgus monkey have been shown to be potentially good surrogates for AO activity in human (37), single species scaling from these species may be a viable approach for prediction of human clearance (38) after appropriate in vitro studies are conducted.
CARBOXYLESTERASE
Carboxylesterases (CE) are ubiquitous enzymes that hydrolyze carboxylesters (RCOOR1) into the corresponding alcohol (R1OH) and carboxylic acids (RCOOH) (39, 40). CEs are expressed in virtually every living organism, although in mammals no bona fide endogenous substrates have been identified. Furthermore, these enzymes tend to be expressed at high levels in tissues that are exposed to xenobiotics (liver, kidney, blood, gut, etc.) and as a consequence, it is thought that CEs may provide an initial defense in the detoxification of such compounds (39–41). Correspondingly, any drugs that contain an ester chemotype are subject to the action of these proteins. The intent of this section of the review will be to highlight the impact of CEs on drug disposition; reaction phenotyping, and how small mammals represent poor models to study such reactions; inter-individual variability due to polymorphisms, and drug-drug interactions. Since very little information exists regarding prediction of human clearance for esterified molecules, this topic will only be briefly discussed.
Drug esterification
Esters are ubiquitous functional groups that appear in many natural products and drugs. Since the oxygen atoms within this moiety are capable of hydrogen bonding, molecules that contain this chemotype trend toward better water solubility. This property has been exploited by nature and medicinal chemists to allow synthesis and generation of structurally diverse organic molecules. For example, compounds such as ecteinascidin-743 and bryostatin, isolated from marine organisms, and many drugs (e.g., oseltamivir (Tamiflu), clopidogrel (Plavix), methylphenidate (Ritalin)) (Fig. 2) contain this functionality. While esterification clearly impacts the bioavailability and solubility, this moiety makes these compounds substrates for CEs. Recent detailed biochemical, molecular and cell biology studies have provided a platform on which CE-mediated drug hydrolysis can be evaluated.
Fig. 2.
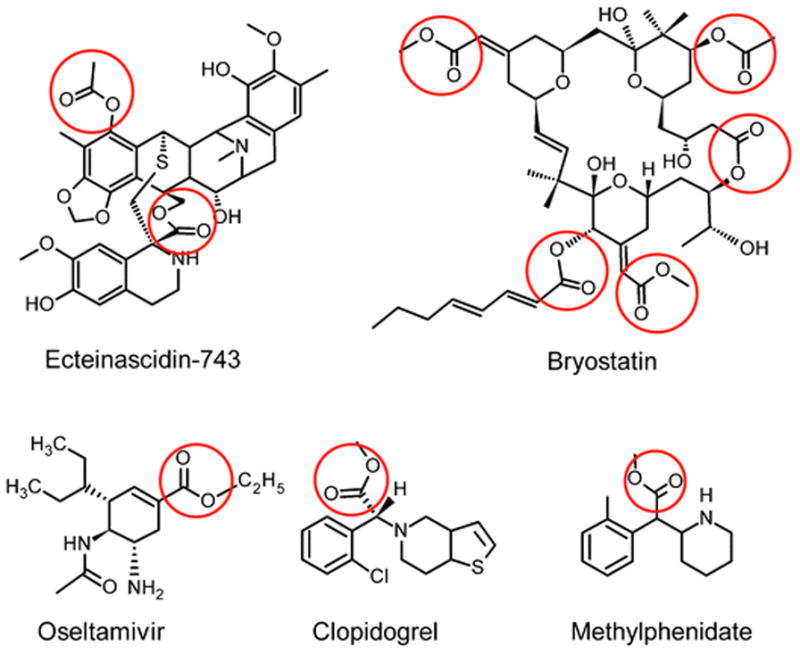
Structures of clinically-approved marine-derived natural products (ecteinascidin-743 and bryostatin) and commonly prescribed drugs that contain ester moieties (indicated by the red circles)
Human carboxylesterases
Sequencing of the human genome has identified 5 potential CE genes, however biochemical data has only been reported for 3 such enzymes. Of these, only two (hCE1 [CES1] and hiCE [CES2]) have been studied to any great extent, and hence, the vast majority of in vitro studies have focused on these two proteins.
hCE1 is a 60kDa enzyme highly expressed in the liver, with much lower levels detected in the lung (42), and is absent in the gut and kidney. The enzyme must be processed within the endoplasmic reticulum for functional activity. Biochemical studies have indicated that, in comparison to hiCE, hCE1 prefers smaller, more planar substrates (e.g., oseltamivir, clopidogrel) (43). Structural studies have demonstrated that this is likely due to physical constraints enforced upon on the active site by two loops of amino acids (44–46). The latter are present at the entrance to the catalytic gorge and are immobile in hCE1 (45, 47). In the highly related rabbit enzyme (rCE), which demonstrates greater than 85% amino acid identity to hCE1, these loops are flexible (44). As a consequence rCE can readily hydrolyze much larger substrates such as CPT-11 (48–50). These studies have also suggested the presence of a ‘side door’ within both hCE1 and rCE that may allow the products of hydrolysis to rapidly exit the active site, hence increasing the rate of substrate turnover. To date, this has not been proven, but similar studies using the structurally related acetylcholinesterase suggest that such a mechanism is plausible (51, 52).
hiCE is also 60kDa and principally expressed in the gut and kidney, with lower, and much more variable levels in the liver (42). High expression is seen in the duodenal epithelium, the area in which the bile duct enters the gut, and hence it is believed this enzyme may provide protection from agents that are metabolized in the liver and excreted via the bile. In contrast to hCE1, hiCE can hydrolyze very large complex molecules such as CPT-11 and cocaine (49, 53). As described above, this is likely due to the enhanced flexibility of the active site gorge as compared to hCE1. Due to the lability of hiCE, to date, no successful, detailed structural studies have been conducted with this enzyme.
Reaction phenotyping
While it is much more difficult to identify other hydrolases involved in drug metabolism, involvement of hCE vs. hiCE can be readily ascertained. Additionally, as methods for the purification to homogeneity for both hCE1 and hiCE have been described (53, 54), incubation of potential substrates with recombinant enzymes provide kinetic parameters than can identify the relevant proteins. For example, the kcat/Km values for CPT-11 hydrolysis by human CEs demonstrated that hiCE was ~100 fold more efficient (42) and likely to be the enzyme involved in CPT-11 hydrolysis in vivo. However, when a panel of human liver microsomes were assessed for their ability to hydrolyze this drug (Table I), a lack of correlation was observed between hiCE expression and SN-38 production. This is exemplified by sample 3, where ~60% of total hydrolysis occurs from enzymes other than hiCE, likely hCE1. Additionally, ~15% of the SN-38 generated arose from non-CE mediated metabolism by other enzymes in liver microsomes. To date, the identity of these hydrolases is unknown, but may include cholinesterases (butyrylcholinesterase has weak activity toward CPT-11 (55, 56)), paraoxonases, and possibly lipases. Hence, in vitro studies to assess hydrolytic metabolism should be undertaken with caution, with numerous independent samples, employing use of different enzyme inhibitors and combinations thereof.
Table I.
CPT-11 Hydrolysis by Human Liver Microsomes in the Presence or Absence of CE Inhibitors
| Sample | SN-38 (pmol/hr/mg) | SN-38 + hiCE inhibitor* (pmol/hr/mg) | Hydrolysis due to enzymes other than hiCE (%) | SN-38 + pan CE inhibitor** (pmol/hr/mg) | Hydrolysis due to enzymes other than CEs (%) |
|---|---|---|---|---|---|
| 1 | 33.9 | 18.5 | 54.6 | 3.9 | 11.6 |
| 2 | 22.2 | 13.4 | 60.3 | 2.8 | 12.6 |
| 3 | 55.3 | 33.6 | 60.7 | 7.8 | 14.0 |
| 4 | 26.6 | 14.1 | 52.8 | 4.3 | 16.1 |
| 5 | 19.5 | 13.7 | 70.2 | 3.9 | 20.0 |
| 6 | 43.8 | 28.5 | 42.3 | 6.0 | 13.6 |
hiCE specific inhibitor N,N′-(2,3,5,6-tetrafluoro-1,4-phenylene)bis(4-bromobenzenesulfonamide)
pan CE inhibitor benzil
Species differences
It is difficult to identify an animal model predictive of CE-mediated metabolism, partly due to the complexity of the esterase genome (20 and 15 CE genes have been identified in mice and rats, respectively; (57, 58)), but also because the enzymes in small mammals tend to be much more efficient at substrate hydrolysis than human proteins (59). This is again exemplified with CPT-11 where a rabbit liver CE (rCE) is ~650-fold more efficient than hCE1 (60). Furthermore, most small mammals express very high levels of CE in plasma, in contrast to humans that are essentially devoid of this activity. Therefore, predicting CE-mediated drug metabolism in humans based upon small mammal studies is difficult.
Recently, two mouse strains (Es1e and Es1e/scid) have been developed that lack plasma esterase activity and may represent more appropriate models for assessing esterase-mediated drug hydrolysis (59, 61). The Es1e mice were originally generated in the 1970’s using mutagen treated sperm and were subsequently found to lack a circulating esterase (62). Two decades later, these mice were retrieved from Jackson labs and plasma obtained from these animals was essentially unable to hydrolyze CPT-11 (59). To assess the impact of the plasma esterase on the antitumor efficacy of CPT-11, the Es1e mice were crossed with a scid strain to develop an immune-deficient mouse that lacked the circulating enzyme (Es1e/scid). Pharmacokinetic experiments indicated that the AUC values for SN-38 were significantly reduced in this strain, and subsequent studies using human tumor xenografts demonstrated that ~4- to 5-fold higher dose of CPT-11 was required to achieve comparable antitumor activity to that seen in wild-type scid mice (61). These mice may therefore represent an appropriate animal model to predict disposition of ester drugs.
Since dog is the more common non-rodent species for preclinical PK studies, different groups have assessed ester drug metabolism by CEs in this species. The results have then been compared to those obtained from monkeys and humans (63, 64). Such studies clearly identify enzyme homologues, patterns of CE expression, and can assist in reaction phenotyping in the species of interest. For example, results by Williams et al., demonstrate that CE tissue expression patterns are comparable between human and monkey, but distinctly different in dogs. Indeed, expression of the intestinal enzyme was absent in the latter, arguing that dogs may not be suitable models for examining the metabolism of orally-dosed ester drugs (64). It would be interesting to conduct retrospective analyses to confirm or disprove this hypothesis.
Challenges in the prediction of human clearance
Recently, a detailed examination of the hydrolysis of a panel of ester drugs by CEs present in liver, intestine and kidney of dogs, monkey and human has been undertaken, and attempts made to correlate these results with oral clearance in human (63). In these experiments, hepatocytes and S9 fractions of the different organs were used as the source of enzymes (hence, representing a mix of the different hydrolases), and data were scaled using known parameters for organ cellularity. Modest levels of agreement were observed when using liver S9 fractions as the source of CEs, especially for agents such as perindopril and oseltamivir. However, there were significant outliers in these studies (e.g., trandolapril and quinapril), even though the chemical structures of these substrates were very similar, and hydrolysis in all cases led to the loss of an ethyl group. Interestingly, results obtained from incubation of these same drugs with human hepatocytes resulted in poorer predictions (the predicted clearance was ~20% of the observed value) as compared to the liver S9 fractions (63). However in these studies, the human, monkey and dog intestinal S9 used were from a single individual and hence unlikely to be representative of the population as a whole. Clearly therefore, there is potential merit to such studies, but it would seem unlikely that such studies could ever be used in a quantitative fashion to predict drug hydrolysis and disposition in patients. This is likely due to numerous factors including: the cadre and levels of different CEs expressed in both target and non-target tissues in different populations; the accuracy of scaling factors; the impact on drug hydrolysis by CEs expressed in the gut epithelia for orally-dosed drugs; as well as the substrate specificity of the different enzymes.
CE polymorphism
With the recent advances in genome and exome sequencing, considerable information on CE polymorphism is being generated. For example, Zhu and coworkers (65) have identified mutations within the mRNA encoding hCE1 that result in either a non-conservative substitution, or a deletion that results in a frame shift and premature termination of the protein coding sequence. The consequences of these alterations are dramatic, with the resulting proteins demonstrating reduced ability to hydrolyze methylphenidate (65), clopidogrel (66) or oseltamivir (67). The loss of this enzymatic activity (slow metabolizers) leads to significantly increased plasma concentrations of the former two active parent drugs. In contrast, with drugs such as CPT-11 and oseltamivir, where hydrolysis is absolutely required to generate the active metabolite, the reduction in apparent enzymatic activity may result in significantly poorer efficacy in these patients. While the incidence of this polymorphism in the general population is relatively low (2.0% – 4.7% (65)), due to the widespread use of ester drugs in clinical practice, a significant number of individuals will be affected. However, without detailed PK studies, coupled with patient-specific DNA/RNA sequencing, the identification of such problems is likely to be missed.
Work by Hu et al., (68) used PBPK modeling to assess oseltamivir metabolism and to simulate prodrug and active metabolite plasma concentrations. They confirmed that in a patient harbouring the previously observed hCE1 polymorphism that affected drug metabolism (67), their model could accurately predict both oseltamivir and oseltamivir carboxylate (the active metabolite) exposures following oral drug dosing. In addition, they also observed that ethanol, a weak hCE1 inhibitor (Ki = 23mM; (68)) resulted in increased oseltamivir exposure that was dose-dependent. Interestingly however, ethanol did not significantly change the levels of oseltamivir carboxylate exposure (a 4% reduction as compared to a 37% increase for oseltamivir). The exact reasons for this discrepancy are unclear, but may be due to residual drug hydrolysis by the mutant enzyme and/or the existence of other endogenous hydrolases with weak activity towards this substrate. Hence, PBPK may allow prediction of disposition of CEs substrates, although considerably more work will be required to validate this methodology for the whole host of different ester agents that are currently in clinical practice.
Drug-drug interactions
In contrast to the work detailing drug-drug interactions with CYP, very little information has been reported with ester drugs. Recent studies describe a comprehensive approach to identify CE inhibitors, to assess the presence of such agents in herbal medicines, natural products and prescribed medicines, and impact on drug metabolism (69–79). A small scale screen identified 1,2-diones as potent CE inhibitors and exhaustive chemical and biological assays allowed generation of a pharmacophore that was used to computationally search natural product libraries. A series of abietane diterpenoids (tanshinones) were identified and demonstrated to be potent human CE inhibitors (80). Furthermore, the tanshinones are present in high quantities in Danshen, a widely used Chinese herbal medicine, and also a component of ‘Danshen Dripping Pill’. The latter has recently been approved by the FDA for clinical trials and individuals who use this material may experience modulation of ester drug metabolism. Both in vitro and in vivo studies to assess the impact of the tanshinones on the hydrolysis of agents such as CPT-11 are currently underway. In addition to the tanshinones, carbamates have been demonstrated to be inhibitors of esterases. Rivastigmine (Exelon), used to treat Alzheimer’s disease, contains this chemotype and is a potent irreversible inhibitor of hiCE in vitro (81). The impact of rivastigmine on ester drug metabolism in vivo needs to be evaluated in detailed animal and clinical studies.
URIDINE DIPHOSPHOGLUCURONOSYL TRANSFERASES
Uridine diphosphoglucuronosyl transferases (UGTs) carry out an enzymatic reaction termed as glucuronidation, which involves coupling of a polar glucuronic acid group to nucleophilic functional groups of endobiotics and xenobiotics. UGTs are located on smooth endoplasmic reticulum in hepatic and some extra-hepatic tissues (kidney, intestine, lung, etc.), such that their active site faces the inner lumen of the reticular membrane (82). Uridine diphospho glucuronic acid (UDPGA) is utilized as a co-substrate and is thought to be cross-transported with N-acetyl glucosamine/UDP-xylose/UDP-glucose across the reticular membrane. Twenty one functional human UGTs have been identified to date. The glucuronidation reaction occurs via an SN2 type reaction, in a compulsory ordered bi bi reaction, where typically a functional group such as -COOH, -OH, -SH, or –NH (primary, secondary or tertiary) of a xenobiotic or endobiotic serves as a nucleophile and UDP (uridine diphosphate) is released (82). Unusual glucuronides, i.e. glucuronides which are S-linked, C-linked, Se-linked, carbamoyl linked, and di-glucuronides, classified as ‘linked’ and ‘discrete’ are seldom observed (83). Application of softer ionization techniques coupled with high resolution accurate mass spectrometry that yields robust fragmentation has made it easier to elucidate putative structures of glucuronide conjugates (84).
Reaction phenotyping
UGT superfamily consists of four distinct families – UGT1, UGT2, UGT3 and UGT8 (85). UGT families are further classified into subfamilies such as UGT1A, UGT2A, and UGT2B. UGT1A and UGT2B enzymes are differentially expressed in liver, kidney, and intestine and possess a common UDPGA binding pocket. In addition, UGTs have overlapping substrates and lack of specific inhibitors. This makes reaction phenotyping for UGTs from these families extremely difficult by way of enzyme inhibition in cellular drug metabolism models derived from hepatic or extra-hepatic tissues. UGT2A family is expressed exclusively in olfactory tissues. UGT3A1 forms N-acetylglucosamidines (86) whereas, UGT3A2 has been shown to form xylosides and glucosides (87). UGT8 has not been documented to form glucuronides till date. Thus, characterizing conjugation reactions specifically by UGT2A enzymes can be accomplished in olfactory tissue derived models. Similarly, utilizing UDP-N-acetylglucosamine and UDP-xylose will enable studying reactions by UGT3A1 and UGT3A2 respectively (note: that other UGTs are capable of forming xylose and N-acetylglucosamine conjugates as well). Thus, formation of xylose and N-acetylglucosamine conjugates should be considered suggestive, rather than absolute, before additional evidence can be obtained from phenotyping studies with recombinant UGTs. Thirteen UGTs – UGTs 1A1, 1A3, 1A4, 1A6, 1A7, 1A8, 1A9, 1A10, 2B4, 2B7, 2B10, 2B15 and 2B17 – are commercially available as recombinant enzymes as Supersomes®. When coupled with protein quantification data such systems (88) can be used for estimation of CL (Tables II and III). When comparing the activities of individual UGT enzymes for a single substrate (isoenzyme screening), it is important to normalize for expression. Western blotting in the UGT1A family with antibodies directed against the UGT1A constant region (UDPGA binding pocket) is another technique of normalizing for protein expression.
Table II.
Challenges Associated with UGT CL Prediction
| System (Model) | Challenge and causes | Effect (Resulting Observation) | Comments and Examples | References |
|---|---|---|---|---|
| In vitro | Glucuronidation as a metabolic pathway may be dependent on a substrate’s entry into the cell and may be rate limiting by a substrate’s exit out of the cell. | Kinetics in microsomes cannot be directly translated to hepatocytes. | IVIVE is further complicated when the UGT substrate is also a substrate for one or more uptake and efflux transporters. | |
| In vitro | Active site of UGTs faces the inner lumen of endoplasmic reticular membrane and therefore faces inward in microsomes. | In vitro incubations need to utilize pore-forming agents in appropriate amounts for reproducible kinetics. | Literature reports include surfactants such as Brij-58, Triton-X, etc. as well as alamethicin, a pore-forming peptide. | (103) |
| In vitro/In vivo | UGTs have overlapping substrates and lack of specific inhibitors. | Extremely difficult to carry out enzyme inhibition and DDI studies in vitro. Multienzyme kinetics needs to be taken into account in systems like microsomes and hepatocytes. | Multiple nucleophilic groups in substrates may present an additional complication. | (103) |
| In vitro | Over expression of UGT enzymes in recombinant systems | Elucidation of atypical kinetics is difficult. Kinetics in recombinant systems cannot be directly translated to microsomes or hepatocytes. | A general UGT substrate, 4-methyl umbelliferone, shows Michaelis-Menten kinetics in HLM, however, atypical kinetics are observed in recombinant enzyme systems. (e.g. Hyperbolic kinetics with UGT1A1, substrate inhibition with UGT1A3, homotropic co-operativity with UGT2B7. | (108, 109) |
| In vitro | Differences in preparation of cell lysates, Supersomes, microsomes | Observed differences in enzyme kinetics for a given substrate. Resultant Km and Vmax values are a range rather than a number (Inter-lot and inter-laboratory variability). | Supersomes, microsomes, hepatocytes, tissue slices, cloned expressed UGTs in HEK cells, sf9 cells are viable in vitro models. | |
| In vitro | Buffer related differences in incubation conditions | Observed differences in enzyme kinetics for a given substrate. Resultant Km and Vmax values are a range rather than a number (Inter-laboratory variability). | In HLM, UGT1A4 and UGT1A9 showed up to 2x higher activity in Tris-HCl buffer, than phosphate buffer. In HLM, UGT2B7: showed higher activity with carbonate buffer/Williams E media. Similarly, higher activity with bicarbonate buffer may be observed depending on type of glucuronidation reaction. | (110–112) |
| In vitro | Differences in incubation conditions arising from different membrane disrupting agents/latency | Observed differences in enzyme kinetics for a given substrate. Resultant Km and Vmax values are a range rather than a number (Inter-laboratory variability). | Alamethicin is better than surfactants. Recommended use: 50 μg/mg of protein (if protein concentration > 0.17mg/mL) or 10 μg alamethicin/mL of incubation. | (110, 113, 114) |
| In vitro | Differences in incubation conditions related to organic solvents | Observed differences in enzyme kinetics for a given substrate. Resultant Km and Vmax values are a range rather than a number (Inter-laboratory variability). | Organic solvent recommended to be below <1%. UGT1A9 and UGT2B17 are not as well tolerant of DMSO as other UGTs. | (115, 116) |
| In vitro | Use of saccharolactone in incubations | Observed differences in enzyme kinetics for a given substrate. Resultant Km and Vmax values are a range rather than a number (Inter-laboratory variability). | It is believed that increased glucuronidation activity in the presence of saccharolactone may be due to decrease in pH and not necessarily inhibition of beta-glucuronidases. | (110, 117) |
| In vitro/In vivo | Expression of UGT isoforms in preclinical species and human is different. | Poor translation from preclinical models to in vivo clearance. | Species differences in expression of UGTs are not well understood. | (103) |
| In vitro/in vivo | Expression of UGT isoforms is tissue-specific in preclinical species and human. | Poor translation of in vitro kinetics to in vivo clearance in preclinical models and human, whenever extrahepatic glucuronidation plays a role. | Species differences in extra-hepatic expression of UGTs are even lesser studied. | (89) |
| In vitro | Microsomal fatty acids are released during in vitro incubations in various concentrations. The concentrations of released fatty acids change over time (upto 120 mins) and vary in rat, monkey and human liver microsomes. | Fatty acids outcompete the substrates, thus yielding artificially high Km values (high μM to low mM). In vitro – In vivo Extrapolation is challenging. | Incubations with BSA, HSA or IFABP (preferred) are carried out to negate the fatty acid effect. Although these have been shown to help IVIVE of UGT1A9 and UGT2B7 substrates, Kms remain unaltered for substrates of UGTs 1A4, 1A1 and 1A6 (low μM). | (98, 118–120) |
| In vitro/In vivo | Demanding syntheses for reference standards | Formation kinetics of glucuronides is resource intensive. Dependency on substrate depletion kinetics even in the presence of competing metabolic pathways. Absolute quantification of the glucuronide metabolite in circulation is challenging. | Accurate kinetics for formation of glucuronides are performed by either using a synthetic reference standard for the glucuronide or by the use of 14C-UDPGA to form the glucuronide. Neither technique is time and cost effective. |
Table III.
Opportunities for UGT CL Prediction
| Method | Advantages | Disadvantages | References |
|---|---|---|---|
| Single species scaling e.g. based on monkey PK | Simple. Based on relative differences in physiological attributes underlying drug disposition (e.g., liver weight, hepatic blood flow, glomerular filtration rate). Extensive modelling (and data generation) not needed. | Coefficients derived for each species should represent an undefined amalgam of such constants. Non-mechanistic in nature. Large animal PK data are needed. | (121–123) |
| Use of Relative Activity Factors (RAFs) | Works well when glucuronide reference standards are available or when glucuronidation is the predominant pathway (ideally one metabolite). Semi-mechanistic, powerful when cross checked with UGT protein concentrations. Applicable when interspecies differences in rate/extent of glucuronidation are observed. Can be applied to other organs for CL estimation. Large animal PK data are not needed. | UGT phenotyping is needed. Some UGTs - 1A9, 2B7 are shown to work better in the presence of BSA. Absence of specific UGT substrates for calculation of RAFs is a complicating factor. e.g. Estradiol, although used for UGT1A1 is also a UGT1A3 substrate; chenodeoxy cholic acid, although used for UGT1A3 is also a UGT1A1 and UGT2B7 substrate; AZT, although used for UGT2B7, is also a UGT2B4 and UGT2B17 substrate. | (124) |
| CL delta: fmCYP and fmUGT approach | Semi-mechanistic. Applicable when interspecies differences in rate/extent of glucuronidation are observed. Can be applied to other organs for CL estimation, e.g. kidney (1A9), intestine (1A8, 1A10). UGT phenotyping is not needed. Large animal PK data are not needed. | Laborious, especially if fu, and Clint with and without BSA are estimated. Some UGTs - 1A9, 2B7 are shown to work better in the presence of BSA. Works in HLMs, sequential and concurrent metabolism in hepatocytes act as limiting factors. | (96–98, 125) |
Species differences
Remarkable species differences are observed for UGTs (82), thus adding a level of complexity to interspecies scaling of substrates which are predominantly glucuronidated. UGTs 1A1, 1A3, 1A4, 1A6, 1A7, 1A8, 1A9, 1A10, 2B4, 2B7, 2B10, 2B11, 2B15, 2B17, 3A1, 3A2, and 8A1 are found in humans. Interestingly, UGT1A1 is expressed across most species. UGT1A2 is expressed in rodents, but is a pseudogene in humans. UGT1A4 is not expressed in rodents and therefore, rodents do not form N-glucuronides well. UGT1A6 is not expressed in cats. UGTs 1B1, 1B2, and 1B3 are expressed in rodents and not humans. UGTs 2B5, 2B13, 2B14, and 2B16 are rabbit isoforms. UGT2B8 is expressed in rats, whereas, UGTs 2B9, 2B18, 2B19, and 2B20 are shown to be present in monkeys. UGT2B21 is expressed in guinea pigs.
UGT Polymorphisms
Many UGTs have documented genetic polymorphisms that impact glucuronidation kinetics (89). The most well studied polymorphism occurs in UGT1A1. In Caucasians, a TA insertion into a TATA box (UGT1A1*28) results in lowered expression of the enzyme and a mild hyperbilirubinemia (Gilbert’s syndrome) with ~15% frequency. Incidence of Grade 2 or greater hyberbilirubinemia is significantly higher in Gilbert’s patients taking atazanavir or indinavir. Gilbert’s patients also experience a significantly higher rate of neutropenia from irinotecan (CPT-11), and the FDA has approved a genotyping test for patients prior to irinotecan therapy. Polymorphisms in UGT1A3 and UGT1A4 have also been identified. UGT1A4*2 and UGT1A4*3 have shown to have differential effect on enzyme kinetics of substrates such as tamoxifen, lamotrigine, dihydrotestosterone and trans-androsterone (90). For a UGT1A6*2 variant, 2-fold higher activity has been documented for several substrates in in vitro recombinant systems (91). It is thought that polymorphisms of UGTs 1A1, 1A6 and 1A9 may play a role in acetaminophen drug interactions. Like UGT1A1, there is also a common TATA box polymorphism in the UGT1A9 gene. Allele frequencies for this gene were 60% in Japanese, 39% in Caucasians, and 44% in African Americans. Variants in UGT1A10 have been shown to possess lower activity for phenolic substrates. For UGT2B7 variants, no difference has been observed for enzyme kinetics of AZT, morphine, or codeine, but, the wild type enzyme has been shown to have higher activity toward aldosterone. Similar to observations for UGT1A4 polymorphs, UGT2B15*2 results in 50% lower activity for xenobiotic substrates, but increased activity toward androgens.
Challenges in prediction of human clearance
Challenges associated with CL prediction when UGTs play a major role in compound’s elimination are manifold. Resultantly the translation from in vitro models to in vivo situation is poor and mostly leads to clearance under-prediction (92–95). Major challenges in this regard are presented in Table II. Some useful approaches, along with their advantages, disadvantages and caveats are described in Table III. Since many of the UGTs are extrahepatic, inclusion of a glucuronidation component from organs such as kidney and intestines have enabled better CL or CL/F prediction for certain UGT substrates that are metabolized in these organs (96–99). In such circumstances, a whole body PBPK based approach may be worth investigating as well (100). Following are some common misconceptions and approaches that are not recommended for in vitro-in vivo extrapolation (IVIVE) (when glucuronidation is the predominant metabolic pathway):
Inhibition by UDP is not a true pan-inhibition method for UGTs. This has been shown to work only for UGTs 1A1, 1A4 in microsomes but the data was not corroborated in Supersomes. It has also been documented for UGT1A9 in sf9 cell lysate supernatants but not validated in Supersomes or microsomes (101, 102). Moreover, there is no evidence yet that this is a valid method of inhibiting hepatic and other extra-hepatic UGTs either in microsomes or recombinant systems. Although it is a tempting method to estimate UGT activity, especially in the absence of specific inhibitors, this approach cannot be generalized or extrapolated to other in vitro systems where differences in protein expression, lipid compositions and functional activity complicate matters.
Use of total substrate concentrations instead of unbound substrate concentrations when BSA, HSA or IFABP is utilized in incubations.
Michealis-Menten kinetics are not absolute and evidence for atypical kinetics such as homotropic/heterotropic activation, needs to be carefully evaluated.
In silico approaches have recently come to prominence for understanding the role of glucuronidation as a clearance pathway and for PK projections in humans. These are either training set based QSAR models or non-training set based models (e.g. molecular interaction fields). Although these models are a step towards the future, at present they are very limited in their applications. In these models, a molecule is always assumed to be a substrate and a model itself is unable to differentiate between substrates, non-substrates and inhibitors. Additionally, the binding modes cannot be separated, e.g. atypical kinetics vs. Michealis-Menten kinetics. Finally, for poly-functional substrates, i.e. substrates with multiple unmasked nucleophilic groups, the models are unable to provide differentiation between multiple glucuronidation sites. It is recommended that these models be utilized with extreme caution and under the guidance of a drug metabolism expert.
Drug-drug interactions
Drug-Drug Interactions involving UGTs are thought to be possible, although documented clinical occurrences point toward low to moderate alterations in pharmacokinetics of victim drugs. There are several instances where co-administration of two or more UGT substrates has resulted in modulation of CL of a victim drug (103). However, the occurrences of clinical DDIs or greater than two fold change in AUCi/AUC are rare (104). Mechanistically DDIs with UGTs and glucuronides can be rationalized in multiple ways. Decrease in CL of victim drug(s) may occur due to direct enzyme inhibition, depletion of UDPGA, inhibition of UDPGA transport, inhibition of renal elimination of glucuronides and inhibition of hepatic/renal uptake of glucuronides. Increase in CL of victim drug(s) may result from induction of one or more UGTs, interruption of enterohepatic recirculation by intestinal microflora and increase in hepatic/renal efflux of glucuronides. Although, it is uncommon for a glucuronide to be inhibitor of a CYP enzyme, there have been reports for drug interactions arising as a result of time dependent inhibition of CYPs, specifically, CYP2C8, by glucuronides of gemfibrozil (105) and clopidogrel (106). However, it should also be noted that transporters from the OATP family also come into play in interactions with gemfibrozil (107), thus presenting evidence that enzyme-transporter interplay may add to the complexity of drug-drug interactions arising from glucuronides.
Assessing drug-drug Interactions arising from UGTs is extremely challenging due to lack of specific probe substrates because UGTs exhibit overlapping substrate specificity (103). Reliable estimation of DDIs via UGTs requires an understanding of mechanism of clearance and a reliable measurement of fm and fm,UGT. Determination of true in vivo fm,UGT is difficult due to possibility of entero-hepatic recirculation. Biliary elimination of glucuronides in humans cannot be determined accurately in a non-invasive manner. Furthermore, amount of glucuronide in urine/bile is not representative of the total glucuronide formed. Marked differences exist in the expression of extrahepatic UGTs, for example, UGTs 1A3, 1A9, 2B7 are expressed in kidney in addition to liver and UGTs 1A1, 1A8, 1A10 are found in the intestinal tract, with differential expression between duodenum, ileum and jejunum. Prediction of extrahepatic glucuronidation from preclinical species to human is extremely challenging. An accurate prediction requires use of appropriate scaling factors associated with relative organ weights, UDPGA concentrations and differential activities and yields between hepatic vs. extra-hepatic enzymes.
It is worth noting that assessing DDIs has substantial experimental limitations as well. Addition of BSA while carrying out in vitro experiments is thought to offset the high Km values (for example, fluconazole-zidovudine, valproic acid-lamotrigine) (103). However, without BSA, the fatty acids may outcompete the perpetrator resulting in over-prediction whereas with BSA, the victim and/or the perpetrator may bind to albumin, resulting in under-prediction of DDIs. The in vitro enzyme kinetics observed for glucuronidation may be system-dependent and is another caveat worthy of consideration. In other words, kinetics of the drugs of interest may be different in rUGTs vs. HLM, due to overexpression of UGTs in the recombinant system and possibility of multi-enzyme kinetics in the microsomal system. Finally, effects of homo- and hetero-dimerization, as well as the surrounding microenvironment, i.e. membrane integrity, accessory proteins, redox states, cellular environment, are not fully understood yet, further complicating the assessment of drug-drug interactions involving UGTs.
CONCLUSIONS
Overall, phenotyping for AO metabolism is fairly straight-forward, since specific inhibitors that can differentiate AO from XO and CYP exist and methods have been published. The key is to evaluate metabolism of a chemical series in an in vitro subcellular fraction or hepatocytes where AO is expressed and active. In addition, it is evident that for AO substrates, a “one-size-fits-all” approach for clearance projection such as 3 or 4-species allometric scaling using empirical scaling correction factors (e.g. maximum lifespan potential, MLP) is not appropriate given the profound species differences in activity. Instead, a more mechanistic approach, utilizing a combination of in vitro metabolism data from pooled cryopreserved hepatocytes (or subcellular fractions) composed of high AO activity donors, as well as pharmacokinetic studies in a species that is representative of human from an AO-metabolism standpoint (e.g. cynomolgus monkey, supported by in vitro data), is the recommended approach for minimizing the noted under-predictions of clearance from the literature.
The role of esterases in drug metabolism is frequently ignored, however, it is likely that the ester function will be increasingly used to improve water solubility and bioavailability. Since a detailed understanding of how esterase-mediated drug hydrolysis in animals compares to that in humans is currently unavailable, the use of such models will likely be uninformative. Therefore, the use of the ester chemotype should be minimized in the development of novel agents, and bioisosteres with similar functionalities should be employed.
A multitude of challenges exist with glucuronidation CL prediction. Although the available techniques have progressed with time, none are close to perfect. Multiple approaches may be utilized to gain confidence in the predicted CL when glucuronidation is the predominant pathway. However, these approaches are not recommended to be used with the mindset that ‘many ordinary approaches can be stacked together to make an exceptional method’. Rather, depending on the information available and the question that needs to be answered, one or more approach may be employed, refined and recalibrated as additional data becomes available. Finally, as the drug candidate progresses through, evidence for entero-hepatic recirculation, induction and regulation of UGTs, patient population based evaluations, will provide added value and will aid in fine-tuning the applied CL prediction methodologies.
Despite the significant progress made in the characterization of these non CYP pathways, better understanding of their extra-hepatic expression and in vitro tools to measure that activity are needed. Understanding of hydrolases beyond CEs and identification of their selective substrates and inhibitors has been challenging. More clinical experience is essential to translate in vitro reaction phenotyping (fm) and inter-subject variability. Until then drug discovery scientists will be skeptical about advancing non-CYP substrates into development.
Acknowledgments
Sincere thanks to Jennifer L. (Bushee) Dumouchel, Mithat Gunduz, Amanda Cirello and Bindi Sohal for critical discussions. Work in the Potter lab is supported in part by NIH grants CA108775 and AT007531, a Cancer Center Core grant CA21765, and by the American Lebanese Syrian Associated Charities (ALSAC) and St. Jude Children’s Research Hospital (SJCRH).
References
- 1.Garattini E, Terao M. Increasing recognition of the importance of aldehyde oxidase in drug development and discovery. Drug Metab Rev. 2011;43(3):374–86. doi: 10.3109/03602532.2011.560606. [DOI] [PubMed] [Google Scholar]
- 2.Terao M, Romao MJ, Leimkuhler S, Bolis M, Fratelli M, Coelho C, et al. Structure and function of mammalian aldehyde oxidases. Arch Toxicol. 2016;90(4):753–80. doi: 10.1007/s00204-016-1683-1. [DOI] [PubMed] [Google Scholar]
- 3.Beedham C. Molybdenum hydroxylases: biological distribution and substrate-inhibitor specificity. Prog Med Chem. 1987;24:85–127. doi: 10.1016/s0079-6468(08)70420-x. [DOI] [PubMed] [Google Scholar]
- 4.Kitamura S, Sugihara K, Ohta S. Drug-metabolizing ability of molybdenum hydroxylases. Drug Metab Pharmacokinet. 2006;21(2):83–98. doi: 10.2133/dmpk.21.83. [DOI] [PubMed] [Google Scholar]
- 5.Sugihara K, Kitamura S, Tatsumi K. Involvement of mammalian liver cytosols and aldehyde oxidase in reductive metabolism of zonisamide. Drug Metab Dispos. 1996;24(2):199–202. [PubMed] [Google Scholar]
- 6.Obach RS, Prakash C, Kamel AM. Reduction and methylation of ziprasidone by glutathione, aldehyde oxidase, and thiol S-methyltransferase in humans: an in vitro study. Xenobiotica. 2012;42(11):1049–57. doi: 10.3109/00498254.2012.683203. [DOI] [PubMed] [Google Scholar]
- 7.Sodhi JK, Wong S, Kirkpatrick DS, Liu L, Khojasteh SC, Hop CE, et al. A novel reaction mediated by human aldehyde oxidase: amide hydrolysis of GDC-0834. Drug Metab Dispos. 2015;43(6):908–15. doi: 10.1124/dmd.114.061804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moriwaki Y, Yamamoto T, Takahashi S, Tsutsumi Z, Hada T. Widespread cellular distribution of aldehyde oxidase in human tissues found by immunohistochemistry staining. Histol Histopathol. 2001;16(3):745–53. doi: 10.14670/HH-16.745. [DOI] [PubMed] [Google Scholar]
- 9.Nishimura M, Naito S. Tissue-specific mRNA expression profiles of human phase I metabolizing enzymes except for cytochrome P450 and phase II metabolizing enzymes. Drug Metab Pharmacokinet. 2006;21(5):357–74. doi: 10.2133/dmpk.21.357. [DOI] [PubMed] [Google Scholar]
- 10.Coelho C, Foti A, Hartmann T, Santos-Silva T, Leimkuhler S, Romao MJ. Structural insights into xenobiotic and inhibitor binding to human aldehyde oxidase. Nat Chem Biol. 2015;11(10):779–83. doi: 10.1038/nchembio.1895. [DOI] [PubMed] [Google Scholar]
- 11.Dalvie D, Zhang C, Chen W, Smolarek T, Obach RS, Loi CM. Cross-species comparison of the metabolism and excretion of zoniporide: contribution of aldehyde oxidase to interspecies differences. Drug Metab Dispos. 2010;38(4):641–54. doi: 10.1124/dmd.109.030783. [DOI] [PubMed] [Google Scholar]
- 12.Akabane T, Tanaka K, Irie M, Terashita S, Teramura T. Case report of extensive metabolism by aldehyde oxidase in humans: pharmacokinetics and metabolite profile of FK3453 in rats, dogs, and humans. Xenobiotica. 2011;41(5):372–84. doi: 10.3109/00498254.2010.549970. [DOI] [PubMed] [Google Scholar]
- 13.Dittrich C, Greim G, Borner M, Weigang-Kohler K, Huisman H, Amelsberg A, et al. Phase I and pharmacokinetic study of BIBX 1382 BS, an epidermal growth factor receptor (EGFR) inhibitor, given in a continuous daily oral administration. Eur J Cancer. 2002;38(8):1072–80. doi: 10.1016/s0959-8049(02)00020-5. [DOI] [PubMed] [Google Scholar]
- 14.Kaye B, Offerman JL, Reid JL, Elliott HL, Hillis WS. A species difference in the presystemic metabolism of carbazeran in dog and man. Xenobiotica. 1984;14(12):935–45. doi: 10.3109/00498258409151492. [DOI] [PubMed] [Google Scholar]
- 15.Zhang X, Liu HH, Weller P, Zheng M, Tao W, Wang J, et al. In silico and in vitro pharmacogenetics: aldehyde oxidase rapidly metabolizes a p38 kinase inhibitor. Pharmacogenomics J. 2011;11(1):15–24. doi: 10.1038/tpj.2010.8. [DOI] [PubMed] [Google Scholar]
- 16.Diamond S, Boer J, Maduskuie TP, Jr, Falahatpisheh N, Li Y, Yeleswaram S. Species-specific metabolism of SGX523 by aldehyde oxidase and the toxicological implications. Drug Metab Dispos. 2010;38(8):1277–85. doi: 10.1124/dmd.110.032375. [DOI] [PubMed] [Google Scholar]
- 17.Strolin Benedetti M, Whomsley R, Baltes E. Involvement of enzymes other than CYPs in the oxidative metabolism of xenobiotics. Expert Opin Drug Metab Toxicol. 2006;2(6):895–921. doi: 10.1517/17425255.2.6.895. [DOI] [PubMed] [Google Scholar]
- 18.Strelevitz TJ, Orozco CC, Obach RS. Hydralazine as a selective probe inactivator of aldehyde oxidase in human hepatocytes: estimation of the contribution of aldehyde oxidase to metabolic clearance. Drug Metab Dispos. 2012;40(7):1441–8. doi: 10.1124/dmd.112.045195. [DOI] [PubMed] [Google Scholar]
- 19.Nirogi R, Kandikere V, Palacharla RC, Bhyrapuneni G, Kanamarlapudi VB, Ponnamaneni RK, et al. Identification of a suitable and selective inhibitor towards aldehyde oxidase catalyzed reactions. Xenobiotica. 2014;44(3):197–204. doi: 10.3109/00498254.2013.819594. [DOI] [PubMed] [Google Scholar]
- 20.Baer BR, Wienkers LC, Rock DA. Time-dependent inactivation of P450 3A4 by raloxifene: identification of Cys239 as the site of apoprotein alkylation. Chem Res Toxicol. 2007;20(6):954–64. doi: 10.1021/tx700037e. [DOI] [PubMed] [Google Scholar]
- 21.Zientek M, Jiang Y, Youdim K, Obach RS. In vitro-in vivo correlation for intrinsic clearance for drugs metabolized by human aldehyde oxidase. Drug Metab Dispos. 2010;38(8):1322–7. doi: 10.1124/dmd.110.033555. [DOI] [PubMed] [Google Scholar]
- 22.Hutzler JM, Yang YS, Albaugh D, Fullenwider CL, Schmenk J, Fisher MB. Characterization of aldehyde oxidase enzyme activity in cryopreserved human hepatocytes. Drug Metab Dispos. 2012;40(2):267–75. doi: 10.1124/dmd.111.042861. [DOI] [PubMed] [Google Scholar]
- 23.Akabane T, Gerst N, Naritomi Y, Masters JN, Tamura K. A practical and direct comparison of intrinsic metabolic clearance of several non-CYP enzyme substrates in freshly isolated and cryopreserved hepatocytes. Drug Metab Pharmacokinet. 2012;27(2):181–91. doi: 10.2133/dmpk.dmpk-11-rg-097. [DOI] [PubMed] [Google Scholar]
- 24.Zientek MA, Youdim K. Reaction phenotyping: advances in the experimental strategies used to characterize the contribution of drug-metabolizing enzymes. Drug Metab Dispos. 2015;43(1):163–81. doi: 10.1124/dmd.114.058750. [DOI] [PubMed] [Google Scholar]
- 25.Kitamura S, Sugihara K, Nakatani K, Ohta S, Ohhara T, Ninomiya S, et al. Variation of hepatic methotrexate 7-hydroxylase activity in animals and humans. IUBMB Life. 1999;48(6):607–11. doi: 10.1080/713803569. [DOI] [PubMed] [Google Scholar]
- 26.Al-Salmy HS. Individual variation in hepatic aldehyde oxidase activity. IUBMB Life. 2001;51(4):249–53. doi: 10.1080/152165401753311799. [DOI] [PubMed] [Google Scholar]
- 27.Fu C, Di L, Han X, Soderstrom C, Snyder M, Troutman MD, et al. Aldehyde oxidase 1 (AOX1) in human liver cytosols: quantitative characterization of AOX1 expression level and activity relationship. Drug Metab Dispos. 2013;41(10):1797–804. doi: 10.1124/dmd.113.053082. [DOI] [PubMed] [Google Scholar]
- 28.Hutzler JM, Yang YS, Brown C, Heyward S, Moeller T. Aldehyde oxidase activity in donor-matched fresh and cryopreserved human hepatocytes and assessment of variability in 75 donors. Drug Metab Dispos. 2014;42(6):1090–7. doi: 10.1124/dmd.114.057984. [DOI] [PubMed] [Google Scholar]
- 29.Barr JT, Jones JP, Oberlies NH, Paine MF. Inhibition of human aldehyde oxidase activity by diet-derived constituents: structural influence, enzyme-ligand interactions, and clinical relevance. Drug Metab Dispos. 2015;43(1):34–41. doi: 10.1124/dmd.114.061192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tayama Y, Sugihara K, Sanoh S, Miyake K, Morita S, Kitamura S, et al. Effect of tea beverages on aldehyde oxidase activity. Drug Metab Pharmacokinet. 2011;26(1):94–101. doi: 10.2133/dmpk.dmpk-10-nt-078. [DOI] [PubMed] [Google Scholar]
- 31.Kestell P, Dunlop IC, McCrystal MR, Evans BD, Paxton JW, Gamage RS, et al. Plasma pharmacokinetics of N-[2-(dimethylamino)ethyl]acridine-4-carboxamide in a phase I trial. Cancer Chemother Pharmacol. 1999;44(1):45–50. doi: 10.1007/s002800050943. [DOI] [PubMed] [Google Scholar]
- 32.Akabane T, Gerst N, Masters JN, Tamura K. A quantitative approach to hepatic clearance prediction of metabolism by aldehyde oxidase using custom pooled hepatocytes. Xenobiotica. 2012;42(9):863–71. doi: 10.3109/00498254.2012.670736. [DOI] [PubMed] [Google Scholar]
- 33.Manevski N, Balavenkatraman KK, Bertschi B, Swart P, Walles M, Camenisch G, et al. Aldehyde oxidase activity in fresh human skin. Drug Metab Dispos. 2014;42(12):2049–57. doi: 10.1124/dmd.114.060368. [DOI] [PubMed] [Google Scholar]
- 34.Kurosaki M, Bolis M, Fratelli M, Barzago MM, Pattini L, Perretta G, et al. Structure and evolution of vertebrate aldehyde oxidases: from gene duplication to gene suppression. Cell Mol Life Sci. 2013;70(10):1807–30. doi: 10.1007/s00018-012-1229-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beedham C, Bruce SE, Critchley DJ, al-Tayib Y, Rance DJ. Species variation in hepatic aldehyde oxidase activity. Eur J Drug Metab Pharmacokinet. 1987;12(4):307–10. doi: 10.1007/BF03189919. [DOI] [PubMed] [Google Scholar]
- 36.Garattini E, Terao M. The role of aldehyde oxidase in drug metabolism. Expert Opin Drug Metab Toxicol. 2012;8(4):487–503. doi: 10.1517/17425255.2012.663352. [DOI] [PubMed] [Google Scholar]
- 37.Hutzler JM, Cerny MA, Yang YS, Asher C, Wong D, Frederick K, et al. Cynomolgus monkey as a surrogate for human aldehyde oxidase metabolism of the EGFR inhibitor BIBX1382. Drug Metab Dispos. 2014;42(10):1751–60. doi: 10.1124/dmd.114.059030. [DOI] [PubMed] [Google Scholar]
- 38.Hosea NA, Collard WT, Cole S, Maurer TS, Fang RX, Jones H, et al. Prediction of human pharmacokinetics from preclinical information: comparative accuracy of quantitative prediction approaches. J Clin Pharmacol. 2009;49(5):513–33. doi: 10.1177/0091270009333209. [DOI] [PubMed] [Google Scholar]
- 39.Cashman J, Perroti B, Berkman C, Lin J. Pharmacokinetics and molecular detoxification. Environ Health Perspect. 1996;104:23–40. doi: 10.1289/ehp.96104s123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Redinbo MR, Potter PM. Mammalian Carboxylesterases: From drug targets to protein therapeutics. Drug Discov Today. 2005;10:313–25. doi: 10.1016/S1359-6446(05)03383-0. [DOI] [PubMed] [Google Scholar]
- 41.Laizure SC, Herring V, Hu Z, Witbrodt K, Parker RB. The role of human carboxylesterases in drug metabolism: have we overlooked their importance? Pharmacotherapy. 2013;33(2):210–22. doi: 10.1002/phar.1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hatfield MJ, Tsurkan L, Garrett M, Shaver T, Edwards CC, Hyatt JL, et al. Organ-specific carboxylesterase profiling identifies the small intestine and kidney as major contributors of activation of the anticancer prodrug CPT-11. Biochem Pharmacol. 2011;81:24–31. doi: 10.1016/j.bcp.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wadkins RM, Morton CL, Weeks JK, Oliver L, Wierdl M, Danks MK, et al. Structural constraints affect the metabolism of 7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecin (CPT-11) by carboxylesterases. Mol Pharmacol. 2001;60:355–62. doi: 10.1124/mol.60.2.355. [DOI] [PubMed] [Google Scholar]
- 44.Bencharit S, Morton CL, Howard-Williams EL, Danks MK, Potter PM, Redinbo MR. Structural insights into CPT-11 activation by mammalian carboxylesterases. Nat Struct Biol. 2002;9:337–42. doi: 10.1038/nsb790. [DOI] [PubMed] [Google Scholar]
- 45.Bencharit S, Morton CL, Xue Y, Potter PM, Redinbo MR. Structural basis of heroin and cocaine metabolism by a promiscuous human drug-processing enzyme. Nat Struct Biol. 2003;10:349–56. doi: 10.1038/nsb919. [DOI] [PubMed] [Google Scholar]
- 46.Wierdl M, Tsurkan L, Hyatt JL, Edwards CC, Hatfield MJ, Morton CL, et al. An improved human carboxylesterase for enzyme/prodrug therapy with CPT-11. Cancer gene therapy. 2008;15(3):183–92. doi: 10.1038/sj.cgt.7701112. [DOI] [PubMed] [Google Scholar]
- 47.Bencharit S, Morton CL, Hyatt JL, Kuhn P, Danks MK, Potter PM, et al. Crystal structure of human carboxylesterase 1 complexed with the Alzheimer’s drug tacrine. From binding promiscuity to selective inhibition. Chem Biol. 2003;10:341–9. doi: 10.1016/s1074-5521(03)00071-1. [DOI] [PubMed] [Google Scholar]
- 48.Humerickhouse R, Lohrbach K, Li L, Bosron W, Dolan M. Characterization of CPT-11 hydrolysis by human liver carboxylesterase isoforms hCE-1 and hCE-2. Cancer Res. 2000;60:1189–92. [PubMed] [Google Scholar]
- 49.Khanna R, Morton CL, Danks MK, Potter PM. Proficient metabolism of CPT-11 by a human intestinal carboxylesterase. Cancer Res. 2000;60:4725–8. [PubMed] [Google Scholar]
- 50.Potter PM, Pawlik CA, Morton CL, Naeve CW, Danks MK. Isolation and partial characterization of a cDNA encoding a rabbit liver carboxylesterase that activates the prodrug Irinotecan (CPT-11) Cancer Res. 1998;52:2646–51. [PubMed] [Google Scholar]
- 51.Gilson MK, Straatsma TP, McCammon JA, Ripoll DR, Faerman CH, Axelsen PH, et al. Open “back door” in a molecular dynamics simulation of acetylcholinesterase. Science. 1994;263:1276–8. doi: 10.1126/science.8122110. [DOI] [PubMed] [Google Scholar]
- 52.Zhou H-X, Wlodek ST, McCammon JA. Conformation gating as a mechanism for enzyme specificity. Proc Natl Acad Sci USA. 1998;95:9280–3. doi: 10.1073/pnas.95.16.9280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hatfield MJ, Tsurkan L, Hyatt JL, Yu X, Edwards CC, Hicks LD, et al. Biochemical and molecular analysis of carboxylesterase-mediated hydrolysis of cocaine and heroin. Br J Pharmacol. 2010;160(8):1916–28. doi: 10.1111/j.1476-5381.2010.00700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morton CL, Potter PM. Comparison of Escherichia coli, Saccharomyces cerevisiae, Pichia pastoris, Spodoptera frugiperda and COS7 cells for recombinant gene expression: Application to a rabbit liver carboxylesterase. Mol Biotechnol. 2000;16:193–202. doi: 10.1385/MB:16:3:193. [DOI] [PubMed] [Google Scholar]
- 55.Guemei AA, Cottrell J, Band R, Hehman H, Prudhomme M, Pavlov MV, et al. Human plasma carboxylesterase and butyrylcholinesterase enzyme activity: correlations with SN-38 pharmacokinetics during a prolonged infusion of irinotecan. Cancer Chemother Pharmacol. 2001;47:283–90. doi: 10.1007/s002800000258. [DOI] [PubMed] [Google Scholar]
- 56.Morton CL, Wadkins RM, Danks MK, Potter PM. The anticancer prodrug CPT-11 is a potent inhibitor of acetylcholinesterase but is rapidly catalyzed to SN-38 by butyrylcholinesterase. Cancer research. 1999;59(7):1458–63. [PubMed] [Google Scholar]
- 57.Williams ET, Wang H, Wrighton SA, Qian YW, Perkins EJ. Genomic analysis of the carboxylesterases: identification and classification of novel forms. Mol Phylogenet Evol. 2010;57(1):23–34. doi: 10.1016/j.ympev.2010.05.018. [DOI] [PubMed] [Google Scholar]
- 58.Holmes RS, Cox LA, VandeBerg JL. Comparative studies of mammalian acid lipases: Evidence for a new gene family in mouse and rat (Lipo) Comp Biochem Physiol Part D Genomics Proteomics. 2010;5(3):217–26. doi: 10.1016/j.cbd.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morton CL, Wierdl M, Oliver L, Ma M, Danks MK, Stewart CF, et al. Activation of CPT-11 in mice: Identification and analysis of a highly effective plasma esterase. Cancer Res. 2000;60:4206–10. [PubMed] [Google Scholar]
- 60.Danks MK, Morton CL, Krull EJ, Cheshire PJ, Richmond LB, Naeve CW, et al. Comparison of activation of CPT-11 by rabbit and human carboxylesterases for use in enzyme/prodrug therapy. Clin Cancer Res. 1999;5:917–24. [PubMed] [Google Scholar]
- 61.Morton CL, Iacono L, Hyatt JL, Taylor KR, Cheshire PJ, Houghton PJ, et al. Metabolism and antitumor activity of CPT-11 in plasma esterase-deficient mice. Cancer Chemother Pharmacol. 2005;56:629–36. doi: 10.1007/s00280-005-1027-y. [DOI] [PubMed] [Google Scholar]
- 62.Soares ER. Identification of a new allele of Es-1 segregating in an inbred strain of mice. Biochem Genet. 1979;17(7/8):577–83. doi: 10.1007/BF00502119. [DOI] [PubMed] [Google Scholar]
- 63.Nishimuta H, Houston JB, Galetin A. Hepatic, intestinal, renal, and plasma hydrolysis of prodrugs in human, cynomolgus monkey, dog, and rat: implications for in vitro-in vivo extrapolation of clearance of prodrugs. Drug Metab Dispos. 2014;42(9):1522–31. doi: 10.1124/dmd.114.057372. [DOI] [PubMed] [Google Scholar]
- 64.Williams ET, Bacon JA, Bender DM, Lowinger JJ, Guo WK, Ehsani ME, et al. Characterization of the expression and activity of carboxylesterases 1 and 2 from the beagle dog, cynomolgus monkey, and human. Drug Metab Dispos. 2011;39(12):2305–13. doi: 10.1124/dmd.111.041335. [DOI] [PubMed] [Google Scholar]
- 65.Zhu HJ, Patrick KS, Yuan HJ, Wang JS, Donovan JL, DeVane CL, et al. Two CES1 gene mutations lead to dysfunctional carboxylesterase 1 activity in man: clinical significance and molecular basis. Am J Hum Genet. 2008;82(6):1241–8. doi: 10.1016/j.ajhg.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhu HJ, Wang X, Gawronski BE, Brinda BJ, Angiolillo DJ, Markowitz JS. Carboxylesterase 1 as a determinant of clopidogrel metabolism and activation. The Journal of pharmacology and experimental therapeutics. 2013;344(3):665–72. doi: 10.1124/jpet.112.201640. [DOI] [PubMed] [Google Scholar]
- 67.Zhu HJ, Markowitz JS. Activation of the antiviral prodrug oseltamivir is impaired by two newly identified carboxylesterase 1 variants. Drug Metab Dispos. 2009;37(2):264–7. doi: 10.1124/dmd.108.024943. [DOI] [PubMed] [Google Scholar]
- 68.Hu ZY, Edginton AN, Laizure SC, Parker RB. Physiologically based pharmacokinetic modeling of impaired carboxylesterase-1 activity: effects on oseltamivir disposition. Clin Pharmacokinet. 2014;53(9):825–36. doi: 10.1007/s40262-014-0160-3. [DOI] [PubMed] [Google Scholar]
- 69.Hatfield MJ, Potter PM. Carboxylesterase inhibitors. Expert opinion on therapeutic patents. 2011;21(8):1159–71. doi: 10.1517/13543776.2011.586339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hicks LD, Hyatt JL, Moak T, Edwards CC, Tsurkan L, Wierdl M, et al. Analysis of the inhibition of mammalian carboxylesterases by novel fluorobenzoins and fluorobenzils. Bioorg Med Chem. 2007;15:3801–17. doi: 10.1016/j.bmc.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hicks LD, Hyatt JL, Stoddard S, Tsurkan L, Edwards CC, Wadkins RM, et al. Improved, selective, human intestinal carboxylesterase inhibitors designed to modulate 7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecin (Irinotecan; CPT-11) toxicity. J Med Chem. 2009;52(12):3742–52. doi: 10.1021/jm9001296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hyatt JL, Moak T, Hatfield JM, Tsurkan L, Edwards CC, Wierdl M, et al. Selective inhibition of carboxylesterases by isatins, indole-2,3-diones. J Med Chem. 2007;50:1876–85. doi: 10.1021/jm061471k. [DOI] [PubMed] [Google Scholar]
- 73.Hyatt JL, Stacy V, Wadkins RM, Yoon KJ, Wierdl M, Edwards CC, et al. Inhibition of carboxylesterases by benzil (diphenylethane-1,2-dione) and heterocyclic analogues is dependent upon the aromaticity of the ring and the flexibility of the dione moiety. J Med Chem. 2005;48:5543–50. doi: 10.1021/jm0504196. [DOI] [PubMed] [Google Scholar]
- 74.Hyatt JL, Tsurkan L, Wierdl M, Edwards CC, Danks MK, Potter PM. Intracellular inhibition of carboxylesterases by benzil: Modulation of CPT-11 cytotoxicity. Mol Cancer Ther. 2006;5:2281–8. doi: 10.1158/1535-7163.MCT-06-0160. [DOI] [PubMed] [Google Scholar]
- 75.Hyatt JL, Wadkins RM, Tsurkan L, Hicks LD, Hatfield MJ, Edwards CC, et al. Planarity and constraint of the carbonyl groups in 1,2-diones are determinants for selective inhibition of human carboxylesterase 1. J Med Chem. 2007;50:5727–34. doi: 10.1021/jm0706867. [DOI] [PubMed] [Google Scholar]
- 76.Parkinson EI, Jason Hatfield M, Tsurkan L, Hyatt JL, Edwards CC, Hicks LD, et al. Requirements for mammalian carboxylesterase inhibition by substituted ethane-1,2-diones. Bioorg Med Chem. 2011;19(15):4635–43. doi: 10.1016/j.bmc.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wadkins RM, Hyatt JL, Yoon KJ, Morton CL, Lee RE, Damodaran K, et al. Identification of novel selective human intestinal carboxylesterase inhibitors for the amelioration of irinotecan-induced diarrhea: Synthesis, quantitative structure-activity relationship analysis, and biological activity. Mol Pharmacol. 2004;65:1336–43. doi: 10.1124/mol.65.6.1336. [DOI] [PubMed] [Google Scholar]
- 78.Wadkins RM, Hyatt JL, Yoon KJ, Morton CL, Lee RE, Damodaran K, et al. Discovery of novel selective inhibitors of human intestinal carboxylesterase for the amelioration of irinotecan-induced diarrhea: synthesis, quantitative structure-activity relationship analysis, and biological activity. Molecular pharmacology. 2004;65(6):1336–43. doi: 10.1124/mol.65.6.1336. [DOI] [PubMed] [Google Scholar]
- 79.Young BM, Hyatt JL, Bouck DC, Chen T, Hanumesh P, Price J, et al. Structure-activity relationships of substituted 1-pyridyl-2-phenyl-1,2-ethanediones: potent, selective carboxylesterase inhibitors. Journal of medicinal chemistry. 2010;53(24):8709–15. doi: 10.1021/jm101101q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hatfield MJ, Tsurkan LG, Hyatt JL, Edwards CC, Lemoff A, Jeffries C, et al. Modulation of Esterified Drug Metabolism by Tanshinones from Salvia miltiorrhiza (“Danshen”) Journal of natural products. 2013;76(1):36–44. doi: 10.1021/np300628a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tsurkan LG, Hatfield MJ, Edwards CC, Hyatt JL, Potter PM. Inhibition of human carboxylesterases hCE1 and hiCE by cholinesterase inhibitors. Chemico-biological interactions. 2013;203(1):226–30. doi: 10.1016/j.cbi.2012.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Remmel RP, Nagar S, Argikar UA. Conjugative Metabolism of Drugs. In: Zhang D, Zhu M, Humphreys WG, editors. Drug Metabolism in Drug Design and Development. 1. Hoboken, NJ: Wiley-Interscience, John Wiley and Sons, Inc; 2007. p. 609. [Google Scholar]
- 83.Argikar UA. Unusual glucuronides. Drug metabolism and disposition: the biological fate of chemicals. 2012;40(7):1239–51. doi: 10.1124/dmd.112.045096. [DOI] [PubMed] [Google Scholar]
- 84.Bushee JL, Argikar UA. An experimental approach to enhance precursor ion fragmentation for metabolite identification studies: application of dual collision cells in an orbital trap. Rapid communications in mass spectrometry: RCM. 2011;25(10):1356–62. doi: 10.1002/rcm.4996. [DOI] [PubMed] [Google Scholar]
- 85.Mackenzie PI, Bock KW, Burchell B, Guillemette C, Ikushiro S, Iyanagi T, et al. Nomenclature update for the mammalian UDP glycosyltransferase (UGT) gene superfamily. Pharmacogenet Genomics. 2005;15(10):677–85. doi: 10.1097/01.fpc.0000173483.13689.56. [DOI] [PubMed] [Google Scholar]
- 86.Mackenzie PI, Rogers A, Treloar J, Jorgensen BR, Miners JO, Meech R. Identification of UDP glycosyltransferase 3A1 as a UDP N-acetylglucosaminyltransferase. J Biol Chem. 2008;283(52):36205–10. doi: 10.1074/jbc.M807961200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mackenzie PI, Rogers A, Elliot DJ, Chau N, Hulin JA, Miners JO, et al. The Novel Udp Glycosyltransferase 3a2: Cloning, Catalytic Properties and Tissue Distribution. Mol Pharmacol. 2011;79(3):472–8. doi: 10.1124/mol.110.069336. [DOI] [PubMed] [Google Scholar]
- 88.Fallon JK, Neubert H, Goosen TC, Smith PC. Targeted precise quantification of 12 human recombinant uridine-diphosphate glucuronosyl transferase 1A and 2B isoforms using nano-ultra-high-performance liquid chromatography/tandem mass spectrometry with selected reaction monitoring. Drug Metab Dispos. 2013;41(12):2076–80. doi: 10.1124/dmd.113.053801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Remmel RP, Zhou J, Argikar UA. Uridine diphospho Glucuronosyl Transferases. In: Pearson P, Wienkers L, editors. Handbook of Drug Metabolism. 2. New York, NY: Informa Health Care; 2009. p. 630. [Google Scholar]
- 90.Zhou J, Argikar UA, Remmel RP. Functional analysis of UGT1A4(P24T) and UGT1A4(L48V) variant enzymes. Pharmacogenomics. 2011;12(12):1671–9. doi: 10.2217/pgs.11.105. [DOI] [PubMed] [Google Scholar]
- 91.Krishnaswamy S, Hao Q, Al-Rohaimi A, Hesse LM, von Moltke LL, Greenblatt DJ, et al. UDP glucuronosyltransferase (UGT) 1A6 pharmacogenetics: II. Functional impact of the three most common nonsynonymous UGT1A6 polymorphisms (S7A, T181A, and R184S) The Journal of pharmacology and experimental therapeutics. 2005;313(3):1340–6. doi: 10.1124/jpet.104.081968. [DOI] [PubMed] [Google Scholar]
- 92.Miners JO, Knights KM, Houston JB, Mackenzie PI. In vitro-in vivo correlation for drugs and other compounds eliminated by glucuronidation in humans: pitfalls and promises. Biochem Pharmacol. 2006;71(11):1531–9. doi: 10.1016/j.bcp.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 93.Boase S, Miners JO. In vitro-in vivo correlations for drugs eliminated by glucuronidation: investigations with the model substrate zidovudine. Br J Clin Pharmacol. 2002;54(5):493–503. doi: 10.1046/j.1365-2125.2002.01669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mistry M, Houston JB. Glucuronidation in vitro and in vivo. Comparison of intestinal and hepatic conjugation of morphine, naloxone, and buprenorphine. Drug Metab Dispos. 1987;15(5):710–7. [PubMed] [Google Scholar]
- 95.Soars MG, Burchell B, Riley RJ. In vitro analysis of human drug glucuronidation and prediction of in vivo metabolic clearance. The Journal of pharmacology and experimental therapeutics. 2002;301(1):382–90. doi: 10.1124/jpet.301.1.382. [DOI] [PubMed] [Google Scholar]
- 96.Cubitt HE, Houston JB, Galetin A. Relative importance of intestinal and hepatic glucuronidation-impact on the prediction of drug clearance. Pharm Res. 2009;26(5):1073–83. doi: 10.1007/s11095-008-9823-9. [DOI] [PubMed] [Google Scholar]
- 97.Cubitt HE, Houston JB, Galetin A. Prediction of human drug clearance by multiple metabolic pathways: integration of hepatic and intestinal microsomal and cytosolic data. Drug Metab Dispos. 2011;39(5):864–73. doi: 10.1124/dmd.110.036566. [DOI] [PubMed] [Google Scholar]
- 98.Gill KL, Houston JB, Galetin A. Characterization of in vitro glucuronidation clearance of a range of drugs in human kidney microsomes: comparison with liver and intestinal glucuronidation and impact of albumin. Drug Metab Dispos. 2012;40(4):825–35. doi: 10.1124/dmd.111.043984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Naritomi Y, Nakamori F, Furukawa T, Tabata K. Prediction of hepatic and intestinal glucuronidation using in vitro-in vivo extrapolation. Drug Metab Pharmacokinet. 2015;30(1):21–9. doi: 10.1016/j.dmpk.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 100.Galetin A. Rationalizing underprediction of drug clearance from enzyme and transporter kinetic data: from in vitro tools to mechanistic modeling. Methods Mol Biol. 2014;1113:255–88. doi: 10.1007/978-1-62703-758-7_13. [DOI] [PubMed] [Google Scholar]
- 101.Fujiwara R, Nakajima M, Yamanaka H, Katoh M, Yokoi T. Product inhibition of UDP-glucuronosyltransferase (UGT) enzymes by UDP obfuscates the inhibitory effects of UGT substrates. Drug Metab Dispos. 2008;36(2):361–7. doi: 10.1124/dmd.107.018705. [DOI] [PubMed] [Google Scholar]
- 102.Manevski N, Yli-Kauhaluoma J, Finel M. UDP-glucuronic acid binds first and the aglycone substrate binds second to form a ternary complex in UGT1A9-catalyzed reactions, in both the presence and absence of bovine serum albumin. Drug Metab Dispos. 2012;40(11):2192–203. doi: 10.1124/dmd.112.047746. [DOI] [PubMed] [Google Scholar]
- 103.Remmel RP, Zhou J, Argikar UA. UDP-Glucuronosyl Transferases. In: Rodrigues DA, editor. Drug-Drug Interactions. 2. New York, NY: Informa Health Care; 2008. p. 744. [Google Scholar]
- 104.Williams JA, Hyland R, Jones BC, Smith DA, Hurst S, Goosen TC, et al. Drug-drug interactions for UDP-glucuronosyltransferase substrates: a pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. Drug Metab Dispos. 2004;32(11):1201–8. doi: 10.1124/dmd.104.000794. [DOI] [PubMed] [Google Scholar]
- 105.Ogilvie BW, Zhang D, Li W, Rodrigues AD, Gipson AE, Holsapple J, et al. Glucuronidation converts gemfibrozil to a potent, metabolism-dependent inhibitor of CYP2C8: implications for drug-drug interactions. Drug Metab Dispos. 2006;34(1):191–7. doi: 10.1124/dmd.105.007633. [DOI] [PubMed] [Google Scholar]
- 106.Tornio A, Filppula AM, Kailari O, Neuvonen M, Nyronen TH, Tapaninen T, et al. Glucuronidation converts clopidogrel to a strong time-dependent inhibitor of CYP2C8: a phase II metabolite as a perpetrator of drug-drug interactions. Clin Pharmacol Ther. 2014;96(4):498–507. doi: 10.1038/clpt.2014.141. [DOI] [PubMed] [Google Scholar]
- 107.Shitara Y, Hirano M, Sato H, Sugiyama Y. Gemfibrozil and its glucuronide inhibit the organic anion transporting polypeptide 2 (OATP2/OATP1B1:SLC21A6)-mediated hepatic uptake and CYP2C8-mediated metabolism of cerivastatin: analysis of the mechanism of the clinically relevant drug-drug interaction between cerivastatin and gemfibrozil. The Journal of pharmacology and experimental therapeutics. 2004;311(1):228–36. doi: 10.1124/jpet.104.068536. [DOI] [PubMed] [Google Scholar]
- 108.Argikar UA, Liang G, Bushee JL, Hosagrahara VP, Lee W. Evaluation of pharmaceutical excipients as cosolvents in 4-methyl umbelliferone glucuronidation in human liver microsomes: applications for compounds with low solubility. Drug metabolism and pharmacokinetics. 2011;26(1):102–6. doi: 10.2133/dmpk.dmpk-10-sh-086. [DOI] [PubMed] [Google Scholar]
- 109.Miners JO, Smith PA, Sorich MJ, McKinnon RA, Mackenzie PI. Predicting human drug glucuronidation parameters: application of in vitro and in silico modeling approaches. Annu Rev Pharmacol Toxicol. 2004;44:1–25. doi: 10.1146/annurev.pharmtox.44.101802.121546. [DOI] [PubMed] [Google Scholar]
- 110.Walsky RL, Bauman JN, Bourcier K, Giddens G, Lapham K, Negahban A, et al. Optimized assays for human UDP-glucuronosyltransferase (UGT) activities: altered alamethicin concentration and utility to screen for UGT inhibitors. Drug Metab Dispos. 2012;40(5):1051–65. doi: 10.1124/dmd.111.043117. [DOI] [PubMed] [Google Scholar]
- 111.Engtrakul JJ, Foti RS, Strelevitz TJ, Fisher MB. Altered AZT (3′-azido-3′-deoxythymidine) glucuronidation kinetics in liver microsomes as an explanation for underprediction of in vivo clearance: comparison to hepatocytes and effect of incubation environment. Drug Metab Dispos. 2005;33(11):1621–7. doi: 10.1124/dmd.105.005058. [DOI] [PubMed] [Google Scholar]
- 112.Gunduz M, Argikar UA, Baeschlin D, Ferreira S, Hosagrahara V, Harriman S. Identification of a novel N-carbamoyl glucuronide: in vitro, in vivo, and mechanistic studies. Drug metabolism and disposition: the biological fate of chemicals. 2010;38(3):361–7. doi: 10.1124/dmd.109.030650. [DOI] [PubMed] [Google Scholar]
- 113.Fisher MB, Campanale K, Ackermann BL, VandenBranden M, Wrighton SA. In vitro glucuronidation using human liver microsomes and the pore-forming peptide alamethicin. Drug Metab Dispos. 2000;28(5):560–6. [PubMed] [Google Scholar]
- 114.Soars MG, Ring BJ, Wrighton SA. The effect of incubation conditions on the enzyme kinetics of udp-glucuronosyltransferases. Drug Metab Dispos. 2003;31(6):762–7. doi: 10.1124/dmd.31.6.762. [DOI] [PubMed] [Google Scholar]
- 115.Uchaipichat V, Mackenzie PI, Guo XH, Gardner-Stephen D, Galetin A, Houston JB, et al. Human udp-glucuronosyltransferases: isoform selectivity and kinetics of 4-methylumbelliferone and 1-naphthol glucuronidation, effects of organic solvents, and inhibition by diclofenac and probenecid. Drug Metab Dispos. 2004;32(4):413–23. doi: 10.1124/dmd.32.4.413. [DOI] [PubMed] [Google Scholar]
- 116.Chauret N, Gauthier A, Nicoll-Griffith DA. Effect of common organic solvents on in vitro cytochrome P450-mediated metabolic activities in human liver microsomes. Drug Metab Dispos. 1998;26(1):1–4. [PubMed] [Google Scholar]
- 117.Oleson L, Court MH. Effect of the beta-glucuronidase inhibitor saccharolactone on glucuronidation by human tissue microsomes and recombinant UDP-glucuronosyltransferases. J Pharm Pharmacol. 2008;60(9):1175–82. doi: 10.1211/jpp.60.9.0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rowland A, Gaganis P, Elliot DJ, Mackenzie PI, Knights KM, Miners JO. Binding of inhibitory fatty acids is responsible for the enhancement of UDP-glucuronosyltransferase 2B7 activity by albumin: implications for in vitro-in vivo extrapolation. J Pharmacol Exp Ther. 2007;321(1):137–47. doi: 10.1124/jpet.106.118216. [DOI] [PubMed] [Google Scholar]
- 119.Rowland A, Knights KM, Mackenzie PI, Miners JO. The “albumin effect” and drug glucuronidation: bovine serum albumin and fatty acid-free human serum albumin enhance the glucuronidation of UDP-glucuronosyltransferase (UGT) 1A9 substrates but not UGT1A1 and UGT1A6 activities. Drug Metab Dispos. 2008;36(6):1056–62. doi: 10.1124/dmd.108.021105. [DOI] [PubMed] [Google Scholar]
- 120.Bushee JL, Liang G, Dunne CE, Harriman SP, Argikar UA. Identification of saturated and unsaturated fatty acids released during microsomal incubations. Xenobiotica; the fate of foreign compounds in biological systems. 2014;44(8):687–95. doi: 10.3109/00498254.2014.884253. [DOI] [PubMed] [Google Scholar]
- 121.Lombardo F, Waters NJ, Argikar UA, Dennehy MK, Zhan J, Gunduz M, et al. Comprehensive assessment of human pharmacokinetic prediction based on in vivo animal pharmacokinetic data, part 1: volume of distribution at steady state. Journal of clinical pharmacology. 2013;53(2):167–77. doi: 10.1177/0091270012440281. [DOI] [PubMed] [Google Scholar]
- 122.Lombardo F, Waters NJ, Argikar UA, Dennehy MK, Zhan J, Gunduz M, et al. Comprehensive assessment of human pharmacokinetic prediction based on in vivo animal pharmacokinetic data, part 2: clearance. Journal of clinical pharmacology. 2013;53(2):178–91. doi: 10.1177/0091270012440282. [DOI] [PubMed] [Google Scholar]
- 123.Deguchi T, Watanabe N, Kurihara A, Igeta K, Ikenaga H, Fusegawa K, et al. Human pharmacokinetic prediction of UDP-glucuronosyltransferase substrates with an animal scale-up approach. Drug Metab Dispos. 2011;39(5):820–9. doi: 10.1124/dmd.110.037457. [DOI] [PubMed] [Google Scholar]
- 124.Gibson CR, Lu P, Maciolek C, Wudarski C, Barter Z, Rowland-Yeo K, et al. Using human recombinant UDP-glucuronosyltransferase isoforms and a relative activity factor approach to model total body clearance of laropiprant (MK-0524) in humans. Xenobiotica. 2013;43(12):1027–36. doi: 10.3109/00498254.2013.791761. [DOI] [PubMed] [Google Scholar]
- 125.Kilford PJ, Stringer R, Sohal B, Houston JB, Galetin A. Prediction of drug clearance by glucuronidation from in vitro data: use of combined cytochrome P450 and UDP-glucuronosyltransferase cofactors in alamethicin-activated human liver microsomes. Drug Metab Dispos. 2009;37(1):82–9. doi: 10.1124/dmd.108.023853. [DOI] [PubMed] [Google Scholar]