

Abstract
Neurogenesis is essential to brain development and plays a central role in the response to brain injury. Stroke and head trauma stimulate proliferation of endogenous neural stem cells (NSCs); however, the survival of young neurons is sharply reduced by postinjury inflammation. Cellular mitochondria are critical to successful neurogenesis and are a major target of inflammatory injury. Mitochondrial protection was shown to improve survival of young neurons. This study tested whether reducing cellular microRNA-210 (miR-210) would enhance mitochondrial function and improve survival of young murine neurons under inflammatory conditions. Several studies have demonstrated the potential of miR-210 inhibition to enhance and protect mitochondrial function through upregulation of mitochondrial proteins. Here, miR-210 inhibition significantly increased neuronal survival and protected the activity of mitochondrial enzymes cytochrome c oxidase and aconitase in differentiating NSC cultures exposed to inflammatory mediators. Unexpectedly, we found that reducing miR-210 significantly attenuated NSC proliferation upon induction of differentiation. Further investigation revealed that increased mitochondrial function suppressed the shift to primarily glycolytic metabolism and reduced mitochondrial length characteristic of dividing cells. Activation of AMP-regulated protein kinase–retinoblastoma signaling is important in NSC proliferation and the reduction of this activation observed by miR-210 inhibition is one mechanism contributing to the reduced proliferation. Postinjury neurogenesis occurs as a burst of proliferation that peaks in days, followed by migration and differentiation over weeks. Our studies suggest that mitochondrial protective miR-210 inhibition should be delayed until after the initial burst of proliferation, but could be beneficial during the prolonged differentiation stage.
SIGNIFICANCE STATEMENT Increasing the success of endogenous neurogenesis after brain injury holds therapeutic promise. Postinjury inflammation markedly reduces newborn neuron survival. This study found that enhancement of mitochondrial function by reducing microRNA-210 (miR-210) levels could improve survival of young neurons under inflammatory conditions. miR-210 inhibition protected the activity of mitochondrial enzymes cytochrome c oxidase and aconitase. Conversely, we observed decreased precursor cell proliferation likely due to suppression of the AMP-regulated protein kinase–retinoblastoma axis with miR-210 inhibition. Therefore, mitochondrial protection is a double-edged sword: early inhibition reduces proliferation, but inhibition later significantly increases neuroblast survival. This explains in part the contradictory published reports of the effects of miR-210 on neurogenesis.
Keywords: AMP kinase, metabolism, microRNA, mitochondria, neurogenesis, stem cell
Introduction
In addition to its central role in normal and pathological brain development, neurogenesis is stimulated in several pathological conditions including stroke (Johansson et al., 1999; Fallon et al., 2000; Magavi et al., 2000). Focal cerebral ischemia causes a transient increase in neural stem cell (NSC) proliferation (Arvidsson et al., 2001; Jiang et al., 2001; Jin et al., 2001; Parent et al., 2002). However, these studies also demonstrated that 80% or more of the newly produced neurons die shortly after migration to the injury site (Arvidsson et al., 2002; Parent et al., 2002). Although postischemic neurogenesis is beneficial for recovery (Leker et al., 2007; Wang et al., 2012; Sun et al., 2013), unfavorable proinflammatory activation of microglia characteristic of injured brain to account for much of the apoptotic death of immature neurons in this setting (Ekdahl et al., 2003; Monje et al., 2003).
Activated microglia produce proinflammatory factors, including IL-6, TNF-α, reactive oxygen species (ROS), and nitric oxide (Pocock and Liddle, 2001; Hanisch, 2002; Rock et al., 2004). Cellular mitochondria are the major target of injury by these proinflammatory molecules. TNF-α induces mitochondrial damage through suppression of mitochondrial complexes I and IV and pyruvate dehydrogenase (Stadler et al., 1992; Zell et al., 1997; Samavati et al., 2008). Exposure to increased ROS leads to impairment of mitochondrial lipids, sulfhydryl groups, and iron–sulfur complexes (Wagner et al., 1990; Halliwell, 2006). Nitric oxide is a potent inhibitor of mitochondrial cytochrome c oxidase (complex IV) (Brown and Borutaite, 2001).
Several studies have indicated that mitochondria play a key role in the proliferative and differentiation potential of stem cells (Voloboueva et al., 2010; Wang et al., 2011; Zhang et al., 2012; Candelario et al., 2013; Pereira et al., 2013). The undifferentiated proliferative state is associated with lower mitochondrial activity and more glycolytic metabolism, whereas increased mitochondrial metabolism is important for successful differentiation (Zhang et al., 2012; Candelario et al., 2013). This is especially true of daughter cell types with high ATP requirements, such as neurons (Spitkovsky et al., 2004; Facucho-Oliveira et al., 2007).
Previously, we observed that mitochondrial inhibition promoted rapid loss of mitochondrial membrane potential and induced selective death of young doublecortin-positive (Dcx+) neurons in differentiating NSC cultures. We demonstrated that mitochondria-protective compounds attenuated inflammation-associated loss of Dcx+ cells both in vivo and in vitro (Voloboueva et al., 2010).
In this study, we explored the effect of reducing microRNA-210 (miR-210) levels to protect mitochondrial function and test the ability of this manipulation to rescue the impairment of neurogenesis caused by microglia associated inflammation in vitro. MicroRNAs (miRNAs) are a large class of genomically encoded short (∼22 nt long) RNAs that control gene expression often by binding target messenger RNAs (mRNAs). Because microRNAs coordinately regulate multiple targets, they are attractive candidates for development of new therapies. miR-210 downregulates mitochondrial function and mediates the metabolic switch to glycolysis (Chen et al., 2010; Favaro et al., 2010; White et al., 2015). In particular, miR-210 decreases expression of subunit D of succinate dehydrogenase mitochondrial complex II (Puisségur et al., 2011), cytochrome c assembly protein (COX10), and iron–sulfur cluster scaffold homolog (ISCU) (Chen et al., 2010). COX10 is involved in the biosynthesis of heme-α, an essential component of cytochrome c oxidase (complex IV) of the respiratory chain. ISCU is essential for the assembly of iron–sulfur clusters that are major functional groups of enzymes in the mitochondrial electron transport chain and tricarboxylic acid cycle (Johnson et al., 2005).
The purpose of this study was to determine whether inhibiting miR-210 would improve mitochondrial function and enhance neuroblast survival during neurogenesis in the setting of inflammation in vitro. We observed mitochondrial enhancement and significantly increased survival of immature neurons during inflammation. However, we unexpectedly found that miR-210 inhibition significantly attenuated NSC proliferation at the early stages of differentiation. Neurogenesis is a complex process of many steps. Our findings emphasize the necessity of a deeper understanding of the role of mitochondria and metabolism during different stages of neurogenesis.
Materials and Methods
Cell culture and miR-210 transfection.
All experiments were performed according to a protocol approved by the Stanford Animal Care and Use Committee. NSCs were isolated from male and female Swiss Webster newborn mice (Charles River Laboratories). The brains were removed, freed of meninges, diced with a sterile razor blade in dissociation buffer (DMEM/F12 medium containing 2.5 U/ml papain, Worthington; 1 U/ml Dispase II, Roche Diagnostics; and 250 U/ml DNase I, Worthington). After a 1 h incubation, the cells were washed once with DMEM/F12 supplemented with 10% fetal bovine serum (FBS) (HyClone) and then twice with DMEM/F12. The cells were resuspended in growth medium, Neurobasal A (Invitrogen), with 2 mm l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen), B-27 without vitamin A (Invitrogen), 20 ng/ml FGF-2 (Peprotech), and 20 ng/ml EGF (Peprotech) and plated at density of 1 brain per 6-well plate. Neural precursor cells proliferated and started to form neurospheres in 2–3 d. To prevent attachment of the neurospheres to the bottom, the medium was pipetted to resuspend the plated neurospheres. The neurospheres were passaged when the diameter of neurospheres reached ∼100 μm. For passaging, the floating neurospheres were collected by centrifugation at 400 × g for 5 min. The supernatant was removed and the cells were gently triturated with a 200 μl pipette tip 20–25 times, resuspended in growth medium, and plated in 6-well plates after 1:2 dilution.
Neural precursor cells were induced to differentiate by plating dissociated neurospheres into laminin-coated 24-well plates. The plates were coated with 10 μg/well laminin (Invitrogen) in H2O for 2–3 h and then rinsed 2 times with dH2O. The floating neurospheres were again collected by centrifugation and the cell pellet was resuspended in a small volume of differentiation medium, Neurobasal A (Invitrogen), B27 without vitamin A (Invitrogen), 1% FBS, 100 nm all-trans retinoic acid (Sigma-Aldrich), 1 ng/ml FGF-2 (Peprotech), 10 ng/ml brain-derived neurotrophic factor (Peprotech), and 10 ng/ml NT3 (Peprotech). After trituration, cells were resuspended in 5 ml of differentiation medium, passed through a 70 μm cell strainer (BD Biosciences), and plated onto laminin-coated 24-well plates at a density of ∼100,000 cells/well. Half of the differentiation medium was changed every 2 d. The cells were transfected with miR-210 inhibitor or negative control on days 1–2 or 4–5 of differentiation, as explained below. Treatment with microglial conditioned media started the day after the transfection and the cells were collected for biochemical assays or fixed for imaging the next day.
BV-2 cells, primary microglia, and conditioned media (CM).
BV-2 murine microglia were plated on uncoated plastic tissue culture plates and grown in DMEM supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. Primary microglia were isolated by shaking off from 2- to 3-week-old murine mixed glial cultures as described previously (Yenari et al., 2006) and plated at 1–2 × 106 cells/well in 24-well plates. BV-2 cells or primary microglial cells were treated with fresh differentiation medium without serum with added lipopolysaccharide (LPS, 1 μg/ml; Sigma-Aldrich) to prepare CM from activated microglia or without LPS for control medium for 3 h. The medium was collected, supplemented with 1% serum, and applied to differentiating neural progenitor cells.
Transient focal cerebral ischemia.
All experiments were performed according to a protocol approved by the Stanford Animal Care and Use Committee. C57BL6 mice were purchased from Charles River Laboratories. Transient focal cerebral ischemia was induced in male mice, 8–10 weeks of age, weighing 25–30 g, by the intraluminal suture method described previously (Han et al., 2009). Briefly, mice were anesthetized with isoflurane and a 6–0 silicon coated suture was inserted into the left common carotid artery and advanced to occlude the opening of the middle cerebral artery for 60 min via a midline neck incision. Reperfusion was obtained by withdrawal of the suture. During all surgical procedures, animals were maintained normothermic (37°C ± 0.5°C) with a servo-controlled heating blanket (Harvard Apparatus) with rectal temperature monitoring. Pulse oximetry, heart rate, and respiratory rate were monitored continuously (STARR Life Sciences).
TNF-α and IL-6 measurements.
For measurements of proinflammatory cytokines in culture medium, the medium from LPS-treated or control DMEM-treated BV-2 cells or primary microglia was collected after 3 h of LPS treatment (1 μg/ml). For ex vivo cytokine measurements, ischemic or sham-operated mice were killed 3 d after ischemia and perfused with 0.9% saline. The brains were removed and the peri-infarct areas and corresponding brain areas in sham control animals were isolated and immediately homogenized in cold neuronal protein extraction reagent (Thermo Scientific) using a ratio of 1 g of tissue to 10 ml of reagent plus protease inhibitor mixture (Roche Diagnostics). The samples were centrifuged at 10,000 × g for 20 min at 4°C and the supernatants were used for cytokine measurements. Levels of proinflammatory cytokines were determined by TNF-α and IL-6 ELISA kits (Invitrogen) and protein concentrations by BCA protein assay (Pierce).
miR-210 inhibitor and mimic transfection.
miR-210 inhibitor (IH-310570-07-0002) and mimic (C-310570-05-0002) with the target sequence 5′CUGUGCGUGUGACAGCGGCUGA3′, as well as negative control (IN-001005-01) and positive fluorescent siGLO control (D-001630-01), were from Dharmacon. NSCs were transfected using Fugene HD (Promega) and 25 pmol of inhibitor, mimic, or controls/well in a 24-well plate according to the manufacturer's instructions.
qRT-PCR of miR-210.
All materials for qRT-PCR were from Applied Biosciences and used as described previously (Ouyang et al., 2013). Briefly, total RNA was isolated with TRIzol. Equal amounts of total RNA (200 ng) were reverse transcribed using the TaqMan microRNA Reverse Transcription kit. PCRs were then conducted using the TaqMan MicroRNA assay Kit at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Each reaction contained 2 μl of the reverse transcription reaction product, 0.5 μl of miR-210 predesigned primers, and 5 μl of TaqMan2X Universal PCR Master Mix in a total volume of 10 μl. Predesigned primer/probes for miR-210 and mouse U6 were from Applied Biosystems. The expression of miR-210 was normalized to the levels of the internal control U6 using the ΔΔCT method (Livak and Schmittgen, 2001). The PCR experiments were repeated three times, each using separate sets of samples.
In vitro immunocytochemistry.
Fluorescence immunocytochemistry was performed on cell cultures in 24-well plates. The cultures were washed with PBS and then fixed in 4% paraformaldehyde for 30 min at room temperature. The cells were then washed twice with PBS and nonspecific binding was blocked with 3% bovine serum albumin and 0.3% Triton X-100 in PBS for 1 h. The cells were subsequently incubated with the Dcx+ primary antibody (1:500, sc-8066; Santa Cruz Biotechnology) or Dcx+ plus PSA-NCAM (1:200, MAB5324; Millipore), markers for immature neurons, diluted in blocking buffer overnight at 4°C. Cells were subsequently incubated with the appropriate Alexa Fluor 488-conjugated secondary antibody (1:200; Invitrogen). The cell nuclei were counterstained with 4′6′-diamidino-2-phenylindole (DAPI, 0.5 μg/ml) (Sigma-Aldrich). Immunofluorescence in the central area of each well in the 24-well plate (±350 μm from the center of the well) was imaged with an epifluorescence microscope (Zeiss Axiovert 200M) as described previously (Voloboueva et al., 2008).
TUNEL staining.
Staining for apoptotic cells was performed using the in situ terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay kit (ab66108; Abcam) according to the manufacturer's instructions. Due to the rapid disappearance of Dcx+ staining accompanying the apoptosis of neuronal precursors (Voloboueva and Giffard, 2011), the levels of apoptosis were evaluated after 8 h of NSC exposure to microglial CM.
Live imaging.
To measure mitochondrial membrane potential, cells were incubated with the mitochondrial membrane potential sensitive dye tetramethylrhodamine ethyl ester (TMRE, 50 nm). Cells were illuminated at 535 nm, and fluorescence emission was observed at 590 nm. The images were processed and quantified using Photoshop CS6, as described previously (Voloboueva et al., 2008). To quantify the morphological structure of mitochondria, ImageJ analytical software (http://rsb.info.nih.gov/ij, NIH, Bethesda, MD) was used. The images were thresholded, and the “Analyze particles” tool was used to estimate the total area covered by mitochondria size ≤3 μm2, or by mitochondria of all sizes. The ratio of the areas was used to estimate the degree of mitochondrial fragmentation. Green fluorescent dye DiBAC4(3) (B438, Invitrogen) was used to investigate changes in plasma membrane potential as described previously (Voloboueva et al., 2007).
CellROX green measurements.
Measurements of ROS levels were performed as described previously (Sun et al., 2015). Briefly, cells were incubated with the ROS-sensitive dye CellROX Green (5 μm final concentration, C10444, Life Technologies, Grand Island, NY). After a 30 min incubation with CellROX Green the cells were rinsed with the differentiation media twice, and the CellROX green fluorescence was visualized with an epifluorescnce microscope (Axiovert LSM510, Carl Zeiss, Gottingen, Germany). Images were obtained with a Macintosh computer in Open lab software (Improvision, Lexington, MA).
Metabolic and biochemical measurements.
Lactate levels were measured after incubating the NSC cultures in serum-free media for 3 h using a Lactate Assay kit (BioVision, Milpitas, CA, #K627-100) as described previously (Voloboueva et al., 2013). To evaluate the rates of glucose consumption the NSCs were placed in Dulbecco's PBS (DPBS) supplemented with calcium chloride, magnesium chloride, and 0.25 g/L d-glucose (Invitrogen, Grand Island, NY), and the glucose levels were determined by Glucose Assay kit (Abcam, Cambrige, MA, #ab6533). For normalization purposes cell lysate protein concentrations were measured with the BCA protein assay reagent kit (Pierce, Rockford, IL). Cytochrome c Oxidase activity was measured using a Cytochrome c Oxidase Activity kit (BioVision, Milpitas, CA, #K287-100), and aconitase activity was measured with an Aconitase Assay kit (Cayman, #705502) after 6 h of exposure to microglial conditioned or control media. AMP-regulated protein kinase alpha (AMPKα, pT172 ELISA kits, KHO0651, Invitrogen) and pSer-811(804) phospho-retinoblastoma (Rb) ELISA kits (ABIN1380692, Antibodies-online) were used to measure the levels of AMPKα activity and phosphorylation of mouse Rb protein, respectively, according to the manufacturers' instructions.
Luciferase reporter assay.
Mouse dual luciferase expressing 3′-UTR target vectors for COX10 (MmiT054543) and ISCU (MmiT033598) and control vector pEZX-MT06 were from GeneCopoeia. NSC cultures cotransfected upon plating for differentiation with dual luciferase-expressing vectors along with miR-210 mimic or inhibitor using Fugene HD reagent as described above. The cells were analyzed 2 d after the transfection using the Luc-Pair Duo-Luciferase HS Assay Kit (GeneCopoeia) according to the manufacturer's instructions.
Statistics.
Statistical differences between two groups were determined using unpaired two-tailed Student's t test. Comparisons between multiple groups were performed with ANOVA followed by Bonferroni test for selected groups. Data in all plots are pooled from three independent experiments and are presented as mean ± SD.
Results
miR-210 inhibition reduces inflammation-associated Dcx+ cell death in vitro
Studies from us and others have demonstrated that treatment with media preconditioned by proinflammatory activated microglia (CM), an in vitro model of inflammation, significantly decreased the number of Dcx+ cells in NSC cultures. We also showed that various mitochondrial protective strategies effectively alleviate CM-induced Dcx+ cell death (Voloboueva et al., 2010). Multiple studies have demonstrated that reducing miR-210 leads to enhanced mitochondrial function through upregulation of mitochondrial electron transport and Krebs cycle protein activities (Chen et al., 2010; Favaro et al., 2010; Colleoni et al., 2013; He et al., 2014; White et al., 2015). Therefore, we transfected NSC cultures with miR-210 inhibitor or negative control on days 4–5 of differentiation and applied CM 24 h later. Transfection with 25 pmol of inhibitor/well resulted in suppression of miR-210 levels to 12% of negative control (p < 0.0001) when evaluated 24 h later by qRT-PCR. Transfections with higher concentrations did not change the levels of suppression significantly (7% of negative control for 50 pmol/well and 10% of negative control for 100 pmol/well). Therefore, we used 25 pmol/well in our experiments, with the levels of miR-210 remaining significantly lower (<25% compared with negative control) for 3 d after transfection.
Although the specificity of miR-210 targeting of ISCU and Cox10 3′-UTRs was confirmed by luciferase activity assays in earlier studies (Chen et al., 2010; Favaro et al., 2010), including a study using a mouse neural cell line (He et al., 2014), we performed luciferase assays in our primary NSC cultures. Transfection with miR-210 mimic promoted significant 45% (p = 0.035 vs negative control transfection) and 41% (p = 0.01) decreases in the levels of luminescence in cultures transfected with ISCU and COX10 3′-UTR constructs, respectively. Transfection with miR-210 inhibitor induced 26% (p = 0.09) and 28% (p < 0.01) increases in the levels of luminescence in cultures transfected with ISCU and COX10 3′-UTR constructs, respectively. No significant changes were induced by miR-210 mimic or inhibitor transfection in cultures transfected with control vectors, thus confirming specificity of miR-210 targeting. Because inhibitor has an effect on luciferase levels in primary NSCs, this confirms that miR-210 is expressed at sufficient levels to suppress at least the specific target COX10.
Differentiating cultures were exposed to CM from BV2 cells for 20–22 h, after which the cells were fixed and stained. Representative pictures of miR-210-inhibited and control cultures with and without CM injury are shown in Figure 1, A–D. Quantification shows that, although CM treatment did not change the total number of cells in NSC cultures significantly (Fig. 1E), the number of immature neurons (Dcx+ cells) was decreased 48% after CM treatment and this decrease was significantly alleviated in cultures treated with miR-210 inhibitor (Fig. 1F). We recognize that it is important to confirm results obtained with BV2 cells using primary microglia, so we evaluated the effect of CM collected from primary microglia on Dcx+ neuroblast survival. Similar to our results with BV2 CM, proinflammatory CM from primary microglia induced significant loss of Dcx+ cells and miR-210 inhibition protected the Dcx+ cells (Fig. 1F).
Figure 1.
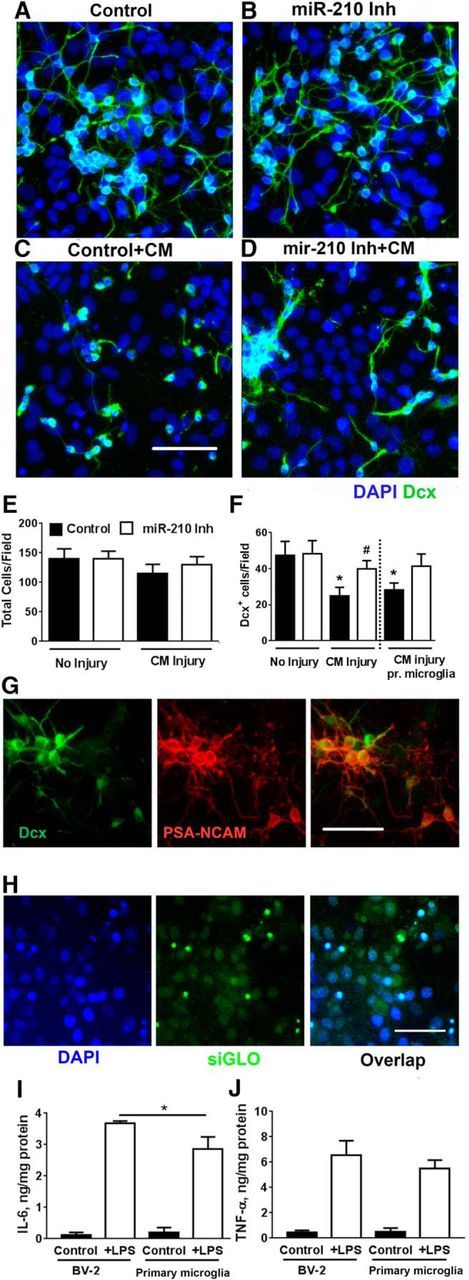
miR-210 inhibition increases Dcx+ cell survival during inflammatory CM injury. NSC cultures were transfected on days 4–5 of differentiation. Representative images of control (A) and miR-210-inhibited (B) NSC cultures without inflammatory injury. Control cultures demonstrate significantly greater loss of Dcx+ cells (C) compared with cultures transfected with miR-210 inhibitor (D) after BV2 CM treatment. Nuclei are labeled with DAPI (blue), Dcx+ cells are green. Quantification of total cell number/field (300 × 400 μm field) (E) and Dcx+ cell number/field (F). Similar effects were seen with CM from BV2 cells and CM made from primary microglia (pr microglia). *p < 0.05 compared with no injury control, #p < 0.05 compared with control-transfected cultures after injury. Immunostaining demonstrated strong overlap in immunoreactivity between Dcx+ and another marker of developing and migrating neurons, PSA-NCAM (G). Transfection of PNC cultures with fluorescent control siGLO shows that nearly all cells are transfected. Scale bar, 50 μm. H, Comparison of LPS induced cytokine levels normalized to protein for BV-2 cells and primary microglia show similar production of IL-6 (I) and TNF-α (J). *p < 0.05 compared with LPS-treated microglia. The data shown represent three independent experiments, with at least four samples per condition in each experiment Scale bar, 50 μm.
Levels of Dcx+ expression can be used as a reliable and specific marker of adult neurogenesis and its modulation by various factors (Couillard-Despres et al., 2005). In our previous study, we demonstrated strong overlap between Dcx+ and the neuronal marker MAP2 (Voloboueva et al., 2010). Here, we observed almost complete overlap (>95%) between Dcx+ and another marker of developing and migrating neurons, PSA-NCAM (Fig. 1G) (Quartu et al., 2008). We also confirmed that transfection was highly efficient, with essentially all the cells showing uptake of the fluorescently tagged positive control siGLO (Fig. 1H).
Mitochondrial function has been shown to play an important role in the maintenance of neuritic processes (Magrané et al., 2009). Consistent with this, we observed that miR-210 inhibition also resulted in better preservation of Dcx+ cell morphology. The average neuritic length of Dcx+ cells after CM injury was significantly higher in miR-210-inhibitor-transfected NSC cultures (18.8 ± 8.3 μm) compared with negative control cultures (10.8 ± 6.3 μm) after CM injury (p < 0.05).
Because the proinflammatory cytokines IL-6 and TNF-α have been shown to play pivotal roles in suppressing neurogenesis (Vallières et al., 2002; Monje et al., 2003; Cacci et al., 2005; Iosif et al., 2006), we compared the levels of these cytokines in CM from BV-2 and primary microglia. Cytokine levels normalized to cellular protein were comparable between primary microglia and BV-2 cells for both IL-6 and TNF-α (Fig. 1I,J). We also investigated the changes in IL-6 and TNF-α levels induced by transient focal ischemia in mice 3 d after injury. A 3.3-fold increase in IL-6 brain levels (39. 5 ± 11.3 vs 12.2 ± 3.9 ng/mg protein) and a 7-fold increase in TNF-α levels (199.4 ± 37.5 vs 28.7 ± 9.5 ng/mg protein) were observed, confirming the proinflammatory milieu in poststroke brain.
miR-210 inhibition protects mitochondrial function and downregulates apoptosis in Dcx+ cells
Activated microglia release a variety of cytokines and reactive species that impair mitochondrial function either directly or through intracellular signaling pathways. We investigated the effect of CM treatment and miR-210 inhibition on mitochondrial function. We measured the activity of two major mitochondrial enzymes, cytochrome c oxidase (electron transport chain) and aconitase (Krebs cycle), which are also targets of miR-210. CM treatment significantly suppressed both cytochrome c and aconitase activities (Fig. 2A,B) and this suppression was attenuated in NSC cultures pretreated with miR-210 inhibitor. In our previous study, we demonstrated that suppression of mitochondrial function induces apoptotic Dcx+ cell death (Voloboueva et al., 2010). CM-treatment-induced apoptotic death of Dcx+ cells was significantly reduced in cultures with miR-210 inhibition (Fig. 2C–G).
Figure 2.

miR-210 inhibition protects mitochondrial function and downregulates apoptosis in Dcx+ cells. miR-210 inhibition attenuated the decrease in mitochondrial cytochrome c oxidase (A) and aconitase activities (B) induced by CM inflammatory exposure. Representative images of apoptotic TUNEL staining (green) in control (C) and miR-210 inhibited (D) NSC cultures without inflammatory injury. Dcx+ cells are immunostained in red. CM inflammatory injury significantly increases the level of TUNEL staining (E) and this is attenuated in NSC cultures transfected with miR-210 inhibitor (F). G, Quantification of TUNEL staining. The data show three independent experiments, with at least four samples per condition in each experiment *p < 0.05 compared with no injury control, #p < 0.05 compared with control-transfected cultures after injury; &p < 0.05, &&p < 0.01 compared with miR-210-inhibitor-transfected cultures without injury. Scale bar, 50 μm.
Increasing levels of miR-210 with mimic reduces NSC survival of CM injury
We investigated the effect of miR-210 mimic on NSC survival during CM injury. As in the case of the inhibitor studies above, we transfected NSC cultures with miR-210 inhibitor or negative control on days 4–5 of differentiation and applied CM 24 h later. As predicted, miR-210 mimic transfection strongly compromised Dcx+ cell survival during CM injury, the opposite of the effect of inhibitor. Photomicrographs demonstrate almost complete loss of neuritic processes (Fig. 3A–D) and quantification of total cell numbers (Fig. 3E) and Dcx+ cell numbers (Fig. 3F) demonstrate a further 60% loss of Dcx+ cells in miR-210 mimic-transfected cultures on top of the 48% loss seen in control-transfected cultures exposed to CM. This is consistent with the notion that increased levels of miR-210 are detrimental for survival during CM-induced injury.
Figure 3.

miR-210 mimic reduces NSC cell survival during CM inflammatory injury. Representative images of control (A) and miR-210-mimic-transfected (B) NSC cultures without inflammatory injury. Control cultures demonstrate significantly less loss of Dcx+ cells (C) compared with cultures transfected with miR-210 mimic (D) after CM treatment. Nuclei are labeled with DAPI (blue), Dcx+ cells are green. Quantification of total cell number/field (E) and Dcx+ cell number/field (F). The data shown represent three independent experiments, with at least four samples per condition in each experiment. *p < 0.05, **p < 0.01, ***p < 0.001 compared with the corresponding conditions without injury; ###p < 0.001 compared with control-transfected cultures after CM injury.
Early miR-210 inhibition reduces NSC density but does alter the proportion of Dcx+ cells
Because reducing levels of miR-210 leads to enhanced mitochondrial function through upregulation of mitochondrial electron transport and Krebs cycle proteins, we sought to maximize mitochondrial energetics and mitogenesis by starting miR-210 inhibition early during NSC culture differentiation. When we transfected with miR-210 inhibitor 1–2 d after the start of differentiation, we unexpectedly observed a significantly lower (28%, p < 0.001) total cell density in the miR-210-inhibitor-transfected cultures by the time of the planned second transfection (days 4–5 of differentiation). Representative images of the miR-210-inhibitor-transfected and control-transfected cultures are shown in Figure 4, A and B. Quantification of cell density shows a significant reduction of the total cell density in cultures with downregulated miR-210 expression, as well as a reduction in Dcx+ cells (Fig. 4C). Although miR-210 suppression promoted a significant decrease in the number of Dcx+ cells (31.8 ± 4.8 vs 23.5 ± 5.2 in miR-210 inhibitor cultures vs control cultures, respectively), the percentage of Dcx+ cells produced in the differentiating cultures was unchanged (26.5 ± 3.2% vs 27.6 ± 4.4% in miR-210 inhibitor cultures vs control cultures, respectively). We did not see any signs of increased cell death in miR-210-inhibitor-transfected cultures, compared with control, at any day of differentiation (e.g., on the second day after the transfection, the percentages of cell death were 6.2 ± 3.6% and 6.5 ± 4.8% in cultures with miR-210 inhibition and negative control, respectively). Several studies indicate that dividing stem cells rely heavily on glycolytic metabolism and demonstrate relatively low levels of mitochondrial oxidative activity (Zhang et al., 2012; Candelario et al., 2013), so we next compared glycolytic and mitochondrial activities of miR-210 inhibitor- and control-transfected cultures.
Figure 4.
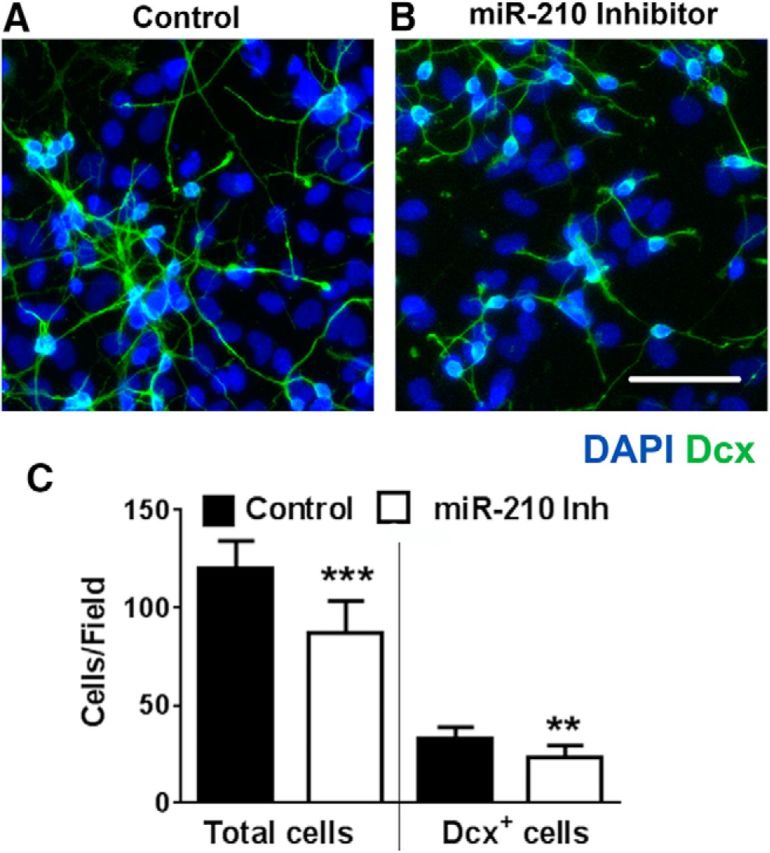
Early (1–2 d after the start of differentiation) miR-210 inhibition reduces cell density but not the proportion of Dcx+ cells. NSC cultures transfected with negative control (A) demonstrate significantly higher cell density compared with miR-210-inhibitor-transfected cultures (B). Quantification of the total cells/field and Dcx+ cells/field (C) after 4–5 d of differentiation. The data shown represent three independent experiments, with at least four samples per condition in each experiment. **p < 0.01, ***p < 0.001 compared with control-transfected. Scale bar, 50 μm.
miR-210 inhibition suppresses glycolytic activity and increases mitochondrial potential
The dividing NSC cultures were transfected with negative control or miR-210 inhibitor 1–2 d after plating and glycolysis and mitochondrial function were assessed the day after transfection (2–3 d after plating). Two measures of glycolytic activity, glucose consumption, and lactate production were suppressed in cultures transfected with miR-210 inhibitor (Fig. 5A,B). Conversely, miR-210 inhibition caused an increase in mitochondrial membrane potential, as assessed with the potentially mitochondria-sensitive dye TMRE (Fig. 5C–E). Interestingly, miR-210 inhibition not only promoted an increase in mitochondrial potential, but also changed the fragmented, more rounded mitochondrial morphology characteristic of dividing cells (Taguchi et al., 2007) to longer tubular structures. Quantification of the morphology of TMRE-stained mitochondria indicates a marked reduction in mitochondria <3 μm2 (Fig. 5F). At days 2–3, few cells had begun to express differentiation markers. At later times, miR-210 inhibition performed either early or late (Fig. 1F, Fig. 4C) did not alter the fraction of Dcx+ cells. Therefore, a contribution from differences in cell differentiation to these measures of mitochondrial function is unlikely. We next sought to investigate the mechanisms underlying the decreased rates of NSC proliferation induced by miR-210 suppression and accompanying metabolic changes.
Figure 5.

miR-210 inhibition induces changes in metabolic activity and mitochondrial function. miR-210 inhibition suppresses glucose consumption (A) and lactate production rates (B) when assessed after 3 h. Glucose consumption and lactate production are both indicators of glycolytic activity. C, D, Representative images of mitochondrial TMRE staining in NSC cultures transfected with negative control (C) and miR-210 inhibitor (D). Quantification of changes in TMRE fluorescence as a measure of mitochondrial potential (E) and mitochondrial morphology (fraction of cell mitochondria <3 μm2 size) (F). The data show three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 compared with control-transfected condition. Scale bar, 25 μm.
miR-210 inhibition suppresses the AMPK–Rb signaling axis
AMPK acts as an intracellular energy sensor, reacting rapidly to changes in cellular AMP:ATP ratio, such as those resulting from increased mitochondrial activity (Wu et al., 2014). Although AMPK works as a negative feedback to maintain cellular energy levels, it has also been shown to exert a profound effect on cellular proliferation (Bonini and Gantner, 2013). The proliferative effects of AMPK activity are highly cell-type specific. AMPK activity was found to be used by growth factors to support proliferation in NSC cultures (Dasgupta and Milbrandt, 2009). AMPK activity was significantly suppressed in NSC cultures transfected with miR-210 inhibitor compared with control (Fig. 6A, left two bars). As a positive control, AMPK was activated by mitochondrial inhibition with antimycin A regardless of miR-210 status (Fig. 6A, right two bars). It has also been shown that, in NSC cultures, AMPK directly phosphorylates Rb protein and that Rb hypophosphorylation results in decreased proliferation of NSCs (Dasgupta and Milbrandt, 2009). Because it has been demonstrated that AMPK phosphorylates Rb at Ser804 (Ser811 in humans) and phosphorylation of this site is important for NSC proliferation, we performed an ELISA assay for Rb-Ser804P. Phosphorylation of Rb was significantly attenuated in cultures transfected with miR-210 inhibitor (Fig. 6B). This demonstrates that miR-210 suppression results in suppression of the AMPK-Rb axis. Using the miRBase.org database, neither AMPK nor Rb are direct targets of miR-210, so the effects are likely due to the observed metabolic changes.
Figure 6.

The effect of miR-210 inhibition on AMPK activity and Rb phosphorylation. A, AMPK activity was significantly suppressed in NSC cultures transfected with miR-210 inhibitor. Mitochondrial inhibition with antimycin A (2 μm) was used as a positive control to activate AMPK. B, Changes in AMPK activity lead to corresponding changes in the levels of Rb-Ser804 phosphorylation. The data are from three independent experiments with at least three samples per condition in each experiment. *p < 0.05 compared with control without antimycin A; #p < 0.05 compared with both conditions without antimycin A treatment.
miR-210 inhibition does not change cell density in NSC cultures with dysfunctional mitochondria
In our next set of experiments, we sought to establish whether the observed changes in NSC density are exclusively associated with the mitochondrial mechanisms of miR-210 action. We used a previously developed model of NSC cultures with dysfunctional mitochondria (Voloboueva and Giffard, 2011). NSC cultures were transfected with miR-210 inhibitor or negative control on day 2 of differentiation and mitochondrial function was inhibited with one of two different mitochondrial inhibitors, antimycin A (2 μm) or oligomycin (2 μm). Either antimycin A or oligomycin was applied at the time of transfection and the treatments were continued until days 4–5 of differentiation. The NSC cultures treated with oligomycin or antimycin A demonstrated cellular proliferation but no signs of neuronal differentiation, as reported previously (Voloboueva and Giffard, 2011). Images of control and miR-210-inhibitor-transfected NSC cultures with and without oligomycin treatment are shown (Fig. 7A–D). Although there was a significant difference in cell density between control and miR-210-inhibitor-transfected cultures under regular differentiating conditions (Fig. 4C, top), mitochondrial inhibition completely eliminated the difference in cell density between the miR-210 inhibitor and control-transfected cultures (90.3 ± 7.8 total cells/field in miR-210 inhibitor cultures vs 92.3 ± 7.1 in control-transfected cultures, both with oligomycin inhibition; or 76.3 ± 7.5 cells/field in miR-210 inhibitor cultures vs 77.4 ± 6.8 in control tranfected cultures both with antimycin A inhibition). To investigate whether ROS levels were modulated by miR-210 inhibition, we used the ROS-sensitive dye CellROX green. Oligomycin treatment significantly increased ROS levels, whereas miR-210 inhibition did not significantly change ROS levels in either control or oligomycin treated cultures (Fig. 7C–E). Because the fluorescence signal of ROS-sensitive dyes can potentially be modulated by mitochondrial or plasma membrane potential changes, we performed measurements of plasma membrane potential using DiBAC4(3) dye, as described in our previous study (Voloboueva et al., 2007), and TMRE measurements of mitochondrial membrane potential. We did not observed significant changes in DiBAC4(3) fluorescence induced by miR-210 inhibition or by prolonged oligomycin treatment. TMRE signal was increased by 23 ± 7% (p = 0.02) in differentiated cultures treated with miR-210 inhibitor under normal conditions. Oligomycin treatment resulted in decreased TMRE signal (74 ± 11% and 77 ± 12% of control in cultures with and without miR-210 inhibition, respectively). Overall, the DiBAC23 and TMRE measurements do not support the notion that the observed increase in CellROX signal is associated with increased plasma or mitochondrial membrane potential. To further validate the CellROX measurements, we used an antioxidant combination from our previous study that efficiently decreased cell ROS levels (Voloboueva et al., 2013). Trolox, a water-soluble vitamin E analog and powerful antioxidant, and the free radical scavenger edaravone (100 μm each) were added to the medium for 3 h before CellROX measurements. This strongly suppressed ROS levels in oligomycin-treated cultures and efficiently attenuated the difference in CellROX fluorescence between normal and oligomycin treated cultures from 1.7-fold to a nonsignificant 1.15-fold. These experiments with mitochondrial inhibitors indicate that mitochondrial function is required for the decreased cellular proliferation seen in miR-210-inhibitor-transfected NSC cultures and the proliferation effect is independent of ROS.
Figure 7.
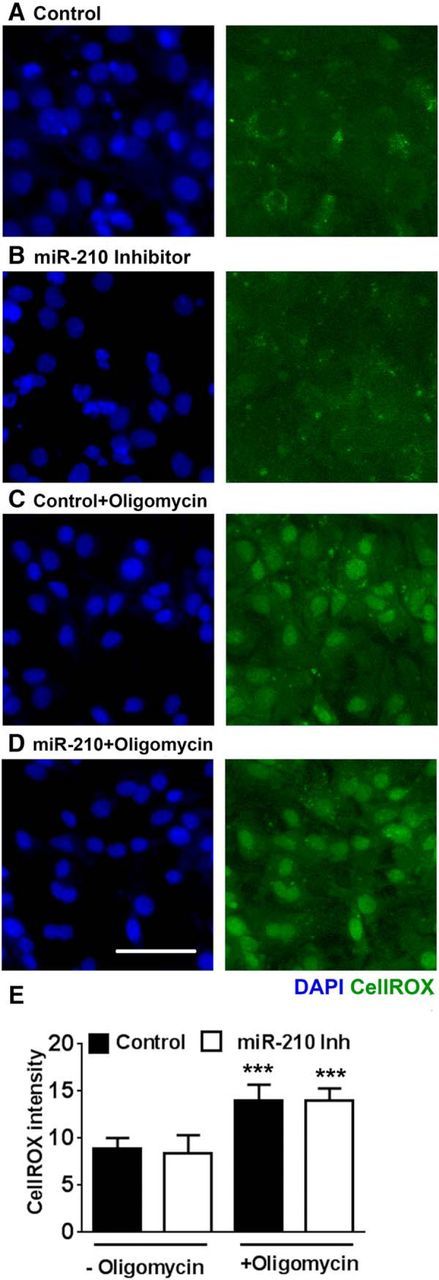
Mitochondrial inhibition by oligomycin eliminates the miR-210 inhibitor-associated decrease in NSC density. Left panels show nuclear DAPI staining, right panels show CellRox fluorescence. miR-210 inhibition leads to decreased cell densities under normal differentiating conditions (A, B), but does not change NSC densities in oligomycin (2 μm)-treated cultures (C, D). See text for quantification of cell density in oligomycin-treated cultures. Quantification shows oligomycin-induced increase in cellular ROS, but no difference in ROS levels between miR-210 inhibitor and control-transfected cultures (E). The data show three independent experiments, with at least six samples per condition in each experiment. ***p < 0.001 compared with the corresponding conditions without oligomycin treatment. Scale bar, 50 μm.
Effect of miR-210 mimic on NCS proliferation
In the last set of experiments, we investigated the effects of early miR-210 mimic transfection on NSC proliferation. miR-210 mimic promoted a small (15%) reduction in mitochondrial potential that did not reach significance (p = 0.07). miR-210 mimic did not significantly change cell density in NSC cultures, but promoted a significant 22% decrease in the number of Dcx+ immature neurons (Fig. 8A–C).
Figure 8.
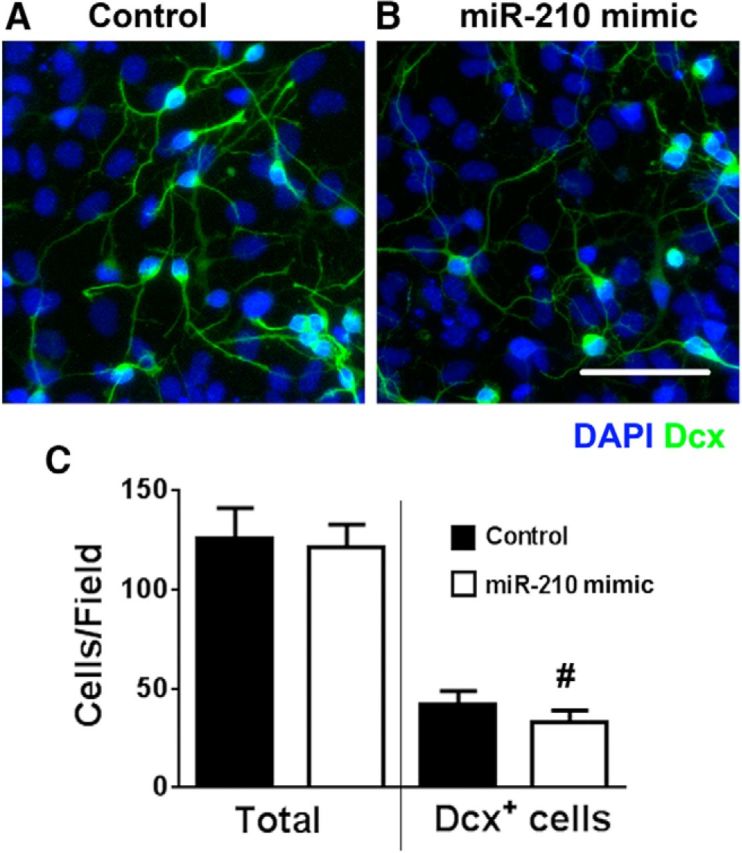
Early (days 1–2 of differentiation) transfection with miR-210 mimic decreases Dcx+ cell density. A, B, NSC cultures transfected with miR-210 mimic demonstrated comparable total cell densities, but decreased Dcx+ cell numbers compared with negative control-transfected cultures on days 4–5 of differentiation (nuclei are labeled with DAPI in blue; Dcx+ cells are green). C, Quantification of total and Dcx+ cell numbers. The data show three independent experiments with at least three samples per condition in each experiment. #p < 0.05 compared with control. Scale bars, 50 μm.
Discussion
Acute brain injury has been shown to stimulate proliferation of endogenous progenitor cells in the brain (Monje et al., 2002; Richardson et al., 2007; Kernie and Parent, 2010; Ohira, 2011). In well studied rodent models of stroke, cerebral ischemia leads to a dramatic burst of NSC proliferation that peaks within several days after the occlusion and declines substantially by the end of the second week after injury (Felling and Levison, 2003; Zhang et al., 2008). Conversely, it takes at least 3–4 weeks for Dcx+ cells to develop the markers and morphology of mature neurons (Liu et al., 1998; Jiang et al., 2001; Arvidsson et al., 2002; Parent et al., 2002). The survival of young neurons is strongly impaired by the concomitant induction of a proinflammatory state (Monje et al., 2002; Winner et al., 2011; Wang and Jin, 2015). Studies have shown that decreased poststroke neurogenesis worsens stroke outcome (Jin et al., 2010; Sun et al., 2013), whereas increased rates of neurogenesis can lead to better functional recovery (Androutsellis-Theotokis et al., 2006; Leker et al., 2007; Schäbitz et al., 2007). Therefore, although there is some overlap between proliferation and neuronal survival and differentiation, the deleterious effects of miR-210 inhibition could be minimized by delaying treatment until after the peak period of proliferation. This will be harder to define in the clinical setting of highly variable strokes.
In the current study, we focused on effects of mitochondrial enhancement on neurogenesis in an isolated in vitro preparation. Metabolism is known to be closely tied to the pattern of inflammatory activation of immune cells (Orihuela et al., 2016), so future in vivo studies of metabolic manipulation as seen with miR-210 inhibition will need to take into account effects on additional cell types. We observed that miR-210 inhibition enhanced mitochondrial oxidative metabolism in NSC cultures. When we transfected NSC cultures on days 4–5 of differentiation with miR-210 inhibitor and exposed them to inflammatory injury the next day, increased survival of Dcx+ cells was observed. miR-210 inhibition during inflammatory CM injury effectively protected mitochondrial function, as evidenced by increased levels of cytochrome c oxidase and aconitase activities. Our previous studies demonstrated that mitochondrial impairment promotes apoptotic cell death of Dcx+ cells (Voloboueva et al., 2010). Here, we found that miR-210 inhibition not only suppressed mitochondrial impairment, but also attenuated inflammation-induced apoptotic death of Dcx+ cells. Our studies indicated that essentially all cells in the NSC culture could be efficiently transfected by miR-210 inhibitor (Fig. 1). Because mitochondria of Dcx+ cells are particularly vulnerable to inhibitors or CM injury (Voloboueva et al., 2010), it is plausible that changes in Dcx+ mitochondrial function contribute significantly to the observed changes in mitochondrial enzyme activities. Glial cells represent another significant fraction of differentiated NSC cells (Voloboueva and Giffard, 2011). Metabolic properties of young neurons are strongly influenced by the function and metabolism of surrounding glial cells (Platel et al., 2010; Gengatharan et al., 2016). In turn, astrocytic mitochondrial function, and even astrocytic survival, are strongly modulated by cocultured neurons and subsequent energetic demands imposed by the neurons (Reichert et al., 2001; Voloboueva et al., 2007). It is thus difficult to separate the contribution of mitochondrial changes in different cell types to the observed changes in mitochondrial enzyme activities due to the complex interplay between different cell types.
Unexpectedly, we found that early miR-210 inhibition resulted in significantly reduced NSC proliferation. Multiple studies demonstrate that dividing progenitor cells, particularly NSCs, crucially rely on glycolytic metabolism and suppress mitochondrial oxidative metabolism compared with differentiated cells (Zhang et al., 2012; Candelario et al., 2013). We observed that glycolytic activity was significantly suppressed in NSC cultures with miR-210 inhibition. One of the major mechanisms uniting metabolic energy sensing, growth factor signaling, and cell proliferation is AMPK activity. AMPK is a highly sensitive cellular energy switch that regulates metabolic pathways generating ATP (Kann and Kovács, 2007). Increased ATP levels, such as those resulting from increased mitochondrial activity, downregulate AMPK activity (Dasgupta and Milbrandt, 2009; Amato and Man, 2011). In our experiments, we observed decreased AMPK activity in NSC cultures transfected with miR-210 inhibitor. AMPK also acts as an important regulator of cell proliferation. Although AMPK was required to maintain proliferation in astrocytic tumors (Ríos et al., 2013) and facilitated stem cell self-renewal in pluripotent cells (Rodríguez-Jimnez et al., 2012), AMPK activation has also been shown to inhibit cell growth in some tumor cells (Shackelford and Shaw, 2009). Overall, the role of AMPK activation in cell proliferation is highly cell and context specific, as reviewed recently (Bonini and Gantner, 2013). Growth factors promote cell proliferation in NSC via AMPK activity (Dasgupta and Milbrandt, 2009). Decreased AMPK activation leads to hypophosphorylation of Rb protein at Ser804, which is required for normal NSC proliferation (Amato and Man, 2011). Suppression of the AMPK-Rb axis leads to a severe impairment of brain growth (Dasgupta and Milbrandt, 2009). In our experiments, decreased AMPK activity in NSC cultures transfected with miR-210 inhibitor also decreased levels of Ser804 phosphorylated Rb. These results indicate that increased mitochondrial activity suppresses AMPK activity/Rb phosphorylation, an important proliferation-regulating mechanism in dividing NSC cultures. The key finding of the stage-specific effects of miR-210 inhibition on proliferation compared with neuronal survival are shown diagrammatically in Figure 9.
Figure 9.

miR-210 inhibition promotes more energized, tubular-shaped mitochondria, resulting in better neuroblast survival during inflammation injury, but suppresses NSC division during early proliferation.
A variety of miR-210 targets have been identified, pointing to its role not only in mitochondrial metabolism, but also in angiogenesis, DNA damage response, cell proliferation, and apoptosis (Huang and Zuo, 2014). Several miR-210 targets associated with cell cycle control and mitosis-related genes have been identified, with opposing effects of miR-210 levels on cell proliferation that appear to be context dependent (Huang et al., 2009; Zhang et al., 2009; Tsuchiya et al., 2011; Chen et al., 2012). Our 3′-UTR luciferase assays confirmed specificity and functionality of miR-210 suppression mechanisms targeting mitochondrial COX 10 and ISCU in NSCs.
Our experiments with mitochondrial inhibitors (Fig. 7) suggest that the observed decrease in NSC proliferation depends on mitochondrial mechanisms of miR-210 action because mitochondrial inhibition eliminates the miR-210 associated decrease in cell proliferation. Mitochondrial dysfunction is closely associated with apoptotic cell death, particularly in Dcx+ cells (Voloboueva et al., 2010). Although we observed significant protection of mitochondrial function resulting from miR-210 inhibition, we cannot exclude a contribution from additional miR-210 targets related to apoptosis and cell survival. Although several apoptosis-related miR-210 targets have been reported, their functional importance in cytoprotection remains largely elusive (Murakami et al., 2006; Chio et al., 2013; Qiu et al., 2013; Huang and Zuo, 2014).
Neurogenesis is a complex cellular process that involves many steps. In some acute brain injuries, neurogenesis starts with intense proliferation and migration of NSCs that peaks several days after injury, followed by a prolonged period of differentiation, survival, and integration (Felling and Levison, 2003; Zhao et al., 2003; Aimone et al., 2010; Kernie and Parent, 2010). Recent studies provide contradictory reports on whether miR-210 action promotes (Zeng et al., 2014) or suppresses (Ma et al., 2016) postischemic neurogenesis in vivo. It is important to note that very early suppression of poststroke inflammation suppresses cell proliferation and neurogenesis (Kim et al., 2009), whereas anti-inflammatory treatment applied at later times (4 d after stroke) promotes increased neurogenesis (Liu et al., 2007). Importantly, the results of our study begin to shed light on the underlying mechanisms of these controversies. Supporting mitochondrial function early reduces NSC proliferation, likely by downregulation of the AMPK/Rb axis, thus reducing the total number of cells produced, including neurons, whereas later treatment increases neuronal survival without markedly impairing production of new cells.
The search for strategies and pharmacological agents to enhance neurogenesis despite a proinflammatory milieu remains a challenge and is the focus of intense investigation. This study shows that, although enhancing mitochondrial metabolism is an effective strategy to protect neurogenesis, particularly survival of Dcx+ cells during inflammation, it is essential to consider the timing of such an intervention because different effects are observed at different stages of neurogenesis. These results suggest that miR-210 inhibition should be delayed until after the initial burst of postinjury proliferation, but could be beneficial during the prolonged survival and final neuronal differentiation stage. A window to support the survival of immature neurons selectively may exist and merits future testing in in vivo models.
Our current study used the novel approach of lowering miR-210 levels to demonstrate that mitochondrial protection effectively supports survival of young Dcx+ neurons in the face of inflammation. Mitochondrial protection induced by miR-210 inhibition thus potentially provides a new strategy to protect neurogenesis in inflammatory settings. Conversely, our findings suggest that mitochondrial protection can be a double-edged sword and should be avoided during the initial proliferative stages of neurogenesis due to the specific metabolic requirements of dividing stem cells.
Footnotes
This work was supported by the National Institutes of Health (Grants NS080177 and NS084396 to R.G.G.).
The authors declare no competing financial interests.
References
- Aimone JB, Deng W, Gage FH (2010) Adult neurogenesis: integrating theories and separating functions. Trends Cogn Sci 14:325–337. 10.1016/j.tics.2010.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amato S, Man HY (2011) Bioenergy sensing in the brain: the role of AMP-activated protein kinase in neuronal metabolism, development and neurological diseases. Cell Cycle 10:3452–3460. 10.4161/cc.10.20.17953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Androutsellis-Theotokis A, Leker RR, Soldner F, Hoeppner DJ, Ravin R, Poser SW, Rueger MA, Bae SK, Kittappa R, McKay RD (2006) Notch signalling regulates stem cell numbers in vitro and in vivo. Nature 442:823–826. 10.1038/nature04940 [DOI] [PubMed] [Google Scholar]
- Arvidsson A, Kokaia Z, Lindvall O (2001) N-methyl-D-aspartate receptor-mediated increase of neurogenesis in adult rat dentate gyrus following stroke. Eur J Neurosci 14:10–18. 10.1046/j.0953-816x.2001.01611.x [DOI] [PubMed] [Google Scholar]
- Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O (2002) Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med 8:963–970. 10.1038/nm747 [DOI] [PubMed] [Google Scholar]
- Bonini MG, Gantner BN (2013) The multifaceted activities of AMPK in tumor progression–why the “one size fits all” definition does not fit at all? IUBMB Life 65:889–896. 10.1002/iub.1213 [DOI] [PubMed] [Google Scholar]
- Brown GC, Borutaite V (2001) Nitric oxide, mitochondria, and cell death. IUBMB Life 52:189–195. 10.1080/15216540152845993 [DOI] [PubMed] [Google Scholar]
- Cacci E, Claasen JH, Kokaia Z (2005) Microglia-derived tumor necrosis factor-alpha exaggerates death of newborn hippocampal progenitor cells in vitro. J Neurosci Res 80:789–797. 10.1002/jnr.20531 [DOI] [PubMed] [Google Scholar]
- Candelario KM, Shuttleworth CW, Cunningham LA (2013) Neural stem/progenitor cells display a low requirement for oxidative metabolism independent of hypoxia inducible factor-1alpha expression. J Neurochem 125:420–429. 10.1111/jnc.12204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WY, Liu WJ, Zhao YP, Zhou L, Zhang TP, Chen G, Shu H (2012) Induction, modulation and potential targets of miR-210 in pancreatic cancer cells. Hepatobiliary Pancreat Dis Int 11:319–324. 10.1016/S1499-3872(12)60168-4 [DOI] [PubMed] [Google Scholar]
- Chen Z, Li Y, Zhang H, Huang P, Luthra R (2010) Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene 29:4362–4368. 10.1038/onc.2010.193 [DOI] [PubMed] [Google Scholar]
- Chio CC, Lin JW, Cheng HA, Chiu WT, Wang YH, Wang JJ, Hsing CH, Chen RM (2013) MicroRNA-210 targets antiapoptotic Bcl-2 expression and mediates hypoxia-induced apoptosis of neuroblastoma cells. Arch Toxicol 87:459–468. 10.1007/s00204-012-0965-5 [DOI] [PubMed] [Google Scholar]
- Colleoni F, Padmanabhan N, Yung HW, Watson ED, Cetin I, Tissot van Patot MC, Burton GJ, Murray AJ (2013) Suppression of mitochondrial electron transport chain function in the hypoxic human placenta: a role for miRNA-210 and protein synthesis inhibition. PLoS One 8:e55194. 10.1371/journal.pone.0055194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couillard-Despres S, Winner B, Schaubeck S, Aigner R, Vroemen M, Weidner N, Bogdahn U, Winkler J, Kuhn HG, Aigner L (2005) Doublecortin expression levels in adult brain reflect neurogenesis. Eur J Neurosci 21:1–14. 10.1111/j.1460-9568.2004.03813.x [DOI] [PubMed] [Google Scholar]
- Dasgupta B, Milbrandt J (2009) AMP-activated protein kinase phosphorylates retinoblastoma protein to control mammalian brain development. Dev Cell 16:256–270. 10.1016/j.devcel.2009.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekdahl CT, Claasen JH, Bonde S, Kokaia Z, Lindvall O (2003) Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci U S A 100:13632–13637. 10.1073/pnas.2234031100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facucho-Oliveira JM, Alderson J, Spikings EC, Egginton S, St John JC (2007) Mitochondrial DNA replication during differentiation of murine embryonic stem cells. J Cell Sci 120:4025–4034. 10.1242/jcs.016972 [DOI] [PubMed] [Google Scholar]
- Fallon J, Reid S, Kinyamu R, Opole I, Opole R, Baratta J, Korc M, Endo TL, Duong A, Nguyen G, Karkehabadhi M, Twardzik D, Patel S, Loughlin S (2000) In vivo induction of massive proliferation, directed migration, and differentiation of neural cells in the adult mammalian brain. Proc Natl Acad Sci U S A 97:14686–14691. 10.1073/pnas.97.26.14686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favaro E, Ramachandran A, McCormick R, Gee H, Blancher C, Crosby M, Devlin C, Blick C, Buffa F, Li JL, Vojnovic B, Pires das Neves R, Glazer P, Iborra F, Ivan M, Ragoussis J, Harris AL (2010) MicroRNA-210 regulates mitochondrial free radical response to hypoxia and Krebs cycle in cancer cells by targeting iron sulfur cluster protein ISCU. PLoS One 5:e10345. 10.1371/journal.pone.0010345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felling RJ, Levison SW (2003) Enhanced neurogenesis following stroke. J Neurosci Res 73:277–283. 10.1002/jnr.10670 [DOI] [PubMed] [Google Scholar]
- Gengatharan A, Bammann RR, Saghatelyan A (2016) The role of astrocytes in the generation, migration, and integration of new neurons in the adult olfactory bulb. Front Neurosci 10:149. 10.3389/fnins.2016.00149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell B. (2006) Oxidative stress and neurodegeneration: where are we now? J Neurochem 97:1634–1658. 10.1111/j.1471-4159.2006.03907.x [DOI] [PubMed] [Google Scholar]
- Han RQ, Ouyang YB, Xu L, Agrawal R, Patterson AJ, Giffard RG (2009) Postischemic brain injury is attenuated in mice lacking the beta2-adrenergic receptor. Anesth Analg 108:280–287. 10.1213/ane.0b013e318187ba6b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch UK. (2002) Microglia as a source and target of cytokines. Glia 40:140–155. 10.1002/glia.10161 [DOI] [PubMed] [Google Scholar]
- He M, Lu Y, Xu S, Mao L, Zhang L, Duan W, Liu C, Pi H, Zhang Y, Zhong M, Yu Z, Zhou Z (2014) MiRNA-210 modulates a nickel-induced cellular energy metabolism shift by repressing the iron-sulfur cluster assembly proteins ISCU1/2 in Neuro-2a cells. Cell Death Dis 5:e1090. 10.1038/cddis.2014.60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Zuo J (2014) Emerging roles of miR-210 and other non-coding RNAs in the hypoxic response. Acta Biochim Biophys Sin 46:220–232. 10.1093/abbs/gmt141 [DOI] [PubMed] [Google Scholar]
- Huang X, Ding L, Bennewith KL, Tong RT, Welford SM, Ang KK, Story M, Le QT, Giaccia AJ (2009) Hypoxia-inducible mir-210 regulates normoxic gene expression involved in tumor initiation. Mol Cell 35:856–867. 10.1016/j.molcel.2009.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iosif RE, Ekdahl CT, Ahlenius H, Pronk CJ, Bonde S, Kokaia Z, Jacobsen SE, Lindvall O (2006) Tumor necrosis factor receptor 1 is a negative regulator of progenitor proliferation in adult hippocampal neurogenesis. J Neurosci 26:9703–9712. 10.1523/JNEUROSCI.2723-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Gu W, Brännström T, Rosqvist R, Wester P (2001) Cortical neurogenesis in adult rats after transient middle cerebral artery occlusion. Stroke 32:1201–1207. 10.1161/01.STR.32.5.1201 [DOI] [PubMed] [Google Scholar]
- Jin K, Minami M, Lan JQ, Mao XO, Batteur S, Simon RP, Greenberg DA (2001) Neurogenesis in dentate subgranular zone and rostral subventricular zone after focal cerebral ischemia in the rat. Proc Natl Acad Sci U S A 98:4710–4715. 10.1073/pnas.081011098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Wang X, Xie L, Mao XO, Greenberg DA (2010) Transgenic ablation of doublecortin-expressing cells suppresses adult neurogenesis and worsens stroke outcome in mice. Proc Natl Acad Sci U S A 107:7993–7998. 10.1073/pnas.1000154107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson CB, Momma S, Clarke DL, Risling M, Lendahl U, Frisén J (1999) Identification of a neural stem cell in the adult mammalian central nervous system. Cell 96:25–34. 10.1016/S0092-8674(00)80956-3 [DOI] [PubMed] [Google Scholar]
- Johnson DC, Dean DR, Smith AD, Johnson MK (2005) Structure, function, and formation of biological iron-sulfur clusters. Annu Rev Biochem 74:247–281. 10.1146/annurev.biochem.74.082803.133518 [DOI] [PubMed] [Google Scholar]
- Kann O, Kovács R (2007) Mitochondria and neuronal activity. Am J Physiol Cell Physiol 292:C641–C657. [DOI] [PubMed] [Google Scholar]
- Kernie SG, Parent JM (2010) Forebrain neurogenesis after focal Ischemic and traumatic brain injury. Neurobiol Dis 37:267–274. 10.1016/j.nbd.2009.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BJ, Kim MJ, Park JM, Lee SH, Kim YJ, Ryu S, Kim YH, Yoon BW (2009) Reduced neurogenesis after suppressed inflammation by minocycline in transient cerebral ischemia in rat. J Neurol Sci 279:70–75. 10.1016/j.jns.2008.12.025 [DOI] [PubMed] [Google Scholar]
- Leker RR, Soldner F, Velasco I, Gavin DK, Androutsellis-Theotokis A, McKay RD (2007) Long-lasting regeneration after ischemia in the cerebral cortex. Stroke 38:153–161. 10.1161/01.STR.0000252156.65953.a9 [DOI] [PubMed] [Google Scholar]
- Liu J, Solway K, Messing RO, Sharp FR (1998) Increased neurogenesis in the dentate gyrus after transient global ischemia in gerbils. J Neurosci 18:7768–7778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Fan Y, Won SJ, Neumann M, Hu D, Zhou L, Weinstein PR, Liu J (2007) Chronic treatment with minocycline preserves adult new neurons and reduces functional impairment after focal cerebral ischemia. Stroke 38:146–152. 10.1161/01.STR.0000251791.64910.cd [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Ma Q, Dasgupta C, Li Y, Bajwa NM, Xiong F, Harding B, Hartman R, Zhang L (2016) Inhibition of microRNA-210 provides neuroprotection in hypoxic-ischemic brain injury in neonatal rats. Neurobiol Dis 89:202–212. 10.1016/j.nbd.2016.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magavi SS, Leavitt BR, Macklis JD (2000) Induction of neurogenesis in the neocortex of adult mice. Nature 405:951–955. 10.1038/35016083 [DOI] [PubMed] [Google Scholar]
- Magrané J, Hervias I, Henning MS, Damiano M, Kawamata H, Manfredi G (2009) Mutant SOD1 in neuronal mitochondria causes toxicity and mitochondrial dynamics abnormalities. Hum Mol Genet 18:4552–4564. 10.1093/hmg/ddp421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monje ML, Mizumatsu S, Fike JR, Palmer TD (2002) Irradiation induces neural precursor-cell dysfunction. Nat Med 8:955–962. 10.1038/nm749 [DOI] [PubMed] [Google Scholar]
- Monje ML, Toda H, Palmer TD (2003) Inflammatory blockade restores adult hippocampal neurogenesis. Science 302:1760–1765. 10.1126/science.1088417 [DOI] [PubMed] [Google Scholar]
- Murakami Y, Yasuda T, Saigo K, Urashima T, Toyoda H, Okanoue T, Shimotohno K (2006) Comprehensive analysis of microRNA expression patterns in hepatocellular carcinoma and non-tumorous tissues. Oncogene 25:2537–2545. 10.1038/sj.onc.1209283 [DOI] [PubMed] [Google Scholar]
- Ohira K. (2011) Injury-induced neurogenesis in the mammalian forebrain. Cell Mol Life Sci 68:1645–1656. 10.1007/s00018-010-0552-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orihuela R, McPherson CA, Harry GJ (2016) Microglial M1/M2 polarization and metabolic states. Br J Pharmacol 173:649–665. 10.1111/bph.13139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang YB, Xu L, Lu Y, Sun X, Yue S, Xiong XX, Giffard RG (2013) Astrocyte-enriched miR-29a targets PUMA and reduces neuronal vulnerability to forebrain ischemia. Glia 61:1784–1794. 10.1002/glia.22556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM, Vexler ZS, Gong C, Derugin N, Ferriero DM (2002) Rat forebrain neurogenesis and striatal neuron replacement after focal stroke. Ann Neurol 52:802–813. 10.1002/ana.10393 [DOI] [PubMed] [Google Scholar]
- Pereira SL, Grãos M, Rodrigues AS, Anjo SI, Carvalho RA, Oliveira PJ, Arenas E, Ramalho-Santos J (2013) Inhibition of mitochondrial complex III blocks neuronal differentiation and maintains embryonic stem cell pluripotency. PLoS One 8:e82095. 10.1371/journal.pone.0082095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platel JC, Dave KA, Gordon V, Lacar B, Rubio ME, Bordey A (2010) NMDA receptors activated by subventricular zone astrocytic glutamate are critical for neuroblast survival prior to entering a synaptic network. Neuron 65:859–872. 10.1016/j.neuron.2010.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocock JM, Liddle AC (2001) Microglial signalling cascades in neurodegenerative disease. Prog Brain Res 132:555–565. 10.1016/S0079-6123(01)32103-9 [DOI] [PubMed] [Google Scholar]
- Puisségur MP, Mazure NM, Bertero T, Pradelli L, Grosso S, Robbe-Sermesant K, Maurin T, Lebrigand K, Cardinaud B, Hofman V, Fourre S, Magnone V, Ricci JE, Pouysségur J, Gounon P, Hofman P, Barbry P, Mari B (2011) miR-210 is overexpressed in late stages of lung cancer and mediates mitochondrial alterations associated with modulation of HIF-1 activity. Cell Death Differ 18:465–478. 10.1038/cdd.2010.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Zhou XY, Zhou XG, Cheng R, Liu HY, Li Y (2013) Neuroprotective effects of microRNA-210 against oxygen-glucose deprivation through inhibition of apoptosis in PC12 cells. Mol Med Rep 7:1955–1959. 10.3892/mmr.2013.1431 [DOI] [PubMed] [Google Scholar]
- Quartu M, Serra MP, Boi M, Ibba V, Melis T, Del Fiacco M (2008) Polysialylated-neural cell adhesion molecule (PSA-NCAM) in the human trigeminal ganglion and brainstem at prenatal and adult ages. BMC Neurosci 9:108. 10.1186/1471-2202-9-108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichert SA, Kim-Han JS, Dugan LL (2001) The mitochondrial permeability transition pore and nitric oxide synthase mediate early mitochondrial depolarization in astrocytes during oxygen-glucose deprivation. J Neurosci 21:6608–6616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson RM, Sun D, Bullock MR (2007) Neurogenesis after traumatic brain injury. Neurosurg Clin N Am 18:169–181, xi. [DOI] [PubMed] [Google Scholar]
- Ríos M, Foretz M, Viollet B, Prieto A, Fraga M, Costoya JA, Senaris R (2013) AMPK activation by oncogenesis is required to maintain cancer cell proliferation in astrocytic tumors. Cancer Res 73:2628–2638. 10.1158/0008-5472.CAN-12-0861 [DOI] [PubMed] [Google Scholar]
- Rock RB, Gekker G, Hu S, Sheng WS, Cheeran M, Lokensgard JR, Peterson PK (2004) Role of microglia in central nervous system infections. Clin Microbiol Rev 17:942–964. 10.1128/CMR.17.4.942-964.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Jimnez FJ, Alastrue-Agudo A, Erceg S, Stojkovic M, Moreno-Manzano V (2012) FM19G11 favors spinal cord injury regeneration and stem cell self-renewal by mitochondrial uncoupling and glucose metabolism induction. Stem Cells 30:2221–2233. 10.1002/stem.1189 [DOI] [PubMed] [Google Scholar]
- Samavati L, Lee I, Mathes I, Lottspeich F, Hüttemann M (2008) Tumor necrosis factor alpha inhibits oxidative phosphorylation through tyrosine phosphorylation at subunit I of cytochrome c oxidase. J Biol Chem 283:21134–21144. 10.1074/jbc.M801954200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäbitz WR, Steigleder T, Cooper-Kuhn CM, Schwab S, Sommer C, Schneider A, Kuhn HG (2007) Intravenous brain-derived neurotrophic factor enhances poststroke sensorimotor recovery and stimulates neurogenesis. Stroke 38:2165–2172. 10.1161/STROKEAHA.106.477331 [DOI] [PubMed] [Google Scholar]
- Shackelford DB, Shaw RJ (2009) The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer 9:563–575. 10.1038/nrc2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitkovsky D, Sasse P, Kolossov E, Böttinger C, Fleischmann BK, Hescheler J, Wiesner RJ (2004) Activity of complex III of the mitochondrial electron transport chain is essential for early heart muscle cell differentiation. FASEB J 18:1300–1302. [DOI] [PubMed] [Google Scholar]
- Stadler J, Bentz BG, Harbrecht BG, Di Silvio M, Curran RD, Billiar TR, Hoffman RA, Simmons RL (1992) Tumor necrosis factor alpha inhibits hepatocyte mitochondrial respiration. Ann Surg 216:539–546. 10.1097/00000658-199211000-00003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Sun H, Wu S, Lee CC, Akamatsu Y, Wang RK, Kernie SG, Liu J (2013) Conditional ablation of neuroprogenitor cells in adult mice impedes recovery of poststroke cognitive function and reduces synaptic connectivity in the perforant pathway. J Neurosci 33:17314–17325. 10.1523/JNEUROSCI.2129-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Voloboueva LA, Stary CM, Giffard RG (2015) Physiologically normal 5% O2 supports neuronal differentiation and resistance to inflammatory injury in neural stem cell cultures. J Neurosci Res 93:1703–1712. 10.1002/jnr.23615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K (2007) Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem 282:11521–11529. 10.1074/jbc.M607279200 [DOI] [PubMed] [Google Scholar]
- Tsuchiya S, Fujiwara T, Sato F, Shimada Y, Tanaka E, Sakai Y, Shimizu K, Tsujimoto G (2011) MicroRNA-210 regulates cancer cell proliferation through targeting fibroblast growth factor receptor-like 1 (FGFRL1). J Biol Chem 286:420–428. 10.1074/jbc.M110.170852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallières L, Campbell IL, Gage FH, Sawchenko PE (2002) Reduced hippocampal neurogenesis in adult transgenic mice with chronic astrocytic production of interleukin-6. J Neurosci 22:486–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voloboueva LA, Giffard RG (2011) Inflammation, mitochondria, and the inhibition of adult neurogenesis. J Neurosci Res 89:1989–1996. 10.1002/jnr.22768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voloboueva LA, Suh SW, Swanson RA, Giffard RG (2007) Inhibition of mitochondrial function in astrocytes: implications for neuroprotection. J Neurochem 102:1383–1394. 10.1111/j.1471-4159.2007.04634.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voloboueva LA, Duan M, Ouyang Y, Emery JF, Stoy C, Giffard RG (2008) Overexpression of mitochondrial Hsp70/Hsp75 protects astrocytes against ischemic injury in vitro. J Cereb Blood Flow Metab 28:1009–1016. 10.1038/sj.jcbfm.9600600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voloboueva LA, Lee SW, Emery JF, Palmer TD, Giffard RG (2010) Mitochondrial protection attenuates inflammation-induced impairment of neurogenesis in vitro and in vivo. J Neurosci 30:12242–12251. 10.1523/JNEUROSCI.1752-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voloboueva LA, Emery JF, Sun X, Giffard RG (2013) Inflammatory response of microglial BV-2 cells includes a glycolytic shift and is modulated by mitochondrial glucose-regulated protein 75/mortalin. FEBS Lett 587:756–762. 10.1016/j.febslet.2013.01.067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner KR, Kleinholz M, Myers RE (1990) Delayed decreases in specific brain mitochondrial electron transfer complex activities and cytochrome concentrations following anoxia/ischemia. J Neurol Sci 100:142–151. 10.1016/0022-510X(90)90025-I [DOI] [PubMed] [Google Scholar]
- Wang B, Jin K (2015) Current perspectives on the link between neuroinflammation and neurogenesis. Metab Brain Dis 30:355–365. 10.1007/s11011-014-9523-6 [DOI] [PubMed] [Google Scholar]
- Wang W, Esbensen Y, Kunke D, Suganthan R, Rachek L, Bjørås M, Eide L (2011) Mitochondrial DNA damage level determines neural stem cell differentiation fate. J Neurosci 31:9746–9751. 10.1523/JNEUROSCI.0852-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Mao X, Xie L, Sun F, Greenberg DA, Jin K (2012) Conditional depletion of neurogenesis inhibits long-term recovery after experimental stroke in mice. PLoS One 7:e38932. 10.1371/journal.pone.0038932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White K, et al. (2015) Genetic and hypoxic alterations of the microRNA-210-ISCU1/2 axis promote iron-sulfur deficiency and pulmonary hypertension. EMBO Mol Med 7:695–713. 10.15252/emmm.201404511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winner B, Kohl Z, Gage FH (2011) Neurodegenerative disease and adult neurogenesis. Eur J Neurosci 33:1139–1151. 10.1111/j.1460-9568.2011.07613.x [DOI] [PubMed] [Google Scholar]
- Wu SB, Wu YT, Wu TP, Wei YH (2014) Role of AMPK-mediated adaptive responses in human cells with mitochondrial dysfunction to oxidative stress. Biochim Biophys Acta 1840:1331–1344. 10.1016/j.bbagen.2013.10.034 [DOI] [PubMed] [Google Scholar]
- Yenari MA, Xu L, Tang XN, Qiao Y, Giffard RG (2006) Microglia potentiate damage to blood-brain barrier constituents: improvement by minocycline in vivo and in vitro. Stroke 37:1087–1093. 10.1161/01.STR.0000206281.77178.ac [DOI] [PubMed] [Google Scholar]
- Zell R, Geck P, Werdan K, Boekstegers P (1997) TNF-alpha and IL-1 alpha inhibit both pyruvate dehydrogenase activity and mitochondrial function in cardiomyocytes: evidence for primary impairment of mitochondrial function. Mol Cell Biochem 177:61–67. 10.1023/A:1006896832582 [DOI] [PubMed] [Google Scholar]
- Zeng L, He X, Wang Y, Tang Y, Zheng C, Cai H, Liu J, Wang Y, Fu Y, Yang GY (2014) MicroRNA-210 overexpression induces angiogenesis and neurogenesis in the normal adult mouse brain. Gene Ther 21:37–43. 10.1038/gt.2013.55 [DOI] [PubMed] [Google Scholar]
- Zhang J, Nuebel E, Daley GQ, Koehler CM, Teitell MA (2012) Metabolic regulation in pluripotent stem cells during reprogramming and self-renewal. Cell Stem Cell 11:589–595. 10.1016/j.stem.2012.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang RL, Zhang ZG, Roberts C, LeTourneau Y, Lu M, Zhang L, Wang Y, Chopp M (2008) Lengthening the G(1) phase of neural progenitor cells is concurrent with an increase of symmetric neuron generating division after stroke. J Cereb Blood Flow Metab 28:602–611. 10.1038/sj.jcbfm.9600556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Sun H, Dai H, Walsh RM, Imakura M, Schelter J, Burchard J, Dai X, Chang AN, Diaz RL, Marszalek JR, Bartz SR, Carleton M, Cleary MA, Linsley PS, Grandori C (2009) MicroRNA miR-210 modulates cellular response to hypoxia through the MYC antagonist MNT. Cell Cycle 8:2756–2768. 10.4161/cc.8.17.9387 [DOI] [PubMed] [Google Scholar]
- Zhao X, Ahram A, Berman RF, Muizelaar JP, Lyeth BG (2003) Early loss of astrocytes after experimental traumatic brain injury. Glia 44:140–152. 10.1002/glia.10283 [DOI] [PubMed] [Google Scholar]