

Abstract
To improve our understanding of properties that confer successful inhibition of chemokines in vivo, we analyzed anti-murine CXCL10 monoclonal antibodies (mAb) having different characteristics. 1B6 displayed potent inhibition of cell recruitment in vitro with an IC50 of 0.5 nm but demonstrated little efficacy in various animal models of human disease. On the contrary, 1F11 showed efficacy in several models of inflammation yet was less potent at inhibiting chemotaxis in vitro with an IC50 of 21 nm. Furthermore, we observed that 1B6 displayed a rapid dose-dependent clearance (t½ 10–60 h) in contrast to 1F11, which presented a dose-proportional pharmacokinetic profile and a half-life of 12 days. Moreover, 1B6 recognized glycosaminoglycan (GAG)-bound CXCL10, resulting in target-mediated clearance, which was corroborated using CXCL10-deficient mice. In contrast to 1B6, 1F11 inhibited the interaction of CXCL10 with GAGs, did not recognize GAG-bound CXCL10, and did not display target-mediated drug disposition. Confirming previous animal studies, 1B6 was poor at reversing glycemia in a model of type 1 diabetes, whereas 1F11 induced early and prolonged control of diabetes. Furthermore, when using 1A4, a subsequently generated anti-mCXCL10 mAb that shares the property with 1F11 of being unable to recognize CXCL10 immobilized on GAG, we observed a similar superior control of diabetes as compared with 1B6. We therefore concluded that targeting chemokines with antibodies such as 1B6 that recognize the more abundant GAG-bound form of the chemokine may not be the optimal strategy to achieve disease control.
Keywords: chemokine, chemotaxis, glycosaminoglycan, monoclonal antibody, pharmacokinetics, therapeutic inhibition
Introduction
In the course of acute inflammation, a massive migration of immune cells toward the site of inflammation occurs. This recruitment is essential to fight bacterial or viral infection and is mediated by chemokines, a family of small cytokines. Chemokines are chemoattractant proteins of 8 to 15 kDa, further subclassified into four families (CC, CXC, CX3C, and XC) (1). Chemokines activate cells by a high affinity interaction with G protein-coupled receptors (GPCR)2 and have a low affinity interaction with glycosaminoglycans (GAG), which is essential for activity in vivo (2). GAGs are a family of sulfated linear polysaccharides that are expressed as proteoglycans on the surface of endothelial cells. The interaction of chemokines with GAGs is required for the formation of haptotactic gradients on cell surfaces, preventing the diffusion of chemokines from their site of production under the conditions of flow in the circulation and providing a migration cue to leukocytes (3). In addition, this chemokine-GAG interaction has been demonstrated to play a role in chemokine oligomerization, transport across endothelia, protection from proteolytic cleavage, and modulation of the binding to chemokine receptors (2, 4–7).
Chemokine and chemokine receptors are up-regulated in many inflammatory diseases such as atherosclerosis, colitis, chronic obstructive pulmonary disease, psoriasis, and rheumatoid arthritis (RA). Their role is mainly to induce cellular recruitment to the inflamed organ, perpetuating the inflammatory response (8, 9). Moreover, the chemokine system has also been demonstrated to be involved in tumor growth and metastasis, with chemokines potentially serving also as growth factors (10). The chemokine system therefore has been pursued by pharmaceutical companies using small molecular weight compounds and, more recently, monoclonal antibodies (mAb). mAbs are highly selective molecules that allow the targeting of a single ligand or receptor without interfering with the other closely related proteins of the chemokine system (11). In addition, they have a long half-life in vivo, an interesting property in the context of chronic inflammatory diseases.
Although many antibodies targeting the chemokine system have been studied in animal models and clinical trials, only two have reached the market. Mogamulizumab (Poteligeo®), a humanized mAb directed against the CC chemokine receptor 4 (CCR4), has been approved in Japan for the treatment of relapsed or refractory adult T-cell leukemia/lymphoma and is currently in late phase development in Europe and the United States (12). Abcream (from Anogen) is a topical application of an anti-CXCL8 mAb marketed in China for psoriasis.
To date, no mAb targeting a chemokine ligand has been approved for systemic application, although several have reached clinical trials (11, 14). However, although some preliminary results were promising, several mAbs have been discontinued due to lack of efficacy in late phase clinical trials. For example, the anti-CCL2 mAbs ABN912 (Novartis) and CNTO 888 (Centocor) did not demonstrate clinical benefit under inflammatory conditions (RA and chronic obstructive pulmonary disease) or in the treatment of solid tumors, respectively (15–17). A similar lack of clinical efficacy was observed for the anti-CXCL8 mAb ABX-IL8 (Abgenix) in the case of RA, psoriasis, and chronic obstructive pulmonary disease and the anti-CCL11 mAb bertilimumab (Immune Pharmaceuticals) in allergic rhinitis and allergic conjunctivis (18, 19). Regarding CXCL10, two mAbs have been tested in clinical trials, NI-0801 (Novimmune) and BMS-936557 (Bristol-Myers Squibb). Both have been reported to be safe and well tolerated in healthy volunteers (14, 20). However, the anti-inflammatory activity of BMS-936557 in phase II clinical trials is limited (21, 22).
Explanations such as the promiscuity of the chemokine system have been proposed for the lack of success in clinical trials (23). Indeed, the majority of chemokines are able to bind to several chemokine receptors, and receptors can often be activated by various ligands, leading to the hypothesis that the inhibition of one single chemokine or receptor is not sufficient to observe beneficial clinical effects. However, data are currently accumulating to demonstrate that the chemokine system is not redundant in terms of signal cascades as well as in the temporal and spatial patterns of expression in vivo (24). Recently it has been shown that the ligands for CXCR3 induce different effects. Although it has been established that CXCL10 and, to a slightly lesser extent, CXCL9 are pro-inflammatory, CXCL11 has been shown to induce the development of Tregs (regulatory T-cells) and thus is anti-inflammatory (25). These findings support therapeutic targeting of a ligand as opposed to the promiscuous receptor, highlighting that the more appropriate question may be to find the right target for a given indication (26). However, another factor may be the abundance of the target in the body especially during disease, as chemokines are sequestered on GAGs, and only when they formed complexes in vivo with mAbs could the true level of target be appreciated (14–17, 27). This recent observation suggests that higher doses of mAb will be required to adequately inhibit the activity of the target (23, 26). Finally, as the active form of the chemokine is immobilized on GAGs, it has been proposed that a therapeutically effective mAb should bind to the GAG-bound form as well as the soluble form of the chemokine (14).
Thus, in an attempt to better understand how to best target a chemokine to achieve a therapeutic benefit, we studied the properties of two anti-mouse (m) CXCL10 mAbs shown to have differing levels of efficacy in certain models of human disease. 1B6, a rat anti-mCXCL10 IgG displaying strong inhibitory properties in chemotaxis assays in vitro, is not very efficient in models of inflammatory diseases (data not shown). On the contrary, 1F11, a hamster anti-mCXCL10 IgG (28), has a much lower potency in in vitro assays but is efficacious in various murine models of disease (28–35). We therefore further dissected the mode of action of 1B6 and 1F11 to determine whether, beyond the in vitro potency in the chemotaxis assay, additional properties might be required for a mAb to be efficacious in vivo. To extend our analysis, we also included a recently generated anti-mCXCL10 antibody, 1A4 (36), which does not bind to chemokines in the context of GAGs, a property that we have discovered differentiates 1F11 from 1B6. The results induced us to propose a new compartment to consider targeting when attempting to neutralize chemokines in patients.
Results
Reformatting and in Vitro Characterization of 1B6 and 1F11
As 1B6 is a rat antibody and 1F11 a hamster antibody, both antibodies were reformatted onto the human IgG1 backbone in order to exclude effects linked to isotype differences. The VH and VL domain sequences of each antibody were isolated from their respective hybridomas and subcloned into a vector encoding the constant domains of the human IgG1 sequence. As 1A4 is a human IgG1, no reformatting was needed. All antibodies were expressed in HEK cells and purified using a CH1-specific affinity resin.
The reformatted antibodies named human 1B6 (h1B6) and human 1F11 (h1F11) were tested in vitro to assess the impact of the reformatting on their function. Using surface plasmon resonance and chemotaxis assays, we observed that h1B6 and h1F11 retained equivalent binding and neutralization to that of the original rat and hamster proteins, respectively (data not shown). The reformatting also allowed us to compare the antibodies using the same experimental format. Binding affinities were characterized using the biolayer interferometry (BLI) technology, and both antibodies showed affinities for mCXCL10 with dissociation constants (KD) in the low nanomolar range, i.e. 2.6 nm for h1B6 and 6.2 nm for h1F11 (Fig. 1, A and B, and Table 1). As observed with the native 1B6 and 1F11 antibodies, h1B6 was a more potent inhibitor than h1F11 in a CXCL10-induced transmigration assay using cells expressing CXCR3 (Fig. 1C and Table 1). 1A4 is a human IgG1 antibody that has an affinity and potency similar to 1B6, as indicated in Table 1 (36).
FIGURE 1.

In vitro characterization of chimeric anti-mCXCL10 mAbs. A, binding of mCXCL10 to h1B6 by BLI using Protein A biosensors. Increasing concentrations of NusA-mCXCL10 (46.2, 23.1, 11.5, 5.77, 2.89, and 1.44 nm) were used to determine the affinity of h1B6 for the chemokine. The experimental data are displayed as a straight line and the corresponding fitting curves as dashed lines. Based on these data, the affinity of h1B6 for mCXCL10 (KD) is 2.60 nm. Data are representative of two independent experiments. B, binding of mCXCL10 to h1F11 by BLI. The experiment is similar to that explained in A. Based on these data, the affinity of h1F11 for mCXCLl0 (KD) is 6.16 nm. Data are representative of two independent experiments. C, inhibition of 5 nm mCXCL10-mediated chemotaxis of L1.2/hCXCR3 transfectants by h1B6, h1F11 and 1A4. Data are presented as the mean of triplicates ± S.E. and are representative of four independent experiments.
TABLE 1.
In vitro characterization of h1B6, h1F11, and 1A4
Kinetic parameters were determined by BLI and inhibition by chemotaxis assay (5 nm mCXCL10).
| ka × 104 | kd × 10−4 | KD | IC50 | |
|---|---|---|---|---|
| m−1s−1 | s−1 | nm | nm | |
| h1B6 | 6.40 ± 2.78 | 1.72 ± 0.97 | 2.60 ± 0.38 | 0.48 ± 0.37 |
| h1F11 | 2.02 ± 0.29 | 1.23 ± 0.06 | 6.16 ± 0.62 | 21.3 ± 7.9 |
| 1A4a | 80.4 | 8.3 | 1.0 | 1.6 |
a Ref. 36.
Characterization of mCXCL10, GAG, and Antibody Interactions
The initial experiment used the GAG heparin to evaluate the ability of the antibodies to recognize GAG-bound chemokine. No binding of mCXCL10-heparin complexes to h1F11 or 1A4 was observed, whereas a robust dose-dependent response was obtained in the case of h1B6 (Fig. 2A and supplemental Fig. S2). As expected, the antibodies could not bind directly to heparin in the absence of chemokine (data not shown).
FIGURE 2.

Recognition of GAG-bound mCXCL10 by h1B6 but not by h1F11 or 1A4. Experimental setups are represented schematically on the right: GAG, black curved lines; mCXCL10, red circles. A, ELISA measuring the binding of mCXCL10-heparin complexes to immobilized antibodies. Data are presented as the mean ± S.E. of triplicates and are representative of three independent experiments. B, binding of antibodies to heparan sulfate displayed mCXCL10. Streptavidin biosensors coated with biotinylated heparan sulfate were dipped into wells containing 100 nm mCXCL10 for 5 min. Dissociation was then recorded for 7 min before the biosensors were transferred to wells containing 1 μg/ml antibody (5 min, association). Data are representative of two independent experiments. C, mCXCL10 was coated on adherent HUVEC. Following the washes, the testing antibody, as well as the detection buffer, was added on the cells. The numbers of fluorescent events were quantified. Data are representative of two independent experiments. The white bar on the images represents 100 μm.
To validate these results with another member of the GAG family, we used BLI to observe the binding of the mAbs to mCXCL10 bound to heparan sulfate. Biotinylated heparan sulfate was coated onto streptavidin biosensors, which were dipped into the chemokine solution and subsequently into the antibody-containing wells. We observed that h1B6 bound to mCXCL10 displayed on heparan sulfate, whereas h1F11 and 1A4 did not (Fig. 2B).
Finally, these findings were assessed in a system closer to the in vivo situation by using human umbilical vein endothelial cells (HUVEC), which express a complex mixture of GAGs on their surface. HUVEC were incubated with an excess of mCXCL10 and then h1B6, h1F11, 1A4 or an isotype control antibody, and the number of fluorescently labeled cells was quantified (Fig. 2C). Binding of h1B6 to the mCXCL10-coated HUVEC was observed, whereas no signal was obtained with h1F11, 1A4, or the control antibody. Thus, using three different experimental approaches and different types of GAGs, the results indicate that only h1B6 binds to mCXCL10 immobilized on GAGs, whereas neither h1F11 nor 1A4 interact with GAG-bound CXCL10.
We then determined whether the antibodies prevented the interaction of the chemokine with GAGs. Using an ELISA format, we observed that when heparin was added onto immobilized mCXCL10-h1F11 or mCXCL10-1A4 complexes, the polysaccharide bound only weakly to mCXCL10, indicating that h1F11 and 1A4 preclude the association between mCXCL10 and GAGs (Fig. 3A). On the contrary, the mCXCL10-h1B6 complexes demonstrated a strong binding to heparin, strengthening the concept of simultaneous interaction of mCXCL10 with h1B6 and GAGs. As a control, using biotinylated chemokine we checked that the chemokine was still present on the plate after incubation with heparin, confirming that mCXCL10 was not released from the antibody-antigen complexes following the addition of heparin (data not shown).
FIGURE 3.

Inhibition of mCXCL10 binding to GAGs by h1F11. A, immobilized antibodies were incubated with 50 nm mCXCL10. FITC-tagged heparin was then added on the antibody-chemokine complexes, and bound heparin was detected with an anti-FITC. Data are presented as the mean ± S.E. of duplicates and are representative of two independent experiments. B, biolayer interferometry. Streptavidin biosensors were loaded with biotinylated heparan sulfate and dipped into a solution containing a constant concentration of 30 nm chemokine and a dose response of h1F11; these were preincubated to allow complex formation. A clear decrease in signal was observed with increasing concentrations of h1F11 (top graph). The dose-dependent inhibition of the maximal response is shown on the bottom graph.
To confirm these findings, the binding was then analyzed by BLI. A constant concentration of chemokine (30 nm) was incubated with increasing amounts of h1F11, and the complexes were loaded on heparan sulfate-coated biosensors. Under these conditions, a clear decrease in the maximum response was observed with increasing h1F11 concentrations, demonstrating that the binding of h1F11 to mCXCL10 inhibits the interaction of the chemokine with heparan sulfate (Fig. 3B). This phenomenon was not observed with h1B6 (data not shown).
Pharmacokinetic Properties of Anti-mCXCL10 Antibodies
Having established that the mAbs differed in their ability to recognize GAG-bound mCXCL10, we studied the in vivo behavior of 1B6 and 1F11. First, a single-dose pharmacokinetic (PK) analysis was performed in mice to determine the elimination kinetic profile. Three doses of each antibody were used (i.e. 1, 5, and 25 mg/kg), and the amount of free mAb in the serum was quantified by ELISA. 1F11 demonstrated a linear PK profile with a half-life of 12 days. In contrast, 1B6 rapidly disappeared from the circulation and displayed a non-linear PK profile, and clearance was dependent on the dose administered (Fig. 4A and Table 2).
FIGURE 4.
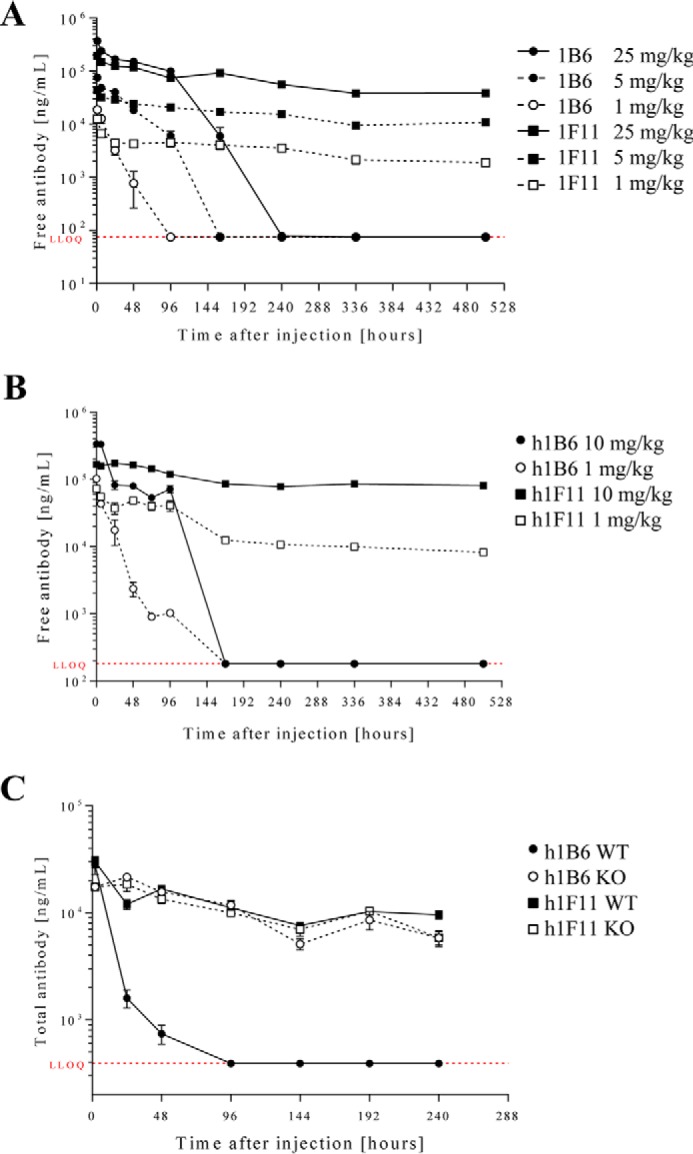
Pharmacokinetic profiles of antibodies in C57BL/6 mice. A single dose of 1B6 and 1F11 (A) or of their humanized version, h1B6 and h1F11 (B), was injected intravenously, and the concentration of free antibody in plasma was determined by ELISA at the indicated time points. Humanized antibodies were also tested in a similar way in C57BL/6 CXCL10 knock-out mice (C). The lower limit of quantification (LLOQ) is indicated in red on each graph. For each experiment, n = 3 or 4 for each time point.
TABLE 2.
Pharmacokinetic properties of 1B6 and 1F11 in C57BL/6 wild type mice
| Antibody | Dose | Clearance | t1/2 |
|---|---|---|---|
| mg/kg | ml/h/kg | h | |
| 1B6 | 1 | 3.33 | 10 |
| 5 | 1.84 | 27 | |
| 25 | 1.29 | 59 | |
| 1F11 | 1 | 0.40 | 313 |
| 5 | 0.40 | 293 | |
| 25 | 0.53 | 246 |
To evaluate whether the difference in the PK profiles of 1B6 and 1F11 was due to the different isotypes, we carried out a single-dose PK study with the antibodies reformatted onto a human IgG1 Fc (h1B6 and h1F11 as well as 1A4) and injected either 1 or 10 mg/kg intravenously into wild type (WT) mice. The results were similar to those obtained in the first experiment, i.e. h1F11 demonstrated a dose-proportional PK profile, whereas h1B6 was rapidly cleared (Fig. 4B and Table 3). To determine whether the rapid disappearance of h1B6 was due to target-mediated clearance, a PK study was performed in CXCL10-deficient mice by injecting each antibody (1 mg/kg) intravenously. The PK profiles of h1F11 in the wild type and knock-out mice were equivalent. However, distinct profiles were observed in WT versus chemokine-deficient mice with h1B6 (Fig. 4C). These results confirmed that the rapid elimination of h1B6 seen in WT mice was due to binding of the antibody to its target, i.e. CXCL10. As anticipated, 1A4 demonstrated a linear PK profile and a half-life comparable with h1F11 in WT mice, correlating with its lack of interaction with GAG-bound mCXCL10 (Table 2).
TABLE 3.
Pharmacokinetic properties of h1B6, h1F11, and 1A4 in C57BL/6 wild type mice
| Antibody | Dose | t1/2 |
|---|---|---|
| mg/kg | h | |
| h1B6 | 1 | 10 |
| 10 | 27 | |
| h1F11 | 1 | 163 |
| 10 | 165 | |
| 1A4 | 1 | 135 |
| 10 | 125 |
It is currently thought that considerably larger amounts of chemokines are present immobilized on endothelial surfaces than in the circulation (14). To confirm this hypothesis for CXCL10, we administrated heparin to naive mice, as free sulfated glycans compete with endogenous GAGs leading to the release of chemokines immobilized on GAGs (37). The level of released chemokine could then be measured in the circulation. Indeed, a massive increase (200-fold) of mCXCL10 was rapidly observed (i.e. within 15 min) after heparin administration (Fig. 5). A similar mobilization was also observed for CXCL9 (data not shown). This result demonstrates that a significant amount of chemokine is sequestered on GAGs even under steady state conditions.
FIGURE 5.
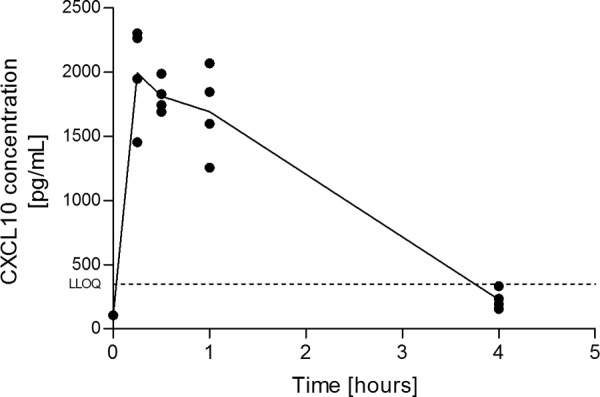
Release of chemokine following heparin administration. C57BL/6 mice were injected with free heparin, and the blood concentration of mCXCL10 was followed for 4 h (n = 4 for each time point). The lower limit of quantification (LLOQ) is indicated on the graph. Data are representative of two independent experiments.
In Vivo Activity of anti-mCXCl10 Antibodies in Murine Models
We had tested 1B6 previously at various doses in murine models of delayed-type hypersensitivity, arthritis, liver fibrosis, and colitis and did not observe significant anti-inflammatory activity (data not shown). In contrast, 1F11 has been reported to ameliorate inflammation in several models of disease (29, 30, 30–33). We, thus, performed a comparison of these two mAbs as well as 1A4, using the CXCL10-dependent model of type 1 diabetes in RIP-LCMV mice. These transgenic mice express the glycoprotein of the lymphocytic choriomeningitis virus (LCMV) in β-cells of the islets of Langerhans driven by the rat insulin promoter (RIP). Infection with LCMV induces the infiltration of glycoprotein-specific T cells into the islets and thus an autoimmune response against β-cells resulting in symptoms that mimic type 1 diabetes in humans. The recently reported combination of anti-CD3 and anti-CXCL10 therapy was used, as it has been shown to be CXCL10-dependent (38).
The mice were infected with LCMV on day 0 and an anti-CD3 antigen-binding fragment (F(ab′)2) was administered on days 10, 11, and 12. This anti-CD3 treatment was followed by administrations of the anti-CXCL10 mAb (100 μg/mouse, three times/week starting at day 13 until day 28). Blood glucose levels were monitored, and mice were considered diabetic if the level rose above 300 mg/dl. Mice that were diabetic at day 15 were monitored until day 112 to determine the percentage of animals with blood glucose levels remaining stably below 300 mg/dl, reflecting control of the disease (Fig. 6). Important fluctuations in glucose levels were observed generally (Fig. 6, F–J); however groups treated with anti-CD3 mAb fragments in combination with h1F11 or 1A4 showed a consistent control of these fluctuation in a majority of the animals after days 60 and 35, respectively (Fig. 6, I and J). When comparing the various treatment groups, the combination of anti-CD3 with either 1F11 or 1A4 with anti-CD3 therapy was the most effective in reducing the percentage of diabetic mice (Fig. 6, D–E). In contrast, the addition of h1B6 to the anti-CD3 treatment brought no benefit over anti-CD3 treatment alone, and fluctuations in glucose levels continued in a majority of the animals (Fig. 6, C and H). Taken together, these results demonstrate that a CXCL10-driven disease is much better controlled using a mAb that has the features of antibodies such as 1F11 and 1A4 rather than 1B6.
FIGURE 6.

Type 1 diabetes model in RIP-LCMV mice and treatment with anti-CXCL10 antibodies. Mice were infected by LCMV at day 0. When indicated, mice were treated with 3 μg/mouse anti-CD3 F(ab′)2 at days 10, 11, and 12 and with 100 μg/mouse anti-CXCL10 IgG (1F11, h1F11, h1B6, or 1A4) from day 13 to day 28. Blood glucose levels were measured throughout the study. A–E, glucose levels for individual animals are represented, with each line corresponding to one animal. The number of animals per group is indicated in the title of each graph. A, isotype control; B, anti-CD3; C, anti-CD3 and 1B6; D, anti-CD3 and 1F11; E, anti-CD3 and 1A4. F, diabetes incidence curves are shown for all treatment groups. G–J, comparison of individual treatment groups with the isotype control group.
Discussion
Despite intensive efforts, therapeutic targeting in the chemokine field for anti-inflammatory therapies has not yet met with much success. Several hypotheses to explain this low rate of success have been proposed (23, 26). The majority of clinical strategies target the receptor, but there have been several trials conducted with antibodies targeting the ligand. Here, we characterized two anti-mCXCL10 mAbs, 1B6 and 1F11, displaying similar in vitro inhibitory activities but showing significantly different in vivo efficacy in different models. We therefore thought to analyze the mode of action of these two antibodies to improve our comprehension of chemokine inhibition in order to define the characteristics that are essential for the activity of anti-chemokine antibodies in vivo.
Surprisingly, and contrary to conventional thinking, the antibody displaying the lower potency in in vitro chemotaxis assays, 1F11, was the more efficient in vivo, whereas the subnanomolar inhibitor, 1B6, was almost ineffective. Looking at the in vivo data obtained with the original rat and hamster antibodies, two hypotheses were advanced to explain the different behaviors of 1B6 and 1F11. First, the considerably longer half-life of 1F11 could have been due to the difference in antibody backbone, i.e. 1B6 being a rat antibody and 1F11 a hamster antibody. In this case, the extended half-life of 1F11 would be the only explanation justifying its increased efficiency in vivo. Second, 1B6, but not 1F11, could be subject to target-mediated drug disposition (TMDD), meaning that the interaction with mCXCL10 accelerates its clearance.
The first hypothesis can be ruled out by comparing the reformatted antibodies that comprise the variable domains of 1B6 or 1F11 linked to the constant domains of human IgG1. These chimeric antibodies displayed PK properties similar to those of the original mAbs. Moreover, analysis of PK profiles in CXCL10 knock-out mice demonstrated that the rapid disappearance of 1B6 from circulation was due exclusively to its interaction with the chemokine rather than to backbone-driven effects, supporting the hypothesis that 1B6 is prone to TMDD, whereas 1F11 is not.
For this second hypothesis to be valid, the interactions of 1B6 and 1F11 with CXCL10 should not be equivalent. This is indeed true, as 1B6 recognized the chemokine when it was displayed on GAG, whereas 1F11 did not. Moreover, 1F11 prevented the binding of CXCL10 to GAGs, which was not the case for 1B6. Epitope mapping studies confirmed these results and revealed that 1B6 and 1F11 binding sites partially overlapped but were not equivalent (data not shown). Interestingly, TMDD is usually observed with antibodies that recognize membrane proteins (39). In a sense, this was also the case here, as 1B6 recognized the “membrane-bound” form of CXCL10, i.e. the GAG-bound chemokine present on endothelial cell surfaces, whereas 1F11 only bound to soluble chemokine.
The difference in the behavior of 1B6 and 1F11 may teach us important concepts of chemokine biology in vivo. The interaction between chemokine and GAGs was shown to be essential for chemokine in vivo activity by using mutants with abrogated GAG-binding properties, which were inactive in a peritoneal recruitment assay (2). In addition, the interaction with GAGs has been demonstrated to mediate chemokine oligomerization on the endothelial surface (5). These results reinforce the concept of haptotaxis proposed previously by Rot (40) that led to the hypothesis that the chemokine gradient may be immobilized on the GAG surface, with the oligomerization of the chemokines increasing their local concentration on GAGs. When the receptor and GAG binding sites overlap partially or entirely, a monomer would be unable to interact with both simultaneously. However, when the chemokine is oligomerized on GAGs, binding to the receptor would be feasible (41), but this three-way interaction has never been formally demonstrated. Recently, direct evidence for the chemokine gradient was provided in an elegant study where endogenous gradients of CCL21 immobilized on heparan sulfate were visualized within the ear dermis; the CCL21 signal peaked on lymphatic vessel walls and decreased with increasing distance from the vessel. Dendritic cells, expressing the CCL21 receptor CCR7, were able to locally detect CCL21 concentration differences and migrated to nearby lymphatic vessels in a directed manner (42).
The results obtained with 1F11 were confirmed with the subsequently created anti-mCXCL10 1A4 mAb, an antibody with properties similar to 1F11, as it does not recognize the GAG-bound form of CXCL10 and does not display an altered pharmacokinetic profile in WT mice. However, 1A4 is more potent in vitro than 1F11 and shows the highest anti-inflammatory properties in vivo, suggesting that high potency and inability to bind to chemokine in the context of GAGs represent superior characteristics to achieve therapeutic effect. The results obtained with this second antibody support the hypothesis that the active, receptor-binding form of CXCL10 is the soluble protein rather than the GAG-displayed chemokine.
Thus, the differences in in vivo activity observed between these two types of antibodies can be explained as follows. Upon administration, 1B6 binds to both GAG-bound and soluble CXCL10. As the amount of chemokine displayed on the endothelial surface is greater than that of free CXCL10, the vast majority of 1B6 rapidly binds to surface-bound CXCL10 and is therefore not detectable in the circulation, resulting in poor pharmacokinetic properties. It is thought that chemokines have a rapid turnover and high synthesis rates; therefore it may be almost impossible to saturate the system even with high doses of an antibody with this type of characteristic. An antibody recognizing GAG-bound chemokines, such as 1B6, will be depleted from the circulation thereby preventing it from inhibiting soluble chemokines released from the endothelial surface effectively. The activity of these chemokines is therefore not inhibited and cell migration occurs. Interestingly, CXCL10 has been reported to be rapidly internalized in vitro following binding to endothelial cells via receptor-independent clathrin-mediated mechanisms (43, 44). If this is also the case in vivo, 1B6 might be concomitantly internalized and degraded, contributing to its rapid clearance. Furthermore, if the GAG-bound 1B6-chemokine complexes are released from the endothelial surface into the circulation, this may lead to the formation of large immune complexes rapidly cleared by phagocytosis, also contributing to the short half-life of 1B6 in circulation.
The case of 1F11 is significantly different. Following administration, 1F11 only binds to the soluble chemokine fraction because 1F11 does not recognize the chemokine displayed on GAGs, and therefore saturation of the antibody will not occur, its half-life is longer, and its pharmacokinetic properties are adequate for clinical settings. During inflammation, active chemokines are released from GAGs but there are sufficient unbound 1F11 molecules available to compete with the binding of free chemokines to their receptor, thereby reducing the number of cells migrating toward the inflamed site and improving the disease.
Based on in vitro data, another possible mode of action of 1F11 would be the inhibition of the binding of chemokine to GAGs. Once they are produced in the tissue, chemokines are thought to be transcytosed from the basal to the apical side of the endothelium. By binding the chemokine in the tissue before its binding to GAGs, 1F11 can prevent the formation of the gradient. However, if this mechanism was the principle mode of action of 1F11, the number of CXCL10 molecules bound by 1F11 would ultimately be very large and the plasma concentration of the free antibody should rapidly decrease, which is not the case (Fig. 4). We therefore assume that this phenomenon may occur in vivo but is probably marginal due to the low penetrance of mAbs into tissues (the concentration of antibodies in tissues is estimated to be ∼10-fold lower than that in the blood (45)). However, the fact that 1F11 inhibits binding to GAGs would prevent the CXCL10 that has been released into solution to rebind to the endothelial surface.
Although data obtained from experiments in mice should be extrapolated with caution, especially in the case of the chemokine system (46, 47), our results demonstrate that therapeutic targeting of chemokines with antibodies recognizing the chemokine immobilized on GAGs may not be an optimal strategy. Even if the development of such antibodies has been proposed as a solution to the failure of anti-chemokine mAbs in clinical trials, the recognition of GAG-bound chemokine appears to have an enormous impact on the half-life of the monoclonal antibody in the circulation. This may be particularly problematic for abundant chemokines such as CXCL10, CCL2, and CCL5. Care should therefore be taken in the selection of antibody candidates to ensure that they have the optimal characteristics for activity in vivo to prevent inflammation. Lastly, this study highlights the complexity of chemokine biology in vivo and warrants further studies in animal models of human inflammatory diseases using these antibodies.
Experimental Procedures
Materials
Chemokines were produced using standard protocols for the expression and purification of native mCXCL10 (48) and the method described elsewhere for the production of mCXCL10-NusA fusion (49). Oligonucleotides were synthesized by Eurofins MWG Operon (Ebersberg, Germany), and sequencing was performed by Fasteris (Geneva, Switzerland). The 1F11 hybridoma was produced as described previously (28). 1B6 was produced by immunizing rats with mouse CXCL10 and cells from the draining lymph nodes fused to the SP2/0 cell line to generate the hybridoma. 1B6 was shown to be specific for mCXCL10 (supplemental Fig. S1).
Reformatting and Expression of 1B6 and 1F11 as Human IgG1
The variable domain sequences of 1B6 and 1F11 were obtained from RNA isolated from hybridoma cells using the RNeasy Plus mini kit (Qiagen). Cells were homogenized initially on a QIAshredder homogenizer (Qiagen), and RNA was subsequently extracted according to the manufacturer's instructions. Different strategies were then applied for the isolation of the 1B6 and 1F11 sequences.
1B6 cDNA was prepared from previously isolated RNA using ThermoScript reverse transcriptase (Life Technologies) and amplified by PCR using a mouse IgG library primer set (Progen, Heidelberg, Germany). Variable heavy or light chain primers were used in combination with a constant region reverse IgG primer, and PCR products were loaded on a 1% agarose EX-Gel (Life Technologies). Reactions leading to the presence of a fragment around 300–400 bp were purified and used for further steps. A poly-A tail was then added to each fragment isolated from the PCR by the use of the NEBNext dA-tailing module (New England Biolabs), and the DNA fragments were cloned into a plasmid vector for sequencing using the TOPO TA cloning kit (Life Technologies). Following transformation into TOP10 Escherichia coli bacteria (Life Technologies), the DNA was amplified with the QIAPrep spin miniprep kit (Qiagen) and sequenced externally.
As we did not expect the mouse library primer set to cross-react with hamster sequences, a different strategy was chosen for the isolation of 1F11 sequences. cDNA was prepared from isolated RNA using the SMARTer RACE (rapid amplification of cDNA ends) kit (Clontech, Saint-Germain-en-Laye, France) according to the manufacturer's instructions. The same kit was used to perform a 5′-RACE. Primers specific for hamster heavy chain (CTT GTC CAC CTT GGT GCT GCT GG), κ chain (CAT TCC TGT TCA GGG TCT TGA CAA TG), or λ chain (GCT CTT CTC CAC AGT TTC CCC TTC ATG GGT AAC TTG GCA GGA AAC C) were used to amplify the variable domain sequences. The protocol described for 1B6 was then followed to add a poly-A tail and to clone the DNA into sequencing plasmid vector.
Once the VH and VL sequences of both antibodies were identified, they were subcloned into a vector containing the constant domains of human IgG1. Antibodies were expressed in PEAKTM cells and purified as described previously (36) to obtain the reformatted h1B6 and h1F11 mAbs.
Determination of Binding and Inhibitory Properties of h1B6 and h1F11
The affinities of h1B6 and h1F11 for mCXCL10 were determined by BLI binding experiments performed on the Octet RED96 system (ForteBio, Portsmouth, United Kingdom). Antibodies diluted to 2.5 μg/ml in kinetics buffer (KB) (PBS containing 0.1% bovine serum albumin, 0.02% Tween 20, and 0.05% sodium azide) were loaded for 2 min on protein A biosensors. The baseline was then allowed to stabilize for 2 min, and biosensors were subsequently dipped into the chemokine solution. As a nonspecific interaction was observed between mCXCL10 and the Protein A biosensors, the NusA-tagged variant of mCXCL10 was used instead of the native chemokine. Chemokine concentrations of 46.2, 23.1, 11.5, 5.77, 2.89, 1.44, and 0.72 nm were used, and association was followed for 10 min. Dissociation was performed for 15 min in KB, and biosensors were then regenerated by three cycles of 5 s in 10 mm glycine, pH 1.7, followed by 5 s in KB. Data were analyzed using Octet software and a global fitting 1:1 model. The kinetic parameters were measured twice for each antibody.
To evaluate the ability of each antibody to inhibit mCXCL10 activity in vitro, chemotaxis assays were performed as described previously (36). Briefly, mCXCL10 was used to induce the migration of murine pre-B L1.2 cells transfected with hCXCR3 in ChemoTx system chemotaxis plates with a pore size of 5 μm (NeuroProbe Inc., Gaithersburg, MD). The cell migration induced by 5 nm mCXCL10, corresponding to the EC80, was inhibited by increasing concentrations of h1B6 or h1F11. Plates were incubated for 4 h at 37 °C, and chemotaxis was evaluated by fluorometric microvolume assay technology.
Recognition of GAG-bound mCXCL10
The ability of h1B6 and h1F11 to recognize GAG-bound mCXCL10 was assessed initially using heparin as the GAG in an ELISA-like format. Maxisorp plates (Nunc) were coated overnight with an anti-human Fcγ (Jackson ImmunoResearch) and blocked on the following day with Dulbecco's phosphate-buffered saline (DPBS, Sigma-Aldrich) containing 3% bovine serum albumin. A dose response of antibodies was then captured onto the plate with incubation for 1 h. mCXCL10 at 5 nm was incubated with 55 nm fluorescein-tagged heparin (Life Technologies) to obtain chemokine-heparin complexes. These complexes were then added to the MicroWell plate. The plates were washed three times, and bound complexes detected with an HRP-coupled anti-fluorescein Fab (Roche Applied Science).
Binding of mAbs to heparan sulfate-displayed mCXCL10 was analyzed by BLI. Heparan sulfate sodium from bovine kidney (Sigma-Aldrich) was biotinylated by incubating EZ-link hydrazide-PEG4-biotin (Thermo Fisher Scientific) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC, GE Healthcare) with 1 mg of heparan sulfate in 200 μl MES buffer (Thermo Fisher Scientific) to final concentrations of 1.25 and 6.5 mm, respectively. The solution was incubated overnight at room temperature, and the excess biotin was removed by centrifugation using a 3K centrifugal filter device (Merck Millipore). For the BLI assay, biotinylated heparan sulfate diluted to2 μg/ml in 200 μl of KB was loaded for 2 min onto streptavidin biosensors. The biosensors were then successively dipped into 100 nm mCXCL10 (association, 5 min), KB (dissociation, 7 min), 1 μg/ml antibody (association, 5 min), and finally KB (dissociation, 8 min). Biosensors coated with an irrelevant biotinylated protein were used to assess nonspecific interactions.
Finally, we investigated whether h1B6 and h1F11 recognized the mCXCL10 displayed on the surface of endothelial cells. HUVEC (Lonza) were kept in culture in EGM-2 medium supplemented with hEGF, VEGF, hFGF-B, R3-IGF, and heparin (all from Lonza) according to the manufacturer's instructions until passage 8. Cells were split at 80–90% confluence (every 7–8 days) and seeded at 8000 cells/cm2. Two days before the experiment, confluent cells were seeded at 20,000 cells/well in black 96-well plates with transparent flat bottoms. On the day of the experiment, cells were washed twice with DPBS and then incubated at 4 °C for 1 h with 100 nm mCXCL10. The chemokine solution was discarded, and cells were again washed twice with DPBS. A solution containing the antibody to test (final concentration 5 nm) and the detection antibody, an Alexa Fluor 647-tagged anti-human Fcγ Fab (Jackson ImmunoResearch), was then added on the cells. Cells were incubated for 1 h at 4 °C, and the signals in the plates were read and quantified by fluorometric microvolume assay technology (FMAT) on an Applied Biosystems 8200 cellular detection system.
Inhibition of mCXCL10 Binding to GAGs
The ELISA-like assay described above used to determine whether h1B6, h1F11, and 1A4 recognize heparin-bound mCXCL10 was modified slightly to investigate whether the two mAbs are able to inhibit the binding of mCXCL10 to heparin. Antibodies (10 μg/ml) were coated onto the plate and blocked as described above before incubation with 50 nm mCXCL10for one hour. Following three washes with PBS containing 0.05% Tween-20, fluorescein-tagged heparin was added into the wells and binding was detected as described above. To ensure that mCXCL10 did not detach from the antibody in presence of heparin, the assay was also performed with biotinylated mCXCL10 which was detected with an HRP-coupled streptavidin (Jackson ImmunoResearch).
The ability of h1B6 and h1F11 to inhibit the binding of mCXCL10 to heparan sulfate was determined by BLI. Biotinylated heparan sulfate was loaded onto streptavidin biosensors as described above. Two hundred microliters of KB containing 30 nm mCXCL10 was preincubated for 30 min with increasing concentrations of antibody (50–0.2 μg/ml) before its addition into the BLI microplate. Biosensors were then dipped into the chemokine/antibody solution. Association and dissociation were followed for 12 min, and biosensors were regenerated by three cycles of 10 s in 5 m NaCl followed by 10 s in KB. Control experiments were also performed to ensure that the chemokine bound to the heparan sulfate biosensors and that the antibodies did not.
Analysis of Pharmacokinetic Profiles
The PK profiles of 1B6 and 1F11 were determined by single-dose PK studies. C75BL/6 female mice were injected intravenously with 1, 5, or 25 mg/kg antibody (1B6 and 1F11) or with 1 or 10 mg/kg antibody (h1B6, h1F11, and 1A4) (n = 3–4 for each group). Throughout the study, the mice were monitored for signs of pain or distress. Blood samples were taken at 0.25, 6, 24 h and 2, 3, 4, 7, 10, 14, and 21 days. To reduce the number of mice used, each animal was bled twice; submandibular samples were obtained initially under anesthesia, and at least 4 days later intracardiac samples were taken under terminal anesthesia prior to sacrifice. The PK properties in CXCL10 knock-out (50) and wild type C57BL/6 mice were also compared to assess CXCL10 influence on the mAb PK profile. Mice were injected intravenously with 1 mg/kg h1B6 or h1F11 (n = 5), and blood samples were taken at 2 and 24 h and at 2, 4, 6, 8, and 10 days.
Plasma samples were prepared and analyzed using two ELISAs. Total IgG levels were determined using a human IgG FastELISA kit (RD Biotech, Besançon, France). The levels of free or semi-occupied IgG were measured using an analytical ELISA developed in our laboratory. Plasma samples were added on biotinylated mCXCL10 (Almac, Craigavon, United Kingdom) captured beforehand on streptavidin microplates (Greiner Bio-One). Bound IgG were detected using a HRP-coupled anti-human Fcγ IgG (Jackson ImmunoResearch).
The pharmacokinetic analyses were conducted using a pharmacokinetic software package (WinNonlin Professional, version 6.3, Pharsight Corp., Mountain View, CA). The apparent terminal elimination half-life (t½) and the total serum clearance were determined from the mean serum concentration-time data of each antibody using non-compartmental procedures. The elimination rate constant (λz) was calculated by least squares linear regression of the terminal portion of the log-transformed serum concentration-time curve, and t½ was calculated according to the following formula.
| (Eq. 1) |
The area under the curve (AUC0 − last) was calculated using the linear/log trapezoidal rule. The total serum clearance (CL) was calculated using the following formula.
| (Eq. 2) |
In Vivo Mobilization of GAG-bound CXCL10
To release CXCL10 immobilized on GAGs, naive C57BL/6 mice were injected intravenously with 1.25 mg of heparin (Sigma-Aldrich). Blood was obtained from the retro-orbital plexus before heparin injection and at 15, 30, 60, and 240 min after injection (four separate mice were used for each time point). Plasma CXCL10 concentration was determined by multiplex ELISA (R&D Systems).
LCMV Model
The RIP-LCMV glycoprotein model of diabetes and the combi-therapy with anti-CD3/anti-CXCL10 has been described recently (38). Briefly, diabetes was induced at day 0 by LCMV infection using LCMV Armstrong clone 53b (51). Anti-murine CD3e antibody (F(ab′)2 fragment) clone 145-2C11 was obtained from Bio X Cell (West Lebanon, NH) and administrated intravenously at days 10, 11, and 12 (3 μg/mouse). Anti-CXCL10 treatment was performed intraperitoneally three times a week with 100 μg/mouse from days 13 to 28. Blood samples were taken from the tail vein and analyzed with a dynaValeo glucometer (dynamiCARE, Pfäffikon, Switzerland). Animals with blood glucose levels > 300 mg/dl were considered diabetic (13). In a minority of the mice, the infection was not successful, and the blood glucose levels never exceeded 300 mg/dl. These mice were therefore excluded from the analysis.
All animal studies were approved by the Geneva Cantonal Office for Veterinary Affairs, Switzerland (PK experiments in wild type mice) or by the local Ethics Animal Review Board of Darmstadt (PK experiments in CXCL10 knock-out mice, LCMV model).
Statistical Analysis
Diabetes incidence curves were analyzed using the Mantel-Cox log-rank test. The inhibition of heparin binding to mCXCL10 immobilized on antibodies was analyzed using the two-way analysis of variance test.
Author Contributions
Experiments were conceived by P. B., N. F., A. P., M. K.-V., F. G., V. B., W. F., and Z. J., and they were performed and analyzed by P. B., F. G., S. L., M. M., V. B., S. L., and M. C.-G. P. B., N. F., A. P., and M. K.-V. wrote the paper with the substantial support from U. C. and A. D. L. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank G. Magistrelli, G. Pontini, and Y. Poitevin for help with antibody expression and purification, P. Malinge for valuable advice on the biolayer interferometry assays, and L. Chatel for help with ELISA development and optimization.
This was supported by Grant HEALTH-F4-2011-281608 (TIMER) from the European Union Seventh Framework Programme (FP7–2007-2013). P. B., F. G., V. B., M. C.-G., Z. J., W. F., M. K.-V., A. P., and N. F. are current or former employees of Novimmune SA.

This article contains supplemental Figs. S1 and S2.
- GPCR
- G protein-coupled receptor
- BLI
- biolayer interferometry
- GAG
- glycosaminoglycan
- HUVEC
- human umbilical vein endothelial cell(s)
- KB
- kinetics buffer
- LCMV
- lymphocytic choriomeningitis virus
- PK
- pharmacokinetic
- RA
- rheumatoid arthritis
- TMDD
- target-mediated drug disposition
- DPBS
- Dulbecco's phosphate-buffered saline
- mCXCL10
- mouse CXCL10.
References
- 1. Zlotnik A., and Yoshie O. (2000) Chemokines: a new classification system and their role in immunity. Immunity 12, 121–127 [DOI] [PubMed] [Google Scholar]
- 2. Proudfoot A. E., Handel T. M., Johnson Z., Lau E. K., LiWang P., Clark-Lewis I., Borlat F., Wells T. N., and Kosco-Vilbois M. H. (2003) Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl. Acad. Sci. U.S.A. 100, 1885–1890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Salanga C. L., and Handel T. M. (2011) Chemokine oligomerization and interactions with receptors and glycosaminoglycans: the role of structural dynamics in function. Exp. Cell Res. 317, 590–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ellyard J. I., Simson L., Bezos A., Johnston K., Freeman C., and Parish C. R. (2007) Eotaxin selectively binds heparin: an interaction that protects eotaxin from proteolysis and potentiates chemotactic activity in vivo. J. Biol. Chem. 282, 15238–15247 [DOI] [PubMed] [Google Scholar]
- 5. Hoogewerf A. J., Kuschert G. S., Proudfoot A. E., Borlat F., Clark-Lewis I., Power C. A., and Wells T. N. (1997) Glycosaminoglycans mediate cell surface oligomerization of chemokines. Biochemistry 36, 13570–13578 [DOI] [PubMed] [Google Scholar]
- 6. Wang L., Fuster M., Sriramarao P., and Esko J. D. (2005) Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat. Immunol. 6, 902–910 [DOI] [PubMed] [Google Scholar]
- 7. Webb L. M., Ehrengruber M. U., Clark-Lewis I., Baggiolini M., and Rot A. (1993) Binding to heparan sulfate or heparin enhances neutrophil responses to interleukin 8. Proc. Natl. Acad. Sci. U.S.A. 90, 7158–7162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Comerford I., Kara E. E., McKenzie D. R., and McColl S. R. (2014) Advances in understanding the pathogenesis of autoimmune disorders: focus on chemokines and lymphocyte trafficking. Br. J. Haematol. 164, 329–341 [DOI] [PubMed] [Google Scholar]
- 9. Koelink P. J., Overbeek S. A., Braber S., de Kruijf P., Folkerts G., Smit M. J., and Kraneveld A. D. (2012) Targeting chemokine receptors in chronic inflammatory diseases: an extensive review. Pharmacol. Ther. 133, 1–18 [DOI] [PubMed] [Google Scholar]
- 10. Mukaida N., Sasaki S., and Baba T. (2014) Chemokines in cancer development and progression and their potential as targeting molecules for cancer treatment. Mediators Inflamm. 2014, 170381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Klarenbeek A., Maussang D., Blanchetot C., Saunders M., van der Woning S., Smit M. J., de Haard H., and Hofman E. (2012) Targeting chemokines and chemokine receptors with antibodies. Drug Discov. Today Technol. 9, e227–e314 [DOI] [PubMed] [Google Scholar]
- 12. Yoshie O., and Matsushima K. (2015) CCR4 and its ligands: from bench to bedside. Int. Immunol. 27, 11–20 [DOI] [PubMed] [Google Scholar]
- 13. Christen U., Wolfe T., Möhrle U., Hughes A. C., Rodrigo E., Green E. A., Flavell R. A., and von Herrath M. G. (2001) A dual role for TNF-α in type 1 diabetes: islet-specific expression abrogates the ongoing autoimmune process when induced late but not early during pathogenesis. J. Immunol. 166, 7023–7032 [DOI] [PubMed] [Google Scholar]
- 14. de Graaf K. L., Kosco-Vilbois M. H., and Fischer N. (2012) Therapeutic targeting of chemokines with monoclonal antibodies. Curr. Immunol. Rev. 8, 141–148 [Google Scholar]
- 15. Pienta K. J., Machiels J. P., Schrijvers D., Alekseev B., Shkolnik M., Crabb S. J., Li S., Seetharam S., Puchalski T. A., Takimoto C., Elsayed Y., Dawkins F., and de Bono J. S. (2013) Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Invest. New Drugs 31, 760–768 [DOI] [PubMed] [Google Scholar]
- 16. Sandhu S. K., Papadopoulos K., Fong P. C., Patnaik A., Messiou C., Olmos D., Wang G., Tromp B. J., Puchalski T. A., Balkwill F., Berns B., Seetharam S., de Bono J. S., and Tolcher A. W. (2013) A first-in-human, first-in-class, phase I study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 in patients with solid tumors. Cancer Chemother. Pharmacol. 71, 1041–1050 [DOI] [PubMed] [Google Scholar]
- 17. Haringman J. J., Gerlag D. M., Smeets T. J., Baeten D., van den Bosch F., Bresnihan B., Breedveld F. C., Dinant H. J., Legay F., Gram H., Loetscher P., Schmouder R., Woodworth T., and Tak P. P. (2006) A randomized controlled trial with an anti-CCL2 (anti-monocyte chemotactic protein 1) monoclonal antibody in patients with rheumatoid arthritis. Arthritis Rheum. 54, 2387–2392 [DOI] [PubMed] [Google Scholar]
- 18. Mahler D. A., Huang S., Tabrizi M., and Bell G. M. (2004) Efficacy and safety of a monoclonal antibody recognizing interleukin-8 in COPD: a pilot study. Chest 126, 926–934 [DOI] [PubMed] [Google Scholar]
- 19. Salib R., Salagean M., Lau L., DiGiovanna I., Brennan N., Scadding G., and Howarth P. (2003) The anti-inflammatory response of anti-eotaxin monoclonal antibody CAT-213 on nasal allergen-induced cell infiltration and activation. J. Allergy Clin. Immunol. 111, S347 [Google Scholar]
- 20. Yellin M., Goldwater R., Blanset D. L., and Cardarelli P. M. (2008) A double-blind, placebo-controlled, dose-escalation, safety and pharmacokinetic study of MDX-1100, a fully human anti-CXCL10 monoclonal antibody, in healthy subjects. Gastroenterology 134, A493–A494 [Google Scholar]
- 21. Yellin M., Paliienko I., Balanescu A., Ter-Vartanian S., Tseluyko V., Xu L. A., Tao X., Cardarelli P. M., Leblanc H., Nichol G., Ancuta C., Chirieac R., and Luo A. (2012) A phase II, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of MDX-1100, a fully human anti-CXCL10 monoclonal antibody, in combination with methotrexate in patients with rheumatoid arthritis. Arthritis Rheum. 64, 1730–1739 [DOI] [PubMed] [Google Scholar]
- 22. Mayer L., Sandborn W. J., Stepanov Y., Geboes K., Hardi R., Yellin M., Tao X., Xu L. A., Salter-Cid L., Gujrathi S., Aranda R., and Luo A. Y. (2014) Anti-IP-10 antibody (BMS-936557) for ulcerative colitis: a phase II randomised study. Gut 63, 442–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Solari R., Pease J. E., and Begg M. (2015) Chemokine receptors as therapeutic targets: why aren't there more drugs? Eur. J. Pharmacol. 746, 363–367 [DOI] [PubMed] [Google Scholar]
- 24. Steen A., Larsen O., Thiele S., and Rosenkilde M. M. (2014) Biased and G protein-independent signaling of chemokine receptors. Front. Immunol. 5, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zohar Y., Wildbaum G., Novak R., Salzman A. L., Thelen M., Alon R., Barsheshet Y., Karp C. L., and Karin N. (2014) CXCL11-dependent induction of FOXP3-negative regulatory T cells suppresses autoimmune encephalomyelitis. J. Clin. Invest. 124, 2009–2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schall T. J., and Proudfoot A. E. (2011) Overcoming hurdles in developing successful drugs targeting chemokine receptors. Nat. Rev. Immunol. 11, 355–363 [DOI] [PubMed] [Google Scholar]
- 27. Fetterly G. J., Aras U., Meholick P. D., Takimoto C., Seetharam S., McIntosh T., de Bono J. S., Sandhu S. K., Tolcher A., Davis H. M., Zhou H., and Puchalski T. A. (2013) Utilizing pharmacokinetics/pharmacodynamics modeling to simultaneously examine free CCL2, total CCL2, and carlumab (CNTO 888) concentration time data. J. Clin. Pharmacol. 53, 1020–1027 [DOI] [PubMed] [Google Scholar]
- 28. Khan I. A., MacLean J. A., Lee F. S., Casciotti L., DeHaan E., Schwartzman J. D., and Luster A. D. (2000) IP-10 is critical for effector T cell trafficking and host survival in Toxoplasma gondii infection. Immunity 12, 483–494 [DOI] [PubMed] [Google Scholar]
- 29. Christen U., McGavern D. B., Luster A. D., von Herrath M. G., and Oldstone M. B. (2003) Among CXCR3 chemokines, IFN-γ-inducible protein of 10 kDa (CXC chemokine ligand (CXCL) 10) but not monokine induced by IFN-γ (CXCL9) imprints a pattern for the subsequent development of autoimmune disease. J. Immunol. 171, 6838–6845 [DOI] [PubMed] [Google Scholar]
- 30. Hintermann E., Bayer M., Pfeilschifter J. M., Luster A. D., and Christen U. (2010) CXCL10 promotes liver fibrosis by prevention of NK cell-mediated hepatic stellate cell inactivation. J. Autoimmun. 35, 424–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hyun J. G., Lee G., Brown J. B., Grimm G. R., Tang Y., Mittal N., Dirisina R., Zhang Z., Fryer J. P., Weinstock J. V., Luster A. D., and Barrett T. A. (2005) Anti-interferon-inducible chemokine, CXCL10, reduces colitis by impairing T helper-1 induction and recruitment in mice. Inflamm. Bowel Dis. 11, 799–805 [DOI] [PubMed] [Google Scholar]
- 32. Zhang Z., Kaptanoglu L., Haddad W., Ivancic D., Alnadjim Z., Hurst S., Tishler D., Luster A. D., Barrett T. A., and Fryer J. (2002) Donor T cell activation initiates small bowel allograft rejection through an IFN-γ-inducible protein-10-dependent mechanism. J. Immunol. 168, 3205–3212 [DOI] [PubMed] [Google Scholar]
- 33. Rashighi M., Agarwal P., Richmond J. M., Harris T. H., Dresser K., Su M. W., Zhou Y., Deng A., Hunter C. A., Luster A. D., and Harris J. E. (2014) CXCL10 is critical for the progression and maintenance of depigmentation in a mouse model of vitiligo. Sci. Transl. Med. 6, 223ra23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Harris T. H., Banigan E. J., Christian D. A., Konradt C., Tait Wojno E. D., Norose K., Wilson E. H., John B., Weninger W., Luster A. D., Liu A. J., and Hunter C. A. (2012) Generalized Lévy walks and the role of chemokines in migration of effector CD8+ T cells. Nature 486, 545–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Campanella G. S., Tager A. M., El Khoury J. K., Thomas S. Y., Abrazinski T. A., Manice L. A., Colvin R. A., and Luster A. D. (2008) Chemokine receptor CXCR3 and its ligands CXCL9 and CXCL10 are required for the development of murine cerebral malaria. Proc. Natl. Acad. Sci. U.S.A. 105, 4814–4819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bonvin P., Venet S., Fontaine G., Ravn U., Gueneau F., Kosco-Vilbois M., Proudfoot A. E., and Fischer N. (2015) De novo isolation of antibodies with pH-dependent binding properties. MAbs 7, 294–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sweeney E. A., and Papayannopoulou T. (2001) Increase in circulating SDF-1 after treatment with sulfated glycans: the role of SDF-1 in mobilization. Ann. N.Y. Acad. Sci. 938, 48–52 [DOI] [PubMed] [Google Scholar]
- 38. Lasch S., Müller P., Bayer M., Pfeilschifter J. M., Luster A. D., Hintermann E., and Christen U. (2015) Anti-CD3/anti-CXCL10 antibody combination therapy induces a persistent remission of type 1 diabetes in two mouse models. Diabetes 64, 4198–4211 [DOI] [PubMed] [Google Scholar]
- 39. Tabrizi M. A., Tseng C. M., and Roskos L. K. (2006) Elimination mechanisms of therapeutic monoclonal antibodies. Drug Discov. Today 11, 81–88 [DOI] [PubMed] [Google Scholar]
- 40. Rot A. (1993) Neutrophil attractant/activation protein-1 (interleukin-8) induces in vitro neutrophil migration by haptotactic mechanism. Eur. J. Immunol. 23, 303–306 [DOI] [PubMed] [Google Scholar]
- 41. Wang X., Sharp J. S., Handel T. M., and Prestegard J. H. (2013) Chemokine oligomerization in cell signaling and migration. Prog. Mol. Biol. Transl. Sci. 117, 531–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Weber M., Hauschild R., Schwarz J., Moussion C., de Vries I., Legler D. F., Luther S. A., Bollenbach T., and Sixt M. (2013) Interstitial dendritic cell guidance by haptotactic chemokine gradients. Science 339, 328–332 [DOI] [PubMed] [Google Scholar]
- 43. Hillyer P., and Male D. (2005) Expression of chemokines on the surface of different human endothelia. Immunol. Cell Biol. 83, 375–382 [DOI] [PubMed] [Google Scholar]
- 44. Mordelet E., Davies H. A., Hillyer P., Romero I. A., and Male D. (2007) Chemokine transport across human vascular endothelial cells. Endothelium 14, 7–15 [DOI] [PubMed] [Google Scholar]
- 45. Wang W., Wang E. Q., and Balthasar J. P. (2008) Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin. Pharmacol. Ther. 84, 548–558 [DOI] [PubMed] [Google Scholar]
- 46. Zlotnik A., Yoshie O., and Nomiyama H. (2006) The chemokine and chemokine receptor superfamilies and their molecular evolution. Genome Biol. 7, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Johnson Z., Power C. A., and Proudfoot A. E. (2012) Pitfalls and solutions for the validation of novel drugs in animal models of disease. Curr. Immunol. Rev. 8, 181–189 [Google Scholar]
- 48. Proudfoot A. E., and Borlat F. (2000) Purification of recombinant chemokines from E. coli. Methods Mol. Biol. 138, 75–87 [DOI] [PubMed] [Google Scholar]
- 49. Magistrelli G., Gueneau F., Muslmani M., Ravn U., Kosco-Vilbois M., and Fischer N. (2005) Chemokines derived from soluble fusion proteins expressed in Escherichia coli are biologically active. Biochem. Biophys. Res. Commun. 334, 370–375 [DOI] [PubMed] [Google Scholar]
- 50. Dufour J. H., Dziejman M., Liu M. T., Leung J. H., Lane T. E., and Luster A. D. (2002) IFN-γ-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J. Immunol. 168, 3195–3204 [DOI] [PubMed] [Google Scholar]
- 51. von Herrath M. G., Dockter J., and Oldstone M. B. (1994) How virus induces a rapid or slow onset insulin-dependent diabetes mellitus in a transgenic model. Immunity 1, 231–242 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.