

Abstract
Solid and hematologic cancers colonized bone produce a number of pathologies. One of the most common complications associated with bone-colonized cancer is bone pain. Cancer-associated bone pain (CABP) is a major cause of increased morbidity and mortality and diminishes the quality of life and affects survival in cancer patients. Current treatments do not satisfactorily control CABP and can elicit serious side effects. Thus, new therapeutic interventions are needed to manage CABP. However, the mechanisms responsible for CABP are poorly understood. The observation that specific osteoclast inhibitors can reduce CABP in patients indicates a critical role of osteoclasts in the pathophysiology of CABP. Osteoclasts create an acidic extracellular microenvironment by secretion of protons via plasma membrane vacuolar proton pumps during bone resorption. In addition, bone-colonized cancer cells also release protons and lactate via plasma membrane pH regulators to avoid intracellular acidification resulting from increased aerobic glycolysis known as Warburg effect, thus exacerbating the acidic microenvironment. Since acidosis is algogenic for primary afferent sensory neurons and bone is densely innervated by sensory neurons that express acid-sensing nociceptors, the acidic bone microenvironments can evoke CABP. Understanding of the cellular and molecular mechanism by which the acidic extracellular microenvironment is created in cancer-colonized bone and the expression and function of these acid-sensing nociceptors are regulated may facilitate the development of novel therapeutic approaches for management of CABP. In this review, the contribution of the acidic extracellular microenvironment created by bone-colonized cancer cells and bone-resorbing osteoclasts to excitation and sensitization of sensory nerves innervating bone and elicitation of CABP and potential therapeutic implications of blocking the development and recognition of acidic extracellular microenvironment will be described.
Keywords: Bone metastasis, osteoclastic bone resorption, Warburg effect, monocarboxylate transporters, TRPV1, ASIC3
Graphical Abstract
1. Introduction
Common solid cancers such as breast cancer, prostate cancer and lung cancer preferentially spread to bone [1]. Rare primary bone malignancies such as osteosarcoma, Ewing's sarcoma, chondrosarcoma also aggressively expand in bone [2]. Multiple myeloma, which is a malignant plasma cell disorder accounting for approximately 10% of all hematologic cancers, exclusively colonizes bone [3]. These bone-colonizing cancers induce the development of either osteolytic, osteosclerotic or mixed bone disease by disrupting the homeostasis of bone environment. In addition, they are associated with skeletal-related events including bone pain, pathologic fractures, hypercalcemia, spinal cord compressions, palliative radiotherapy to bone and surgery to bone to treat or prevent a fracture during the clinical course of the disease [4]. Of these, bone pain is one of the most common and detrimental complications associated with cancer colonization in bone [5]. Cancer-associated bone pain (CABP) profoundly diminishes quality of life, impairs host immune surveillance and delays recovery from the illness, leading to increased secondary death. Accordingly, control of bone pain is a major goal for medical oncologists to achieve in the management of cancer patients. However, current treatments for CABP are not satisfactory and adequate and have serious side effects. Thus, new effective therapeutic interventions for CABP with reduced adverse effects need to be developed. Despite these circumstances, little is known about the mechanism of CABP.
Although the mechanism of CABP is poorly understood, accumulating clinical studies have shown that the specific inhibitors of osteoclasts, bisphosphonate and denosumab, significantly reduce CABP [6, 7]. Osteoclasts are the principal bone resorbing cells in physiological and pathological conditions associated with increased bone resorption [8]. They play a central role in the pathophysiology of cancer colonization in bone [1]. These results collectively suggest that factors released at the tumor-bone interface during osteoclastic bone resorption may be an important mechanism of CABP. However, it should be noted that suppression of osteoclastic bone resorption fails to prevent the progression of CABP as the disease advances [7], confirming that not only osteoclasts but also cancer cells contribute to the pathophysiology of CABP.
In cancer-colonized bone microenvironment, osteoclasts, cancer cells, and cancer-associated stromal cells and inflammatory immune cells produce varieties of algogenic mediators that can excite and sensitize peripheral nociceptive sensory neurons and evoke pain through binding to their cognitive receptors present on the sensory neurons [9–11]. Protons are one of these algogenic mediators [9–11]. Of note, osteoclasts release protons via the plasma membrane (a3 isoform) vacuolar-H+-ATPase coupled with chloride channels to secrete hydrochloric acid to degrade bone minerals during bone resorption [8, 12]. In addition, it has been well-recognized that aggressive cancer cells secrete substantial levels of protons/lactate into the extracellular environments to avoid intracellular acidification due to elevated aerobic glycolysis known as Warburg effect [13]. Thus, protons released from osteoclasts and cancer cells co-operatively create an acidic extracellular microenvironment in cancer-colonized bone.
Here we will overview the role of the acidic microenvironment created by protons/lactate released from bone-resorbing osteoclasts and bone-colonizing cancer cells in the pathophysiology of CABP with our recent experimental observations.
2. Acidic extracellular microenvironment in cancer-colonized bone
2.1. Osteoclastogenesis
Osteoclasts are multinucleated giant cells formed by the fusion of mononuclear progenitors of the monocyte/macrophage lineage [8]. They are the principal bone resorbing cells and play a central role in the formation of the skeleton and regulation of its mass. For osteoclast formation from the osteoclast precursors, macrophage colony-stimulating factor (M-CSF) and receptor for activation of nuclear factor kappa B (NF-κB) (RANK) ligand (RANKL) [14] produced in neighboring osteoblasts or stromal cells are essential [8] (Figure 1). RANKL is a member of tumor necrosis factor family and primarily a membrane-bound cytokine. Therefore, osteoclast precursors that express receptors for RANKL, RANK, need to contact with osteoblasts or stromal cells to differentiate into mature osteoclasts. Osteoprotegerin (OPG) is a natural soluble decoy receptor that competes with RANK for RANKL and thus inhibits RANKL-induced osteoclast formation and bone resorption [8]. The balance between the expression of RANKL and OPG (RANKL/OPG ratio) controls osteoclastogenesis and the degree of resulting bone resorption. Mice lacking M-CSF, RANKL or RANK showed osteopetrosis due to decreased osteoclastogenesis and dysfunction of mature osteoclasts [14]. On the other hand, mice deficient of OPG exhibited severe osteopenia due to increased osteoclastogenesis and bone resorption [14]. The mutations in the signal peptide region of the RANK protein cause familial expansile osteolysis, a rare autosomal dominant disorder characterized by focal areas of enhanced bone resorption, and familial Paget s disease [15]. OPG deficiency due to homozygous loss-of-function mutations within the TNFRSF11B gene is a cause of Juvenile Paget's disease [16]. Thus, osteoclasts are evidently the principal causative player in diverse bone disorders.
Figure 1. Proton secretion by bone-resorbing osteoclasts.

To dissolve bone minerals, mature osteoclasts release protons (H+) and chloride ions (Cl−) into the resorption lacunae via the plasma membrane (a3 isoform) vacuolar H+-ATPase proton pump [23] and chloride ion-proton anti-porter ClC-7 [24], acidifying the resorption lacunae to a pH of 4.5 [7]. Concomitantly, the lysosomal cysteine peptidase cathepsin K [25] degrades bone matrix including type I collagen.
RANKL stimulates osteoclastogenesis and bone resorption and prolongs survival by inhibiting apoptosis. CAII: Carbonic anhydrase II, ClC7: Plasma membrane chloride ion-proton anti-porter, RANK: receptor activation of NF-κB, RANKL: receptor activation of NF-κB ligand, V-H+-ATPase: Plasma membrane (a3 isoform) vacuolar H+-ATPase proton pump,
2.2. Role of osteoclasts in cancer colonization in bone
In cancer-colonized bone and bone metastasis, osteoclasts are increased and activated to destroy bone by factors produced by cancers [1, 17, 18]. Bone destruction, in turn, further stimulates the colonization of cancer cells in bone via the release of bone-stored growth factors including transforming growth factor-β (TGF-β) and insulin-like growth factors (IGFs). This interactive process between bone-colonizing cancer cells and bone-resorbing osteoclasts is called “the vicious cycle” (Figure 2). Thus, osteoclasts are a central regulatory player in the pathophysiology of cancer colonization in bone and bone metastasis. However, their role in CABP remains poorly understood.
Figure 2. Vicious cycle between osteoclasts and cancer cells in bone.

Bone-derived growth factors (GFs) such as insulin-like growth factors (IGF) and transforming growth factor-β (TGF-β), promote proliferation and inhibit apoptosis and stimulate epithelial-mesenchymal transition (EMT) and production of bone-modifying cytokines such as parathyroid hormone-related protein (PTH-rP), prostaglandin E2 (PGE2) and interleukin-11 (IL-11) in bone-colonizing cancer cells, representing the concept of “Seed and Soil” theory proposed by Paget [81]. These bone-modifying factors further stimulate osteoclastic bone resorption via activation of receptor activator of nuclear factor-κB (RANKL)/RANK pathway in osteoblasts and osteoclasts, thereby further increasing release of bone-stored growth factors, thus establishing “vicious cycle” between bone-resorbing osteoclasts and bone-colonizing cancer cells [1, 17, 18]. Bone-colonizing cancer cells reside in stromal cell niche via cell-cell contact that is mediated by cell adhesion molecules (CAMs) and stay dormant or undergo EMT and acquire further aggressiveness. Role of osteocytes in bone metastasis and CABP needs to be elucidated. CAM: cell adhesion molecule, EMT: Epithelial-mesenchymal transition, RANK: receptor activation of NF-κB, RANKL: receptor activation of NF-κB ligand,
2.3. Bone resorption and proton release by mature osteoclasts
Significant reduction of bone pain by the specific inhibitors of osteoclastic bone resorption, bisphosphonates and denosumab, in patients with multiple myeloma and solid cancers [6, 7, 19, 20] indicates a critical role of osteoclasts in the pathophysiology of CABP. Consistent with these clinical observations, Honore et al [21] reported that OPG, which inhibits osteoclast formation and bone resorption through interfering RANKL binding to RANK [8], suppressed CABP using an experimental animal model. We also showed that the most potent bisphosphonate zoledronic acid significantly reduced CABP [22]. It is therefore important to understand how osteoclasts resorb bone to gain better insights into the mechanism underlying CABP.
Bone resorption by mature osteoclasts is a dynamic multi-step process [8]. First, osteoclasts migrate and attach tightly to the bone surface targeted for degradation and removal via the αvβ3 integrin, thereby forming a tight “sealing zone”. Plasma membrane then polarizes to form the resorption organelle, called “ruffled border”. The ruffled border is a unique folded highly permeable membrane facing to the resorbing bone surface. To dissolve bone minerals, protons (H+) and chloride ions (Cl−) is released via the plasma membrane (a3 isoform) vacuolar H+-ATPase proton pump [23] and chloride ion-proton anti-porter ClC-7 [24] clustered in the ruffled border into the resorption lacunae, acidifying the resorptive lacunae to a pH of 4.5 [8]. Concomitantly, the cysteine peptidase cathepsin K [25] degrades bone matrix. The degraded bone matrix is trans-endocytosed from the resorption lacunae to the “functional secretory domain” and released into the extracellular environment [26]. Finally, osteoclasts gone through bone resorption detach from the bone surface and undergo apoptosis [8].
Our RT-PCR and immuno-fluorescent analysis confirmed that bone-resorbing osteoclasts strongly express a3 vacuolar-H+-ATPase on their plasma membrane, while another acid-secreting tissue, the gastric epithelium exhibited marginal expression of a3 vacuolar-H+-ATPase (unpublished data). On the other hand, the gastric epithelium displayed high expression of p-type-H+-ATPase [27], while the expression of p-type-H+-ATPase in osteoclasts was undetectable. These data suggest that algogenic protons released from bone-resorbing osteoclasts via a3 vacuolar-H+-ATPase into the extracellular microenvironment likely impact pH-sensitive peripheral nerve fibers and directly contribute to bone pain. In support of this notion, we reported that the non-selective vacuolar-H+-ATPase inhibitor, bafilomycin A1 reduces inflammatory bone pain [28]. Moreover, we recently found that bafilomycin A1 blocked the development of acidic environment in cancer-colonized bone determined by acridine orange staining [29] and significantly reduced CABP (unpublished data). Of interest, the p-type-H+-ATPase inhibitor, rabeprazole [30] that is widely used for the treatment of gastric pain failed to suppress CABP. The pathophysiology of CABP may be thus unique compared with that of other types of pain because of the contribution of osteoclast a3 vacuolar-H+-ATPase. Many classes of inhibitors of osteoclast proton pump for the treatment of osteoporosis are currently developed and tested in pre-clinical and clinical settings. It is expected that these agents not only inhibit bone resorption but also alleviate CABP.
2.4. Proton and lactate release by cancer cells
The failure of inhibitors of osteoclastic bone resorption to block the progression of CABP in patients [7, 20] and tumor-bearing animals [21, 22] indicates that osteoclasts are not the only cells contributing to CABP. Undoubtedly, cancer cells, inflammatory cells and immune cells contribute to the pathophysiology of CABP as well. Cancer cells produce varieties of nociceptive substances including protons, proteases, glutamate, lysophosphatidic acid, prostaglandins, nerve growth factors, endothelin, bradykinin, extracellular adenosine triphosphate and pro-inflammatory cytokines and chemokines such as interleukins, tumor necrosis factors and macrophage inflammatory protein-1 [9–11]. Among these, the contribution of protons released from cancer cells, in addition to osteoclasts, to the pathophysiology of CABP will be discussed here.
Some regions of cancer environment are hypoxic due to inadequate oxygen delivery, which limits oxidative phosphorylation that efficiently produces ATP via glucose metabolism in the mitochondria in normal cells [13]. To meet increased requirements for ATP and metabolic intermediates and precursors to maintain their aggressiveness, hypoxic cancer cells shift their energy metabolism to glycolysis that is much less energy-efficient but independent of oxygen. The elevated aerobic glycolysis, known as Warburg effect, consumes high glucose and produce substantial amounts of lactate that lowers intracellular pH [13]. To avoid intracellular acidification, cancers actively extrude lactate and protons via plasma membrane pH regulators such as monocarboxylate transporter 1 and 4 (MCT1 and MCT4), Na+/H+ exchangers, anion exchangers, carbonic anhydrases, V-H+-ATPase, Na+/HCO3− co-transporters and HCO3−transporter, creating acidic extracellular cancer microenvironment [31]. We showed high-metastatic B16 mouse melanoma cells expressed increased levels of the a3 isoform V-H+-ATPase compared to the low-metastatic B16 parental cells. Knockdown of a3 V-H+-ATPase suppressed invasiveness and migration and significantly decreased lung and bone metastases [32], suggesting that a3 V-H+-ATPase promotes distant metastasis of B16 melanoma cells by creating acidic environments via proton secretion. From these results, we propose that inhibition of the development of cancer-associated acidic environments by suppressing a3 V-H+-ATPase could be a novel therapeutic approach for the treatment of cancer metastasis. The potential of the inhibitors of aerobic glycolysis (Warburg effect) [33] and these proton pumps [31] as anti-cancer drugs has been extensively explored. However, the effects of these inhibitors on CABP are unknown. Determination of the effects of these inhibitors on CABP may facilitate to the development of drugs with both anti-cancer and analgesic actions.
MCT1 and MCT4 are two of the major proton-coupled lactate symporters mediating bidirectional lactate transport across the plasma membrane [34]. Many cancer cells express MCT1 and MCT4 through which substantial amounts of lactate resulting from Warburg effect are released [35]. Of note, MCT expression in cancer is correlated with outcome of breast cancer patients [36]. Thus extracellular lactate levels are likely elevated in cancer environment. Recent studies reported that lactate released via MCTs from astrocytes is not a waste of glycolysis but rather an energy source shuttling between astrocytes and neurons [37, 38], suggesting that lactate is a critical regulator of neuronal function. Therefore, nociceptive sensory neurons can be also activated by lactate released via MCTs from aggressive cancer cells to evoke CABP. We have recently found that MCT1 and MCT4 are expressed in multiple myeloma cells isolated from patients and several lines of breast cancer cells and multiple myeloma cells (unpublished data). These cancer cells secreted lactate via MCTs and lower extracellular pH. Furthermore, dorsal root ganglion (DRG) sensory neurons also expressed MCT1 and MCT4. Acidic environment (pH 6.5) created by lactate stimulated an excitation of DRG sensory neuronal cells determined by intracellular Ca2+ mobilization using Fura-2 calcium imaging assay. More importantly, we found that the non-specific MCT antagonist, α-cyano-4-hydroxycinnamate (CHC) [39] reduced CABP in an animal model. MCT inhibitors such as CHC are also shown to have anti-cancer effects and some of them are in Phase I clinical trials for advanced solid tumors and for diffuse large B-cell lymphoma [35]. Our results together with these earlier results suggest that MCT inhibitors could be developed as drugs that inhibit tumor growth and CABP.
3. Acid-sensing machinery in bone
3.1. Innervation of nociceptive sensory neurons in bone
It has been proposed that the densely innervated periosteum is the primary site from which bone pain arises. However, patients often complain of bone pain even if cancer localizes to bone marrow or mineralized bone and in the absence of evident periosteal involvement [40], indicating the presence of sensory nerve fibers in non-periosteal sites in bone. An early study using a transmission electron microscopy described by Cooper demonstrated that nerve fibers are present in cortical bone [41]. Furthermore, data are accumulating that bone is densely innervated [40, 42–44]. Of note, Mach et al reported that the bone marrow had the greatest total number of sensory fibers, followed by mineralized bone and then periosteum [40]. Consistent with these earlier studies, we also showed that calcitonin gene-related peptide (CGRP)-positive sensory neurons innervate the mineralized bone and bone marrow. CGRP has been widely used as a marker for sensory neurons and implicated in migraine [45]. Mice lacking CGRP showed attenuated responses to nociceptive chemicals and inflammation [46].
The sensory neurons innervating bone are distal nerve fibers from a number of subpopulations of primary afferent sensory neurons associated with lumbar DRG (L3–L5) [40, 47]. These cell populations are largely subdivided by the presence of neurofilament (NF) 200-positive (A-δ) and CGRP-positive (A-δ or peptidergic C fibers). Bone can be further distinguished by the general lack of innervation by the non-peptidergic subpopulation of neurons that bind isolectin B4 (IB4) [47]. Though functional distinction of these neurons is not fully understood, CGRP-positive peptidergic C-fibers detect noxious stimuli and are classified as nociceptors [48]. Recent studies demonstrated that CGRP-positive sensory neurons exhibited the pathological sprouting in the presence of bone cancer [49]. Nerve growth factor (NGF) derived from cancer cells and stromal cells is likely responsible for the sprouting of sensory neurons, since anti-NGF neutralizing antibody blocked the sprouting and reduced CABP [49]. This pathological remodeling of the peripheral sensory fibers may exacerbate CABP, making control of CABP difficult. We observed similar sprouting of CGRP-positive sensory neurons in bone colonized by breast cancer cells and multiple myeloma cells (unpublished data). These results suggest that the products in cancer-colonized bone environment promote the sensory neurons innervating bone to remodel and progress CABP.
3.2. Acid-sensing receptor
Based on previous and our results, it is likely that the pathological acidic microenvironments created by protons/lactate secreted from bone-resorbing osteoclasts and bone-colonizing cancer cells up-regulate and activate acid-sensing nociceptors expressed on sensory neurons to evoke CABP. Two of the specific and representative pH-sensitive acid-sensing nociceptors that are related to acid-induced bone pain are the transient receptor potential channel-vanilloid subfamily member 1 (TRPV1) and the acid-sensing ion channel 3 (ASIC3) [48].
3.2.1. Transient receptor potential channel-vanilloid subfamily member 1
The TRPV1/vanilloid receptor 1 (VR1)/capsaicin receptor, which was cloned from vertebrates by Caterina et al [50], is a family member of transient receptor potential (TRP) ion channels expressed on a subset of nociceptive sensory neurons. It is activated by heat (>42C) and acid (<pH 6.0) and is the only channel that is excited by the vanilloid capsaicin [50, 51]. In bone TRPV1 is found to express in osteoblasts and osteoclasts and regulate their differentiation and function [52].
Using a mouse model of inflammatory pain [53], we showed that TRPV1 was co-expressed on CGRP-positive sensory neurons in DRG. Mice with inflammation in their hind-paw exhibited a nociceptive behavior (thermal hyperalgesia) with elevated CGRP mRNA expression in the DRG. Treatment by acid (pH 5.5) excited primary DRG sensory neuron cells and increased CGRP mRNA expression and protein production in these cells. The specific antagonist of TRPV1, 5 -iodoresiniferatoxin (I-RTX), blocked the excitation and the acid-increased CGRP expression and production. Further, primary DRG cells isolated from TRPV1 −/− mice failed to show an increase in CGRP expression upon treatment with acid. Of importance, inflammatory pain was markedly diminished in TRPV1 −/− mice. These results collectively suggest that activation of TRPV1 in inflammatory acidic environment leads to an up-regulation of CGRP expression, which in turn further increases inflammation and pain. Thus, blockade of TRPV1, CGRP or both could be an effective pharmacological intervention for acid-associated inflammatory pain.
TRPV1 has been implicated in the pathophysiology of CABP [9, 11, 54]. In preclinical models of cancer pain, TRPV1 expression was increased in the ipsi-lateral DRG in the presence of cancer in bone [55, 56]. In support of the important role of TRPV1 in CABP, TRPV1 gene disruption or TRPV1 antagonist reduces CABP [57]. Similarly, the specific TRPV1 antagonist I-RTX also suppressed CABP [56]. Of interest, the TRPV1 antagonist SB366791 was shown to potentiate analgesic effects of morphine on bone cancer pain [58]. These data suggest that suppression of TRPV1 activation under the acidic cancer environment is a promising therapeutic approach to attenuate CABP. However, preclinical and clinical studies revealed that TRPV1 antagonists induce hyperthermia as an adverse effect [59]. Attempts to develop or identify antagonists devoid of temperature effects have not been successful. In this context, it is noted that recent studies have shown that the expression and activity of TRPV1 on the DRG sensory neurons are up-regulated by transforming growth factor-β1 (TGF-β1) [60] and insulin-like growth factor-1 (IGF-1) [61]. Since TGF-β and IGF-1 are abundantly stored in bone and released as a consequence of bone resorption by cancer-activated osteoclasts, expression and activation of TRPV1 on the sensory neurons innervating bone may be modulated by these bone-derived growth factors. In fact, these studies showed that inhibition of TGF-β [60] or IGF receptor signaling [61] reduced CABP due to suppression of TRPV1 activation. Suppression of TRPV1 activity and expression by blocking the activity of growth factors released from bone may be an alternative approach to attenuate CABP.
3.2.2. Acid-sensing ion channel 3
ASIC3 is a pH sensor predominantly expressed in primary afferent sensory neurons [62, 63]. In DRG, ASIC3 expression was increased in inflammatory acidic conditions and co-expressed with CGRP in the sensory neurons innervating the knee joint, indicating ASIC3 contribution to acute arthritis pain [64]. ASIC3 was also detected on the CGRP-positive sensory neurons innervating periosteal surface of bone [65]. In bone ASIC3 is present in monocytes, osteoclasts and osteoblasts [66]. However, its functional role in bone homeostasis is unclear.
A recent study that showed that an injection of the synthetic ASIC3 agonist, 2-guanidine-4-methylquinazoline into mouse paw induced noxious behaviors in wild-type mice but not ASIC3−/− mice [67] provided the evidence that ASIC3 activation is sufficient to evoke pain [68]. However, the role of ASIC3 in CABP still remains elusive. We reported that ASCI3 mRNA expression was up-regulated in DRG associated with CABP in a model of rat mammary tumor [55]. In support of our results, Qiu et al [69] recently showed ASIC3 expression was elevated in DRG using a different model of rat mammary tumor. Although these results suggest that ASIC3 is involved in the pathophysiology of CABP, it needs to be shown that suppression of increased ASIC3 expression in DRG leads to reduced CABP and that CABP is alleviated in ASIC3 knockout mice. We have recently found that, upon acid treatment (pH 6.5), primary sensory neuron cells isolated from DRG displayed Ca2+ influx (unpublished data), which is a widely-used early indicator for sensory neuron excitation [70]. Of note, the specific ASIC3 antagonist APETx2 [71] inhibited Ca2+ influx, indicating a critical role of ASIC3 in sensory neuron excitation. Of interest, APETx2 was shown to reduce acid-induced and inflammatory pain due to complete Freund's adjuvant in rat [72] and slower the progression of the disease in a rat model of osteoarthritis [73]. It is intriguing to examine the effects of APETx2 on CABP.
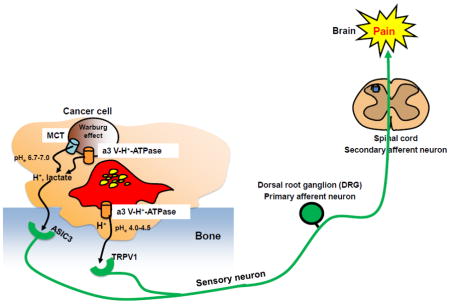
ASIC3 senses mild extracellular acidification (pH 6.8–7.0) [63], while TRPV1 is activated at the pH lower than 6.0 [50, 51]. We reported TRPV1 was activated at pH 5.5 [53]. The pH of the acidic environment created by cancer cells is shown to be 6.5–7.0 [31] and that by bone-resorbing osteoclasts is assumed to be 4.0–4.5 [23]. Sensory neurons innervating bone may be excited and sensitized by discriminating mild and strong acidic extracellular microenvironment through ASIC3 and TRPV1 in the pathophysiology of CABP, respectively (Figure 3).
Figure 3. Acid-evoked cancer-associated bone pain (CABP).

Bone-resorbing osteoclasts secrete protons via plasma membrane a3 V-H+-ATPase to degrade bone minerals. Bone-colonizing cancer cells also release protons/lactate via MCT, a3 V-H+-ATPase and other proton pumps including Na+/H+ exchangers, anion exchangers, carbonic anhydrases, Na+/HCO3- co-transporters and HCO3-transporter to avoid intracellular acidification due to Warburg effect. The pH of the acidic environment created by cancer cells and bone-resorbing osteoclasts is assumed to be 6.5–7.0 [31] and 4.0–4.5 [23], respectively. The acidic microenvironment directly excites and sensitizes sensory neurons innervating bone via activation of the acid-sensing nociceptors, TRPV1 and ASIC3, transducing noxious signals to via DRG (primary afferent neuron) and spinal cord (secondary afferent neuron) and evoke bone pain in brain.
TRPV1 activation promotes eCa2+ influx into cytoplasm and induces propagation of intracellular signaling molecules including calmodulin kinase II (CaMKII) and the transcription factor cAMP responsive element-binding protein (CREB), leading to transcriptional activation of target molecules [52]. TRPV1 activation also propagates Erk and Akt presumably to prevent apoptosis of neuron cells. Blockade of TRPV1 activation under the acidic cancer environments may be a promising therapeutic approach to alleviate CABP.
ASIC3: Acid-sensing ion channel 3, CAMKII: Calmodulin kinase II, CGRP: calcitonin gene-related peptide, CREB: cAMP responsive element-binding protein, DRG: Dorsal root ganglion, MCT: Monocarboxylate transporter, TRPV1: Transient receptor potential channel-vanilloid subfamily member 1
3.3 Other acid-sensing machineries potentially contributing to CABP
Transient receptor potential (TRP) ion channels are the large families of cellular sensors mediating taste perception, thermo-sensation, mechano-sensing and osmolality sensing by transducing various physical stimuli into neuronal signals in predominantly C and Aδ nociceptors in primary sensory neurons [74]. Further, TRP ion channels also mediate transduction of peripheral nociceptive stimuli into pain. Of interest, recent studies described that the members of TRP channels are involved in the regulation of skeletal homeostasis through affecting intestinal calcium absorption (TRPV6), renal calcium reabsorption (TRPV5), and differentiation of osteoclasts (TRPV1, TRPV2, TRPV4, TRPV5), chondrocytes (TRPV4) and osteoblasts (TRPV1) [52]. It is thus tempting to propose that these family members of TRP channels play a role in acid-induced CABP. However, current available evidence suggests that acid unlikely activates these TRPVs except for TRPV1.
TRP Ankyrin 1 (TRPA1) is a non-selective Ca2+ permeable cation channel that uniquely possesses 17 ankyrin repeat domains [74]. TRPA1 is predominantly expressed C and Aδ nerve fibers. Although still controversial, TRPA1 is proposed to be activated by noxious cold temperatures (< 18° C) and mechanical force and some studies reported that TRPA1 deficient mice showed impaired behavioral responses to cold plate and mechanical stimuli. Expression of TRPA1 in bone cells has been unknown but a recent study demonstrated the expression of TRPA1 in human odontoblasts [75]. It is unclear whether TRPA1 is activated by acid and plays a role in CABP. Notably, however, most neurons that express TRPA1 also express TRPV1, raising the possibility that TRPA1 may modulate or partially share the acid-sensing functions of TRPV1. In case this is proved to be the case, hyperthermia, which is a major obstacle in the development of TRPV1 antagonists, may not be a serious problem with TRPA1 antagonists.
TRP Melastatin 8 (TRPM8) is expressed in a subset of C and Aδ nerve fibers in DRG, trigeminal ganglion and nodose ganglion [74]. In bone, TRPM8 expression was shown in osteoblasts, however, its functional role is unknown [76]. TRPM8 is activated by cold temperatures (< 15 °C) and cooling compounds such as menthol and icilin and peppermint oil. In contrast, it is unclear whether TRPM8 is activated by acid. TRPM8 was initially cloned as a molecule that has high homology to a TRP-like channel that was identified in prostate cancers [74]. Later, it was found that increased expression of TRPM8 is correlated with the aggressiveness of a variety of cancers including prostate, lung, breast, gastric, ovarian and liver cancer and melanoma [77]. Since prostate, breast and lung cancers preferentially spread to bone and are frequently associated with CABP, it is plausible to speculate that TRPM8 contributes to the pathophysiology of CABP. The role of TRPM in CABP needs to be elucidated.
4. Conclusion
Bone is a unique organ in which multinucleated giant osteoclasts continuously secrete protons to maintain bone homeostasis in physiological conditions [8]. The secretion of protons by osteoclasts is increased in response to the bone-modifying factors produced by bone-colonizing cancer cells [1, 17, 18]. Protons and lactate are also secreted by bone-colonizing cancer cells as a consequence of elevated oxygen-independent glycolysis (Warburg effect) [13, 31]. In addition to autonomous secretion of protons, osteoclasts and cancer cells may further increase proton and lactate secretion in bone microenvironment that is relatively hypoxic [78]. Hypoxia is shown to up-regulate the expression and function of the plasma membrane proton/lactate transporters [31]. Therefore, it is likely that cancer-colonized bone microenvironment readily falls into pathological acidosis and that the contribution of acid is more specific and critical in the pathophysiology of CABP than that of other types of pain. In addition, accumulating evidence indicates that the acidic extracellular microenvironments critically influence malignant behaviors of cancer including invasiveness, metastasis, and chemo- and radio-resistance and thus are associated with poor prognosis [79, 80]. Accordingly, suppression of the creation of acidic extracellular cancer-colonized bone microenvironment through inhibition of the function of the proton/lactate transporters in osteoclasts and cancer cells seems likely to simultaneously inhibit cancer aggressiveness and CABP. Furthermore, pharmacological agents that interfere with the activation of acid-sensing nociceptors in sensory neurons associated with bone will be an effective and selective therapeutic means for CABP. Finally, development of new analgesic drugs for CABP is expected to reduce dosing of opioids that are currently the primary agent in the management of cancer pain but cause unwanted adverse effects. Control of CABP will be a challenging goal in the management of patients with cancers in bone.
Highlights.
Osteoclasts secrete protons via membrane a3 V-H+-ATPase.
Cancer releases proton/lactate via proton pumps and transporters.
Extracellular environment of cancer-colonized bone is acidic.
Sensory neurons expressing acid-sensing nociceptors innervate bone.
Acidic environment evokes CABP by activating the nociceptors TRPV1 and ASIC3.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Weilbaecher KN, Guise TA, McCauley LK. Cancer to bone: a fatal attraction. Nat Rev Cancer. 2011;11:411–425. doi: 10.1038/nrc3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Driel M, van Leeuwen JP. Cancer and bone: A complex complex. Arch Biochem Biophys. 2014 doi: 10.1016/j.abb.2014.07.013. [DOI] [PubMed] [Google Scholar]
- 3.Terpos E, Berenson J, Raje N, Roodman GD. Management of bone disease in multiple myeloma. Expert Rev Hematol. 2014;7:113–125. doi: 10.1586/17474086.2013.874943. [DOI] [PubMed] [Google Scholar]
- 4.Brown JE, Cook RJ, Lipton A, Costa L, Coleman RE. Prognostic factors for skeletal complications from metastatic bone disease in breast cancer. Breast Cancer Res Treat. 2010:767–779. doi: 10.1007/s10549-010-0981-1. [DOI] [PubMed] [Google Scholar]
- 5.Mercadante S. Malignant bone pain: pathophysiology and treatment. Pain. 1997;69:1–19. doi: 10.1016/S0304-3959(96)03267-8. [DOI] [PubMed] [Google Scholar]
- 6.Kohno N, Aogi K, Minami H, Nakamura S, Asaga T, Iino Y, Watanabe T, Goessl C, Ohashi Y, Takashima S. Zoledronic acid significantly reduces skeletal complications compared with placebo in Japanese women with bone metastases from breast cancer: a randomized, placebo-controlled trial. J Clin Oncol. 2005;23:3314–3321. doi: 10.1200/JCO.2005.05.116. [DOI] [PubMed] [Google Scholar]
- 7.Cleeland CS, Body JJ, Stopeck A, von Moos R, Fallowfield L, Mathias SD, Patrick DL, Clemons M, Tonkin K, Masuda N, Lipton A, de Boer R, Salvagni S, Oliveira CT, Qian Y, Jiang Q, Dansey R, Braun A, Chung K. Pain outcomes in patients with advanced breast cancer and bone metastases: Results from a randomized, double-blind study of denosumab and zoledronic acid. Cancer. 2013;119:832–838. doi: 10.1002/cncr.27789. [DOI] [PubMed] [Google Scholar]
- 8.Teitelbaum SL. Bone resorption by osteoclasts. Science. 2000;289:1504–1508. doi: 10.1126/science.289.5484.1504. [DOI] [PubMed] [Google Scholar]
- 9.Mantyh PW. Cancer pain and its impact on diagnosis, survival and quality of life. Nat Rev Neurosci. 2006;7:797–809. doi: 10.1038/nrn1914. [DOI] [PubMed] [Google Scholar]
- 10.Schmidt BL, Hamamoto DT, Simone DA, Wilcox GL. Mechanism of cancer pain. Mol Interv. 2010;10:164–178. doi: 10.1124/mi.10.3.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lozano-Ondoua AN, Symons-Liguori AM, Vanderah TW. Cancer-induced bone pain: Mechanisms and models. Neurosci Lett. 2013;557(Pt. A):52–59. doi: 10.1016/j.neulet.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qin A, Cheng TS, Pavlos NJ, Lin Z, Dai KR, Zheng MH. V-ATPases in osteoclasts: structure, function and potential inhibitors of bone resorption. Int J Biochem Cell Biol. 2012;44:1422–1435. doi: 10.1016/j.biocel.2012.05.014. [DOI] [PubMed] [Google Scholar]
- 13.Parks SK, Chiche J, Pouysségur J. Disrupting proton dynamics and energy metabolism for cancer therapy. Nat Rev Cancer. 2013;13:611–623. doi: 10.1038/nrc3579. [DOI] [PubMed] [Google Scholar]
- 14.Lacey DL, Boyle WJ, Simonet WS, Kostenuik PJ, Dougall WC, Sullivan JK, San Martin J, Dansey R. Bench to bedside: elucidation of the OPG-RANK-RANKL pathway and the development of denosumab. Nat Rev Drug Discov. 2012;11:401–419. doi: 10.1038/nrd3705. [DOI] [PubMed] [Google Scholar]
- 15.Hughes AE, Ralston SH, Marken J, Bell C, MacPherson H, Wallace RG, van Hul W, Whyte MP, Nakatsuka K, Hovy L, Anderson DM. Mutations in TNFRSF11A, affecting the signal peptide of RANK, cause familial expansile osteolysis. Nat Genet. 2000;24:45–48. doi: 10.1038/71667. [DOI] [PubMed] [Google Scholar]
- 16.Saki F, Karamizadeh Z, Nasirabadi S, Mumm S, McAlister WH, Whyte MP. Juvenile paget's disease in an Iranian kindred with vitamin D deficiency and novel homozygous TNFRSF11B mutation. J Bone Miner Res. 2013;28:1501–1508. doi: 10.1002/jbmr.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nature Rev Cancer. 2002;2:584–593. doi: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- 18.Yoneda T, Tanaka S, Hata K. Role of RANKL/RANK in primary and secondary breast cancer. World J Orthop. 2013;4:178–185. doi: 10.5312/wjo.v4.i4.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terpos E, Morga G, Dimopoulos MA, Drake MT, Lentzsch S, Raje N, Sezer O, García-Sanz R, Shimizu K, Turesson I, Reiman T, Jurczyszyn A, Merlini G, Spencer A, Leleu X, Cavo M, Munshi N, Rajkumar SV, Durie BG, Roodman GD. International myeloma working group recommendations for the treatment of multiple myeloma-related bone disease. J Clin Oncol. 2013;31:2347–2357. doi: 10.1200/JCO.2012.47.7901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.von Moos R, Body JJ, Egerdie B, Stopeck A, Brown JE, Damyanov D, Fallowfield LJ, Marx G, Cleeland CS, Patrick DL, Palazzo FG, Qian Y, Braun A, Chung K. Pain and health-related quality of life in patients with advanced solid tumours and bone metastases: integrated results from three randomized, double-blind studies of denosumab and zoledronic acid. Support Care Cancer. 2013;21:3497–3507. doi: 10.1007/s00520-013-1932-2. [DOI] [PubMed] [Google Scholar]
- 21.Honore P, Luger NM, Sabino MA, Schwei MJ, Rogers SD, Mach DB, O'keefe PF, Ramnaraine ML, Clohisy DR, Mantyh PW. Osteoprotegerin blocks bone cancer-induced skeletal destruction, skeletal pain and pain-related neurochemical reorganization of the spinal cord. Nat Med. 2000;6:521–528. doi: 10.1038/74999. [DOI] [PubMed] [Google Scholar]
- 22.Nagae M, Hiraga T, Yoneda T. Acidic microenvironment created by osteoclasts causes bone pain associated with tumor colonization. J Bone Miner Metab. 2007;25:99–104. doi: 10.1007/s00774-006-0734-8. [DOI] [PubMed] [Google Scholar]
- 23.Qin A, Cheng TS, Pavlos NJ, Lin Z, Dai KR, Zheng MH. V-ATPases in osteoclasts: structure, function and potential inhibitors of bone resorption. Int J Biochem Cell Biol. 2012;44:1422–1435. doi: 10.1016/j.biocel.2012.05.014. [DOI] [PubMed] [Google Scholar]
- 24.Supanchart C, Wartosch L, Schlack C, Kühnisch J, Felsenberg D, Fuhrmann JC, de Vernejoul MC, Jentsch TJ, Kornak U. ClC-7 expression levels critically regulate bone turnover, but not gastric acid secretion. Bone. 2014;58:92–102. doi: 10.1016/j.bone.2013.09.022. [DOI] [PubMed] [Google Scholar]
- 25.Costa AG, Cusano NE, Silva BC, Cremers S, Bilezikian JP. Cathepsin K: its skeletal actions and role as a therapeutic target in osteoporosis. Nat Rev Rheumatol. 2011;7:447–456. doi: 10.1038/nrrheum.2011.77. [DOI] [PubMed] [Google Scholar]
- 26.Zhao H. Membrane trafficking in osteoblasts and osteoclasts: new avenues for understanding and treating skeletal diseases. Traffic. 2012;13:1307–1314. doi: 10.1111/j.1600-0854.2012.01395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morth JP, Pedersen BP, Buch-Pedersen MJ, Andersen JP, Vilsen B, Palmgren MG, Nissen P. A structural overview of the plasma membrane Na+,K+-ATPase and H+-ATPase ion pumps. Nat Rev Mol Cell Biol. 2011;12:60–70. doi: 10.1038/nrm3031. [DOI] [PubMed] [Google Scholar]
- 28.Nagae M, Hiraga T, Wakabayashi H, Wang L, Iwata K, Yoneda T. Osteoclasts play a part in pain due to the inflammation adjacent to bone. Bone. 2006;39:1107–1115. doi: 10.1016/j.bone.2006.04.033. [DOI] [PubMed] [Google Scholar]
- 29.Henriksen K, Sørensen MG, Jensen VK, Dziegiel MH, Nosjean O, Karsdal MA. Ion transporters involved in acidification of the resorption lacuna in osteoclasts. Calcif Tissue Int. 2008;83:230–242. doi: 10.1007/s00223-008-9168-8. [DOI] [PubMed] [Google Scholar]
- 30.Marelli S, Pace F. Rabeprazole for the treatment of acid-related disorders. Expert Rev Gastroenterol Hepatol. 2012;6:423–435. doi: 10.1586/egh.12.18. [DOI] [PubMed] [Google Scholar]
- 31.Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov. 2011;10:767–777. doi: 10.1038/nrd3554. [DOI] [PubMed] [Google Scholar]
- 32.Nishisho T, Hata K, Nakanishi M, Morita Y, Sun-Wada GH, Wada Y, Yasui N, Yoneda T. The a3 isoform vacuolar type H+-ATPase promotes distant metastasis in the mouse B16 melanoma cells. Mol Cancer Res. 2011;9:845–855. doi: 10.1158/1541-7786.MCR-10-0449. [DOI] [PubMed] [Google Scholar]
- 33.Lincet H, Icard P. How do glycolytic enzymes favour cancer cell proliferation by nonmetabolic functions? Oncogene. 2014 Sep 29;0 doi: 10.1038/onc.2014.320. [DOI] [PubMed] [Google Scholar]
- 34.Halestrap AP. Monocarboxylic acid transport. Compr Physiol. 2013;3:1611–1643. doi: 10.1002/cphy.c130008. [DOI] [PubMed] [Google Scholar]
- 35.Doherty JR, Cleveland JL. Targeting lactate metabolism for cancer therapeutics. J Clin Invest. 2013;123:3685–3692. doi: 10.1172/JCI69741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Doyen J, Trastour C, Ettore F, Peyrottes I, Toussant N, Gal J, Ilc K, Roux D, Parks SK, Ferrero JM, Pouysségur J. Expression of the hypoxia-inducible monocarboxylate transporter MCT4 is increased in triple negative breast cancer and correlates independently with clinical outcome. Biochem Biophys Res Commun. 2014 doi: 10.1016/j.bbrc.2014.07.050. [DOI] [PubMed] [Google Scholar]
- 37.Bergersen LH. Is lactate food for neurons? Comparison of monocarboxylate transporter subtypes in brain and muscle. Neuroscience. 2007;145:11–19. doi: 10.1016/j.neuroscience.2006.11.062. [DOI] [PubMed] [Google Scholar]
- 38.Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, Alberini CM. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell. 2011;144:810–823. doi: 10.1016/j.cell.2011.02.018. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Q, Morris ME. The role of monocarboxylate transporter 2 and 4 in the transport of gamma-hydroxybutyric acid in mammalian cells. Drug Metab Dispos. 2007;35:1393–1399. doi: 10.1124/dmd.107.014852. [DOI] [PubMed] [Google Scholar]
- 40.Mach DB, Rogers SD, Sabino MC, Luger NM, Schwei MJ, Pomonis JD, Keyser CP, Clohisy DR, Adams DJ, O’Leary P, Mantyh PW. Origins of skeletal pain: sensory and sympathetic innervation of the mouse femur. Neuroscience. 2002;113:155–166. doi: 10.1016/S0306-4522(02)00165-3. [DOI] [PubMed] [Google Scholar]
- 41.Cooper RR. Nerves in cortical bone. Science. 1968;160:327–328. doi: 10.1126/science.160.3825.327. [DOI] [PubMed] [Google Scholar]
- 42.Serre CM, Farlay D, Delmas PD, Chenu C. Evidence for a dense and intimate innervation of the bone tissue, including glutamate-containing fibers. Bone. 1999;25:623–629. doi: 10.1016/S8756-3282(99)00215-X. [DOI] [PubMed] [Google Scholar]
- 43.Irie K, Hara-Irie F, Ozawa H, Yajima T. Calcitonin gene-related peptide (CGRP)-containing nerve fibers in bone tissue and their involvement in bone remodeling. Microsc Res Tech. 2002;58:85–90. doi: 10.1002/jemt.10122. [DOI] [PubMed] [Google Scholar]
- 44.Fukuda T, Takeda S, Xu R, Ochi H, Sunamura S, Sato T, Shibata S, Yoshida Y, Gu Z, Kimura A, Ma C, Xu C, Bando W, Fujita K, Shinomiya K, Hirai T, Asou Y, Enomoto M, Okano H, Okawa A, Itoh H. Sema3A regulates bone-mass accrual through sensory innervations. Nature. 2013;497:490–493. doi: 10.1038/nature12115. doi: 10.1038/nature12115 2013. [DOI] [PubMed] [Google Scholar]
- 45.Benemei S, Nicoletti P, Capone JG, Geppetti P. CGRP receptors in the control of pain and inflammation. Curr Opin Pharmacol. 2009;9:9–14. doi: 10.1016/j.coph.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 46.Salmon AM, Damaj MI, Marubio LM, Epping-Jordan MP, Merlo-Pich E, Changeux JP. Altered neuroadaptation in opiate dependence and neurogenic inflammatory nociception in alpha CGRP-deficient mice. Nat Neurosci. 2001;4:357–358. doi: 10.1038/86001. [DOI] [PubMed] [Google Scholar]
- 47.Mantyh P. Bone cancer pain: Causes, consequences, and therapeutic opportunities. Pain. 2013;154(Suppl 1):S54–S62. doi: 10.1016/j.pain.2013.07.044. [DOI] [PubMed] [Google Scholar]
- 48.Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mantyh WG, Jimenez-Andrade JM, Stake JI, Bloom AP, Kaczmarska MJ, Taylor RN, Freeman KT, Ghilardi JR, Kuskowski MA, Mantyh PW. Blockade of nerve sprouting and neuroma formation markedly attenuates the development of late stage cancer pain. Neuroscience. 2010;171:588–598. doi: 10.1016/j.neuroscience.2010.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- 51.Ho KW, Ward NJ, Calkins DJ. TRPV1: a stress response protein in the central nervous system. Am J Neurodegener Dis. 2012;1:1–14. [PMC free article] [PubMed] [Google Scholar]
- 52.Lieben L, Carmeliet G. The involvement of TRP channels in bone homeostasis. Front Endocrinol (Lausanne) 2012;3:99. doi: 10.3389/fendo.2012.00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakanishi M, Hata K, Nagayama T, Sakurai T, Nishisho T, Wakabayashi H, Hiraga T, Ebisu S, Yoneda T. Acid activation of Trpv1 leads to an up-regulation of calcitonin gene-related peptide expression in dorsal root ganglion neurons via the CaMK-CREB cascade: a potential mechanism of inflammatory pain. Mol Biol Cell. 2010;21:2568–2577. doi: 10.1091/mbc.E10-01-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mantyh P. Bone cancer pain: Causes, consequences, and therapeutic opportunities. Pain. 2013;154(Suppl. 1):S54–S62. doi: 10.1016/j.pain.2013.07.044. [DOI] [PubMed] [Google Scholar]
- 55.Nagae M, Hiraga T, Yoneda T. Acidic microenvironment created by osteoclasts causes bone pain associated with tumor colonization. J Bone Miner Metab. 2007;25:99–104. doi: 10.1007/s00774-006-0734-8. [DOI] [PubMed] [Google Scholar]
- 56.Niiyama Y, Kawamata T, Yamamoto J, Omote K, Namiki A. Bone cancer increases transient receptor potential vanilloid subfamily 1 expression within distinct subpopulations of dorsal root ganglion neurons. Neuroscience. 2007;148:560–572. doi: 10.1016/j.neuroscience.2007.05.049. [DOI] [PubMed] [Google Scholar]
- 57.Ghilardi JR, Röhrich H, Lindsay TH, Sevcik MA, Schwei MJ, Kubota K, Halvorson KG, Poblete J, Chaplan SR, Dubin AE, Carruthers NI, Swanson D, Kuskowski M, Flores CM, Julius D, Mantyh PW. Selective blockade of the capsaicin receptor TRPV1 attenuates bone cancer pain. J Neurosci. 2005;25:3126–3131. doi: 10.1523/JNEUROSCI.3815-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Niiyama Y, Kawamata T, Yamamoto J, Furuse S, Namiki A. SB366791, a TRPV1 antagonist, potentiates analgesic effects of systemic morphine in a murine model of bone cancer pain. Br J Anaesth. 2009;102:251–258. doi: 10.1093/bja/aen347. [DOI] [PubMed] [Google Scholar]
- 59.Brederson JD, Kym PR, Szallasi A. Targeting TRP channels for pain relief. Eur J Pharmacol. 2013;716:61–76. doi: 10.1016/j.ejphar.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 60.Xu Q, Zhang XM, Duan KZ, Gu XY, Han M, Liu BL, Zhao ZQ, Zhang YQ. Peripheral TGF-β1 signaling is a critical event in bone cancer-induced hyperalgesia in rodents. J Neurosci. 2013;33:19099–19111. doi: 10.1523/JNEUROSCI.4852-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li Y, Cai J, Han Y, Xiao X, Meng XL, Su L, Liu FY, Xing GG, Wan Y. Enhanced function of TRPV1 via up-regulation by insulin-like growth factor-1 in a rat model of bone cancer pain. Eur J Pain. 2014;18:774–784. doi: 10.1002/j.1532-2149.2013.00420.x. [DOI] [PubMed] [Google Scholar]
- 62.de la Rosa AD, Zhang P, Shao D, White F, Canessa CM. Functional implications of the localization and activity of acid-sensitive channels in rat peripheral nervous system. Proc Natl Acad Sci USA. 2002;99:2326–2331. doi: 10.1073/pnas.042688199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu WL, Cheng CF, Sun WH, Wong CW, Chen CC. Targeting ASIC3 for pain, anxiety, and insulin resistance. Pharmacol Ther. 2012;134:127–138. doi: 10.1016/j.pharmthera.2011.12.009. [DOI] [PubMed] [Google Scholar]
- 64.Ikeuchi M, Kolker SJ, Sluka KA. Acid-sensing ion channel 3 expression in mouse knee joint afferents and effects of carrageenan-induced arthritis. J Pain. 2009;10:336–342. doi: 10.1016/j.jpain.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Olson TH, Riede MS, Vulchanova L, Ortiz-Gonzalez XR, Elde R. An acid sensing ion channel (ASIC) localizes to small primary afferent neurons in rats. NeuroReport. 1998;9:1109–1113. doi: 10.1097/00001756-199804200-00028. [DOI] [PubMed] [Google Scholar]
- 66.Jahr H, van Driel M, van Osch GJ, Weinans H, van Leeuwen JP. Identification of acid-sensing ion channels in bone. Biochem Biophys Res Commun. 2005;337:349–354. doi: 10.1016/j.bbrc.2005.09.054. [DOI] [PubMed] [Google Scholar]
- 67.Yu Y, Chen Z, Li WG, Cao H, Feng EG, Yu F, Liu H, Jiang H, Xu TL. A nonproton ligand sensor in the acid-sensing ion channel. Neuron. 2010;68:61–72. doi: 10.1016/j.neuron.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 68.Wemmie JA, Taugher RJ, Kreple CJ. Acid-sensing ion channels in pain and disease. Nat Rev Neurosci. 2013;14:461–471. doi: 10.1038/nrn3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Qiu F, Wei X, Zhang S, Yuan W, Mi W. Increased expression of acid-sensing ion channel 3 within dorsal root ganglia in a rat model of bone cancer pain. Neuroreport. 2014;25:887–893. doi: 10.1097/WNR.0000000000000182. [DOI] [PubMed] [Google Scholar]
- 70.Feldman P, Due MR, Ripsch MS, Khanna R, White FA. The persistent release of HMGB1 contributes to tactile hyperalgesia in a rodent model of neuropathic pain. J Neuroinflammation. 2012;9:180. doi: 10.1186/1742-2094-9-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Diochot S, Baron A, Rash LD, Deval E, Escoubas P, Scarzello S, Salinas M, Lazdunski M. A new sea anemone peptide, APETx2, inhibits ASIC3, a major acid-sensitive channel in sensory neurons. EMBO J. 2004;23:1516–1525. doi: 10.1038/sj.emboj.7600177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Karczewski J, Spencer RH, Garsky VM, Liang A, Leitl MD, Cato MJ, Cook SP, Kane S, Urban MO. Reversal of acid-induced and inflammatory pain by the selective ASIC3 inhibitor, APETx2. Br J Pharmacol. 2010;161:950–960. doi: 10.1111/j.1476-5381.2010.00918.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Izumi M, Ikeuchi M, Ji Q, Tani T. Local ASIC3 modulates pain and disease progression in a rat model of osteoarthritis. J Biomed Sci. 2012;19:77. doi: 10.1186/1423-0127-19-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Premkumar LS, Abooj M. TRP channels and analgesia. Life Sci. 2013;92:415–424. doi: 10.1016/j.lfs.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Egbuniwe O, Grover S, Duggal AK, Mavroudis A, Yazdi M, Renton T, Di Silvio L, Grant AD. TRPA1 and TRPV4 activation in human odontoblasts stimulates ATP release. J Dent Res. 2014;93:911–917. doi: 10.1177/0022034514544507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Abed E, Labelle D, Martineau C, Loghin A, Moreau R. Expression of transient receptor potential (TRP) channels in human and murine osteoblast-like cells. Mol Membr Biol. 2009;26:146–158. doi: 10.1080/09687680802612721. [DOI] [PubMed] [Google Scholar]
- 77.Chen J, Luan Y, Yu R, Zhang Z, Zhang J, Wang W. Transient receptor potential (TRP) channels, promising potential diagnostic and therapeutic tools for cancer. Biosci Trends. 2014;8:1–10. doi: 10.5582/bst.8.1. doi: http://dx.doi.org/10.5582/bst.8.1. [DOI] [PubMed] [Google Scholar]
- 78.Schipani E, Maes C, Carmeliet G, Semenza GL. Regulation of osteogenesis-angiogenesis coupling by HIFs and VEGF. J Bone Miner Res. 2009;24:1347–1353. doi: 10.1359/jbmr.090602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gillies RJ, Verduzco D, Gatenby RA. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer. 2012;12:487–493. doi: 10.1038/nrc3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Peppicelli S, Bianchini F, Calorini L. Extracellular acidity, a “reappreciated” trait of tumor environment driving malignancy: perspectives in diagnosis and therapy. Cancer Metastasis Rev. 2014;33:823–832. doi: 10.1007/s10555-014-9506-4. [DOI] [PubMed] [Google Scholar]
- 81.Paget S. The distribution of secondary growths in cancer of the breast. Lancet. 1889;8:98–101. [PubMed] [Google Scholar]