


Abstract
The inhibitory potencies of several hairpins comprising DNA, RNA and 2′,5′-linked RNA segments were assessed against the RNase H activity of the human immunodeficiency virus reverse transcriptase (HIV-1 RT), an indispensable enzyme for HIV genomic replication. The hairpin library was constructed via diversity-oriented nucleic-acid synthesis (DONAS), an approach inspired from traditional split-pool synthesis. DONAS provided access to an array of oligonucleotide hairpins possessing distinct conformational, structural and biological properties. The inhibitory potency of these compounds was highly specific towards HIV-1 RT RNase H and strongly depended on the structure of both the stem and tetraloop. Hairpins that have an overall A-type geometry are better inhibitors of RNase H activity than hairpins with ‘intermediate’ or B-type conformations, although interestingly, the inhibitory activity is quite sensitive to the nucleotide sequence in both the stem and loop regions of the hairpin. The most potent hairpins bear a 3′,5′-linked rather than 2′,5′-linked RNA loop, but the latter is necessary for activity of hairpins consisting of DNA stems. Inhibitory activity was essentially independent of hairpin thermal stability. The potent hairpins also demonstrated high nuclease resistance in biological media, particularly those bearing a 2′,5′-linked tetraloop. These studies collectively bring into light a new class of nucleic acid aptamers that act exclusively upon the retroviral RNase H domain in vitro, and thus represent novel lead compounds for the development of specific and potent HIV-1 RT inhibitors.
INTRODUCTION
The Acquired Immunodeficiency Syndrome (AIDS) is a devastating disease that has captivated the attention of the general public, interest groups, health professionals, scientists and governments for more than two decades (1–3). The etiological agent of AIDS is the human immunodeficiency virus (HIV), a transmittable retrovirus. HIV Reverse Transcriptase (RT) is an essential enzyme for viral replication, and therefore represents a major target for the development of antiretroviral agents (4–9). This enzyme possesses DNA polymerase activities on both RNA and DNA templates as well as ribonuclease H (RNase H) activity. Both the RT-associated DNA polymerase and RNase H activities are required to convert the single-stranded RNA genome into double-stranded DNA that is later integrated into the host chromosome.
Of the numerous lead compounds studied, only those that specifically target HIV-1 RT or HIV-1 protease enzyme, and, more recently, the entry process, have been approved for HIV therapy. The common nucleoside reverse transcriptase inhibitors (NRTIs), e.g. AZT, 3TC and d4T, and non-nucleoside reverse transcriptase inhibitors (NNRTIs), e.g. nevirapine, efavirenz and delavirdine are effective at blocking viral DNA replication and slowing the onset and progression of AIDS (10–12). Despite the tremendous success associated with antiretroviral combination therapy, which may also include inhibitors of the viral protease, the emergence of drug resistance cannot be prevented and is a major cause for treatment failure. Vaccination against HIV-1 is still a long-term goal. Thus, the development of novel antiretroviral agents with potency against resistant HIV variants is of highest priority. Ideally, these novel compounds should act synergistically with existing drugs.
Most of the antiviral compounds inhibiting HIV replication in cell culture, such as AZT, 3TC and NNRTIs act primarily by inhibiting the DNA polymerase function of the RT. NRTIs are competitive inhibitors and act as chain terminators, while NNRTIs bind to a hydrophobic pocket close to the active site and block the catalytic step.
Mutations in the RNase H domain of RT lead to a marked decrease in the level of virus proliferation, an indication of the crucial role of RNase H during the retroviral cycle (13). Point mutations at the active site can completely block replication of the virus. Surprisingly, very few inhibitors of RNase H have been described, and no clear specificity for HIV over the mammalian enzyme has yet emerged, probably reflecting the structural similarities between the retrovirally associated RNase H and other cellular RNases H. In addition, the majority of HIV-1 RNase H inhibitors developed to date simultaneously inhibit both polymerase and RNase H activity. This is likely because the two subdomains are in close proximity; therefore binding within either site can restrict access to the other as well (i.e. via allosteric effects). For example, a product isolated from Juglans mandshurica has recently been shown to inhibit both the DNA polymerase (IC50 = 40 nM) and RNase H functions of HIV-1 RT (IC50 = 39 μM) (14). Parniak and co-workers have shown that indiscriminate inhibition of both activities by N-(4-tert-butylbenzoyl)-2-hydroxy-1-naphthaldehyde hydrazone (BBNH) also occurs (IC50 ∼4.0 μM) (15). Certain analogues of naphthalene sulfonic acid exhibit non-selective inhibition of both DNA polymerase and RNase H catalytic functions of HIV-1 RT, with IC50 values in the 15–28 μM range for the most potent compounds (16).
Recently, new inhibitors of HIV-1 RT have been identified using the SELEX approach (systematic evolution of ligands by exponential enrichment). These oligonucleotides, termed ‘aptamers’, are emerging as a class of molecules that may be useful both as selective inhibitors of HIV-1 RT and for structural studies of the enzyme (17–21). The first results using SELEX (20,21) allowed the selection of RNA guanosine-rich ‘pseudoknots’ that bind specifically to HIV-1 RT (22). The researchers noted that the DNA polymerase activity of the RT enzyme was also inhibited by aptamers inhibiting the retroviral RNase H. They speculated that these RNA aptamers (comprising >40 nt residues in length) were occupying different positions in the RNase H and DNA polymerase active sites of HIV-1 RT. Most recently, a diketoacid and a hydrazone (DABNH) derivative have been shown to inhibit RNase H activity (IC50 ∼ 4.0 μM) with only modest effect on DNA polymerization (IC50 > 50 μM). However, the specificity of these compounds for retroviral over the human enzyme is unknown (23,24).
Preliminary work from our laboratory has demonstrated that RNA-hairpin and RNA-dumbbell molecules containing the unique UUCG structural loop motif are potent and selective inhibitors of the RNase H activity of HIV-1 RT (25). These RNAs effectively inhibited the RNase H activity of HIV-1 RT without affecting human or E.coli RNases H. Furthermore, the DNA polymerase activity, an intrinsic property of HIV-1 RT, was not inhibited by these compounds, a property not previously observed for any nucleic acid aptamer directed against RT RNase H. These results have prompted us to evaluate the inhibitory potential of a larger number of RNAs in order to assess the structural requirements for recognition by HIV-RT RNase H. Towards this end, we describe here a ‘diversity-oriented’ solid-phase synthesis of structurally and conformationally diverse hairpin molecules, as well as the apparent ability of these compounds to inhibit the RNase H activity of HIV-1 RT in a site-specific manner (Figure 1). The combined results indicate that the stem-length and conformation are both important factors in designing potent inhibitors of HIV-1 RT RNase H. RNA hairpin molecules which adopted global A-type helices were the most potent inhibitors. Finally, these studies indicate that HIV-1 RT distinguishes and recognizes the unusually folded 3′,5′- or 2′,5′-linked rUUCG loop structure as a signal for binding to its substrate.
Figure 1.

Schematic representation of the mode of inhibition of HIV-1 RT RNase H-mediated degradation of viral RNA by small-molecule hairpin aptamers. The hairpins bind to the RNase H domain, found in the C-terminus of the p66 subunit of RT.
MATERIALS AND METHODS
Materials and general methods
5′-O-Dimethoxytrityl-3′-O-(2-cyanoethyl)-N,N′-diisopropylphosphoramidite monomers for normal DNA and RNA synthesis were purchased from Dalton Chemical Laboratories (Ontario, Canada). The corresponding ribonucleoside 2′-O-phosphoramidites (2′,5′-RNA synthesis) were obtained from ChemGenes Corp. (Watham, MA). These reagents were stored at −20°C and dried in vacuo (under P2O5) for 24 h prior to use. E.coli RNase H, gamma-ATP and 32P-labeling kit were all purchased from Amersham Pharmacia. The RNA and DNA strands utilized in RNase H inhibition assays were synthesized using standard phosphoramidite chemistry protocols as described below. Similarly, the RNA and DNA templates as well as the primer (from PBS region of HIV-1) utilized in the polymerase assays were also synthesized in the lab via the same method (see assay for sequence). Rabbit reticulocytes lysate was purchased from Sigma-Aldrich. 5′ end 32P-labeling and circular dichroism (CD) spectroscopy of hairpins were conducted as described in the Supplementary Material (Part A).
Hairpin synthesis and purification
The hairpin oligonucleotides were synthesized with slight modifications of our previously published procedures (26,27). Library synthesis was achieved via using a Perceptive Biosystems (Expedite) synthesizer on a 1-μmol scale and utilizing LCAA-controlled pore glass (500 Å) as solid support. Monomer coupling times were 10 min (RNA or 2′,5′-RNA monomers), and 2 min (DNA monomers). Extended coupling times were used for riboG 2′- or 3′-O-phosphoramidite (15 min) and dG monomers (3 min). Concentration of monomers was 0.15–0.17 M (RNA) and 0.1 M (DNA). The activator solution consisted of 0.5 M 4,5-dicyanoimidazole/acetonitrile for DNA, 3′,5′-RNA and 2′,5′-RNA synthesis (28). We found that this reagent works best for 2′,5′-RNA synthesis. Following chain assembly, the CPG support was treated with aqueous ammonia/ethanol (3:1—1.5 ml total volume) for 48 h at room temperature. After centrifugation, the supernatant was collected and the solid support was washed with 3 × 1 ml ethanol. The supernatant and ethanol washings were combined and evaporated to dryness under vacuum. The pellet obtained was treated with NEt3.3HF (200 μl) at room temperature for 48 h (29). The reaction was quenched by addition of deionized double distilled water and the resulting solution was evaporated to dryness under vacuum. The oligomers were purified by anion-exchange HPLC (Protein Pak DEAE-5PW column—Waters, 7.5 mm × 7.5 cm) using a linear gradient of 0–20% LiClO4 in H2O (1 M) with a flow rate 1 ml/min at 55°C over a period of 50 min. The oligonucleotides were then desalted by using reversed-phase chromatography on a Sep-Pak cartridge (26) and evaporated to dryness. Prior to any studies, the purity of all purified desalted oligonucleotides was checked by using either analytical 24% denaturing acrylamide gels or analytical ion-exchange HPLC, and was found to be >92% in most cases. The molecular weights of the purified oligonucleotides were also confirmed by MALDI-TOF mass spectrometry (for representative examples on all of the above characterizations, see Supplementary Material B). Molar extinction coefficients of hairpins were calculated from those of mononucleotides and dinucleotides using the nearest-neighbor approximation method (30).
HIV-1 RT RNase H inhibition assay
The p66- and p51-kDa subunits of HIV-1 RT were cloned into a pBAD/HisB prokaryotic expression vector (Invitrogen) between the XhoI and HindIII sites. RT p66/p51 heterodimers and p51/p51 homodimers were purified as described previously by Fletcher and co-workers (31). This assay measured the ability of various hairpin oligomers to inhibit the RNase H-mediated degradation of a 5′-[32P]-labeled RNA:DNA 18 bp substrate. The sequence of the 5′-[32P]-RNA used is 5′-GAU CUG AGC CUG GGA GCU-3′, and was prepared by the transfer of 32P from [γ-32P]ATP in a reaction catalyzed by bacteriophage T4 polynucleotide kinase (see Supplementary Material A). This 5′-[32P]RNA oligonucleotide was annealed to a complementary unlabeled DNA strand of the sequence: 5′-AGC TCC CAG GCT CAG ATC-3′ to form the [32P]-RNA/DNA hybrid duplex substrate. In separate microtubes, aliquots of 10 μl of 50 mM Tris–HCl (pH 7.8, 37°C), containing 60 mM KCl and 2.5 mM MgCl2, 150 nM p51/p66 RT were mixed with variable amounts of hairpins and incubated at 37°C for 15 min. The reactions were initiated by addition of [32P]-RNA/DNA hybrid duplex substrate (final concentration 50 nM), and the assay tubes were then incubated at 37°C. After 15 min, the reactions were stopped by the addition of an equal volume of denaturing gel loading buffer (98% deionized formamide, 10 mM EDTA, 1 mg/ml bromophenol blue and 1 mg/ml xylene cyanol). The reaction products were denatured by heating at 100°C for 5 min, and were then loaded onto a 16% polyacrylamide sequencing gel containing 7 M urea and the products resolved by electrophoresis. The resolved reaction products were then visualized by autoradiography, and the extent of RNase H inhibition was determined quantitatively by densitometric analysis as judged from the disappearance of the full-length RNA substrate and/or the appearance of the smaller degradation products. In this experiment, the band corresponding to the undegraded 18 nt 5′-[32P]-RNA was quantified by densitometry, using the software UN-SCAN-IT (Silk Scientific, Orem, UT). The IC50 values for hairpin inhibition of HIV-1 RT-associated RNase H activity were calculated from plots of the residual undegraded 5′-[32P]-RNA versus oligomer/hairpin concentration.
Biostability assays
Cell lysates
These assays aimed at assessing the biological stability of the various hairpin inhibitors. The individual aptamers were 5′-32P-end-labeled and then dissolved in 60 mM Tris (pH 7.8), 60 mM KCl, 5 mM MgCl2 and 5 mM K2HPO4 buffer. After the addition of rabbit reticulocytes lysate (Promega), the reaction mixture was incubated for 18 h at 37°C. The degradation assay was stopped by the addition of an equal volume of loading dye (98% deionized formamide, 10 mM EDTA, 1 mg/ml bromophenol blue and 1 mg/ml xylene cyanol) followed by heating to 95°C. It was then loaded onto 16% (w/v) polyacrylamide denaturing gels (7 M urea) and then exposed on an X-ray film. The individual lanes were quantitated using the software UN-SCAN-IT by measuring the amount of remaining undegraded hairpin after 18 h.
Nuclease P1 assays
These were conducted at 37°C on a Varian CARY 1 UV-VIS spectrophotometer equipped with a multi-cell holder and a Peltier temperature controller. Hairpin samples were dissolved in 30 mM NaOAc buffer (pH 5.30) to a final concentration of 6 μM, and then annealed by heating rapidly to 95°C for 10–15 min, followed by cooling slowly to room temperature and subsequent equilibration at 37°C. Reactions were initiated by the addition of nuclease P1 (0.001 U), and the UV absorbance of the hairpins was monitored with time over a range of 180 min at 260 nm with 0.5 min increments and an average time of 0.1 s (signal band width = 1 nm). The data was processed using the Enzyme Kinetics Software provided by Varian.
RESULTS AND DISCUSSION
Library generation via diversity-oriented nucleic acid synthesis
Several reports describing synthetic oligonucleotide libraries have been documented (32,33). In this study, we utilize a ‘split and pool’ method to generate a large library of structurally and conformationally diverse compounds (Figure 2). A schematic representation of our ‘diversity-oriented nucleic-acid synthesis’ (DONAS) approach is shown in Figure 3. The advantages of this method are many: (a) a library can be built using established solid-phase phosphoramidite synthesis; (b) any number of diverse building blocks can be used in synthesis (ribonucleoside 2′- or 3′-phosphoramidites, etc.); (c) unlike SELEX, which requires rounds of enzymatic amplification and selection from a large (randomized) mixture of compounds, DONAS generates solid-supports each containing a unique nucleic acid sequence that is isolated and identified by standard techniques (e.g. HPLC and mass spectrometry); and (d) it offers the opportunity to introduce virtually any modification on the sugar-phosphate backbone and obtain significant amounts of material (mg–g scale). During the synthesis of the 27-member library (Figure 3), the base sequence was maintained while varying the sugar (2′-deoxyribose, ribose) and phosphate backbone (3′,5′- and 2′,5′-linkages). The loop sequences 3′,5′-C(UUCG)G and 2′,5′-C(UUCG)G were selected as they have been shown in (23,30,31) to adopt distinct conformations that provide extra stability to the helical stem (see melting temperature, Tm, values in Table 1).
Figure 2.
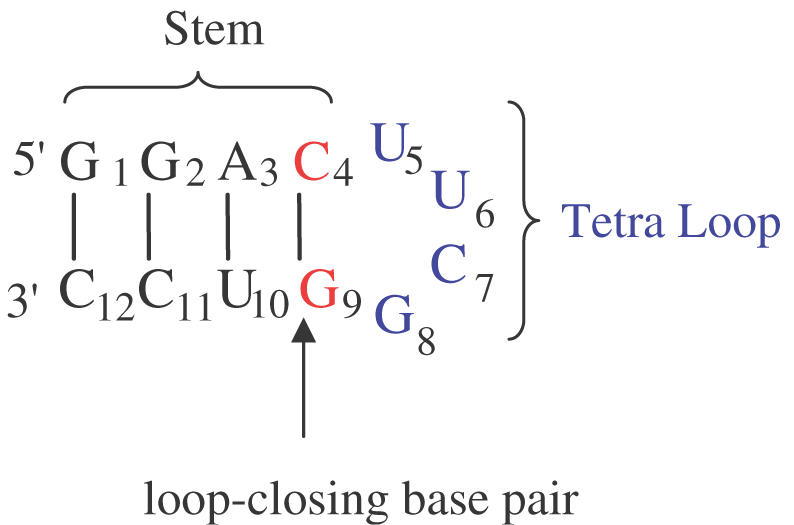
Secondary structure and base sequence of the hairpin constituting the building core for library generation. The structure consists of a loop (U5U6C7G8), and a stem region (defined by G1–C4 and G9–C12 residues) that includes the ‘loop-closing base pair’ C·G. During diversity-oriented nucleic acid synthesis (DONAS) of the library, the stem can be either DNA:DNA ‘DD’, RNA:RNA ‘RR’, 2′,5′-RNA:2′,5′-RNA ‘RR’, DNA:RNA ‘DR’, DNA:2′,5′-RNA ‘DR’, or RNA:2′,5′-RNA ‘RR’, while the loop was modified with DNA ‘D’, RNA ‘R’ and 2′,5′-RNA ‘R’ residues.
Figure 3.

Protocol for the diversity-oriented nucleic acid synthesis (DONAS) of a 27-member hairpin library. The synthesis proceeds from the 3′ to the 5′ end. Starting with the 3′-stem, (GUCC) is synthesized as DNA, RNA or 2′,5′-RNA fragments via parallel combinatorial synthesis. The columns (containing the solid support, CPG) are then equally arranged ‘without mixing’ into three identical equivalent pools. Subsequent combinatorial synthesis would yield the loop region in again three different morphologies (DNA, RNA and 2′,5′-RNA). The same approach (arranging into pools followed by combinatorial synthesis) installs the 5′-stem, thus yielding 27 hairpin members.
Table 1. Inhibition of RNase H activity of HIV-1 RT by a nucleic-acid hairpin librarya.
| Entry | Code | 5′-Sequence-2′/3′b | IC50 (μM) | Tm (°C) |
|---|---|---|---|---|
| 1 | DDD | ggac(uucg)gtcc | — | 56.2 |
| 2 | DTD | ggac(tttt)gtcc | — | 54.7 |
| 3 | DRD | ggac(UUCG)gtcc | — | 54.6 |
| 4 | DRD | ggac(UUCG)gtcc | 96 | 61.4 |
| 5 | DR1D | ggac(UACG)gtcc | — | 56.7 |
| 6 | DR2D | ggac(UUUG)gtcc | 69.1 | 62.0 |
| 7 | DR3D | ggac(UUUU)gtcc | 97.2 | 54.5 |
| 8 | RDR | GGAC(uucg)GUCC | — | 63.4 |
| 9 | RRR | GGAC(UUCG)GUCC | 25.8 | 71.8 |
| 10 | RRR | GGAC(UUCG)GUCC | 68.9 | 69.3 |
| 11 | RR1R | GGAC(UACG)GUCC | — | 62.3 |
| 12 | RcRgR | GGAc(UUCG)gUCC | >100 | 60.0 |
| 13 | RcRgR | GGAc(UUCG)gUCC | 39.4 | 57.6 |
| 14 | RcRR | GGAc(UUCG)GUCC | — | 67.3 |
| 15 | RRgR | GGAC(UUCG)gUCC | 46 | 66.6 |
| 16 | RRUR | GGAC(UUCG)GUCC | 50.1 | 60.2 |
| 17 | RCRR | GGAC(UUCG)GUCC | >100 | 62.6 |
| 18 | RRGR | GGAC(UUCG)GUCC | 98 | 58.0 |
| 19 | RCRR | GGAC(UUCG)GUCC | — | 61.5 |
| 20 | DCRGD | ggaC(UUCG)Gtcc | — | 59.5 |
| 21 | DCUGD | ggaC(UUUU)Gtcc | — | 51.6 |
| 22 | DCRGD | ggaC(UUCG)Gtcc | — | 52.3 |
| 23 | DCRGD | ggaC(UUCG)Gtcc | — | 57.0 |
| 24 | DDR | ggac(uucg)GUCC | — | n.d. |
| 25 | DRR | ggac(UUCG)GUCC | — | 56.5 |
| 26 | DRR | ggac(UUCG)GUCC | — | 56.7 |
| 27 | RDD | GGAC(uucg)gtcc | — | n.d. |
| 28 | RRD | GGAC(UUCG)gtcc | — | 48.1 |
| 29 | RRD | GGAC(UUCG)gtcc | 47 | 52.8 |
| 30 | DRR | ggac(UUCG)GUCC | >100 | 24.1 |
| 31 | DRR | ggac(UUCG)GUCC | 71.4 | 30.2 |
| 32 | RDR | GGAC(uucg)GUCC | — | n.d. |
| 33 | RRR | GGAC(UUCG)GUCC | — | 62.6 |
| 34 | RRR | GGAC(UUCG)GUCC | 58.9 | 62.4 |
| 35 | RRR | GGAC(UUCG)GUCC | — | 54.1 |
| 36 | RRR | GGAC(UUCG)GUCC | 42.8 | 58.1 |
| 37 | RRR | GGAC(UUCG)GUCC | 26.2 | 45.2 |
| 38 | RRR | GGAC(UUCG)GUCC | 88.5 | 54.8 |
| 39 | TRT | tttt(UUCG)tttt | — | — |
| 40 | R6RR6 | GUGGAC(UUCG)GUCCAC | 7.8 | n.d. |
| 41 | R6RR6 | GUGGAC(UUCG)GUCCAC | 29.7 | n.d. |
| 42 | R R R | GGAC(UUCG)GUCC | ∼300 | 81.0 |
aIC50 is the hairpin concentration required to inhibit 50% RNase H activity of HIV-1 RT and was determined as described in Materials and Methods. Dashes ‘—’ mean no inhibition observed up to 100 μM. ‘>100 μM’ means some inhibition was detected at this concentration. Values represent the average of 2–3 independent measurements. Errors in IC50 values represent SD and were within ±3 μM.
bCapital letters represent RNA residues; capital italicized letters represent 2′-OMe ribonucleotide residues (entry 42); underlined letters are 2′,5′-RNA residues (e.g. UC = U2′p5′C2′p); DNA residues are represented by small letters; bold letters represent specific point mutations in loop base sequence (entries 5–7 and 11) or sugar/phosphodiester composition (entries 11–22). (n.d. = Tm was not determined).
Rather than conducting 27 independent syntheses (27 × 11 base additions = 297 couplings), the solid-supports were divided into ‘pools’ every time a tetranucleotide segment (3′-stem, loop, 5′-stem) was completed, reducing the number of coupling steps by ∼50% (156 couplings). Synthesis of this particular library took advantage of the presence of only three bases in each of the 5′-stem, 3′-stem and loop regions, reducing the number of monomer bottle changes to one. At the end of synthesis, each solid support carried a unique, single oligonucleotide sequence (Figure 3). Eighteen other hairpins were prepared by the same approach (Table 1).
The individual oligomers were cleaved off the solid support, purified by ion-exchange HPLC, and subsequently characterized by MALDI-TOF mass spectrometry (Supplementary Material B). The formation of unimolecular hairpin species for each library member was verified by thermal denaturation studies. Thus, the Tm values of the different hairpins were found to be independent of oligonucleotide concentration over at least a 30-fold concentration range (Supplementary Material C).
Conformational diversity of library members
By introducing DNA and RNA sugars during solid-phase synthesis, DONAS offered rapid access to a broad range of hairpin conformations. All library members were studied by CD spectroscopy, and as expected, their collective structures spanned a wide spectrum of helical conformations (see Figure 4 for representative examples). A-form helices were identified from the characteristic strong positive CD band at ∼265 nm and a negative band at ∼212 nm (34). B-form hairpins were identified from the red-shifted CD band at ∼278 nm (34). Other structures exhibited strong positive CD bands that were mixtures of the 265 and 278 nm bands (35). Depending on the extent of their resemblance to the typical A- or B-spectra, they were classified as either ‘A-like’ or ‘B-like’. In cases when this could not be inferred, the structures were classified as ‘I-form’ or ‘intermediate’ form. For example, the hairpins RRR and DDD (Table 1) exhibit typical A-form and B-form conformations, respectively. By contrast, the conformations of RRR, DRD and DRD are, respectively, A-like, I-form and B-like (Figure 4). This is fully consistent with high-resolution NMR data (36–38,39), which supports the qualitative conformational assignment of hairpins via CD spectroscopy (Figure 4). The I-form helical conformation of hairpin DRD arises from the heterogeneous conformation of the deoxynucleotide residues, that is, a dynamic equilibrium between the C2′-endo and C3′-endo puckers for the stem DNA residues (38), whereas DRD exhibits B-like features due to the C2′-endo conformation of its DNA residues.
Figure 4.

Conformation spectrum (CD spectroscopy) generated by DONAS.
Library screening and biological evaluation
The DONAS-generated hairpin molecules were consequently screened for their ability to inhibit the RNase H activity of HIV-1 RT (Figure 5A). They showed various IC50 values varying with hairpin stem and loop composition and ranging from 7.8 to 100 μM. The hairpin molecules were also screened for their ability to interfere with HIV-RT's RNA- and DNA-dependent polymerase activities (Figure 5B). Of the several hairpins tested, none inhibited the polymerase activities even at 80 μM.
Figure 5.

(A) Gel autoradiogram illustrating representative RNase H inhibitory activity resulting from screening DONAS hairpin library members. A [32P]-RNA:DNA hybrid was incubated with HIV-1 RT in the presence of varying amounts of hairpins (0–50 μM). (B) Inhibition assay of DNA synthesis catalyzed by (a) DNA-dependent DNA polymerase and (b) RNA-dependent DNA polymerase activities of HIV-1 RT. Unlabeled hairpin (80 μM) was pre-incubated with RT for 20 min at room temperature prior to initiating the polymerization reaction by adding the proper DNA or RNA template hybridized to a 5′-[32P]-labeled primer in the presence of dNTPs (see Materials and Methods). The various lanes show full-length DNA products synthesized after 15 min in the absence (−) and presence (+) of unlabeled hairpins.
To exclude the possibility that the hairpins themselves were substrates for the enzyme (which in turn would be responsible for the decrease in the observed rate of RNase H degradation of the natural RNA:DNA substrate), the most potent hairpins (R6RR6, RRR, RRR, RcRgR) were 5′-[32P]-labeled and then incubated with HIV-1 RT in the absence of RNA:DNA substrate (1 h, 37°C; Supplementary Material D). Under these conditions, none of the hairpins were cleaved by RNase H. This is not surprising given the small length of the hairpins' helical stem (4–6 bp) and the fact that RNA duplexes are not generally cleaved by RNase H. When the hybrid hairpins DRR and DRR were subjected to HIV-1 RT, no degradation was observed, again reflecting the short nature of the helical stem. In contrast, the control 18 bp DNA:RNA heteroduplex was completely cleaved within 20 min (no hairpin inhibitor).
We recently reported a UV cross-linking experiment between R6RR6 and either the HIV-1 RT heterodimer (p66/p51) or homodimer (p51/p51). The former contains both the DNA polymerase and RNase H domains, whereas the latter consists of only a functional DNA polymerase domain. R6RR6 and other related compounds were found to form a covalent complex only with the p66/p51 homodimer, indicating that they are highly specific toward the RNase H domain of the enzyme (25).
Furthermore, the two most potent hairpins R6RR6 and RRR did not have any effect on the bacterial or human RNase H-mediated degradation of the RNA template strand, indicating an effect remarkably specific toward the retroviral RNase H domain.
Structure–activity relationships (SARs)
From the data obtained (Table 1), the following conclusions can be drawn:
A folded helical structure is recognized by HIV-1 RT RNase H
This is evident from the data shown in Table 1, and the finding that neither the linear (unpaired) sequences TRT (Table 1, entry 39) nor 5′-UUCGGUCC-3′ and 5′-UUCGGUCC-3′ display inhibitory activity against HIV RT RNase H (Supplementary Material E). The same was true for a 1:1 mixture of (5′-GGAC-3′) and either 5′-UUCGGUCC-3′, 5′-UUCGGUCC-3′ or (5′-GUCC-3′) (data not shown), supporting the notion that HIV RT RNase H recognizes the folded structure of hairpin molecules.
Helix type and loop structure are both important determinants of RNase H inhibition
Inhibitory potency generally increases with increasing A-form helicity; among the 4 bp hairpins, the following order of activity was observed: RRR, RRR > RRD, RRR > RRR >>> RDR, DDD (Figure 6). Thus, B-type hairpins show only modest or no inhibitory activity. This is fully consistent with the preferred A-helical geometry for effective RNase H binding within the minor groove (40,41). The slightest perturbations within the stem structure significantly affected inhibitory potency. For example, replacement of the loop-closing C residue with dC has a detrimental effect on inhibitory activity (compare RRR entry 9 versus RCRR entry 14, Table 1). Given the significant drop in melting temperature (ΔTm ∼ 5°C) and the small but detectable changes in the CD spectra (Figure 4), this mutation probably disrupts the hairpin structure at the stem–loop junction. Similar effects were observed when a single 2′,5′-internucleotide linkage was incorporated at the same position (entries 9 versus 17; Table 1). Furthermore, introduction of 2′-OMe ribonucleotide residues within the stem significantly reduced activity (IC50 300 versus 25.8 μM; Table 1), implying that key HIV-RT–hairpin contacts are disrupted upon methylation of the helix minor groove. Consistent with this notion, substitution with a smaller 2′-substituent (2′-fluoro) within the stem does not affect inhibitory activity relative to the unmodified RRR hairpin (A. Wahba and M.J. Damha, unpublished results).
Figure 6.

(A) Three-dimensional graph showing % inhibition (at 40 μM [aptamer]) versus helical conformation for hairpins with various loops and having the same stem compositions. More pronounced inhibitory activity (‘hits’) against HIV-1 RT RNase H is observed for hairpins containing 2′,5′-RNA loops, although the most potent member contains a 3′,5′-RNA loop. Hairpins containing DNA loops, regardless of their stem composition, do not show any degree of inhibition. (B) Effects of loop base sequence, point insertions and stem length.
When the stem was increased to 6 bp (R6RR6 versus RRR; R6RR6 versus RRR), a 2- to 3-fold enhancement in potency was evident, suggesting that longer helices are better accommodated in the binding domain (Table 1; Figure 6A and B). The results are consistent with previous data demonstrating that RNA dumbbells, containing the identical 3′,5′-rUUCG loop motif, but with a 10 bp stem, were more effective at inhibiting enzymatic function (25). Neither of these compounds inhibit the human RNase H (class II, type 1—major enzyme) or bacterial RNase H activities suggesting a remarkably specific effect towards the retroviral enzyme (HIV-1 RT; Supplementary Material). It should be noted, however, that specificity towards the retroviral RNase H enzyme is compromised with longer duplexes, e.g. 18-bp RNA:RNA, as these have been shown to inhibit the bacterial RNase H enzyme as well (41).
Replacement of the native loop structure with a 2′,5′-UUCG loop leads to a ∼3-fold decrease in potency [IC50 RRR (25.8 μM) versus RRR (68.9 μM); R6RR6 (7.8 μM) versus R6RR6 (29.7 μM)]. The data also show that while the 3′,5′-linked loop gave rise to potent inhibitors, the inhibitory activity was more prevalent for hairpins containing the 2′,5′-loops; in other words, hairpins with 2′,5′-linked loops afforded the most ‘hits’ (Figure 6A). For example, whereas DRD, DR2D and DR3D (entries 4, 6 and 7, Table 1) display modest inhibitory activity (IC50 69–97 μM), DRD does not interfere with RNase H function. Although the origin for these observations is unclear, we note that the 2′,5′-UUCG loop exhibits a higher degree of flexibility relative to the native 3′,5′-UUCG (38), and so may better tolerate adaptive conformational changes to the enzyme surface upon binding, ultimately enhancing the integrity of the resulting protein–hairpin complex.
Two more observations are consistent with the notion that HIV-1 RT recognizes the loop conformation: (a) changing the loop composition from RNA to DNA (hairpin RRR versus RDR, Table 1) significantly diminishes inhibitory potency. This is likely because the DNA loop, unlike the corresponding 3′,5′-rUUCG and 2′,5′-rUUCG loops, does not fold into a unique rigid structure; rather it is very flexible and undefined (39); and (b) the slightest perturbations within or near the loop moiety significantly affect the inhibitory potency (Figure 6B). For example, introducing a single base mutation within the loop of RRR hairpin (UUCG to UACG) completely abolishes inhibitory activity (Table 1; Figure 6B). As reported in a recent NMR study, this mutation is expected to disrupt loop conformation because the U residue undergoes extensive base–base stacking interactions with other loop residues (38).
Hairpin inhibitory activity is largely independent of thermal stability
This is evident from Figure 7, which plots %inhibition of RNase H versus the melting temperature for some of the hairpin molecules. For example, hairpin RcRgR is more thermally stable than RcRgR (ΔTm ∼ 2.4°C), yet the latter has a lower IC50 value. Hairpins RRUR, RCRR and RRGR are of almost the same order of thermal stability; however, RRUR shows good inhibitory potency while RCRR and RRGR do not. Likewise, RRR is significantly more thermally stable than RRR, yet it displays a lower inhibitory activity. Few of the hairpins do show some correlation of inhibitory potency with melting temperature (e.g. DRR versus DRR and RRR versus RRR), though this general trend appears to be absent for the majority of compounds evaluated (Figure 7).
Figure 7.

Variation of inhibitory constant (IC50) with melting temperature. The hairpin aptamers are shown in order of decreasing potency (increasing IC50). Bars represent the IC50 plot and the line represents the Tm plot.
Hairpin inhibitory activity is dependent on stem base composition
To assess the effect of stem base composition on the inhibitory properties of hairpins, the compounds listed in Table 2 were examined. Based on the preliminary data obtained, substitution of the second and/or third base pairs is well tolerated (RRR versus R2RR2 and R3RR3, Table 2). While it remains for us to test the full extent of stem sequence variability, it is interesting to note that the most active hairpins contain the sequence G-N-R-C at their 5′ end. Together, the results suggest that RNase H recognizes the fold of the tetraloop, the shape of the helix (A- or A-like), and certain nucleotides in the stem.
Table 2. Probing HIV-1 RT specificity towards stem base sequence.
| Code | 5′-Sequence-2′/3′b | IC50 (μM) |
|---|---|---|
| RRR | GGAC(UUCG)GUCC | 25.8 |
| ARU | AAAC(UUCG)GUUU | — |
| R1RR1 | UCGC(UUCG)GCGA | — |
| R2RR2 | GAGC(UUCG)GCUC | 28.5 |
| R3RR3 | GCAC(UUCG)GUGC | 43.4 |
IC50 is the oligomer concentration required to inhibit 50% RNase H activity of HIV-1 RT. Capital letters represent RNA residues. Measurements for the inhibition assay were repeated twice and were run in the same way as described in Materials and Methods section.
Nuclease stability of hairpin aptamers
The susceptibility of various hairpins towards enzymatic degradation was first examined in a nuclease-rich rabbit reticulocyte lysate. Regardless of their composition, hairpins remained fully intact after 6 h incubation with the lysate (at 0.5× dilution, i.e. equal volume of water added to lysate). Under the same conditions, a control linear oligomer of the same length underwent significant degradation (∼50%; data not shown). This is probably due to the highly stacked and relatively inaccessible structure of the hairpins (42). In general, hairpin biostability does not parallel hairpin thermal stability. For instance, RRR and RRR exhibit similar thermal stabilities, yet RRR is more resistant to enzymatic hydrolysis (Figure 8A). In fact, the presence of 2′,5′-linkages within the loop region enhances the biological stability of the molecules relative to the 3′,5′-RNA and DNA loops (compare RRD versus RRD; RRR versus RRR and RDR; Figure 8A).
Figure 8.

(A) Biological stability of hairpin aptamers after treatment with 0.5× reticulocyte lysate for 18 h at 37°C. The 2′,5′-RNA loop enhances hairpin biostability. TRT does not form any hairpin structure and serves as a control. Equal amounts of 5′-[32P]-labeled oligomers were prepared in separate tubes containing 60 mM Tris (pH 7.8), 60 mM KCl, 5 mM MgCl2 and 5 mM K2HPO4. After addition of rabbit reticulocytes lysate, the reaction mixtures were incubated at 37°C for 18 h. Analysis was done on 16% denaturing polyacrylamide gels. (B) Plots of absorbance (at 260 nm) versus time of exposure of hairpins to nuclease P1. The time necessary to cause 50% degradation of the hairpin structure (t1/2) was calculated at absorbance = 0.5. (C) Gel degradation assay showing the specific cuts induced by nuclease P1 in hairpin DRR. The degradation ladder is formed via alkaline hydrolysis of the hairpin.
The stability of several hairpins towards nuclease P1 (NP1) was also assessed using a combination of UV spectroscopy and gel electrophoresis. NP1 is an endonuclease that cleaves specifically single-stranded DNA and RNA, although it can hydrolyze double-stranded duplexes albeit slowly (43,44). The hairpins (6 μM) were suspended in appropriate buffer (30 mM NaOAc, pH 5.3) and then the reaction was initiated by the addition of NP1. The increase in UV absorbance with time was monitored at 260 nm. After 150–180 min, the absorbance of all hairpins leveled off indicating that no more degradation was taking place (Figure 8B). The relative half-life time of various hairpins was calculated relative to the more susceptible hairpins (DD′D and RDR; Table 3). Again similar to the trend seen when suspended in nuclease-rich reticulocyte lysate, the hairpins containing a 2′,5′-RNA loop show increased resistance towards nuclease P1 relative to hairpins with 3′,5′-RNA and DNA loops. Gel electrophoresis of for example, DRR+NP1 and DRR+NP1 mixtures revealed a very interesting distribution of products (Figure 8C). In both cases, NP1 (endonuclease) made specific ‘cuts’ within the loop (position 6) as well as the loop closing base pairs (position 3 or 4; Figure 8C; data for DRR+NP1 not shown). No other degradation products (or ‘ladder’ of products) were observed as would be expected if strand dissociation occurred after the first cleavage reaction. Although this raised the interesting possibility that the hairpins maintained their folded structure after enzymatic cleavage, incubation of the appropriate fragments (e.g. GGAC + (UUCG)GUCC) at various concentrations and under different buffer conditions did not lead to duplex formation (data not shown).
Table 3. Relative half-life times of representative hairpins towards nuclease P1 digestion.
| Entry | Hairpin | t1/2 (min) | Trel |
|---|---|---|---|
| 1 | DD′D | 8.0 | 1.0 |
| 2 | DDD | 9.8 | 1.2 |
| 3 | DRD | 8.0 | 1.0 |
| 4 | DRD | 16.1 | 2.0 |
| 5 | RRR | 21.2 | 1.4 |
| 6 | RRR | 27.2 | 1.9 |
| 7 | RDR | 14.7 | 1.0 |
The hairpins (6 μM) were incubated with nuclease P1 (0.001 U) in 30 mM NaOAc buffer, pH = 5.3 and their UV absorbance monitored at 260 nm for 150 min. The half-life time (t1/2) was calculated from the obtained curve at absorbance = 0.5. Trel is the relative half-life time value compared to the fastest degrading (least nuclease stable) hairpin oligomer in each set (DD′D in the first set and RDR in the second set). DD′D is a control of the following sequence: 5′-ggacttcggtcc-3′. Values were obtained from three different measurements. Errors in values of t1/2 are within ±1 min.
CONCLUSIONS
Diversity-oriented nucleic acid synthesis furnished a collection of hairpin molecules that differed in the nature of the sugar (DNA versus RNA), phosphodiester linkages (2′,5′- versus 3′,5′-), stem lengths as well as conformations. Four hairpins were identified as potent inhibitors of the activity of HIV-1 RT RNase H in degrading the RNA strand in RNA:DNA hybrids. The hairpin R6RR6 was found to be the most potent inhibitor with an IC50 = 7.8 μM, while RRR, RRR and R6RR6 displayed IC50 in the ∼26–30 μM range. Known RNase H inhibitors are few, and generally do not discriminate between host and viral RNase H activity. R6RR6 is ‘first in class’ with experiments to date indicating high selectivity (if not specificity) toward HIV-1 RT RNase H over the mammalian enzyme. Furthermore, the ‘hairpin’ architecture of this and other hairpins studied here makes them exceedingly resistant to degradation by ubiquitous cellular nucleases.
The vast majority of compounds that inhibit the RNase H activity of HIV-1 RT in cell-free assays also block the RT-associated polymerase activity, and, as a consequence, these compounds diminish the beneficial effects of both classes of existing RT inhibitors. Our studies have shown that small hairpin molecules (e.g. R6RR6) are remarkably specific toward RNase H inhibition. These hairpins do not appear to occupy the DNA polymerase binding site (A. Wahba, B. Marchand, M. Götte and M.J. Damha, unpublished results), and thus have the potential to act synergistically with both NRTIs and NNRTIs. These studies as well as cell culture assays with sugar-modified hairpins are in progress.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We thank M. M. Mangos for helpful discussions and proofreading the manuscript. This work was supported by a grant from the Canadian Institute of Health Research (CIHR) and the National Science and Research Council of Canada (NSERC). R.N.H. is recipient of NSERC postgraduate and postdoctoral scholarships. M.J.D. is recipient of a James McGill scholarship (McGill University).
REFERENCES
- 1.Coffin J., Hasse,A., Levy,J.A., Montagnier,L., Oroszlan,S., Teich,N., Temin,H., Toyoshmia,K., Varmus,H., Vogt,P. and Weiss,R. (1986) Human immunodeficiency viruses. Science, 232, 697–697. [DOI] [PubMed] [Google Scholar]
- 2.Gallo R.C. and Montagnier,L. (1988) AIDS in 1988. Sci. Am., 259, 40–48. [PubMed] [Google Scholar]
- 3.Arts E.J. and Wainberg,M.A. (1998) Human immunodeficiency virus type 1 reverse transcriptase and early events in reverse transcription. Adv. Virus Res., 46, 97–163. [DOI] [PubMed] [Google Scholar]
- 4.De Clercq E. (1986) Chemotherapeutic approaches to the treatment of the acquired immune deficiency syndrome (AIDS). J. Med. Chem., 29, 1561–1569. [DOI] [PubMed] [Google Scholar]
- 5.Krug M.S. and Berger,S.L. (1991) Reverse transcriptase from human immunodeficiency virus: a single template-primer binding site serves two physically separable catalytic functions. Biochemistry, 30, 10614–10623. [DOI] [PubMed] [Google Scholar]
- 6.Kedar P.S., Abbotts,J., Kovacs,T., Lesiak,K., Torrence,P. and Wilson,S.H. (1990) Mechanism of HIV reverse transcriptase: enzyme–primer interaction as revealed through studies of a dNTP analog, 3′-azido-dTTP. Biochemistry, 29, 3603–3611. [DOI] [PubMed] [Google Scholar]
- 7.Gilboa E., Mitra,S.W., Goff,S. and Baltimore,D. (1979) A detailed model of reverse transcription and tests of crucial aspects. Cell, 18, 93–100. [DOI] [PubMed] [Google Scholar]
- 8.Smerdon S., Jager,J., Wang,J., Kohlstaedt,L., Chirino,A., Friedman,J., Rice,P. and Steitz,T. (1994) Structure of the binding site for nonnucleoside inhibitors of the reverse transcriptase of human immunodeficiency virus type 1. Proc. Natl Acad. Sci. USA, 91, 3911–3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsu M. and Wainberg,M.A. (2000) Interactions between human immunodeficiency virus type 1 reverse transcriptase, tRNA primer, and nucleocapsid protein during reverse transcription. J. Hum. Virol., 3, 16–26. [PubMed] [Google Scholar]
- 10.Mitsuya H., Yarchoan,R. and Broder,S. (1990) Molecular targets for AIDS therapy. Science, 249, 1533–1544. [DOI] [PubMed] [Google Scholar]
- 11.Kohlstaedt L.A., Wang,J., Friedman,J.M., Rice,P.A. and Steitz,T.A. (1992) Crystal structure at 3.5 Å resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science, 256, 1783–1790. [DOI] [PubMed] [Google Scholar]
- 12.Merluzzi V.J., Hargrave,K.D., Labadia,M., Grozinger,K., Skoog,M., Wu,J.C., Shih,C.-K., Eckner,K.; Hattox,S.; Adams,J. et al. (1990) Inhibition of HIV-1 replication by a nonnucleoside reverse transcriptase inhibitor. Science, 250, 1411–1413. [DOI] [PubMed] [Google Scholar]
- 13.O.Schatz F.V., Cromme,T., Naas,D., Lindemann,J., Mous,S.F. and LeGrice,J. (1990) Inactivation of the RNase H domain of HIV-1 reverse transcriptase blocks viral infectivity. In T. Papas (ed.), Gene Regulation and AIDS. Portfolio, Tex, pp. 293–404. [Google Scholar]
- 14.Min B.S., Nakamura,N., Miyashiro,H., Kim,Y.H. and Hattori,M. (2000) Inhibition of human immunodeficiency virus type 1 reverse transcriptase and ribonuclease H activities by constituents of Juglans mandshurica. Chem. Pharm. Bull., 48, 194–200. [DOI] [PubMed] [Google Scholar]
- 15.Borkow G., Fletcher,R.S., Barnard,J., Arion,D., Motakis,D., Dmitrienko,G.I. and Parniak,M.A. (1997) Inhibition of the ribonuclease H and DNA polymerase activities of HIV-1 reverse transcriptase by N-(4-tert-butylbenzoyl)-2-hydroxy-1-naphthaldehyde hydrazone. Biochemistry, 36, 3179–3185. [DOI] [PubMed] [Google Scholar]
- 16.Mohan P., Loya,S., Avidan,O., Verma,S., Dhindsa,G.S., Wong,M.F., Huang,P.P., Yashiro,M., Baba,M. and Hizi,A. (1994) Structure activity relationship studies with symmetric naphthalenesulfonic acid derivatives. Synthesis and influence of spacer and naphthalenesulfonic acid moiety on anti-HIV activity. J. Med. Chem., 37, 2513–2519.7520080 [Google Scholar]
- 17.Majumdar C., Stein,C.A., Cohen,J.S., Broder,S. and Wilson,S.H. (1989) Stepwise mechanism of HIV reverse transcriptase: primer function of phosphorothioate oligodeoxynucleotide. Biochemistry, 28, 1340–1346. [DOI] [PubMed] [Google Scholar]
- 18.Dirani-Diab R.E., Sarih-Cottin,L., Delord,B., Dumon,B., Moreau,S., Toulme,J.J., Fleury,H. and Litvak,S. (1997) Phosphorothioate oligonucleotides derived from human immunodeficiency virus type 1 (HIV-1) primer tRNALys3 are strong inhibitors of HIV-1 reverse transcriptase and arrest viral replication in infected cells. Antimicrob. Agents Chemother., 41, 2141–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tuerk C., MacDougal,S. and Gold,L. (1992) RNA pseudoknots that inhibit human immunodeficiency virus type 1 reverse transcriptase. Proc. Natl Acad. Sci. USA, 89, 6988–6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tuerk C. and Gold,L. (1990) Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science, 249, 505–510. [DOI] [PubMed] [Google Scholar]
- 21.Ellington A.D. and Szostak,J.W. (1990) In vitro selection of RNA molecules that bind specific ligands. Nature, 346, 818–822. [DOI] [PubMed] [Google Scholar]
- 22.Andreola M.L., Pileur,F., Calmels,C., Ventura,M., Tarrago-Litvak,L., Toulme,J.J. and Litvak,S. (2001) DNA aptamers selected against the HIV-1 RNase H display in vitro antiviral activity. Biochemistry, 40, 10087–10094. [DOI] [PubMed] [Google Scholar]
- 23.Shaw-Reid C.A., Munshi,V., Graham,P., Wolfe,A., Witmer,M., Danzeisen,R., Olsen,D.B., Carroll,S.S., Embrey,M., Wai,J.S. et al. (2003) Inhibition of HIV-1 ribonuclease H by a novel diketo acid, 4-[5-(benzoylamino)thien-2-yl]-2,4-dioxobutanoic acid. J. Biol. Chem., 278, 2777–2780. [DOI] [PubMed] [Google Scholar]
- 24.Sluis-Cremer N., Arion,D. and Parniak,M.A. (2002) Destabilization of the HIV-1 reverse transcriptase dimer upon interaction with N-acyl hydrazone inhibitors. Mol. Pharm., 62, 398–405. [DOI] [PubMed] [Google Scholar]
- 25.Hannoush R.N., Carriero,S., Min,K.-L. and Damha,M.J. (2004) Selective inhibition of HIV-1 reverse transcriptase (HIV-1 RT) RNase H by small hairpin and dumbbell RNA. ChemBioChem., 5, 527–533. [DOI] [PubMed] [Google Scholar]
- 26.Damha M.J. and Ogilvie,K.K. (1993) Oligoribonucleotide synthesis—the silyl-phosphoramidite method. In Agrawal,S. (ed.), Methods in Molecular Biology: Protocols for Oligonucleotides and Analogues—Synthesis and Properties. The Humana Press, NJ, Vol. 20, pp. 81–114. [DOI] [PubMed] [Google Scholar]
- 27.Hannoush R.N. and Damha,M.J. (2001) Remarkable stability of hairpins containing 2′,5′-linked RNA loops. J. Am.Chem. Soc., 123, 12368–12374. [DOI] [PubMed] [Google Scholar]
- 28.Vargeese C., Carter,J., Yegge,J., Krivjansky,S., Settle,A., Kropp,E., Peterson,K. and Pieken,W. (1995) Efficient activation of nucleoside phosphoramidites with 4,5-dicyanoimidazole during oligonucleotide synthesis. Nucleic Acids Res., 26, 1046–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gasparutto D., Livache,T., Bazin,J., Duplaa,A.-M., Guy,A., Khorlin,A., Molko,D., Roget,A. and Teoule,R. (1992) Chemical synthesis of a biologically active natural tRNA with its minor bases. Nucleic Acids Res., 20, 5159–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Puglisi J.D. and Tinoco,I.,Jr. (1989) Absorbance melting curves of RNA. In Dahlberg,J.E. and Abelson,J.N (eds), Methods in Enzymology: RNA Processing. Academic Press, NY, Vol. 180, pp. 304–332. [DOI] [PubMed] [Google Scholar]
- 31.Fletcher R.S., Holleshak,G., Nagy,E., Arion,D., Borkow,G., Gu,Z., Wainberg,M.A. and Parniak,M.A. (1996) Single-step purification of recombinant wild-type and mutant HIV-1 reverse transcriptase. Protein Expr. Purif., 7, 27–32. [DOI] [PubMed] [Google Scholar]
- 32.Jin Y., Chen,X., Cote,M.-E., Roland,A., Korba,B., Mounir,S. and Iyer,R.P. (2001) Parallel solid-phase synthesis of nucleoside phosphoramidate libraries. Bioorg. Med. Chem. Lett., 11, 2057–2060. [DOI] [PubMed] [Google Scholar]
- 33.Zhou W., Roland,A., Jin,Y. and Iyer,R.P. (2000) Combinatorial synthesis using nucleic acid-based (NAB) scaffold: parallel solid-phase synthesis of nucleotide libraries. Tetrahedron Lett., 41, 441–445. [Google Scholar]
- 34.Hung S.-H., Yu,Q., Gray,D.M. and Ratliff,R.L. (1994) Evidence from CD spectra that of d(purine).r(pyrimidine) and r(purine).d(pyrimidine) hybrids are in different structural classes. Nucleic Acids Res., 22, 4326–4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ratmeyer L., Vinayak,R., Zhong,Y.Y., Zon,G. and Wilson,W.D. (1994) Sequence specific thermodynamic and structural properties for DNA.RNA duplexes. Biochemistry, 33, 5298–5304. [DOI] [PubMed] [Google Scholar]
- 36.Cheong C., Varani,G. and Tinoco,I.J. (1990) Solution structure of an unusually stable RNA hairpin, 5′GGAC(UUCG)GUCC. Nature, 346, 680–682. [DOI] [PubMed] [Google Scholar]
- 37.Varani G., Cheong,C. and Tinoco,I.J. (1991) Structure of an unusually stable RNA hairpin. Biochemistry, 30, 3280–3289. [DOI] [PubMed] [Google Scholar]
- 38.Denisov A., Hannoush,R.N., Gehring,K. and Damha,M.J. (2003) A novel RNA motif based on structure of unusually stable 2′,5′-linked r(UUCG) loops. J. Am.Chem. Soc., 125, 11525–11531. [DOI] [PubMed] [Google Scholar]
- 39.James J.K. and Tinoco,I.,Jr. (1993) The solution structure of a d[C(TTCG)G] DNA hairpin and comparison to the unusually stable RNA analogue. Nucleic Acids Res., 21, 3287–3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lima W.F. and Crooke,S.T. (1997) Binding affinity and specificity of Escherichia coli RNase H1: impact on the kinetics of catalysis of antisense oligonucleotide–RNA hybrids. Biochemistry, 36, 390–398. [DOI] [PubMed] [Google Scholar]
- 41.Wasner M., Arion,D., Borkow,G., Noronha,A.M., Uddin,A.H., Parniak,M.A. and Damha,M.J. (1998) Physicochemical and biochemical properties of 2′,5′-linked RNA and 2′,5′-RNA:3′,5′-RNA ′hybrid′ duplexes. Biochemistry, 37, 7478–7486. [DOI] [PubMed] [Google Scholar]
- 42.Khan I.M. and Coulson,J.M. (1993) A novel method to stabilise antisense oligonucleotides against exonuclease degradation. Nucleic Acids Res., 21, 2957–2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Allen R.J.L. (1940) The estimation of phosphorus. Biochem. J., 34, 858–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Furuichi Y. and Miura,K. (1975) A blocked structure at the 5′ terminus of mRNA from cytoplasmic polyhedrosis virus. Nature, 253, 374–375. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.