

Abstract
Despite major advances in first-line treatment, a significant proportion of patients with diffuse large B-cell lymphoma (DLBCL) will experience treatment failure. Prognosis is particularly poor for relapses occurring less than one year after the end of first-line treatment (early relapses/ER) compared to those occurring more than one year after (late relapses/LR). To better understand genomic alterations underlying the delay of relapse, we identified copy number variations (CNVs) on 39 tumor samples from a homogeneous series of patients included in the Collaborative Trial in Relapsed Aggressive Lymphoma (CORAL) prospective study. To identify CNVs associated with ER or LR, we devised an original method based on Significance Analysis of Microarrays, a permutation-based method which allows control of false positives due to multiple testing. Deletions of CDKN2A/B (28%) and IBTK (23%) were frequent events in relapsed DLBCLs. We identified 56 protein-coding genes and 25 long non-coding RNAs with significantly differential CNVs distribution between ER and LR DLBCLs, with a false discovery rate < 0.05. In ER DLBCLs, CNVs were related to transcription regulation, cell cycle and apoptosis, with duplications of histone H1T (31%), deletions of DIABLO (26%), PTMS (21%) and CK2B (15%). In LR DLBCLs, CNVs were related to immune response, with deletions of B2M (20%) and CD58 (10%), cell proliferation regulation, with duplications of HES1 (25%) and DVL3 (20%), and transcription regulation, with MTERF4 deletions (20%). This study provides new insights into the genetic aberrations in relapsed DLBCLs and suggest pathway-targeted therapies in ER and LR DLBCLs.
Keywords: diffuse large B-cell lymphoma, early relapse, late relapse, genomics, copy number variations
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common subtype of non-Hodgkin lymphoma in adults [1]. The addition of the anti-CD20 monoclonal antibody rituximab to the chemotherapy CHOP (cyclophosphamide, adriamycin, vincristine and prednisone) has dramatically improved the outcome of patients with DLBCL [2, 3]. Despite this major advance a significant number of patients will experience treatment failure. At such a time, treatment is a real challenge and options are to be discussed regarding the eligibility or not for transplant and prognostic factors at relapse [4, 5]. One of the most important prognostic parameters in this context is the time of relapse [6, 7]. Two groups of patients are identified. The first group of early relapse (ER) is defined as relapse occurring within 1 year after the initial treatment. The second group of late relapse (LR) is defined as relapse occurring more than 1 year after the initial treatment. In the ER group, the overall survival (OS) is estimated at less than 17% at 3 years, while in the LR group, the OS is estimated at about 50% at 5 years [4].
The heterogeneous prognosis of patients with DLBCL reflects a deep biological diversity. DLBCL is composed of at least 2 molecular subtypes that differ in gene expression profile (GEP): germinal center B-cell like (GCB) and activated B-cell like (ABC), each with different prognosis [8]. The prognostic impact of this molecular classification has been reported in several independent series and different types of treatment at diagnosis as well as at relapse [9-11].
Recent advances in genomic technology have provided further understanding in the biology of DLBCL. Constitutive activation of the NF-κB pathway, owing to oncogenic mutations activating BCR (B-Cell Receptor) and toll-like receptor signaling, leads to cell proliferation and resistance to apoptosis. These constitutive activations, characterizing ABC DLBCLs, are related to somatic gain of function mutations and/or duplications affecting positive regulators of this pathway such as CD79a and CD79b [12], MYD88 [13], CARD11 [14], TRAF2, TRAF5, MAP3K7/TAK1 and TNFRSF11A/RANK [15] or somatic inactivating mutations and/or deletions of negative regulators such as TNFAIP3/A20 [15,16]. The GCB subtype is associated with abnormalities of epigenetic modifiers, with frequent bi-allelic point mutations leading to the loss of function of the histone lysine methyl-transferase MLL2 [17,18], high frequency of gain-of-function mutations in the histone lysine methyl-transferase EZH2 [19], inactivating mutations and deletions of the histone lysine acetyltransferases CREBBP and EP300 [18], and mutations of their cofactor, ME2FB [17]. Constitutive activation of the PI3K/AKT/mTOR signaling pathway through PTEN deletions, MiR17-92 locus amplifications and mutations of PIK3CD, PIK3R1 and mTOR is another oncogenic mechanism commonly observed in GCB DLBCLs [20, 21]. Dysregulation of cell cycle is a common event in all DLBCLs, potentially owing to an increased expression of the anti-apoptotic factor BCL2 [22-24], mono and bi-allelic deletions affecting the CDKN2A/B locus [22,25], point mutations and allele deletions leading to the loss of function of TP53 [26], or dysfunction of the transcription factor MYC, which has been described in 5 to 11% of de novo DLBCLs [27] and 17% of relapsed DLBCLs [28]. Constitutive activation of the NOTCH pathway through NOTCH1 and NOTCH2 somatic gain-of-function mutations or gain of copy numbers [29-31] cause an increased cell proliferation in B-cell lymphoma. BCL6 expression beyond the germinal center reaction leads to increased tolerance to DNA breaks and blockade of plasmablastic differentiation [32, 33]. Importantly, escape to immune response through biallelic inactivations of B2M leading to defects of surface HLA class I molecules assembly [34], downregulation of surface HLA class II molecules expression due to breakpoints and mutations of CIITA [35] or deletions and truncating mutations of the CD58 locus [34] represent another major player in lymphomagenesis in DLBCLs.
All these genomic studies have been conducted on de novo DLBCLs at diagnosis. Cellular functions and signaling pathway disruptions associated with relapsed DLBCLs remain largely unknown. Here we describe the number and the type of genetic alterations present in relapsed DLBCLs and make a comparison of Copy Number Variations (CNVs) between ER and LR DLBCLs to identify genetic alterations that are associated with the delay of relapse. To achieve this outcome, we designed an original method allowing the statistical assessment with the high level of confidence required when dealing with a very large set of candidates. Finally we evaluated the impact of relevant CNVs on gene expression.
Results
Landscape of large copy number variations in relapsed DLBCL
CNVs ≥ 2 megabases (Mb) were counted to evaluate the complexity of structural aberrations of relapsed DLBCL (Figure 1). For the whole group, average CNVs per sample was 15.50 (ranging from 0 to 67). We found an equivalent number of duplications and deletions with an average 8.30 duplications per sample (0-43) and 7.20 deletions per sample (0-34; p = 0.60, Student's t-test). Chromosomes 1, 2, 3, 6, 12 and 18 were the most frequently altered, with 1.38 CNV per chromosome per sample on average while other chromosomes presented with an average of 0.45 CNV per chromosome per sample (p = 6x10-4).
Figure 1. Landscape of large copy number variations in relapsed DLBCL.
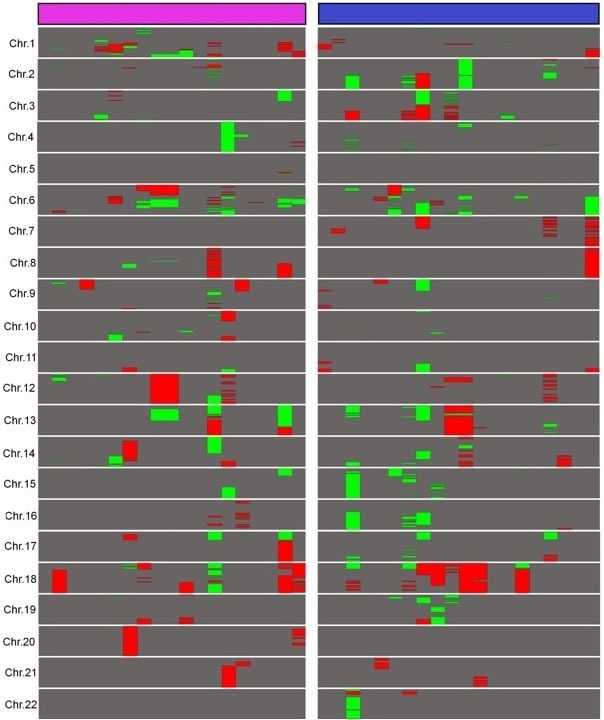
A comparative representation of abnormalities longer than 2 Mb in ER and LR DLBCLs was built. Duplications are represented in red and deletions in green. ER DLBCLs (n = 19) are represented in purple and LR DLBCLs (n = 20) in blue. Chromosomes X and Y are not shown because of gender differences in copy numbers. This figure was obtained using Java Treeview 3.0.
In the ER group, the average CNVs per sample was 14.89. There was a wide heterogeneity within this group as samples presented with 0 to 67 CNVs ≥ 2 Mb. Proportions of duplications and deletions were comparable with an average 8.15 duplications (0-34) and 6.73 deletions per sample (0-33; p = 0.61). Chromosomes 1, 2, 6, 8, 12 and 18 were the most frequently altered, with an average of 1.58 CNV per chromosome per sample, whereas other chromosomes presented with an average of 0.34 CNV per chromosome per sample (p = 6x10-3).
In the LR group, the average CNVs per sample was 16.1. CNVs varied from 0 to 50 per sample within this group. Duplications and deletions were present in equivalent proportions with an average of 8.45 duplications per sample (0-43) and 7.65 deletions per sample (0-34), respectively (p = 0.81). Chromosomes 2, 3, 6, 7, 15, 18 were the most frequently altered, with an average of 1.55 CNV per chromosome, whereas remaining chromosomes presented with 0.42 CNV per chromosome on average (p = 7x10-4).
ER and LR DLBCLs samples were affected by an equivalent total CNVs number with an average of 14.89 and 16.11 CNVs per sample, respectively (p = 0.81). Comparing ER and LR for each chromosome revealed differential signatures on chromosome 1 only, with an average of 2.63 CNVs per sample in the ER group and 0.70 CNV per sample in the LR group (p = 6x10-3).
Identification of genes differentially altered among the ER and the LR subgroups
After smoothing all the copy number probes using the sliding window technique, we observed that 0.38% of the values were below 1 and 1.53% above 3. This means that almost all losses and gains of copy numbers corresponded to heterozygous deletions and duplications, respectively.
The use of Significance Analysis of Microarrays (SAM), a permutation-based method built on discriminating scores and their significance [36] identified 148 genes with significantly differential CNVs distribution between ER and LR DLBCLs with a false discovery rate (FDR) < 0.05. Among these genes were 56 protein coding genes (Table 1; Figure 2), 25 Long non-coding RNA (LncRNA), 22 antisense RNAs, 2 Small Nucleolar RNAs, 1 sense overlapping RNA, 1 miscellaneous RNA and 41 pseudogenes (Supplemental Table 1).
Table 1. Detailed list of the 56 protein coding genes presenting with differential CNVs distribution between ER and LR.
| Chr. Band |
Gene | Gene full name | Description/function |
|---|---|---|---|
| 1p21 | AMY2A | Amylase, Alpha 2A | Member of the alpha-amylase family |
| 1q44 | HNRPU | Heterogeneous Nuclear Ribonucleoprotein U | pre-mRNA processing |
| 1q44 | OR2T34 | Olfactory receptor, family 2, subfam. T, mb 34 | G-protein-coupled olfactory receptor |
| 1q44 | OR2AK2 | Olfactory receptor, family 2, subfam. AK, mb 2 | G-protein-coupled olfactory receptor |
| 2p11 | IGKV3D-7 | Immunoglobulin kappa variable 3D-7 | BCR assembly |
| 2q24 | GALNT5 | N-Acetylgalactosaminyltransferase 5 | O-linked oligosaccharide biosynthesis |
| 2q31 | NFE2L2 | Nuclear factor (erythroid-derived 2)-like 2 | Transcriptionnal regulator |
| 2q33 | SNORA1 | Small nucleolar RNA, H/ACA box 1 | Novel snoRNA/Small nucleolar RNA |
| 2q37 | MTERF4 | Mitochondrial Transcription Termination Factor 4 | Regulator of mitochondrial gene expression |
| 2q37 | UBE2F-SCLY | UBE2F-SCLY Readthrough | MHC class I mediated antigen processing |
| 2q37 | C2orf82 | Chromosome 2 Open Reading Frame 82 | --- |
| 3q22 | PRR23B | Proline Rich 23B | --- |
| 3q25 | AADAC | Arylacetamide deacetylase | Microsomal arylacetamide deacetylase |
| 3q27 | DVL3 | Dishevelled, dsh homolog 3 | Regulation of cell proliferation |
| 3q29 | CPN2 | Carboxypeptidase N, Polypeptide 2 | Carboxypeptidase |
| 3q29 | SENP5 | SUMO1/sentrin specific peptidase 5 | Post-translational modification of proteins |
| 3q29 | MUC20 | Mucin 20, Cell Surface Associated | Glycoprotein of mucous barrier |
| 3q29 | HES1 | Hairy enhancer of split 1 | Transcription factor |
| 6p21 | CSNK2B/CK2B | Casein kinase 2, beta subunit | Regulation of programmed cell death |
| 6p21 | BTNL2 | Butyrophilin-Like 2 | Negative regulation of T-cell proliferation |
| 6p21 | CCHCR1 | Coiled-Coil Alpha-Helical Rod Protein 1 | Regulation of keratinocyte proliferation |
| 6p21 | HIST1H1T | Histone Cluster 1, H1t | Epigenetic regulation of transcription |
| 6p21 | HLA-DRA | MHC complex, Class II, DR Alpha | Antigen presentation |
| 6p22 | HIST1H2BC | Histone Cluster 1, H2bc | Epigenetic regulation of transcription |
| 6p22 | HIS1H2AK | Histone Cluster 1, H2ak | Epigenetic regulation of transcription |
| 6p22 | KAAG1 | Kidney Associated Antigen 1 | --- |
| 6p25 | SERPINB1 | Serpin Peptidase Inhibitor, Class B , Mb1 | Proteinase inhibition |
| 6q13 | KHDC1 | KH homology domain containing 1 | Predicted membrane protein |
| 7p22 | GPR146 | G Protein-Coupled Receptor 146 | G protein coupled receptor |
| 7q11 | ABHD11 | Abhydrolase domain containing 11 | --- |
| 7q11 | STX1A | Syntaxin 1A | Docking of synaptic vesicles |
| 11p15 | ART5 | ADP-Ribosyltransferase 5 | ARG-specific ADP-ribosyltransferase |
| 12p13 | PRH1 | Proline-Rich Protein HaeIII Subfamily 1 | Inhibition of crystal calcium phosphates |
| 12p13 | C1R | Complement Component 1, R Subcomponent | Complement activation |
| 12p13 | PTMS | Parathymosin | Inhibition of linkage of H1 to chromatin |
| 12q24 | DIABLO | Diablo, IAP-Binding Mitochondrial Protein | Apoptosis promotion |
| 12q24 | LRRC43 | Leucine Rich Repeat Containing 43 | --- |
| 13q13 | EXOSC8 | Exosome Component 8 | Transcriptionnal regulation |
| 14q11 | OR4Q3 | Olfactory receptor, family 4, subfam. Q, mb 3 | G-protein-coupled olfactory receptor |
| 14q11 | TRAV-26-1 | T Cell Receptor Alpha Variable 26-1 | TCR assembly |
| 14q11 | TRAJ10 | T Cell Receptor Alpha Joining 10 | TCR assembly |
| 14q11 | TRAJ13 | T Cell Receptor Alpha Joining 13 | TCR assembly |
| 14q12 | RP11-468E2.6 | --- | --- |
| 14q32 | IGHVIII-2-1 | Immunoglobulin Heavy Variable (III)-2-1 | BCR assembly |
| 14q32 | IGHVIII-16-1 | Immunoglobulin Heavy Variable (III)-16-1 | BCR assembly |
| 14q32 | DCAF11 | DDB1 And CUL4 Associated Factor 11 | Assembly of the DDB1–CUL4A ubiquitin ligase |
| 15q15 | CATSPER2 | Cation Channel, Sperm Associated 2 | Sperm motility and male fertility |
| 16p13 | NARFL | Nuclear prelamin A recognition factor-like | Modulation of hypoxia-inducible factor-1α activity |
| 16q22 | HAS3 | Hyaluronan Synthase 3 | Synthesis of glycosaminoglycan hyaluronan |
| 16q22 | CDH16 | Cadherin 16 | Ca2+-dependent glycoprotein |
| 16q22 | CLEC18B | C-Type Lectin Domain Family 18, Member B | --- |
| 17p11 | LGALS9C | Lectin, Galactoside-Binding, Soluble, 9C | Regulation of immune response |
| 19p13 | LRG1 | Leucine-rich alpha-2-glycoprotein 1 | Signal transduction, and cell adhesion |
| 19p13 | ZNF414 | Zinc Finger Protein 414 | Transcriptional regulation |
| 19p13 | AC008686.1 | --- | Uncharacterized protein |
| 22q13 | ADSL | Adenylosuccinate lyase | De novo purine biosynthesis |
The SAM method allowed identification of 148 genes with differentially distributed genomic alterations between ER and LR DLBCLs. The 56 protein coding genes are presented in this table.
Figure 2. Genes differentially altered between ER DLBCLs and LR DLBCLs.
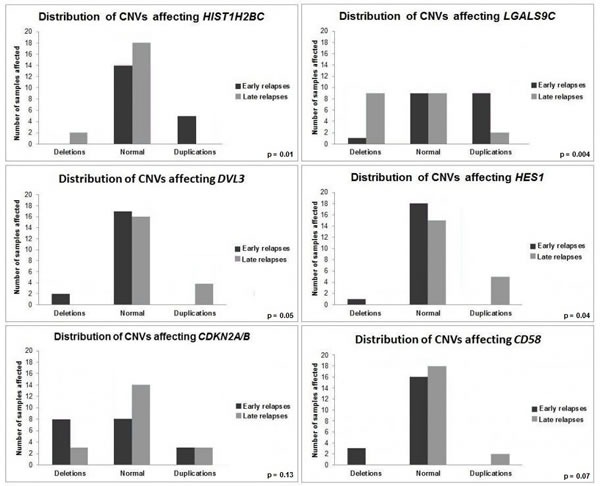
Comparison of CNV profiles between ER and LR DLBCLs has been performed at a genomic scale. HIST1H2BC, LGALS9C, DVL3, HES1, CDKN2A/B and CD58 are genes with structural aberrations differentially distributed between ER and LR DLBCLs. ER DLBCLs (n = 19) are represented in dark grey and LR DLBCLs (n = 20) in light grey.
ER DLBCLs were characterized by frequent duplications affecting HIST1H1T (6/19 samples, 31%), HIST1H2BC (5/19 samples, 26%), HIST1H2AK (5/19 samples, 26%), and PTMS (4/19 samples, 21%), implicated in histone-related regulation of transcription. We found a high number of duplications of AIRN (4/19 samples, 21%), a LncRNA interacting with the chromatin remodeling complex G9a. Deletions of HNRNPU (4/19 samples, 21%), implicated in pre-mRNA processing and alternative splicing, were also present. In addition, deletions of programmed-cell-death implicated genes DIABLO (5/19 samples, 26%) and CSNK2B/CK2B (3/19 samples, 15%), were identified. Duplications of LGALS9C (9/19 samples, 47%), deletions of BTNL2 (5/19 samples, 26%) and HLA-DRA (4/19 samples, 21%) were also revealed. A significant fraction of samples was affected by duplications of CCHCR1 (6/19 samples, 31%), a regulator of keratinocyte proliferation.
LR DLBCLs were characterized by a high number of duplications affecting DVL3 (4/20 samples, 20%), a regulator of cell proliferation. We found frequent loss of copy number for LGALS9C (9/20 samples, 45%), GALNT5 (4/20 samples, 21%), implicated in O-linked oligosaccharide biosynthesis, ADSL (3/20 samples, 15%), involved in de novo purine biosynthesis and ART5 (4/20 samples, 20%), an ADP-ribosyltransferase. Genes implicated in BCR (IgHV, IgKV) and T-Cell Receptor (TCR) assembly (TRAJ and TRAV) and transcriptional regulation, such as NFE2l2, MTERF4, HES1, EXOSC8, NARFL and ZNF414, were affected by either duplication or deletion in at least 20% of LR DLBCL samples.
Among the subset of 36 genes selected for their implication in lymphomagenesis, the 10 most frequently altered were CDKN2A/B (43%), IBTK (33%), PRDM1 (33%), FOXO1 (25%), BCL2 (25%), TP53 (20%), TNFAIP3 (20%), XPO1 (15%), B2M (15%) and CD58 (12%)(Figure 3). Cell cycle regulation and apoptosis were the most represented of affected pathways, with a high number of alterations encompassing CDKN2A/B, PRDM1, BCL2, TP53 and XPO1. Deletions of CDKN2A/B and IBTK were identified in 28% and 23% of DLBCL samples, respectively.
Figure 3. Genomic alterations of the 36 genes set involved in lymphomagenesis.
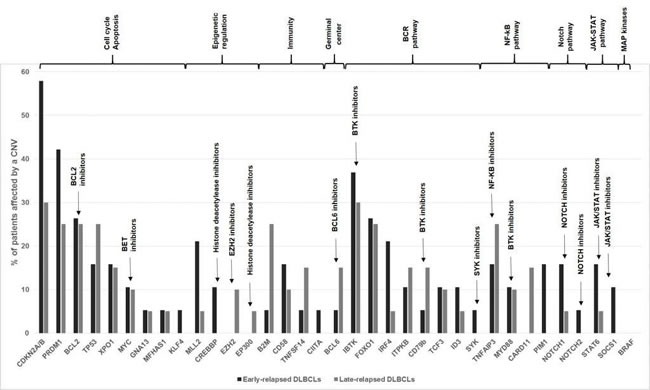
Comparative distribution of CNVs among ER and LR DLBCLs is shown. Genes are grouped according to key dysregulated pathways in DLBCLs. ER DLBCLs (n = 19) are colored in dark grey and LR DLBCLs (n = 20) in light grey. CNV: Copy Number Variation.
ER DLBCLs harbored frequent deletions of CDKN2A/B (8/19 ER samples, 42% versus 3/20 LR samples, 15%), PIM1 (3/19 ER samples, 15% versus 0/20 LR sample, 0%), MLL2 (4/19 ER samples, 21% versus 1/20 LR sample, 5%) and CREBBP (2/19 ER samples, 10% versus 0 LR sample). In LR DLBCLs, we identified CNVs affecting B2M (5/20 LR samples, 25% versus 1/19 ER samples, 5.26%) (p = 0.1), deletions of B2M being restricted to LR DLBCLs, and CD58 (deletions in 2/20 LR samples, 10% versus duplications in 3/19 ER samples, 15.7%) (p = 0.07).
When we compared CNVs distribution among ER and LR samples, we did not find any statistically significant difference. We also did not observe any correlation between CNVs distribution and the GC/ABC subtype (data not shown).
Evidence on recurrent events in relapsed DLBCLs
We performed a hierarchical clustering on 56 protein-coding genes and 25 LncRNA CNVs to identify events recurrently associated with ER and LR DLBCLs (Figure 4). The resulting heatmap shows recurrent CNVs in DLBCLs and defines three groups of patients.
Figure 4. Evidence for recurrent events in relapsed DLBCLs.
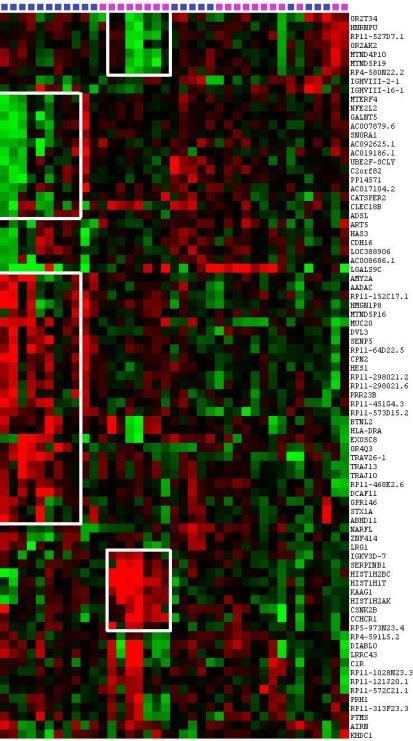
Hierarchical clustering of 56 protein-coding genes CNVs and 25 LncRNAs CNVs identifies events associated with ER and LR DLBCLs. The heatmap shows recurrent CNVs in DLBCL patients and gives evidence of genetic aberration occurrences in ER and LR DLBCLs. In ER DLBCLs, HIST1H2BC, HIST1H1T, HIS1H2AK, CSNK2B/CK2B, KAAG1, CCHCR1, and IgKV3D-7 are co-amplified in the same patients. Accordingly, in LR DLBCLs, AMY2A, AADAC, MUC20, DVL3, SENP5, CPN2, and HES1 are co-amplified in the same patients. This figure was obtained using Cluster 3.0 and Java Treeview 3.0.
The first group is homogeneous and comprises 7 ER samples presenting with correlated amplifications of HIST1H2BC, HIST1H1T, HIS1H2AK, CSNK2B/CK2B, KAAG1, CCHCR1, SERPINB1 and IgKV, as well as correlated deletions of HRNPU and four LncRNAs: RP11-527D7.1, MTND4P10, MTND5P19 and RP4-580N22.2.
The second group is composed of 9 LR DLBCLs showing correlated amplifications of AMY2A, AADAC, MUC20, DVL3, SENP5, CPN2, and HES1 as well as correlated deletions of MTERF4, NFE2L2, GALNT5, ADSL and three LncRNAs: AC007879.6, AC092625.1, and AC019186.1.
Remaining ER and LR samples constituted a third group without specific CNV profiles and no recurrent events.
Impact of CNVs on gene expression
We examined the impact of CNVs on average gene expression level (AGEL) (Figure 5).
Figure 5. Consequences of relevant copy number variations on gene expression.
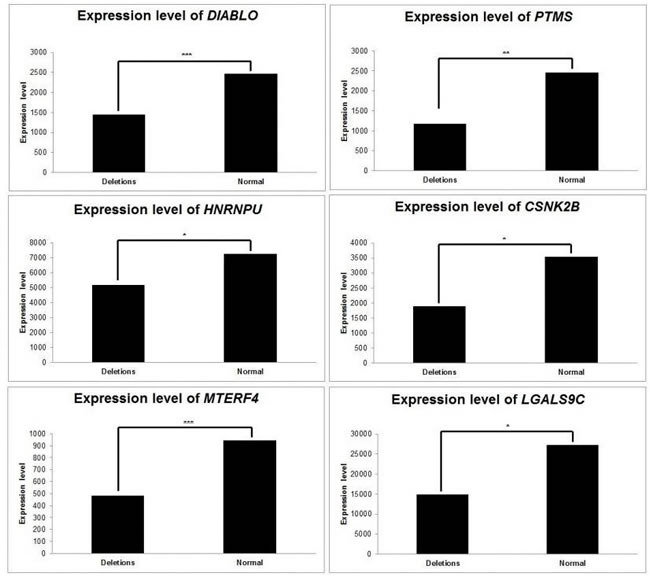
Average gene expression was computed, according to relevant CNVs. CNVs affecting DIABLO, PTMS, HNRNPU, CSNK2B, MTERF4 and LGLAS9C have subsequent consequences on the expression levels of those genes. *: p < 5 x10-2; **: p < 10-2; ***: p < 10-3.
In ER DLBCLs, deletions of DIABLO were related to a decrease in its AGEL, with a fold change of 0.58 between deleted and normal samples (p = 5x10-4, Student's t-Test). We observed the same correlation between deletions of the genes and decrease in their expression level for PTMS, HNRPU, and CSNK2B. Deletions of these three genes resulted in a decrease in their respective AGELs, with a fold change of 0.47, 0.71, and 0.53, respectively (p = 0.01, 0.02 and 0.03).
In LR DLBCLs, deletions of MTERF4 were related to a decrease in its AGEL, with a fold change of 0.51 between deleted and normal samples (p = 8x10-7). Accordingly, deletions of LGALS9C led to a decrease in AGEL compared to normal samples (fold change = 0.54; p = 0.05).
In the set of 36 genes, deletions of CDKN2A/B were related to a decrease in AGEL (fold change = 0.31; p = 0.07). Deletions of CD58 were also related to a decrease in AGEL (fold change=0.65; p = 0.08).
Discussion
Understanding the relationship between tumor biology and outcome is important for identifying molecular targets and may yield more effective therapies for DLBCLs. Almost all genomic studies reported to date were performed on de novo DLBCLs at diagnosis and little is known about structural aberrations of refractory/relapsed DLBCLs. Here we analyzed one of the few series of relapsed DLBCLs published to date [37, 38], composed of 39 samples from the Collaborative Trial in Relapsed Aggressive Lymphoma (CORAL), a unique and well-documented prospective study on relapsed DLBCLs [7]. Studying this thirty-nine-patient cohort allowed us to make strong statements since we devised and used for the first time a robust, comprehensive, unsupervised analysis method to analyze CNVs. This method makes use of SAM, a statistical technique originally designed for gene expression data analysis [36]. This permutation-based method allowed the discovery of a list a gene containing less than 5% of false positives despite multiple testing, independent from data distribution. Applied to oligonucleotide microarrays covering the entire genome, the method allowed us to delineate statistically validated CNVs with great precision and locate structural aberrations linked with ER and LR DLBCLs. Other published algorithms such as GISTIC [39], GEDI [40], or ComFocal [25] are designed to identify recurrent regions of deletions or duplications, which is not the purpose of this work. Here we aimed at identifying CNVs strongly associated with ER or LR, which is the main reason motivating the development of this original method.
ER and LR DLBCLs display a comparable CNV landscape, with a high degree of heterogeneity within each group. In relapsed DLBCLs, the most frequently altered genes were related to cell cycle regulation and apoptosis, regulation of transcription, immune response and NF-κB pathway (Figure 6). Among genes associated with lymphomagenesis, CNVs affecting CDKN2A/B locus (42% of samples) were the most frequent, with a majority of deletions (28% of samples). CDKN2A is a cycline-dependent kinase restraining cellular proliferation. These results are consistent with previous reports describing 30-35% of CDKN2A deletions in primary DLBCLs, predominantly in ABC subtype, with subsequent increase in cell proliferation and poorer outcome [22, 41, 42]. We also observed a high rate of IBTK aberrations (33% of samples), mainly deletions (23% of samples). IBTK (Inhibitor of Burton Tyrosine Kinase) is a BTK binding protein which inhibits BTK's kinase activity and disrupts BTK-mediated calcium mobilization leading to a decrease in BTK-induced activation of the anti-apoptotic NF-κB pathway [43]. A recent study reported a high rate of complete or partial responses and long-lasting remissions with ibrutinib treatment in relapsed/refractory DLBCLs, essentially in ABC subtype [44]. In this work, we did not observe any correlation between CNVs and the GC/ABC subtype. These results are in keeping with a recently published study showing (i) an absence of correlation between known driver mutations in lymphomagenesis and response to therapy, and (ii) an equivalent distribution of genomic events predicting survival across GC and ABC subtypes [37].
Figure 6. Key oncogenic pathways in relapsed DLBCLs.
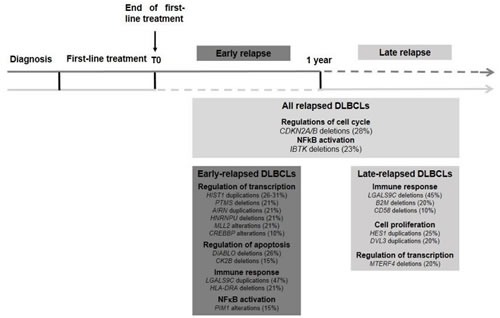
Structural abnormalities related with relapsed DLBCLs are shown, with distinction between ER and LR DLBCLs. ER DLBCLs are preferentially associated with dysregulation of gene transcription, cell cycle and apoptosis. LR DLBCLs are rather associated with dysregulation of immune response, cell proliferation and regulation of transcription.
When we compared ER and LR DLBCLs at the gene scale, aberrant genomic profiles were distinct on 56 protein-coding genes, with a subsequent effect on gene expression levels. In some cases, we were unable to demonstrate a statistically significant effect on gene expression. This can be explained by intronic localization of CNVs.
First, structural aberrations of cell cycle and apoptosis regulators were rather associated with ER DLBCLs, with deletions of DIABLO/Smac, a promoter of apoptosis which allows activation of caspases by binding to inhibitor of apoptosis proteins [45], and deletions of CSNK2B/CK2B, the regulatory subunit of protein kinase CK2, involved in a wide number of cellular processes, such as regulation of programmed cell death, cell division and proliferation, DNA damage repair, gene transcription and protein translation. CK2 increased protein expression and enzymatic activity has been described to positively regulate the PI3K pathway and STAT3 and NF-κB signaling in hematological malignancies [46]. We hypothesize that deletions of the CSNK2B/CK2B regulatory subunit with subsequent decrease in gene expression level result in increased enzymatic activity. LR DLBCLs were associated with duplications of DVL3, a member of the disheveled protein family which has been described to promote cell growth in ALK-positive anaplastic large cell lymphoma [47].
Second, CNVs encompassing genes related to immune response included duplications of LGALS9C identified in 47% of ER DLBCLs. LR DLBCLs were associated with partial deletions and duplications of TRAJ and TRAV, both implicated in TCR assembly. This suggests infiltration of the tumor by T-cell lymphocytes as part of the immune response. Deletions of B2M (20%) and CD58 (10%) were restricted to LR DLBCLs. Deletions and duplications of HLA related genes, were observed frequently in our cohort and are most likely related to HLA system polymorphism. Losses in IgHV and IgKV clusters are presumably a sign of BCR rearrangement and cannot be considered as recurrent lesion in tumor samples. We made the choice to consider all CNVs, including those that are described in databases of genomic variants. Limiting our analysis to acquired lesions may avoid constitutional abnormalities that are prone to foster lymphoma development.
Third, dysregulation of transcription through altering events in genes encoding regulators of chromatin compaction and DNA replication were associated with ER DLBCLs. Duplications of the linker histone H1 gene and/or deletions of its regulatory partner PTMS [48] were found in 21 to 31% of cases. This observation is consistent with a previous report, where mutations of the linker histone H1 gene have been described as a key evolutionary process governing transformation of follicular lymphoma into DLBCL [49]. LR DLBCLs were linked with deletions of MTERF4, a mitochondrial transcription regulator [50], in 20% of cases. LncRNA are nucleic acids often longer than 2 kb involved in the regulation of transcription through epigenetic modifications by recruiting chromatin remodeling complexes to various genomic loci [51]. AIRN (Air noncoding RNA) was of particular interest as it was deleted in 20% of ER DLBCLs. This LncRNA plays a role in epigenetic regulation of transcription by cooperating with the histone methyl transferase G9a [52]. Duplications of HES1, a downstream effector of the NOTCH pathway associated with Fanconi anemia [53] and blast crisis transformation in chronic myelogenous leukemia [54] were restricted to LR samples (25%).
Even though DLBCL is a heterogeneous disease, our method allowed the identification of structural aberrations that are statistically associated with ER and LR. Moreover, hierarchical clustering of CNVs identified three groups of patients. Two groups (the first one composed of ER samples and the second one of LR samples) were well separated in homogeneous clusters with common deletions and duplications. The remaining samples formed a mixed group of ER and LR samples with no recurrent CNV. This result gives evidence of a continuum between ER and LR DLBCLs. Identifying two groups of samples with correlated CNVs profiles demonstrates the statistical significance of our results, since these correlated groups could not be identified by chance.
Our original method unraveled genomic aberrations typical of the “LR signature”, which includes genomic aberrations encompassing genes related to immune response, cell proliferation and regulation of transcription. The “ER signature” includes events leading to disruption of cell cycle, apoptosis and gene transcription and provides a structural basis for selective growth advantage and chemotherapy resistance leading to a worse prognosis. This is of great importance since it may be a starting point for further studies and supports the rationale for targeted therapy. Even if this study needs to be confirmed on a larger cohort of patients, it provides new insights into the genetic complexity and dysregulated pathways in relapsed DLBCLs, suggesting the possibility of individualized targeted therapy for those patients with poor prognosis.
Materials and methods
Patient cohort
The phase III CORAL prospective study included 396 patients presenting with relapsed/refractory DLBCL after first-line treatment with R-CHOP (rituximab, cyclophosphamide, adriamycin, vincristine and prednisone). These patients were randomly assigned to receive a salvage chemotherapy regimen, either rituximab, ifosfamide, carboplatin and etoposide (R-ICE) or rituximab, dexamethasone, cytarabine, and cisplatin (R-DHAP). Patients who responded to the chemotherapy were submitted to high dose therapy and autologous stem cell transplant (ASCT). After ASCT, the trial compared rituximab treatment every 2 months for 1 year with observation alone. The initial results revealed no significant difference in outcome. Several factors did affect survival, including ER, the international prognostic index at relapse, and prior exposure to rituximab [7].
From these 396 patients with relapsed/refractory DLBCL, frozen lymph node biopsies were available from a homogeneous group of 39 patients. A part of tumors were sampled at diagnosis, before first-line treatment, and the others were sampled at relapse, before salvage chemotherapy. Thus, among the 19 tumor samples composing the ER group, 11 were sampled at diagnosis, and 8 at relapse. Among the 20 tumor samples from the LR group, 9 were sampled at diagnosis and 11 at relapse. Molecular characterization regarding GCB DLBCLs and ABC DLBCLs could be determined with GEP in 30 cases. Characteristics of the patients are listed in Table 2. Procedures used were in accordance with the Helsinki Declaration of 1975 (as revised in 1983).
Table 2. Characteristics of the cohort of 39 relapsed DLBCLs.
| N° | Sex | Age at diagnosis | IPI at diagnosis | First-line chemotherapy regimen | Delay of relapse (months) | ER/LR | Subtype | Rearrangements | Sample | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MYC | BCL2 | BCL6 | |||||||||
| 1 | Male | 62 | 3 | CHOP | 11 | ER | GC | No | 1 | 2 | Diagnosis |
| 2 | Female | 20 | 1 | CHOP | 6 | ER | ABC | No | 2 | 2 | Diagnosis |
| 3 | Female | 60 | 4 | CHOP | 6 | ER | ABC | No | 1 | 2 | Diagnosis |
| 4 | Female | 60 | 4 | ACVB | 8 | ER | NA | No | 2 | 2 | Diagnosis |
| 5 | Male | 59 | 0 | CHOP | 3 | ER | GC | No | 1 | 2 | Relapse |
| 6 | Male | 28 | 0 | ACVB | 9 | ER | NA | No | 2 | 2 | Relapse |
| 7 | Male | 43 | 1 | CHOP | 6 | ER | ABC | NA | NA | NA | Diagnosis |
| 8 | Male | 41 | 1 | CHOP | 11 | ER | NP | NA | NA | NA | Relapse |
| 9 | Female | 38 | 3 | CHOP | 5 | ER | ABC | No | 2 | 2 | Diagnosis |
| 10 | Male | 46 | 2 | CHOP | 0 | ER | GC | No | 1 | NA | Diagnosis |
| 11 | Male | 34 | 2 | CHOP | 2 | ER | GC | NA | NA | NA | Relapse |
| 12 | Male | 54 | 0 | ACVB | 9 | ER | GC | No | 1 | 2 | Relapse |
| 13 | Male | 57 | 1 | CHOP | 0 | ER | GC | Yes | 1 | 2 | Diagnosis |
| 14 | Male | 57 | 1 | CHOP | 0 | ER | GC | Yes | 1 | 2 | Relapse |
| 15 | Male | 57 | 2 | ACVB | 2 | ER | NP | No | 2 | NA | Diagnosis |
| 16 | Male | 32 | 2 | CHOP | 0 | ER | GC | No | 2 | 2 | Relapse |
| 17 | Female | 35 | 2 | ACVB | 7 | ER | ABC | NA | NA | NA | Diagnosis |
| 18 | Female | 35 | 2 | ACVB | 7 | ER | ABC | No | 2 | 2 | Relapse |
| 19 | Male | 33 | 0 | ACVB | 11 | ER | GC | No | 2 | 2 | Diagnosis |
| 20 | Male | 29 | 2 | ACVB | 82 | LR | GC | NA | NA | NA | Relapse |
| 21 | Male | 41 | 0 | CHOP | 148 | LR | NA | NA | NA | NA | Relapse |
| 22 | Male | 42 | 2 | ACVB | 29 | LR | NA | Yes | 2 | 2 | Diagnosis |
| 23 | Male | 41 | 1 | ACVB | 155 | LR | ABC | No | 2 | 2 | Relapse |
| 24 | Male | 59 | 0 | ACVB | 20 | LR | GC | No | 1 | 2 | Relapse |
| 25 | Male | 53 | 0 | ACVB | 50 | LR | GC | No | 2 | 2 | Diagnosis |
| 26 | Male | 53 | 0 | ACVB | 50 | LR | GC | No | 2 | 2 | Relapse |
| 27 | Female | 58 | 2 | ACVB | 60 | LR | ABC | NA | 1 | 1 | Relapse |
| 28 | Male | 46 | 0 | ACVB | 40 | LR | ABC | NA | NA | NA | Diagnosis |
| 29 | Male | 33 | 0 | ACVB | 109 | LR | ABC | No | 2 | 2 | Relapse |
| 30 | Male | 43 | 0 | ACVB | 103 | LR | ABC | No | 2 | 2 | Diagnosis |
| 31 | Female | 38 | 1 | CHOP | 62 | LR | NA | No | 2 | NA | Diagnosis |
| 32 | Male | 43 | 0 | CHOP | 158 | LR | GC | Yes | 1 | NA | Diagnosis |
| 33 | Male | 62 | 2 | CHOP | 21 | LR | NA | No | 1 | 2 | Diagnosis |
| 34 | Male | 56 | 0 | ACVB | 29 | LR | GC | No | 2 | 2 | Diagnosis |
| 35 | Male | 56 | 0 | ACVB | 29 | LR | NA | No | 2 | 2 | Relapse |
| 36 | Male | 45 | 0 | CHOP | 153 | LR | GC | No | 1 | 2 | Relapse |
| 37 | Male | 26 | 0 | CHOP | 116 | LR | GC | No | 2 | 2 | Relapse |
| 38 | Male | 58 | 2 | CHOP | 19 | LR | NA | NA | NA | NA | Diagnosis |
| 39 | Male | NA | NA | NA | NA | LR | NA | No | 2 | 1 | Relapse |
IPI: International prognostic index; CHOP: cyclophosphamide, adriamycin, vincristine and prednisone; ACVB: adriamycin, cyclophosphamide, vindesine, bleomycin; ER: Early Relapse; LR: Late Relapse; GC: Germinal Center B; ABC: Activated B-cell; NA: Non Available; NP: Non Predictable.
Copy number analysis
CNVs of the 39 samples were determined using the Affymetrix SNP 6.0 platform (Affymetrix, Santa Clara, CA, USA), which interrogates 1 800 000 copy number probes distributed evenly along the human genome (mean interval: 1.8 kilobases) [55]. The 1 800 000 probes were annotated with home-made tools querying the Ensembl 77th version with Human Genome HG37 MYSQL database available at ftp.ensembl.org/pub/release-77/mysql/. Total copy numbers were computed using the Bioconductor crlmm package [56]. A sliding window (length = 11 probes) representing the signal median value was used to smooth artifactual data.
This strategy allowed CNV discovery at high resolution and identification of genes frequently altered in relapsed DLBCLs. Duplications were defined by a copy number ≥2.5, deletions were defined by a copy number ≤1.5 and copy numbers between 1.5 and 2.5 were considered normal, according to an already published method [22]. We used Java Treeview 3.0 [57] to visualize the results at a genomic scale.
Identification of differential genomic alterations between ER and LR
We aimed at identifying genes affected by differentially distributed CNVs between ER and LR DLBCL patients, ie cases where deletions and duplications are associated preferentially with ER or LR. Given the large number of statistical tests, we used a permutation-based analysis to reduce false-positive results due to multiple testing. Our method was based on the widely used technique called Significance Analysis of Microarrays (SAM) [36]. SAM is a non-parametric test that avoids unjustified assumptions over gene distribution and gene independence. SAM was originally designed to determine whether changes in gene expression are experimentally significant. As claimed by the authors, SAM can be adapted for other experimental types of data. For our purpose, one needs to define a measure of the strength of the relationship between the CNV distribution (loss of copy number, normal copy number and gain of copy number) and ER or LR. Many statistics such as Pearson's chi-squared test, and other measures such as the Kullback-Leibler divergence, are only defined in case of non-null frequencies. Therefore we preferred to select the Hellinger distance H(P,Q) which quantifies the difference between probability distributions P = (p1,…, pk) and Q = (q1,…, qk). The Hellinger distance is defined as:
The lower bound of H(P,Q) = 0 corresponds to P = Q and its upper bound H(P,Q) = 1 implies that piqi = 0 " i Î [1,k]. In our method, P and Q represent the probability distributions over the ER and LR DLBCL copy numbers, expressed in three discrete states (deletion, normal, duplication).
Genes were considered positive when the FDR was inferior to 5%. For a FDR of 5%, our algorithm can be stated as: (i) compute the observed Hellinger distances for all the probes mapping to gene gi. The distance associated to gi is defined as the mean of the probe distances. (ii) form an observed vector Hi by ordering the observed gene distances. (iii) perform B = 20 random permutations over the patient labels and compute the corresponding expected null gene distances. Order the B distance vectors and compute the corresponding mean expected vector HEi. (iv) plot the observed vector against the expected null vector (Supplemental Figure 1). (v) set a threshold D = max(Hi - HEi) (vi) define the significant genes verifying Hi - HEi 3 D (vii) estimate the FDR. This value can be computed as the average of genes called significant from all the B permutations. (viii) if FDR > 0.05, decrease the threshold D and repeat steps 6 and 7.
Then, the aim of this method is not to provide an individual p-value for each gene of the genome but to identify of a list of genes which CNVs distribution is differential between ER and LR, with systematic correction of multiple testing and less than 5% of false positives.
SNP microarray data acquired on Affymetrix SNP 6.0 platform can be accessed online on Gene Expression Omnibus (GSE73791).
Selected set of genes implicated in lymphomagenesis
We also explored a subset of 36 genes selected from literature and already known to play a role in lymphomagenesis through the dysregulation of key cellular pathways [58]. CDKN2A, CDKN2B, PRDM1, TP53, BCL2, XPO1, MYC, GNA13, MFHAS1 and KLF4 were chosen for their implication in cell cycle and apoptosis; MLL2, CREBBP, EZH2 and EP300 for their role in epigenetic regulation; B2M, CD58, TNFSF14 and CIITA for their implication in dysregulation of immunity and escape to immune response; BCL6 for its role in germinal center reaction; IBTK, FOXO1, IRF4, ITPKB, CD79b, TCF3, ID3 and SYK for their implication in the BCR pathway; TNFAIP3, MYD88, CARD11 and PIM1, for their implication in constitutive activation of NF-κB pathway; NOTCH1 and NOTCH2 for their association with NOTCH pathway disruption; STAT6 and SOCS1 implicated in JAK-STAT pathway; BRAF for its role in MAP kinase pathway.
CNV impact on gene expression through integrative genomics
Gene expression profiles were available for 30 out of the 39 patients. These data were acquired on an Agilent Whole Human Genome microarray 4x44K platform (Agilent technologies, Santa Clara, CA, USA). Gene expression levels for each sample were integrated with CNVs. The impact of deletions and duplications of the selected genes on their own expression level was evaluated by comparing the average gene expression levels of deleted, duplicated and normal samples using Student's t-test.
SUPPLEMENTARY MATERIALS FIGURE AND TABLE
Acknowledgments
The authors would like to thank fondation Nelia et Amedeo Barletta, Paris, France.
We thank the LYSARC for providing the clinical data; Fabienne Morand, and the project leaders from the different countries; the patients and their families; all investigators and pathologists.
Footnotes
CONFLICTS OF INTEREST
Professor C. Gisselbrecht: Research Funding by Roche. The other authors declare no competing financial interests.
GRANT SUPPORT
The authors would like to thank fondation Nelia et Amedeo Barletta, Paris, France.
We thank the Regional Council of Lorraine for funding through the “Chercheurs d’Excellence” program
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget”
REFERENCES
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Coiffier B, Lepage E, Briere J, Herbrecht R, Tilly H, Bouabdallah R, Morel P, Van Den Neste E, Salles G, Gaulard P, Reyes F, Lederlin P, Gisselbrecht C. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med. 2002;346:235–42. doi: 10.1056/NEJMoa011795. [DOI] [PubMed] [Google Scholar]
- 3.Feugier P, Van Hoof A, Sebban C, Solal-Celigny P, Bouabdallah R, Fermé C, Christian B, Lepage E, Tilly H, Morschhauser F, Gaulard P, Salles G, Bosly A, et al. Long-term results of the R-CHOP study in the treatment of elderly patients with diffuse large B-cell lymphoma: a study by the Groupe d’Etude des Lymphomes de l’Adulte. J Clin Oncol. 2005;23:4117–26. doi: 10.1200/JCO.2005.09.131. [DOI] [PubMed] [Google Scholar]
- 4.Thieblemont C, Gisselbrecht C. Second-line treatment paradigms for diffuse large B-cell lymphomas. Curr Oncol Rep. 2009;11:386–93. doi: 10.1007/s11912-009-0052-0. [DOI] [PubMed] [Google Scholar]
- 5.Sehn LH, Gascoyne RD. Diffuse large B-cell lymphoma: optimizing outcome in the context of clinical and biologic heterogeneity. Blood. 2015;125:22–32. doi: 10.1182/blood-2014-05-577189. [DOI] [PubMed] [Google Scholar]
- 6.Guglielmi C, Gomez F, Philip T, Hagenbeek A, Martelli M, Sebban C, Milpied N, Bron D, Cahn JY, Somers R, Sonneveld P, Gisselbrecht C, Van Der Lelie H, et al. Time to relapse has prognostic value in patients with aggressive lymphoma enrolled onto the Parma trial. J Clin Oncol. 1998;16:3264–9. doi: 10.1200/JCO.1998.16.10.3264. [DOI] [PubMed] [Google Scholar]
- 7.Gisselbrecht C, Glass B, Mounier N, D Singh Gill, Linch DC, Trneny M, Bosly A, Ketterer N, Shpilberg O, Haqberg H, Ma D, Brière J, Moskowitz CH, Schmitz N. Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J Clin Oncol. 2010;28:4184–90. doi: 10.1200/JCO.2010.28.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, Powell JI, Yang L, Marti GE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–11. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 9.Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, Gascoyne RD, Muller-Hermelink HK, Smeland EB, Giltnane JM, Hurt EM, Zhao H, Averett L, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002;346:1937–47. doi: 10.1056/NEJMoa012914. [DOI] [PubMed] [Google Scholar]
- 10.Thieblemont C, Briere J, Mounier N, Voelker HU, Cuccuini W, Hirchaud E, Rosenwald A, Jack A, Sundstrom C, Coqliatti S, Trougouboff P, Boudova L, Ysebaert L, et al. The germinal center/activated B-cell subclassification has a prognostic impact for response to salvage therapy in relapsed/refractory diffuse large B-cell lymphoma: a bio-CORAL study. J Clin Oncol. 2011;29:4079–87. doi: 10.1200/JCO.2011.35.4423. [DOI] [PubMed] [Google Scholar]
- 11.Molina TJ, Canioni D, Copie-Bergman C, Recher C, Brière J, Haioun C, Berger F, Fermé C, Copin MC, Casasnovas O, Thieblemont C, Petrella T, Leroy K, et al. Young patients with non-germinal center B-cell-like diffuse large B-cell lymphoma benefit from intensified chemotherapy with ACVBP plus rituximab compared with CHOP plus rituximab: analysis of data from the Groupe d’Etudes des Lymphomes de l’Adulte/lymphoma study association phase III trial LNH 03-2B. J Clin Oncol. 2014;32:3996–4003. doi: 10.1200/JCO.2013.54.9493. [DOI] [PubMed] [Google Scholar]
- 12.Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, Kohlhammer H, Lamy L, Zhao H, Yang Y, Xu W, Shaffer AL, Wright G, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463:88–92. doi: 10.1038/nature08638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, Kohlhammer H, Xu W, Yang Y, Zhao H, Shaffer AL, Romesser P, Wright G, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470:115–9. doi: 10.1038/nature09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright GW, Dave SS, Zhao H, Xu W, Rosenwald A, Ott G, Muller-Hermelink HK, Gascoyne RD, et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science. 2008;319:1676–9. doi: 10.1126/science.1153629. [DOI] [PubMed] [Google Scholar]
- 15.Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, Bertoni F, Ponzoni M, Scandurra M, Califano A, Bhagat G, Chadburn A, Dalla-Favera R, et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature. 2009;459:717–21. doi: 10.1038/nature07968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kato M, Sanada M, Kato I, Sato Y, Takita J, Takeuchi K, Niwa A, Chen Y, Nakazaki K, Nomoto J, Asakura Y, Muto S, Tamura A, et al. Frequent inactivation of A20 in B-cell lymphomas. Nature. 2009;459:712–6. doi: 10.1038/nature07969. [DOI] [PubMed] [Google Scholar]
- 17.Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, Johnson NA, Severson TM, Chiu R, Field M, Jackman S, Krzywinski M, Scott DW, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, Kasper LH, Lerach S, Tang H, Ma J, Rossi D, Chadburn A, Murty VV, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471:189–95. doi: 10.1038/nature09730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, Yap D, Humphries RK, Griffith OL, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42:181–5. doi: 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roschewski M, Staudt LM, Wilson WH. Diffuse large B-cell lymphoma-treatment approaches in the molecular era. Nat Rev Clin Oncol. 2014;11:12–23. doi: 10.1038/nrclinonc.2013.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang J, Grubir V, Love CL, Banerjee A, Richards KL, Mieczkowski PA, Dunphy C, Choi W, Au WY, Srivastava G, Lugar PL, Rizzieri DA, Lagoo AS, et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc Natl Acad Sci USA. 2013;110:1398–1403. doi: 10.1073/pnas.1205299110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lenz G, Wright GW, Emre NC, Kohlhammer H, Dave SS, Davis RE, Carty S, Lam LT, Shaffer AL, Xiao W, Powell J, Rosenwald A, Ott G, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci USA. 2008;105:13520–5. doi: 10.1073/pnas.0804295105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schuetz JM, Johnson NA, Morin RD, Scott DW, Tan K, Ben-Nierah S, Boyle M, Slack GW, Marra MA, Connors JM, Brooks-Wilson AR, Gascoyne R. BCL2 mutations in diffuse large B-cell lymphoma. Leukemia. 2012;26:1383–90. doi: 10.1038/leu.2011.378. [DOI] [PubMed] [Google Scholar]
- 24.Iqbal J, Neppalli VT, Wright G, Dave BJ, Horsman DE, Rosenwald A, Lynch J, Hans CP, Weisenburger DD, Greiner TC, Gascoyne R, Campo E, Ott G, et al. BCL2 expression is a prognostic marker for the activated B-cell-like type of diffuse large B-cell lymphoma. J Clin Oncol. 2006;24:961–8. doi: 10.1200/JCO.2005.03.4264. [DOI] [PubMed] [Google Scholar]
- 25.Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, Wells VA, Grunn A, Messina M, Elliot O, Chan J, Bhagat G, Chadburn A, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet. 2011;43:830–7. doi: 10.1038/ng.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu-Monette ZY, Wu L, Visco C, Tai YC, Tzankov A, Liu WM, Montes-Moreno S, Dybkaer K, Chiu A, Orazi A, Zu Y, Bhagat G, Richards KL, et al. Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with R-CHOP: report from an International DLBCL Rituximab-CHOP Consortium Program Study. Blood. 2012;120:3986–96. doi: 10.1182/blood-2012-05-433334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Savage KJ, Johnson NA, Ben-Neriah S, Connors JM, Sehn LH, Farinha P, Horsman DE, Gascoyne RD. MYC gene rearrangements are associated with a poor prognosis in diffuse large B-cell lymphoma patients treated with R-CHOP chemotherapy. Blood. 2009(114):3533–7. doi: 10.1182/blood-2009-05-220095. [DOI] [PubMed] [Google Scholar]
- 28.Cuccuini W, Briere J, Mounier N, Voelker HU, Rosenwald A, Sundstrom C, Cogliatti S, Hirchaud E, Ysebaert L, Bron D, Soulier J, Gaulard F, Houlgatte R, et al. MYC+ diffuse large B-cell lymphoma is not salvaged by classical R-ICE or R-DHAP followed by BEAM plus autologous stem cell transplantation. Blood. 2012;119:4619–24. doi: 10.1182/blood-2012-01-406033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lohr J, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, Cruz-Gordillo P, Knoechel B, Asmann YW, Slager SL, Novak AJ, Dogan A, Ansell SM, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci USA. 2012;109:3879–84. doi: 10.1073/pnas.1121343109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rossi D, Trifonov V, Fangazio M, Bruscaggin A, Rasi S, Spina V, Monti S, Vaisitti T, Arruga F, Famà R, Ciardullo C, Greco M, Cresta S, et al. The coding genome of splenic marginal zone lymphoma: activation of NOTCH2 and other pathways regulating marginal zone development. J Exp Med. 2012;209:1537–51. doi: 10.1084/jem.20120904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee SY, Kumano K, Nakazaki K, Sanada M, Matsumoto A, Yamamoto G, Nannya Y, Suzuki R, Ota S, Ota Y, Izutsu K, Sakata-Yanagimoto M, Hangaishi A, et al. Gain-of-function mutations and copy number increases of Notch2 in diffuse large B-cell lymphoma. Cancer Sci. 2009;100:920–6. doi: 10.1111/j.1349-7006.2009.01130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pasqualucci L. The genetic basis of diffuse large B-cell lymphoma. Curr Opin Hematol. 2013;20:336–44. doi: 10.1097/MOH.0b013e3283623d7f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iqbal J, Greiner TC, Patel K, Dave BJ, Smith L, Ji J, Wright G, Sanger WG, Pickering DL, Jain S, Horsman DE, K ShenY Fu, et al. Distinctive patterns of BCL6 molecular alterations and thier functionnal consequences in different subgroups of diffuse large B-cell lymphoma. Leukemia. 2007;21:2332–43. doi: 10.1038/sj.leu.2404856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Challa-Malladi M, Lieu YK, Califano O, Holmes AB, Bhagat G, Murty VV, Dominguez-Sola D, Pasqualucci L, Dalla-Favera R. Combined genetic inactivation of 2-Microglobulin and CD58 reveals frequent escape from immune recognition in diffuse large B cell lymphoma. Cancer Cell. 2011;20:728–40. doi: 10.1016/j.ccr.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steidl C, Shah SP, Woolcock BW, Rui L, Kawahara M, Farinha P, Jonhson NA, Zhao Y, Telenius A, Neriah SB, PcPherson A, Meissner B, Okoye UC, et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature. 2011;471:377–81. doi: 10.1038/nature09754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarray applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Novak AJ, Asmann YW, Maurer MJ, Wang C, Slager SL, Hodge LS, Manske M, Price-Troska T, Yang ZZ, Zimmermann MT, Nowakowski GS, Ansell SM, Witzig TE, et al. Whole-exome analysis reveals novel somatic genomic alterations associated with outcome in immunochemotherapy-treated diffuse large B-cell lymphoma. Blood Cancer J. 2015;28(5):e346. doi: 10.1038/bcj.2015.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xian RR, Crane GM, Haley LM, Gocke CD, Lin MT, Pallavajjalla A, Borowitz MJ, Swinnen LJ, Duffield AS. Late-Relapse Diffuse Large B-Cell Lymphoma Frequently Represents Recurrence of the Original Disease, and Demonstrates Evidence of Superimposed Clonal Heterogeneity and Clonal Evolution. Blood. 2014;124:2955. [Google Scholar]
- 39.Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fujita A, Sato JR, Ferreira CE, Sogayar MC. GEDI: a user-friendly toolbox for analysis of large-scale gene expression data. BMC Bioinformatics. 2007;8:457. doi: 10.1186/1471-2105-8-457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Monti S, Chapuy B, Takeyama K, Rodig SJ, Hao Y, Yeda KT, et al. Integrative analysis reveals an outcome-associated and targetable pattern of p53 and cell cycle deregulation in diffuse large B-cell lymphoma. Cancer Cell. 2012;22:359–372. doi: 10.1016/j.ccr.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jardin F, Jais JP, Molina TJ, Parmentier F, Picquenot JM, Ruminy P, Tilly H, Bastard C, Salles GA, Feugier P, Thieblemont C, Gisselbrecht C, De Reynies A, et al. Diffuse large B-cell lymphomas with CDKN2A deletion have a distinct gene expression signature and a poor prognosis under R-CHOP treatment: a GELA study. Blood. 2010;116:1092–104. doi: 10.1182/blood-2009-10-247122. [DOI] [PubMed] [Google Scholar]
- 43.Janda E, Palmieri C, Pisano A, Pontoriero M, Iaccino E, Falcone C, Fiume G, Gaspari M, Nevolo M, Di Salle E, Rossi A, De Laurentiis A, Greco A, et al. Btk regulation in human and mouse B cells via protein kinase C phosphorylation of IBtk. Blood. 2011;117:6520–31. doi: 10.1182/blood-2010-09-308080. [DOI] [PubMed] [Google Scholar]
- 44.Wilson WH, Young RM, Schmitz R, Yang Y, Pittaluga S, Wright G, Lih CJ, Williams PM, Shaffer AL, Gerecitano J, De Vos S, Goy A, Kenkre VP, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med. 2015;21:922–6. doi: 10.1038/nm.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 46.Piazza F, Manni S, Ruzzene M, Pinna LA, Gurrieri C, Semenzato G. Protein kinase CK2 in hematologic malignancies: reliance on a pivotal cell survival regulator by oncogenic signaling pathways. Leukemia. 2012;26:1174–1179. doi: 10.1038/leu.2011.385. [DOI] [PubMed] [Google Scholar]
- 47.Hegazy SA, Alshareef A, Gelebart P, Anand M, Armanious H, Ingham RJ, Lai R. Disheveled proteins promote cell growth and tumorigenicity in ALK-positive anaplastic large cell lymphoma. Cell Signal. 2013;25:295–307. doi: 10.1016/j.cellsig.2012.09.027. [DOI] [PubMed] [Google Scholar]
- 48.Martic G, Karetsou Z, Kefala K, Politou AS, Clapier CR, Straub T, Papamarcaki T. Parathymosin affects the binding of linker histone H1 to nucleosomes and remodels chromatin structure. J Biol Chem. 2005;280:16143–50. doi: 10.1074/jbc.M410175200. [DOI] [PubMed] [Google Scholar]
- 49.Okosun J, Bödör C, Wang J, Araf S, Yang CY, Pan C, Boller S, Cittaro D, Bozek M, Iqbal S, Matthews J, Wrench D, Marzec J, et al. Integrated genomics identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat Genet. 2014;46:176–81. doi: 10.1038/ng.2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cámara Y, Asin-Cayuela J, Park CB, Metodiev MD, Shi Y, Ruzzenente B, Kukat C, Habermann B, Wibom R, Hultenby K, Franz T, Erdjument-Bromage H, Tempst P, et al. MTERF4 regulates translation by targeting the methyltransferase NSUN4 to the mammalian mitochondrial ribosome. Cell Metab. 2011;13:527–39. doi: 10.1016/j.cmet.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 51.Saxena A, Carninci P. Long non-coding RNA modifies chromatin: epigenetic silencing by long non-coding RNAs. Bioessays. 2011;33:830–9. doi: 10.1002/bies.201100084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nagano T, Mitchell JA, Sanz LA, Pauler FM, Ferguson-Smith AC, Feil R, Fraser P. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science. 2008;322:1717–20. doi: 10.1126/science.1163802. [DOI] [PubMed] [Google Scholar]
- 53.Tremblay CS, Huang FF, Habi O, Huard CC, Godin C, Lévesque G, Carreau M. HES1 is a novel interactor of the Fanconi anemia core complex. Blood. 2008;112:2062–70. doi: 10.1182/blood-2008-04-152710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakahara F, Sakata-Yanagimoto M, Komeno Y, Kato N, Uchida T, Haragushi K, Kumano K, Harada Y, Harada H, Kitaura J, Ogawa S, Kurokawa M, Kitamura T, et al. Hes1 immortalizes committed progenitors and plays a role in blast crisis transition in chronic myelogenous leukemia. Blood. 2010;115:2872–81. doi: 10.1182/blood-2009-05-222836. [DOI] [PubMed] [Google Scholar]
- 55.McCarroll SA, Kuruvilla FG, Korn JM, Cawley S, Nemesh J, Wysoker A, Shapero MH, De Bakker PI, Maller JB, Kirby A, Elliott AL, Parkin M, Hubbel E, et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet. 2008;40:1166–74. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- 56.Scharpf RB, Irizarry RA, Ritchie ME, Carvalho B, Ruczinski I. Using the R Package crlmm for Genotyping and Copy Number Estimation. J Stat Softw. 2011;40:1–32. [PMC free article] [PubMed] [Google Scholar]
- 57.Saldanha AJ. Java Treeview-extensible visualization of microarray data. Bioinformatics. 2004;20:3246–8. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
- 58.Jardin F. Next generation sequencing and the management of diffuse large B-cell lymphoma: from whole exome analysis to targeted therapy. Discov Med. 2014;18:51–65. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.