

Abstract
We have identified a novel structure-specific nuclease in highly fractionated extracts of the thermophilic archaeon Methanothermobacter thermautotrophicus (Mth). The 71 kDa protein product of open reading frame mth1090 is a nuclease with ATPase activity, which we call Nar71 (Nuclease-ATPase in Repair, 71 kDa). The nar71 gene is located in a gene neighbourhood proposed by genomics to encode a novel DNA repair system conserved in thermophiles. The biochemical characterization of Nar71 presented here is the first analysis from within this neighbourhood, and it supports the insight from genomics. Nuclease activity of Nar71 is specific for 3′ flaps and flayed duplexes, targeting single-stranded DNA (ssDNA) regions. This activity requires Mg2+ or Mn2+ and is greatly reduced in ATP. In ATP, Nar71 displaces ssDNA, also with high specificity for 3′ flap and flayed duplex DNA. Strand displacement is weak compared with nuclease activity, but in ATPγS it is abolished, suggesting that Nar71 couples ATP hydrolysis to DNA strand separation. ATPase assays confirmed that Nar71 is stimulated by ssDNA, though not double-stranded DNA. Mutation of Lys-117 in Nar71 abolished ATPase and nuclease activity, and we describe a separation-of-function mutant (K68A) that has lost ATPase activity but retains nuclease activity. A model of possible Nar71 function in DNA repair is presented.
INTRODUCTION
Thermophilic organisms are adapted to overcome the denaturing effects of high temperatures on biological macromolecules. Thermostable enzymes have been characterized from many metabolic pathways in hyperthermophilic bacteria and archaea, [for example see (1)]. Understanding the structural basis of thermophily in these proteins has progressed by identifying structural characteristics that stabilize protein folds, including enlarged hydrophobic cores and extensive networks of hydrogen bonds and salt bridges (2,3). Archaeal lipids are also structurally modified against thermal stress, most radically as cell membranes arranged in monolayers of ether-linked phospholipids (4).
In contrast, the chemical composition and structure of DNA molecules is not modified as an adaptation to life at high temperature. There is no correlation between thermophily and high G-C content in genomes (5). DNA molecules are highly susceptible to chemical damage through cytosine deamination, depurination and strand breaks. Rates of these reactions are elevated at high temperatures and are a direct cause of genome instability (6). Thermophiles must therefore be adept at dealing with genome damage, tolerating elevated mutation rates, packaging DNA to protect against damage and efficiently repairing DNA lesions prior to replication. The molecular basis of how thermophiles perform these processes is unclear. In archaea, what is known about responses to genome instability indicates differences between the two major sub-divisions of archaea: crenarchaea and euryarchaea. For example, the hyperthermophilic euryarchaeon Pyrococcus shows extreme resistance to DNA damage by γ-irradiation compared with Escherichia coli (7,8), but the crenarchaeaon Sulfolobus acidocaldarius has a hypermutation phenotype after γ-irradiation or exposure to a range of DNA cross-linking agents (9). However, S.acidocaldarius grown in its natural conditions (75°C, pH 3.5) has a very low rate of base pair substitutions, suggesting specific mechanisms of DNA repair at least in this organism (10).
Packaging proteins may also protect DNA against chemical instability, and they vary between crenarchaea and euryarchaea. Histones lacking N- and C-terminal tails package double-strand DNA (dsDNA) in euryarchaea and can protect against DNA damage (11,12). Genome packaging by highly abundant dsDNA-binding proteins Alba and Sac7a is found in other archaeal species, most notably those of crenarchaea (13,14). In common with all forms of life, archaea have evolved proteins that bind single-stranded DNA (ssDNA) arising during genome disruption. In crenarchaea, single-strand binding protein (SSB) has unique characteristics compared with bacterial SSB and eukaryotic RPA (15). The topology of genomic DNA may also counter DNA damage through supercoiling (16), or formation of DNA toroids (17).
Despite apparent species differences in how thermophiles attenuate genome instability, they share some commonly known potential DNA repair mechanisms. Although known proteins of non-homologous end joining are largely absent from bacterial and archaeal thermophiles, RecA or RadA strand exchange enzymes are found (18–20), as are Holliday junction resolvases (RuvC, RusA or Hjc) (21–23). This suggests that homologous recombination may be important for DNA repair in thermophilic archaea and bacteria, as it is in all other organisms. Genetics has shown that homologous recombination is active in archaea (24), but many enzymes remain to be unearthed in this system (25). Our knowledge of homologous recombination in these organisms lags behind the information on base excision repair and nucleotide excision repair (26,27).
Two unique features of thermophiles, both bacterial and archaeal, are the presence of reverse gyrase and the predicted existence of a novel DNA repair system. Reverse gyrase introduces negative supercoiling into DNA molecules and is found only in hyperthermophiles (28), and has been characterized biochemically and structurally (29). Far less well characterized is the identification through genomics of a putative DNA repair system specific for thermophiles (30). This system comprises 10–20 genes, including proposed RecB-like nucleases, a DEAD-box helicase, a DNA polymerase and several repair-associated mysterious proteins. We identified members of this putative system during screening of purified cell extracts of the euryarchaeon Methanothermobacter for helicase and nuclease activities. In this paper, we describe a novel nuclease-ATPase activity from the product of open reading frame (ORF) 1090, which is located in this proposed DNA repair gene neighbourhood.
MATERIALS AND METHODS
M.thermautotrophicus biomass
M.thermautotrophicus (DSM 1053) cultures were grown in mineral salt media at 58°C sparged with a H2/CO2 (5:1) gas mixture at a rate of 240 ml/min under strictly anaerobic conditions in a 2.5 l fermenter batch culture (Applikon). Exponentially growing cells were harvested aerobically into 250 ml centrifuge bottles and rapidly cooled in an ice bath. The cells were pelleted by centrifugation at 7500 g for 10 min at 4°C, the supernatant was discarded and the pellets were frozen at −80°C.
The fractionation scheme outlined below was used to follow unwinding of Holliday junction DNA from Mth biomass. As described in the results, neither recombinant Nar71 (Nuclease-ATPase in Repair, 71 kDa) nor Mth1088 is catalytically active on Holliday junctions, therefore details of assays used to dictate how fractionation proceeded will be described elsewhere. Briefly, 20 g of Mth biomass was thawed and mixed in 10 ml buffer C (20 mM Tris pH 8.0, 1 mM EDTA, 10% glycerol, 1 mM DTT, 100 mM KCl and 0.1 mM PMSF) for re-freezing with liquid nitrogen in a pestle and mortar. Re-frozen mass was pulverized by hand into a fine powder and allowed to thaw at room temperature. The thawed mixture was sonicated and clarified (20 000 r.p.m., 30 min) and the resulting supernatant treated with polyethylenimine. Soluble proteins were recovered by centrifugation (20 000 r.p.m., 30 min) and loaded in buffer C onto tandemly arranged 5 ml HiTrap SP and Q-sepharose columns. Pooled fractions were desalted into buffer C by a PD-10 column and then loaded onto a 1 ml HiTrap heparin column, collecting 0.3 ml fractions. Pooled fractions were treated for SDS–PAGE by adding an equal volume of 50% (w/v) trichloroacetic acid on ice for 15 min. Proteins were recovered by centrifugation (13 000 r.p.m. for 5 min at 4°C) and then washed with ice-cold acetone and centrifuged again. Acetone was removed and protein pellets air-dried before resuspending in a minimal volume of buffer for SDS–PAGE.
Proteins
Nar71 and Mth1088 were identified from SDS–PAGE analysis of mixtures of proteins from Methanothermobacter biomass. Single polypeptides running between 62 and 83 kDa (Nar71) or close to the 50 kDa marker (Mth1088) were excised from the gel after Coomassie staining, and digested with trypsin. Quadrupole time-of-flight (Q-TOF) and database searching identified the products of Methanothermobacter Orf1090, which we have called Nar71 and of Mth1088. Genomic DNA used as PCR template was obtained from Mth biomass as follows: 5 g of frozen biomass was diluted with 20 ml lysis buffer (10 mM Tris–HCl pH 7.4, 10 mM EDTA, 0.25 M sucrose) and fresh proteinase K was added to 20 μg/ml followed by SDS (to 1% w/v final). This mixture was incubated at 55°C for 30 min, and NaCl added to 0.5 M. The mixture was placed on ice for 2 h before clarifying by centrifugation (20 000 r.p.m., 15 min) and the supernatant was mixed with an equal volume of isopropyl alcohol. Precipitated nucleic acid was recovered immediately by centrifugation (20 000 r.p.m., 15 min) and the pellet dissolved in 200 μl TE. DNA was then purified by phenol–chloroform treatment and ethanol precipitation. RNA was removed by treatment of the resuspended precipitate with RNase A.
Pure Mth genomic DNA was used as a template in PCR reactions to clone nar71 into pT7-7 via NdeI and SmaI sites, giving pEB367. The following primers were used: 5′-GGAATTCCATATGATCTACGAATTCAGACG-3′ and 5′-TCCCCCGGGTTATCTGCCTCCTAAAAATCC-3′. Gene mth1088 was cloned similarly using NdeI and EcoRI sites into pT7-7 giving pEB376 from the following primers: 5′-GGAATTCCATATGAAAATGTCAAGGTAC-3′ and 5′-GGAATTCCCTACTAATCCCCTGAACCTTTCC-3′. Restriction sites are underlined. Full sequencing of Nar71 and Mth1088 in pEB365 and pEB376, respectively, revealed no discrepancies with database gene sequences (http://www.biosci.ohio-state.edu/~genomes/mthermo/).
Nar71 was produced in CodonPlus E.coli BL21 CodonPlus (DE3) (Stratagene) in Luria–Bertani broth supplemented with 0.5 g/l NaCl and ampicillin (50 μg/ml) and chloramphenicol (25 μg/ml). The cells were grown at 37°C to OD650 of 0.5 and then induced with isopropyl-β-d-thiogalactopyranoside (1 mM) at 20°C for 18 h. Harvested cells were resuspended in buffer C (20 mM Tris pH 8.0, 1 mM EDTA, 10% glycerol, 1 mM DTT, 100 mM KCl and 0.1 mM PMSF) and flash frozen in liquid nitrogen prior to thawing and sonication at 4°C. Purification of Nar71 was by fast performance liquid chromatography (FPLC) at 4°C and was followed by SDS–PAGE.
Soluble proteins were isolated by centrifugation (20 000 r.p.m., 30 min) and passed through a HiPrep heparin column (Amersham-Pharmacia), Nar71 eluting between 600 and 770 mM KCl. Fractions containing Nar71 were pooled and diluted 2-fold with buffer C for loading onto a 20 ml cellulose phosphate (Sigma-Aldrich) column. Nar71 in fractions eluting between 700 and 900 mM KCl was pooled and dialysed into buffer C, and loaded onto a monoQ column (Amersham-Pharmacia) from which Nar71 eluted at 300–400 mM KCl. Pooled Nar71 was dialysed for long-term storage into buffer C containing 30% glycerol and 200 mM KCl. The identity of Nar71 was verified by Q-TOF mass spectrometry.
Nar71 with an N-terminal histidine tag was prepared after sub-cloning nar71 from pEB367 into pET14b via NdeI and ClaI sites giving pEB389. Tagged Nar71 was over-expressed as described above, but harvested cells were resuspended in binding buffer (5 mM imidazole, 500 mM NaCl, 20 mM Tris–HCl pH 7.9). Freeze-thawed cells were loaded onto a Ni2+-charged 5 ml HiTrap chelating column by FPLC, and eluted with a linear gradient of increasing imidazole. His-tagged Nar71 eluted at 1 M imidazole, and these fractions were pooled and dialysed into buffer C lacking DTT, which was added after extensive dialysis. His-tagged Nar71 was further purified through a 5 ml HiTrap heparin column, eluting like untagged Nar71, before storage in the same way as untagged protein.
Mth1088 protein was produced in the same way as Nar71, but biomass was resuspended in buffer C containing 20% (w/v) ammonium sulphate. Cells were lysed by sonication, clarified (20 000 r.p.m., 30 min) and passed into a 5 ml HiTrap phenyl sepharose column, through which Mth1088 protein flowed through without binding. These fractions were dialysed into buffer C and passed through a HiPrep heparin column from which Mth1088 eluted at 630–790 mM KCl. These fractions were pooled and dialysed against buffer C before loading onto tandem 1 ml HiPrep SP-Q sepharose columns, Mth 1088 eluting at between 500 and 740 mM KCl. Mth1088 was then dialysed for storage as for Nar71.
Variant Nar71 proteins
Point mutations were introduced into Nar71 using PCR catalysed by Pfx polymerase (Invitrogen) on pEB367 template. Complementary mutagenic primers were used to introduce point mutations into target codons. After PCR, reactions were treated with DpnI to digest template DNA and the remaining nascent DNA was transformed into E.coli DH5α. Transformants were used to extract plasmid DNA and the desired mutations were verified by sequencing. Variant Nar71 proteins were produced as described for wild-type protein. K68A and K117A were recovered as soluble proteins on cell lysis though most of K117A was present in insoluble cell debris. There was no difference in the purification of soluble K117A compared with wild-type Nar71, and K68A purified similarly, except at the cellulose phosphate step, where it eluted at 1–1.2 M KCl. All protein concentrations are expressed in moles of monomer.
DNA substrates
DNA substrates were end-labelled with 32P using T4 polynucleotide kinase (New England Biolabs). Un-incorporated [γ-32P]ATP was separated from labelled DNA by batch gel filtration through Bio-Spin 6 columns (Bio-Rad). The labelled oligonucleotide was used to form DNA substrates by annealing with unlabelled homologous strands (95°C, 2 min, then 6–18 h cooling to 25°C). Fully annealed substrate was purified by electrophoresis through 10% TBE gels, followed by exposure of the gel to photographic film that was used as a template to excise the desired DNA molecule. DNA was eluted by diffusion for 18–24 h at 4°C in 200–300 μl of 10 mM Tris–HCl, pH 7.5, 0.2 mM EDTA, 50 mM NaCl.
Holliday junction, fork, 3′ flap and 5′ flap substrates in Table 1 correspond to J8, fork 1, fork 3 (here called 3′ flap-1) and fork 5 of Table 1 in (31).
Table 1. Relative activity of Nar71 on DNA substrates.

Mg2+ denotes conditions for detecting nuclease activity, and Mg2+:ATP for ATP-dependent strand displacement activity.
The following substrates were also used as indicated in results; 3′ flap-2: Strand 1: 5′-ATCGATAGTCTCTAGACAGCATGTCCTAGCAAGCCAGAATTCGGCAGCGT-3′. Strand 2: 5′-GGACATGCTGTCTAGAGACTATCGAT-3′. Strand 3: 5′-ACGCTGCCGAATTCTGGCTTGCTAGGACATCTAGAGCTTGACTTTGAC-3′. Flayed duplex: Strand 1; as Strand 1 in 3′ flap. Strand 2: GACGCTGCCGAATTCTGGCTTGCTATGTAACTCTTTGCCCACGTTGACCC-3′. The biotinylated flayed duplex (Figure 5D) was the same as above but with biotin attached to the 3′ OH of strand 1. Linear duplex: Strand 1; as strand 1 above. Strand 2: 5′-ACGCTGCCGAATTCTGGCTTGCTAGGACATGCTGTCTAGAGACTATCGAT-3′. 3′ Tail-duplex: Strand 1 as above. Strand 2: GGACATGCTGTCTAGAGACTATCGAT-3′. 5′ Tail-duplex: Strand 1 as above. Strand 2: ACGCTGCCGAATTCTGGCTTGCTAGG-3′.
Figure 5.

Catalytically inactive mutants of Nar71. (A) Neutral gel showing strand displacement activity of wild-type (lanes a–e), K117A (lanes f–j) and K68A (lanes k–o) Nar71 (0–250 nM) on 3′ flap-1 DNA in 10 mM Mg2+and 15 mM ATP. Labelled partial duplex generated by wtNar71 is indicated on the right of the first panel. Arrows next to the third panel indicate nuclease products of K68A Nar71. (B) Denaturing gel showing nuclease activities of wild-type, K117A and K68A Nar71 (100 nM) in 10 mM Mg2+ on 5 nM 3′ flap-2 DNA. The substrate is labelled (asterisk) only on the strand that forms the 3′ ssDNA region, indicated by a grey line. (C) ATPase activity of 100 nM wild-type, K117A and K68A Nar71 on [γ-32P]ATP, converting it to free phosphate. (D) Effect of 3′ OH biotin on Nar71 nuclease activity. The top panel (lanes a–e) shows cleavage of flayed duplex (5 nM) in 10 mM Mg2+ by 0–240 nM Nar71 next to a radiolabelled ssDNA marker (lane f). The bottom panel (lanes g–k) shows the same reactions but in which the substrate was modified with biotin at the 3′ ssDNA end (as indicated on the substrate to the right of the panel). Lane l contains radiolabelled ssDNA marker.
Marker DNA for mapping Nar71 cleavage sites on flayed duplex and 3′ flap-1 was generated by treating the appropriate 32P-radiolabelled oligonucleotide with pyridinium formate and piperidine in strand cleavage reactions as described previously (32).
Gel filtration
Pure Nar71 (500 μl of 1 mg/ml) was loaded onto a superose-12 column (Amersham-Pharmacia) in buffer C and its elution compared with known molecular weight standards (500 μl of 1.8 mg total protein; Bio-Rad) also in buffer C.
DNA-binding assays
Typical reactions (20 μl) contained 0–250 nM protein and 5 nM DNA at 37°C for 10 min. Reaction mixtures were loaded directly onto 4 or 7% acrylamide gels (5 mM sodium acetate, 10 mM Tris–HCl pH 7.5) containing either 2 mM EDTA, 2 mM MgCl2 or 2 mM MgCl2 and 2 mM ATP. Gels were run at 125 V for 2 h, dried and exposed to phosphorimaging screens.
Nuclease/strand displacement reactions
Standard nuclease reactions (20 μl) were in 10 mM Tris–HCl, pH 8.0, 100 μg/ml BSA, 6% glycerol, containing 10 mM MgCl2 unless stated. Reactions were carried out at 45°C for 20 min, and were stopped by adding 5 μl of stop solution (5 mg/ml proteinase K, 10% SDS, 200 mM EDTA). For neutral gels, ficoll–xylene cyanol gel loading dye was added and reactions were run for 1 h at 200 V on 10% acrylamide, TBE gels. For denaturing gels, DNA was precipitated by adding dH2O (75 μl), 4 M ammonium acetate (30 μl), 1 μl glycogen (100 mg/ml) and 300 μl of 100% ethanol. Reactions were placed at −20°C for 1 h and a DNA pellet recovered by centrifugation (13 000 r.p.m., 5 min) and washing in 70% ethanol. The pellet was resuspended in formamide loading dye and electrophoresis was for 1 h at 2000 V on a Sequi-Gen (Bio-Rad) 12% acrylamide gel containing 7.6 M urea buffered by TBE.
Standard ATP assays were essentially the same as nuclease assays, but magnesium containing reaction buffer was supplemented with 15 mM ATP or ATPγS prior to adding Nar71. Time course reactions were in 200 μl and were started by the addition of protein and stopped at the required time point by removing a 20 μl aliquot into stop solution.
Immunodepletion of Nar71
Standard strand displacement assays were used but Nar71 was pre-incubated with 1 μg of His-tag monoclonal antibody (Novagen) for 10 min at 37°C, prior to adding radiolabelled DNA. Reactions then proceeded at 37°C for 30 min.
ATPase assays
Malachite green assays were used to measure ATP hydrolysis through the detection of liberated free phosphate from ATP (33). Reactions contained 3.3 mM MgCl2 and 5 mM ATP and either zero, 50 nM or 100 nM Nar71 protein, and were at 45°C for 1 h. ssDNA (ØX174 virion) or dsDNA (ØX174 RFI), both from New England Biolabs, were added as required to 100 ng. Measuring absorbance at 660 nm and correcting against the zero protein blank quantified liberation of phosphate. Control reactions contained RecG at 100 nM.
Hydrolysis of [γ-32P]ATP to 32P was visualized from 20 μl strand displacement reactions that were modified to contain 1 mM MgCl2 and 2 mM ATP, supplemented with [γ-32P]ATP. Samples of each reaction were run on a 20% acrylamide gel for 30 min at 125 V.
Protein–protein interactions
These reactions were carried out at room temperature. Pure Nar71 with an N-terminal histidine tag (400 μg) was immobilized on iminodiacetic acid coated membranes that had been charged with Ni2+ within a mini-column, according to the manufacturer's instructions (VivaScience). Nar71 bound tightly to the membrane and was washed with buffer C (minus DTT) before adding purified recombinant Mth1088, Mth1086 or Mth1085 (each 400 μg), also in buffer C without DTT. After incubation for 5 min, the membrane was washed sequentially with buffer C lacking DTT, 2.5 M NaCl, buffer C containing 2 M imidazole and finally 1% SDS. Eluted protein was retained at each step, and analysed by SDS–PAGE. In each case, Nar71 eluted from the column on washing with imidazole. All the other proteins flowed through the mini-column. Untagged Nar71 was used in the way but was covalently coupled to an epoxy membrane mini-column (VivaScience) before treating as described above.
RESULTS
We sought to identify helicase and nuclease enzymes specific for branched DNA substrates in the thermophilic archaeon Mth. To this end, we fractionated Mth biomass, assayed for enzyme activities and used peptide sequence obtained from mass spectrometry to search within the fully sequenced genome of Mth (34). One fractionation scheme (see Materials and Methods) yielded a protein mixture with helicase and nuclease activities on a variety of branched DNA molecules (data not shown). We further investigated the identities of the component proteins.
Identification of Nar71
The product of ORF mth1090 was identified in the protein mixture described above (Figure 1A) from peptide sequence corresponding to 17.7% of its predicted amino acid sequence (Figure 1B). This protein, herein called Nar71, gave no significant matches to any other protein in database searches on overall sequences, though searches allowing short nearly matches showed high homology to a highly conserved region of ATP-dependent DNA ligases (Figure 1B).
Figure 1.

Nar71 protein. (A) SDS–PAGE gel from which Nar71 was identified in a mixture of proteins from Mth biomass, including the product of neighbouring Orf mth1088. (B) The amino acid sequence of Nar71. The underlined region is the only significant database match of Nar71 to other proteins, resembling the active site of ATP-dependent DNA ligases. Italicized sequences correspond to peptides identified by mass spectrometry. Residues in bold were subjected to site-directed mutagenesis. (C) SDS–PAGE of pure recombinant Nar71. (D) Nar71 migrates in gel filtration consistent with a monomer. The elution volume of Nar71 (Ve = 8.6 ml) inserted into the trend-line equation (y = −2.982 + 13.66) gave a size estimate of 50 kDa. (E) Binding of Nar71 to Holliday junction DNA (5 nM) in 2 mM Mg2+ and Mg2+:ATP (2 mM each). Nar71 was titrated at 0–250 nM, and the radiolabelled DNA strand is shown with asterisk.
Over-expressed recombinant Nar71 was soluble in E.coli but purified in low yields (typically 0.2 mg/l). Its properties on SDS–PAGE were consistent with 71.3 kDa molecular mass reported by Q-TOF (Figure 1C), and gel filtration showed Nar71 to elute as a monomer in solution (Figure 1D).
Nar71 is a nuclease and weak ATPase
The protein mixture containing Nar71, obtained from fractionating Mth biomass, could unwind Holliday junctions to flayed duplex (data not shown). Therefore initial assays focussed on trying to reconstitute this activity using recombinant Nar71. Nar71 bound Holliday junctions in both Mg2+ and Mg2+:ATP, though different protein–DNA complexes were observed (Figure 1E). However, no helicase or nuclease activity was detected on Holliday junctions (Table 1) (data not shown). We then mixed Nar71 in 10 mM Mg2+ with a variety of DNA substrates in turn, and catalytic activity was detected on flayed duplex and 3′ flap DNA only (Table 1) (Figure 2A). Reaction products migrated through neutral gels more slowly than labelled ssDNA oligo alone (Figure 2A, c.f. lanes g and h), consistent with Nar71 generating products that can remain base paired but have one or more DNA strands truncated by nuclease activity. The two observed products suggest distinct sites of cleavage by Nar71 on these substrates. This is examined further in the following sections. When we added ATP to reactions containing these substrates the activity of Nar71 changed dramatically (Figure 2B). Neutral gels showed reduced nuclease activity, and a different product appeared. Flayed duplex substrates gave a product that migrated identically to ssDNA (Figure 2B, lanes g and h and Figure 2C lanes a–c), and 3′flap DNA was unwound to product migrating like partial duplex (Figure 2C lanes d–f). This single-strand DNA displacement activity was abolished when poorly hydrolysable ATPγS was substituted for ATP, indicating a requirement for ATP hydrolysis (Figure 2D). In these conditions, some nuclease activity of Nar71 was restored. Mn2+ also supported nuclease activity, and in the absence of ATP the pattern of products was altered in Mn2+ compared with Mg2+ (Figure 2E c.f. lanes c and f). None of Zn2+, Co2+ or Ni2+ supported nuclease activity (data not shown). Displacement of ssDNA from 3′ flap-1 and flayed duplexes by Nar71 is weak compared with nuclease activity (Figure 3A) but was specific for these substrates in either Mg2+ or Mg2+:ATP (Figure 3A and Table 1) (data not shown). Titration of ATP into reactions containing flayed duplex and Nar71 in 5 mM Mg2+ showed clearly the appearance of ssDNA concomitant to severe reduction in nuclease activity, but only when ATP:Mg2+ ratios exceeded one (Figure 3B).
Figure 2.

Neutral gels showing catalysis by Nar71 on flayed duplex and 3′ flap DNA. (A) Reactions in 10 mM Mg2+ contained 5 nM radiolabelled flayed duplex or 3′ flap-1 and 0–250 nM Nar71 (lanes a–f). Lane g shows a reaction of Nar71 in 10 mM Mg2+, with flayed duplex substrate. Catalytic products are compared with labelled ssDNA marker (M). Substrate DNA and likely products are shown to the right of each panel, and the labelled DNA strand is marked with asterisk. (B) Reactions were the same as in (A), but contained 15 mM ATP. Lane g shows a reaction of Nar71 in ATP, with flayed duplex substrate. The catalytic product is compared with labelled ssDNA marker (M). Labelled DNA strands are marked with asterisk. (C) A total of 15% acrylamide neutral gel distinguishing unwinding products of Nar71 on flayed duplex (lanes a and b) and 3′ flap (lanes e and f). Reaction conditions were as in (B), but contained either zero (−) or 240 nM (+) Nar71. Lane c contains ssDNA marker (ss) and lane d contains partial duplex (PD) marker. (D) Nar71 titrated into 3′ flap-1 DNA (5 nM) from 0–250 nM in 10 mM Mg2+ and 15 mM ATPγS. Likely nuclease products are shown on the right. (E) Nar71 activity on flayed duplex in 10 mM Mg2+ or Mn2+, containing 15 mM ATP as indicated (+/−). Reactions contained 5 nM 3′ flap-1 DNA and Nar71 at 100 nM, except in lanes a and d that are controls lacking Nar71.
Figure 3.

(A) Time courses of DNA cleavage and strand displacement by Nar71 (100 nM). Symbols relating to each substrate are labelled except for fully base paired fork substrates, which are shown as filled triangles. (B) Titration of ATP (0–20 mM) into pairs of reactions containing 5 mM Mg2+, zero or 100 nM Nar71 and 5 nM flayed duplex DNA. (C) Denaturing gel analysis of strand cleavage by Nar71 in 10 mM Mg2+ and 15 mM ATP or ATPγS as indicated. Reactions contained 5 nM DNA substrate and zero (−) or 100 nM Nar71 (+) as indicated The labelled strand in each substrate is indicated with asterisk above the panel. (D) Mapping of Nar71 cleavage sites on flayed duplex and 3′ flap-1 substrates used in (C). The 32P-labelled strand is marked with asterisk, and black arrowheads indicate major cleavage sites. Strands numbering on 3′ flap-1 are referred to in the discussion.
Nar71 targets ssDNA
Neutral gels showed Nar71 to be highly specific for 3′ flaps and flayed duplex DNA. Denaturing gels determined which DNA strands are targeted by Nar71 nuclease activity in these substrates (Figure 3C). Nuclease activity was detected on only one strand of flayed duplex DNA (Figure 3C, c.f. lanes b and c with e and f), that which formed the 3′ ssDNA flap. This nuclease activity was altered in poorly hydrolysable ATPγS compared with ATP (Figure 3C, lanes e and f). The same strand was also targeted in 3′ flap-1 DNA, and in this case cleavage was almost abolished in ATP (lanes g–l). No activity was detected on single-stranded regions of 5′ flap DNA, 3′-tailed partial duplex or ssDNA (Figure 3C, lanes p–r) (data not shown). We mapped the sites cleaved by Nar71 in flayed duplex and 3′ flap-1 DNA (Figure 3C, lanes a–l and Figure 3D). Cleavage is concentrated at two sites: at the ss-dsDNA branch point, most significantly in flayed duplex, and at a cytosine triplet distal from the branch point.
ATP hydrolysis by Nar71 was measured through the liberation of free phosphate from ATP (33). Nar71 alone hydrolyses ATP very poorly but is stimulated 5- to 10-fold in ssDNA but not in dsDNA (Figure 4A, i and ii). However, even in ssDNA, ATP hydrolysis by Nar71 is weak compared with known DNA repair helicases like RecG (Figure 4A, iii). Using 3′ flap or flayed duplex DNA substrates did not enhance stimulation of ATPase activity by ssDNA, and hydrolysis of GTP by Nar71 was not detected (data not shown). Time course reactions showed that the ATPase activity of Nar71 was stable for up to 1 h under standard ATPase assay conditions (Figure 4B).
Figure 4.

(A) ATPase activity of Nar71 measured in malachite green assays. Reactions contained (i) 50 nM or (ii) 100 nM Nar71 and (iii) 3.3 mM Mg2+ and 5 mM ATP. dsDNA or ssDNA (100 ng) was added as indicated. RecG (50 nM) was used as a comparison in 5 mM Mg2+and 5 mM ATP. In each panel, data are a mean of reactions in duplicate. (B) Time course reaction on 100 nM Nar71 containing 100 nM Nar71 and 100 ng ssDNA in 3.3 mM Mg2+and 5 mM ATP. Data are a mean of reactions in duplicate. (C) Effect of pre-incubating His-tag monoclonal antibody with tagged Nar71 (250 nM) prior to mixing with 5 nM 3′ flap DNA in 10 mM Mg2+ and 15 mM ATP.
Nar71 modified with an N-terminal histidine tag also displaced ssDNA from flap DNA in ATP (Figure 4C, lane b). This activity was abolished by immunodepletion of tagged Nar71 using monoclonal antibody specific for the histidine tag (Figure 4C, lane c). Pull-down experiments using tagged Nar71 with this antibody coupled to protein-A sepharose were ineffective as control reactions lacking antibody showed high non-specific binding between Nar71 and protein-A sepharose (data not shown).
Catalytically inactive variants of Nar71
Nar71 nuclease and ATPase activities were probed further by introducing amino acid substitutions shown in Figure 1B. The lack of overall sequence homologues of Nar71 led us to target lysine residues for mutagenesis to alanine, based on their known catalytic roles in ATPases and nucleases. Lys68, Lys70, Lys117 and Lys376 were targeted because each precedes a serine or threonine residue typical of ATP-binding motifs, or is part of a sequence resembling the E/DxK nuclease motif of type II restriction endonucleases (35). K70A protein was insoluble and we were unable to obtain the mutation for K376A despite several attempts. However, K117A and K68A Nar71 proteins were purified. K117A was inactive in both Mg2+ and Mg2+:ATP (Figure 5A, lanes f–j and Figure 5B). K68A retained some nuclease activity but did not form ssDNA product in ATP (Figures 5A, lanes k–o). The nature of nuclease activity by K68A was compared with wild-type and inactive K117A by denaturing gels on 3′ flap-2 DNA labelled in the cleavable strand (Figure 5B). The separation-of-function mutation in Lys-68 shows that ATP hydrolysis and nuclease activity can be uncoupled in Nar71. In agreement with this, we were unable to detect any ATPase activity from K68A or K117A variants of Nar71 (Figure 5C).
The minimal substrate for Nar71 is a flayed duplex, which is cleaved in ssDNA at positions 3′ to the ds/ss DNA junction (Table 1 and Figure 3). We modified a flayed duplex by covalently attaching biotin to the 3′ OH group at the ssDNA end as a means of potential steric hindrance to Nar71 nuclease activity (Figure 5D). Cleavage of the unmodified flayed duplex (Figure 5D, lanes a–f) was abolished when biotinylated (Figure 5D, lanes g–l).
The gene neighbourhood of Nar71: a proposed novel DNA repair system
The gene encoding Nar71 (mth1090) is located within a neighbourhood proposed to constitute a novel DNA repair system in thermophiles (30). This system is predicted to encode several proteins of unknown function including Mth1088 and Mth1086 (Figure 6A), which are both conserved in thermophiles as COG1857 and COG1203, respectively. Nar71 and Mth 1088 co-purify after several fractionation steps from Mth biomass (Figure 1A) and this led us to investigate possible concerted biochemical activities. Purified recombinant Mth 1088 binds to 3′ flap-2 DNA like Nar71 (Figure 6B and C), but showed no catalytic activity in Mg2+ or Mg2+:ATP (Figure 6D, lane f). Mth1088 inhibits cleavage by Nar71 but only when in molar excess (Figure 6D, lanes b–e). This effect was the same whether Nar71 was added prior to, or after Mth1088, or if they were added simultaneously, as in Figure 6D. Immobilized Nar71, both untagged and carrying an N-terminal histidine-tag, was used as bait to probe for interactions with purified protein products of Mth1088 and Mth1086, but no evidence of interactions was detected (data not shown).
Figure 6.

(A) Cartoon of the proposed DNA repair neighbourhood containing nar71 in Mth (modified from Genome Information Broker; http://gib.genes.nig.ac.jp). (B) Purified recombinant Mth1088 protein. (C) Binding of Nar71 (0–250 nM) and Mth1088 (0–250 nM) to 3′ flap DNA (5 nM) in 2 mM EDTA. (D) Representative gel of the effects of Mth1088 protein at 10, 100 and 400 nM (lanes c–e) on cleavage of 3′ flap-2 by Nar71 (100 nM, lanes b–e) in 10 mM Mg2+.
DISCUSSION
Thermophilic bacteria and archaea may have evolved enzyme systems dedicated to dealing with DNA damage incurred at high temperatures (25). Genomics has highlighted a gene neighbourhood specific to thermophiles that is predicted to encode DNA processing enzymes (30). However, these insights lack supporting biochemical or genetic evidence. We report catalytic activities from a member of this gene neighbourhood in the thermophilic archaeon Mth. Nar71, the gene product of Mth Orf1090, was identified from Mth biomass and is active as a structure-specific nuclease, targeting ssDNA in 3′ flaps and flayed duplex DNA substrates. This is the first biochemical analysis offering evidence to support core DNA repair functions within the proposed DNA repair gene neighbourhood.
Nuclease activity of Nar71
The amino acid sequence of Nar71 gave little clue of its possible function. Sequence analysis of Nar71 is hindered by it lacking homology to any known enzymes. It seems to be a singleton, absent from any group of Conserved Orthologous Genes (COGs). This contrasts with some other members of the gene neighbourhood, including a predicted polymerase (COG1353) and superfamily 2 helicase (COG1203), both of which are conserved in most species of thermophilic bacteria and archaea. However, the positioning of Nar71 in this neighbourhood from Methanothermobacter replaces that of a missing predicted nuclease (COG1517) that is present in some other species. Like Methanothermobacter Nar71, there is a unique protein in this position in most thermophiles (30). Despite its overall sequence novelty, a short region of Nar71 comprising Ala-84 to Ser-120 (Figure 1B) resembles an essential region of DNA ligases containing an ATP-binding motif (KxDG). In particular, the Nar71 sequence ENKTES (residues 115–120) interested us because it resembled both the ligase ATP-binding loop and a common motif (ExK) found in type II restriction endonucleases. Wild-type Nar71 gave no detectable ligase activity, but introducing the mutation K117A in its ExK motif abolished nuclease activity (Figure 5A and B), suggesting a possible common role for this lysine in Nar71 and type II restriction endonucleases.
The importance of a 3′ ssDNA end for Nar71 nuclease activity was highlighted by modifying a flayed duplex substrate with biotin at the 3′ OH in ssDNA. This abolished catalysis (Figure 5D), and may be attributed to steric hindrance of the enzyme active site by biotin. This hindrance could be through blockage of Nar71 ‘threading’ over the 3′ end of ssDNA. However, this does not easily account for the structure specificity of Nar71 for 3′ flaps and flayed duplexes and its cleaving ssDNA up to 25 nt from the 3′ end. Perhaps Nar71 gains access to cleavage sites by first recognizing a 3′ ssDNA end and then moving along ssDNA with 3′ to 5′ polarity, before recognition of a branch-point triggers cleavage. Alternatively, Nar71 may recognize branch points, but then requires 3′ ss DNA end capture by the nuclease active site before cleavage can occur. In either scenario, the nuclease activity of Nar71 seems to be inhibited by steric hindrance arising from the 3′ biotin group.
Catalysis by Nar71 in ATP
The substrate specificity of Nar71 for flayed duplex and 3′ flaps was unchanged in reactions containing ATP. However, ATP caused nuclease activity to be curtailed and products of DNA unwinding appeared (Figures 2 and 3B). Flayed duplex was unwound to ssDNA by Nar71, whereas 3′ flaps were unwound to partial duplex (Figure 2B and C). These results indicate that Nar71 tracks along ssDNA with 3′ to 5′ polarity, e.g. along strand 2 in 3′ flap-1 (Figure 3D). At the branch point, Nar71 continues in a 3′ to 5′ direction along the same strand, eventually dissociating it from the substrate. Positioning 32P-radiolabel at the 5′ end of strand 3 in this flap (as in Figure 2) would manifest as a partial duplex on neutral gels. Similarly, unwinding of a flayed duplex along the same strand would liberate radiolabelled ssDNA.
We confirmed that Nar71 is multifunctional as a nuclease and helicase by generating a separation-of-function mutant. K68A protein retains nuclease activity but lacks DNA unwinding and ATPase activity (Figure 5), suggesting that Lys-68 has a key role in responding to ATP, either independently from the nuclease active site, or to transmit ATP binding and/or hydrolysis to the site of nuclease activity. We have no direct physical measurement that K68A is folded correctly, but its purification like wild-type Nar71, and retention of nuclease activity, suggests that it is structurally conserved compared with wild type. Nar71 lacks canonical Walker A/B motifs or an obvious Bergerat fold, both well-known ATP-binding folds, but it is intriguing that Lys-68 is next to a serine residue, typical in enzymes that use lysine to interact with ATP. It may also be significant that Nar71 interacts with DNA differently in ATP compared with Mg2+:ATP (Figure 1E). Changes in Nar71-DNA contacts brought about by ATP binding may alter the enzyme active site sufficiently to disengage residues that would otherwise catalyse nuclease activity. ATP was more potent at reducing nuclease activity of Nar71 compared with poorly hydrolysable ATPγS, suggesting that energy derived from ATP hydrolysis can act as the switch to modulate Nar71 nuclease function.
Possible functions of Nar71 in cells
Since Nar71 is multifunctional, the possible cellular roles of each catalytic activity need to be considered. Two other proteins, yeast Dna2 and WRN (a human RecQ-like helicase), are well characterized as having nuclease and helicase activities residing on the same polypeptide (36,37). In each case, helicase and nuclease activities are structure specific, acting on ssDNA flaps. In WRN, ATP is required for nuclease activity, but ATP inhibits the nuclease activity of Dna2, akin to Nar71 (36). Both of these proteins help to restore replication forks at sites of DNA damage. We initially identified Nar71 (and Mth1088) in protein fractions that unwound Holliday junction DNA (Figure 1A), suggesting that they may associate with enzymes active on Holliday junctions or similar DNA repair intermediates. Nar71 may function in late stages of recombination dependent repair (RDR) of stalled DNA replication in thermophiles (Figure 7). Models of RDR have been described for viruses, bacteria and eukaryotes [reviewed in (38–40)]. They are characterized by the formation of D-loops or Holliday junction DNA molecules in the strand invasion step. Nuclease activity of Nar71 may remove extraneous 3′ flaps formed after synthesis of nascent strands by a DNA polymerase in a 5′ to 3′ direction at the site of DNA damage (Figure 7C). An alternative model to Nar71 functioning in RDR arises from the general absence in thermophiles of PolY-family polymerases (41). This has led to suggestions that a novel translesion repair (TR) system, including a novel polymerase, may exist in these organisms (30). We cannot discount the possibility that Nar71 processes 3′ flaps generated by DNA replication during TR. In either scenario, RDR or TR, activities of a putative DNA polymerase (COG1353) within the Nar71 gene neighbourhood may be significant.
Figure 7.
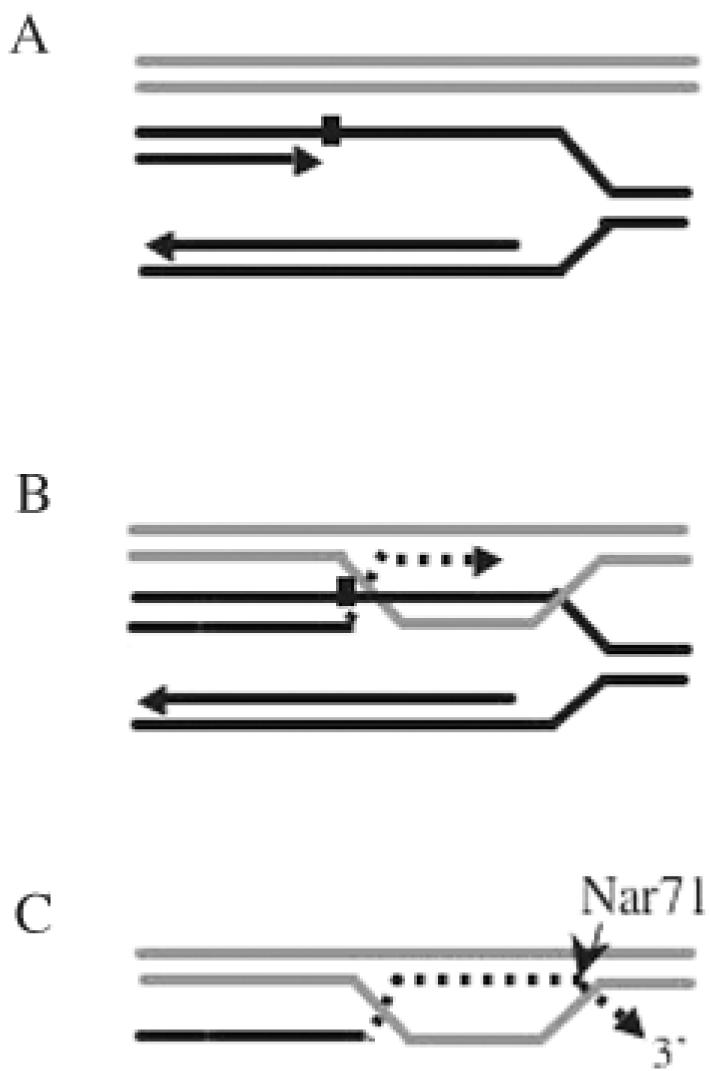
A possible role for Nar71 in DNA repair. The model implies Nar71 function during recombination dependent replication. We do not rule out the same enzymatic role for Nar71 in other repair systems (detailed in the text). (A) Synthesis of nascent DNA on the leading strand of a replication fork is blocked by DNA damage (black square). (B) ssDNA from the gapped fork stimulates pairing with homologous DNA sequence, leading to strand exchange. DNA replication can proceed (dotted line) using a sister DNA template. (C) DNA replication continues beyond the damage and Nar71 trims extraneous, un-annealed 3′ flaps prior to ligation or resumption of replication at the original fork. For clarity, only the nascent DNA strand and its template are shown.
It is unclear exactly how the nuclease activity of Nar71 could be regulated by its exposure to ATP in cells. However, in our in vitro assays Nar71 also showed reduced nuclease activity when mixed with Mth1088 protein. Association of Nar71 with Mth1088 may provide a mechanism by which cells can control Nar71 nuclease activity. A deeper understanding of this requires thorough characterization of Mth1088 protein, which as yet has no known catalytic activity. Co-operative protein complexes are noted in DNA processing systems in all organisms to protect DNA and ensure the appropriate hand-off of substrates between enzymes [for example see (42)]. A clearer picture of DNA repair and/or replication functions of this biologically important gene neighbourhood in thermophilic archaea and bacteria awaits further biochemical analysis. A bottom-up engineering approach is needed to first assign functions to each protein component of this system. The activities of the COG1353 family predicted polymerase and superfamily 2 helicase should be of particular interest.
Acknowledgments
ACKNOWLEDGEMENTS
E.L.B. is a Wellcome Trust Research Career Development Fellow and J.P.J.C. is a BBSRC David Phillips Research Fellow. We gratefully acknowledge Prof. Bob Lloyd FRS, for support in the early stages of this work through an MRC programme grant. We also acknowledge Kevin Bailey at the University of Nottingham mass spectrometry facility and the efforts of Ria Mamujee during a summer placement with E.L.B., funded by The Nuffield Foundation.
REFERENCES
- 1.Vieille C. and Zeikus,G.J. (2001) Hyperthermophilic enzymes: sources, uses, and molecular mechanisms for thermostability. Microbiol. Mol. Biol. Rev., 65, 1–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cambillau C. and Claverie,J.M. (2000) Structural and genomic correlates of hyperthermostability. J. Biol. Chem., 275, 32383–32386. [DOI] [PubMed] [Google Scholar]
- 3.Saunders N.F.W. (2003) Mechanisms of thermal adaptation revealed from the genomes of the Antarctic Archaea Methanogenium frigidum and Methanococcoides burtonii. Genome Res., 13, 1580–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sprott G.D. (1992) Structures of archaebacterial membrane lipids. J. Bioenerg. Biomembr., 24, 555–566. [DOI] [PubMed] [Google Scholar]
- 5.Grogan D.W. (1998) Hyperthermophiles and the problem of DNA instability. Mol. Microbiol., 28, 1043–1049. [DOI] [PubMed] [Google Scholar]
- 6.Lindahl T. and Wood,R.D. (1999) Quality control by DNA repair. Science, 286, 1897–1905. [DOI] [PubMed] [Google Scholar]
- 7.DiRuggiero J., Santangelo,N., Nackerdien,Z., Ravel,J. and Robb,F.T. (1997) Repair of extensive ionizing-radiation DNA damage at 95 degrees C in the hyperthermophilic archaeon Pyrococcus furiosus. J. Bacteriol., 179, 4643–4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peak M.J., Robb,F.T. and Peak,J.G. (1995) Extreme resistance to thermally induced DNA backbone breaks in the hyperthermophilic archaeon Pyrococcus furiosus. J. Bacteriol., 177, 6316–6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reilly M.S. and Grogan,D.W. (2002) Biological effects of DNA damage in the hyperthermophilic archaeon Sulfolobus acidocaldarius. FEMS Microbiol. Lett., 208, 29–34. [DOI] [PubMed] [Google Scholar]
- 10.Grogan D.W., Carver,G.T. and Drake,J.W. (2001) Genetic fidelity under harsh conditions: analysis of spontaneous mutation in the thermoacidophilic archaeon Sulfolobus acidocaldarius. Proc. Natl Acad. Sci. USA, 98, 7928–7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sandman K., Bailey,K.A., Pereira,S.L., Soares,D., Li,W.T. and Reeve,J.N. (2001) Archaeal histones and nucleosomes. Meth. Enzymol., 334, 116–129. [DOI] [PubMed] [Google Scholar]
- 12.Isabelle V., Franchet-Beuzit,J., Sabattier,R., Laine,B., Spotheim-Maurizot,M. and Charlier,M. (1993) Radioprotection of DNA by a DNA-binding protein: MC1 chromosomal protein from the archaebacterium Methanosarcina sp. CHTI55. Int. J. Radiat. Biol., 63, 749–758. [DOI] [PubMed] [Google Scholar]
- 13.Wardleworth B.N., Russell,R.J., Bell,S.D., Taylor,G.L. and White,M.F. (2002) Structure of Alba: an archaeal chromatin protein modulated by acetylation. EMBO J., 21, 4654–4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McAfee J.G., Edmondson,S.P., Datta,P.K., Shriver,J.W. and Gupta,R. (1995) Gene cloning, expression, and characterization of the Sac7 proteins from the hyperthermophile Sulfolobus acidocaldarius. Biochemistry, 34, 10063–10077. [DOI] [PubMed] [Google Scholar]
- 15.Wadsworth R.I. and White,M.F. (2001) Identification and properties of the crenarchaeal single-stranded DNA binding protein from Sulfolobus solfataricus. Nucleic Acids Res., 29, 914–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lopez-Garcia P. and Forterre,P. (2000) DNA topology and the thermal stress response, a tale from mesophiles and hyperthermophiles. Bioessays, 22, 738–746. [DOI] [PubMed] [Google Scholar]
- 17.Englander J., Klein,E., Brumfeld,V., Sharma,A.K., Doherty,A.J. and Minsky,A. (2004) DNA toroids: framework for DNA repair in Deinococcus radiodurans and in germinating bacterial spores. J. Bacteriol., 186, 5973–5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kil Y.V., Baitin,D.M., Masui,R., Bonch-Osmolovskaya,E.A., Kuramitsu,S. and Lanzov,V.A. (2000) Efficient strand transfer by the RadA recombinase from the hyperthermophilic archaeon Desulfurococcus amylolyticus. J. Bacteriol., 182, 130–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Komori K., Miyata,T., DiRuggiero,J., Holley-Shanks,R., Hayashi,I., Cann,I.K., Mayanagi,K., Shinagawa,H. and Ishino,Y. (2000) Both RadA and RadB are involved in homologous recombination in Pyrococcus furiosus. J. Biol. Chem., 275, 33782–33790. [DOI] [PubMed] [Google Scholar]
- 20.Seitz E.M., Brockman,J.P., Sandler,S.J., Clark,A.J. and Kowalczykowski,S.C. (1998) RadA protein is an archaeal RecA protein homolog that catalyzes DNA strand exchange. Genes Dev., 12, 1248–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharples G.J., Ingleston,S.M. and Lloyd,R.G. (1999) Holliday junction processing in bacteria: insights from the evolutionary conservation of RuvABC, RecG, and RusA. J. Bacteriol., 181, 5543–5550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bond C.S., Kvaratskhelia,M., Richard,D., White,M.F. and Hunter,W.N. (2001) Structure of Hjc, a Holliday junction resolvase, from Sulfolobus solfataricus. Proc. Natl Acad. Sci. USA, 98, 5509–5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Komori K., Sakae,S., Shinagawa,H., Morikawa,K. and Ishino,Y. (1999) A Holliday junction resolvase from Pyrococcus furiosus: functional similarity to Escherichia coli RuvC provides evidence for conserved mechanism of homologous recombination in Bacteria, Eukarya, and Archaea. Proc. Natl Acad. Sci. USA, 96, 8873–8878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woods W.G. and Dyall-Smith,M.L. (1997) Construction and analysis of a recombination-deficient (radA) mutant of Haloferax volcanii. Mol. Microbiol., 23, 791–797. [DOI] [PubMed] [Google Scholar]
- 25.Grogan D.W. (2000) The question of DNA repair in hyperthermophilic archaea. Trends Microbiol., 8, 180–185. [DOI] [PubMed] [Google Scholar]
- 26.Sartori A.A. and Jiricny,J. (2003) Enzymology of base excision repair in the hyperthermophilic archaeon Pyrobaculum aerophilum. J. Biol. Chem., 278, 24563–24576. [DOI] [PubMed] [Google Scholar]
- 27.Roberts J.A., Bell,S.D. and White,M.F. (2003) An archaeal XPF repair endonuclease dependent on a heterotrimeric PCNA. Mol. Microbiol., 48, 361–371. [DOI] [PubMed] [Google Scholar]
- 28.Forterre P. (2002) A hot story from comparative genomics: reverse gyrase is the only hyperthermophile-specific protein. Trends Genet., 18, 236–237. [DOI] [PubMed] [Google Scholar]
- 29.Rodriguez A.C. (2002) Studies of a positive supercoiling machine. nucleotide hydrolysis and a multifunctional “latch” in the mechanism of reverse gyrase. J. Biol. Chem., 277, 29865–29873. [DOI] [PubMed] [Google Scholar]
- 30.Makarova K.S., Aravind,L., Grishin,N.V., Rogozin,I.B. and Koonin,E.V. (2002) A DNA repair system specific for thermophilic Archaea and bacteria predicted by genomic context analysis. Nucleic Acids Res., 30, 482–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bolt E.L. and Lloyd,R.G. (2002) Substrate specificity of RusA resolvase reveals the DNA structures targeted by RuvAB and RecG in vivo. Mol. Cell, 10, 187–198. [DOI] [PubMed] [Google Scholar]
- 32.Maxam A.M. and Gilbert,W. (1980) Sequencing end-labeled DNA with base-specific chemical cleavages. Meth. Enzymol., 65, 499–560. [DOI] [PubMed] [Google Scholar]
- 33.Bird L.E., Hakansson,K., Pan,H. and Wigley,D.B. (1997) Characterization and crystallization of the helicase domain of bacteriophage T7 gene 4 protein. Nucleic Acids Res., 25, 2620–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith D.R., Doucette-Stamm,L.A., Deloughery,C., Lee,H., Dubois,J., Aldredge,T., Bashirzadeh,R., Blakely,D., Cook,R., Gilbert,K. et al. (1997) Complete genome sequence of Methanobacterium thermoautotrophicum deltaH: functional analysis and comparative genomics. J. Bacteriol., 179, 7135–7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aggarwal A.K. (1995) Structure and function of restriction endonucleases. Curr. Opin. Struct. Biol., 5, 11–19. [DOI] [PubMed] [Google Scholar]
- 36.Budd M.E., Choe,W. and Campbell,J.L. (2000) The nuclease activity of the yeast DNA2 protein, which is related to the RecB-like nucleases, is essential in vivo. J. Biol. Chem., 275, 16518–16529. [DOI] [PubMed] [Google Scholar]
- 37.Shen J.C., Gray,M.D., Oshima,J., Kamath-Loeb,A.S., Fry,M. and Loeb,L.A. (1998) Werner syndrome protein. I. DNA helicase and dna exonuclease reside on the same polypeptide. J. Biol. Chem., 273, 34139–34144. [DOI] [PubMed] [Google Scholar]
- 38.Mosig G. (1998) Recombination and recombination-dependent DNA replication in bacteriophage T4. Annu. Rev. Genet., 32, 379–413. [DOI] [PubMed] [Google Scholar]
- 39.Kraus E., Leung,W.Y. and Haber,J.E. (2001) Break-induced replication: a review and an example in budding yeast. Proc. Natl Acad. Sci. USA, 98, 8255–8262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kogoma T. (1997) Stable DNA replication: interplay between DNA replication, homologous recombination, and transcription. Microbiol. Mol. Biol. Rev., 61, 212–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boudsocq F., Iwai,S., Hanaoka,F. and Woodgate,R. (2001) Sulfolobus solfataricus P2 DNA polymerase IV (Dpo4): an archaeal DinB-like DNA polymerase with lesion-bypass properties akin to eukaryotic poleta. Nucleic Acids Res., 29, 4607–4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stauffer M.E. and Chazin,W.J. (2004) Structural mechanisms of DNA replication, repair, and recombination. J. Biol. Chem., 279, 30915–30918. [DOI] [PubMed] [Google Scholar]