

Abstract
Androgen receptor (AR) is a member of the steroid receptor family and a therapeutic target for all stages of prostate cancer. AR is activated by ligand binding within its C-terminus ligand-binding domain (LBD). Here we show that overexpression of the AR NTD to generate decoy molecules inhibited both the growth and progression of prostate cancer in castrated hosts. Specifically, it was shown that lentivirus delivery of decoys delayed hormonal progression in castrated hosts as indicated by increased doubling time of tumor volume, prolonged time to achieve pre-castrate levels of serum prostate-specific antigen (PSA) and PSA nadir. These clinical parameters are indicative of delayed hormonal progression and improved therapeutic response and prognosis. Decoys reduced the expression of androgen-regulated genes that correlated with reduced in situ interaction of the AR with androgen response elements. Decoys did not reduce levels of AR protein or prevent nuclear localization of the AR. Nor did decoys interact directly with the AR. Thus decoys did not inhibit AR transactivation by a dominant negative mechanism. This work provides evidence that the AR NTD plays an important role in the hormonal progression of prostate cancer and supports the development of AR antagonists that target the AR NTD.
Introduction
The prostate is an androgen-dependent tissue that requires androgen for the growth and survival of epithelial cells. Androgen receptor (AR) is a transcription factor that mediates the effects of androgen. It is composed of functional domains that include a C-terminal ligand-binding domain (LBD) that contains transactivation function-2 (AF-2), a DNA-binding domain (DBD), and an N-terminal domain (NTD) that harbors AF-1 with two transcriptional activation regions [1]. All current approved hormonal therapies for prostate cancer aim at preventing activation of AR through chemical or surgical castration and intervention with antiandrogens that competitively bind to the LBD of the receptor. These hormonal therapies include LHRH analogues, enzalutamide and other antiandrogens, and abiraterone. Initially, prostate cancer responds to these therapies. However, inevitably the disease will become lethal castration-recurrent disease. AR is suspected to continue to drive castration recurrent disease. The transcriptional activity of AR is dependent upon functional AF-1 [1] thereby providing rationale to develop approaches that inhibit AR by targeting its NTD.
In 2007, the first in vivo proof-of-concept for AR NTD as a novel therapeutic target was provided using copies (decoys) of the AR NTD residues 1–558 (AR1-558) [2]. In non-castrated hosts, these decoys reduced tumor incidence, decreased tumor growth and serum PSA levels [2]. Here we provide evidence that lentiviral delivery of decoys to mice bearing established prostate cancer xenografts inhibited hormonal progression to castration-recurrence as well as investigated possible mechanisms through which decoys exert their activity.
Materials and methods
Animals and cell culture
Male NOD-SCID mice were obtained from the Animal Research Center of the British Columbia Cancer Agency. All animal studies conformed to the relevant regulatory and ethical standards. Analgesic (Metacam) and anaesthesia (isoflurane) were used and all efforts were made to minimize suffering. The University of British Columbia Animal Care Committee approved all experiments involving animals (Permit Number A03-0260). LNCaP cells (from Dr. Leland Chung, Cedars-Sinai Medical Center, Los Angeles, CA) were routinely maintained in RPMI 1640 supplemented with 5% (v/v) FBS (HyClone, Logan, UT). LNCaP cells that stably express decoy AR1–558 have been described [2]. The synthetic androgen (R1881) was purchased from Perkin–Elmer (Wellesley, MA) and forskolin was purchased from Calbiochem (La Jolla, California, USA).
Plasmids
His-tag expression plasmids for AR1-558, AR1-233, and AR392-558 plasmids were made by polymerase chain reaction (PCR) amplification of the nucleotides of the cDNA corresponding to the amino acids 1–558, 1–233, and 392–558 of the human AR and cloning the products into the BamHI site of pcDNA3.1/His©A plasmid (Invitrogen, Carlsbad, CA). The human AR1-558 decoys and lentivirus plasmids have been described [2]. The PSA (-630/+12)-luciferase reporter contains the promoter region with two well-characterized AREs [3,4].
Lentivirus delivery and castration in mice
LNCaP xenografts were established subcutaneously (s.c.) in the flanks of 6-week-old male NOD-SCID mice [2]. The lentivirus particles were prepared by using the ViraPower expression system (Invitrogen) as previously described [2]. When tumors averaged approximately 50–100 mm3 in size, the animals were randomly divided into four groups (Mock media, GFP, GFP-AR1–558, and AR1–558). Treatment consisted of injections every 5 days with 1–2 x 107 particles for GFP-AR1–558 and AR1–558 and 1x108 particles for GFP for the duration of the experiment. Tumors were measured weekly. Castration was performed under anesthesia by making a small incision in the scrotum to remove each testicle after ligation of the cord. At 5 days after the last inoculation, mice were sacrificed, and the tumors and major organs were excised and prepared for immunohistochemistry and Western blot analyses.
Serum PSA levels
Blood samples were obtained from mice weekly before and after castration. Serum PSA levels were determined by an enzymatic immunoassay kit with a lower limit of sensitivity of 0.2 μg/liter (Abbott IMX, Montreal, QC, Canada).
Immunohistochemistry
Tissue sections (5 μm) were blocked in immunohistochemistry solution (Immunovision Technologies, Brisbane, CA) and immunostained with anti-AR LBD (C-19, Santa Cruz Biotechnology, Santa Cruz, CA), or anti-AR NTD (441, Santa Cruz). The Vectastain® ABC Kit (Vector Laboratories, Burlingame, CA) was used for detection. Peroxidase activity was localized with 3,3-diaminobenzidine, and the sections were counterstained with hematoxyline before dehydration and mounting. The slides were visualized using a Zeiss Axioplan 2 Microscope (Carl Zeiss, Toronto, ON, Canada).
Western blot analysis
Whole cell protein lysates were obtained from frozen xenografts that were ground up in N2 (liquid) then homogenized using a polytron homogenizer in ice-cold RIPA buffer (40mM Tris-HCl, pH7.0/ 1mM EDTA/4%glycerol/10mM DTT/0.2%SDS/20mM Na Molybdate/50mM NaF/Complete™ protease inhibitors (Roche, Mississauga, ON, Canada). The protein lysates were treated with Albumin Depletion kit (Millipore). Protein concentrations were determined by RC DC assay (BioRad). Protein samples (40 ug) were loaded on a 10% polyacrylamide gel, transferred on Immobilon™ PVDF membranes (Millipore, Billerica, MA), and probed with anti-GFP (Santa Cruz), or anti-AR (PG21, Upstate) antibodies. Whole cell lystes from LNCaP cells transfected with the His-Tag plasmids were loaded on a 12.5% SDS PAGE and analyzed by Western blot analysis using anti-His-probe (H-15; Santa Cruz Biotechnology).
GFP-AR transfection and microscopy
LNCaP cells (2.5 x 104) that stably express vector or AR1-558 [2] were plated in 4 well chamber slides and transfected with 0.25ug GFP-AR plasmid DNA per well using lipofectin and treated with R1881 (10 nM) for 30 minutes. The cells were fixed in 4% paraformaldehyde (EM Sciences) for 30 minutes at room temperature and then washed several times with PBS. The slides were mounted with Vectasheild mounting medium (vector labs) and examined using a Zeiss Axioplan-2 Fluorescent microscope (Zeiss).
Quantitative real-time PCR (qPCR)
Levels of expression of androgen-regulated genes were analyzed in LNCaP cells that stably express vector or decoy as well as in xenografts derived from mice. LNCaP cells were treated with R1881 (10 nM) for 24 hours and total RNA isolated in Trizol® Reagent (Life Technologies). For androgen-repressed genes, cDNA was generated using the high Capacity RNA-cDNA kit (Applied Biosystems). The appropriate cDNA dilution was mixed with gene-specific primers and Platinum SYBR Green qPCR Supermix–UDG with ROX. Transcript levels were measured using ABI 7900HT qPCR machine. Xenografts from mice with injected empty lentivirus vector, lentivirus vectors for AR1–558, GFP, and GFP-AR1–558 were flash frozen with liquid nitrogen prior to isolation of total RNA also with Trizol®. Oligo-d(T)-primed total RNAs (5 μg per sample) were reverse-transcribed with SuperScript III (Invitrogen). An appropriate dilution of cDNA and gene-specific primers were combined with iQ SYBR Green Supermix (Bio-Rad) and amplified in an iCycler iQ real-time PCR machine (Bio-Rad, Hercules, CA). All qPCR reactions were performed in triplicate. Ct (threshold cycle number) and expression values with standard deviations were calculated using the Gene Expression Macro for Excel (Bio-Rad). Real-time amplification was performed with initial denaturation at 95°C for 2 min, followed by 40 cycles of two-step amplification (95°C for 15 sec, 55°C for 30 sec). The results were normalized for GAPDH as housekeeping gene. Primer sequences for real-time PCRs were as listed in Table 1.
Table 1. Primer sequences for gene expression analysis using real-time PCR.
| Gene | Forward (from 5' to 3') | Reverse (from 5' to 3') |
|---|---|---|
| CAM2KN1 | ATTCTGTATGTTGCACCTTG | TTGAGACACAGGAACAATTC |
| GAPDH | CTGACTTCAACAGCGACACC | TGCTGTAGCCAAATTCGTTG |
| HMGCR | TCCCTGGGAAGTCATAGTGG | AGGATGGCTATGCATCGTGT |
| KLK2 | TGTGTGCTAGAGCTTACTCTGA | CCACTTCCGGTAATGCACCA |
| KLK3 | CCAAGTTCATGCTGTGTGCT | CCCATGACGTGATACCTTGA |
| MAF | CCGTCCTCTCCCGAGTTTTTC | ACACTGGTAAGTACACGATGCT |
| MMP16 | ACCCTCATGACTTGATAAC | TCTGTCTCCCTTGAAGAAATAG |
| RHOU | CCCGTGAGACTCCAACTCTG | TGAAGCAGAGCAGGAAGATG |
| SAT1 | CACTGGACTCCGGAAGGTAA | TCATTGCAACCTGGCTTAGA |
| SESN2 | CTGACTACTTTACCAGCTTC | TACCAGGTAAGAACACTGATG |
| SLC44A1 | TCAGTAAATCGCCTTATTCG | TTTTCCTTTCCTTTGAGCTG |
| ST7 | CTGCTTATATTCTCTTGGCTG | GTCTATGTTGGGCTTCATAC |
| TMEM144 | TTTCCAATAATCACTGCTGG | ATAAGGCTCCAGTCAAGATG |
| UGTB15 | GCAAATCTCTACTTGACACATGG | CTTCTTGGTCATCCCAAAAC |
| UGTB17 | ATTCTGCTCAAAATGAAGCC | CTGAGCTTCCTTATGTTTCAC |
Chromatin Immunoprecipitation Assay (ChIP)
LNCaP (1x106 cells) stably transfected with vector or AR1-558 were plated in 10cm plates (Nunc, Rochester, NY). The next day the media was replaced with 9 ml of serum-free RPMI. The cells were treated for 30 mins with R1881 (10nM), cross-linked with 1% formaldehyde and harvested. The cells were lysed in SDS lysis buffer (1%SDS, 50nM Tris (pH 8.0), 10mM EDTA, Complete™ protease inhibitors), and sonicated. The extracts were used for immunoprecipitation with anti-AR antibody AR (C-19; Santa Cruz Biotechnology). PSA primers used for real time PCR with Sybr green qPCR kit from (Invitrogen) were: 5’-GCCTGGATCTGAGAGATATCATC-3’ (forward) and 5’-ACACCTTTTTTTTCTGGA TTGTTG-3’ (reverse) [5].
Coimmunoprecipitation
LNCaP cells that stably express vector or AR1-558 [2] were cultured in RPMI 1640 culture medium supplemented with 5% FBS (v/v), 100 units/mL penicillin, and 100ug/uL streptomycin. For treatment with androgen, approximately 3x106 cells were plated individually into 15cm tissue culture dishes. After 24 hours, LNCaP cells were incubated in serum-free medium for 48 hours, treated with 1nM R1881 for 3 hours and harvested. Proteins were extracted from harvested LNCaP cells and lysed in buffer containing 10mM Tris-HCl (pH 7.4), 1% NonidetP-40, 150 mM NaCl, 0.5% sodium deoxycholate, 1 mM EDTA, 10ug/ml leupeptin, 10ug/ml aprotinin, 1mM AEBSF, 2mM Na3VO4, 10mM beta-glycerophosphate, and Complete™ protease inhibitor cocktail (Roche). Whole cell lysates were centrifuged at 14,000rpm for 20min at 4°C and supernatant was used for immunoprecipitation. Pre-clearance was done by incubation with rabbit IgG (Santa Cruz Biotechnology) and Protein A/G-agarose beads (Santa Cruz Biotechnology, sc-2003) for 1hour at 4°C with rotation. Protein complexes were incubated with AR antibody(C-19) (Santa Cruz Biotechnology, sc-815) for 90 minutes at 4°C and pulled down with Protein A/G agarose beads. After overnight incubation, protein complexes were washed with lysis buffer three times, eluted with SDS-PAGE sample loading buffer and separated by SDS-PAGE. Western blotting was performed using anti-AR(441) (Santa Cruz Biotechnology, sc-7305) and the protein bands were visualized by chemiluminescence (Amersham Biosciences).
Luciferase assay
LNCaP cells (3x105 cells/well) were plated on 6-well plates (Falcon) containing RPMI with 5% FBS. After 24 hours, the medium was removed and transfection was performed by using Lipofectin® Reagent (Invitrogen). The total amount of plasmid DNA was normalized to 3 μg/ well by the addition of pRc/CMV as the control plasmid with no promoter insert. The cells were cotransfected with PSA promoter-luciferase reporter gene and various portions of AR NTD or His-tag. Cells were treated with or without forskolin (50 μM) for 48 h. Cells were harvested in 1×Passive Lysis Buffer (Promega, Madison, WI, USA) and luciferase activity in the cell lysates was measured using the Luciferase Assay Reagent (Promega). The luciferase activity was normalized to the protein concentration by Bradford assay. The inhibitory effect of each ARN decoy molecule was calculated from the normalized luciferase expression to that of the His-tag control.
Results
Lentivirus delivery of decoy AR1-558 inhibits hormonal progression of established LNCaP tumors in castrated mice
To determine the effects of decoy on the time to castration-recurrence, we delivered AR1-558 decoys by lentivirus to established LNCaP xenografts in combination with androgen ablation therapy (castration). Once the tumors were 50–100 mm3 in volume, the mice were randomly assigned into 4 groups that were: mock injection (vehicle only); decoy AR1-558; GFP; and GFP-AR1-558. Animals were castrated 5 days after the first injection. Upon castration, there were significant differences in the percent decrease in serum PSA levels (nadir) between animals treated with decoys as compared to the controls. In mock-injected animals, serum levels of PSA dropped by 87% in comparison to precastrate levels, while in decoy AR1-558 injected animals the serum levels of PSA dropped by 99% (p = 0.0063). A similar result was obtained when GFP-AR1-558 chimera proteins were used. GFP injected animals showed a 94% drop in serum levels of PSA compared to GFP-AR1-558 that dropped 100% (p = 0.0271) (Fig 1A). For patients treated with androgen deprivation, the PSA nadir (lowest PSA reading) is prognostic of the time to develop castration-recurrent disease [6]. Therefore, based upon this data showing that decoys significantly lower the PSA levels (nadir) it is possible that decoys that block AR transcriptional activity may improve prognosis.
Fig 1. Lentivirus delivery of decoy AR1-558 delays the time to castration-recurrence.

A, PSA nadir or percent drop in serum PSA levels in response to castration of mice bearing LNCaP xenografts and treated with lentivirus for mock (vehicle control), decoy AR1-558 (ARN), GFP, or GFP-AR1-558 (GFP-ARN). B, The time to reach pre-castration levels of serum PSA was doubled in animals injected with decoys. C, The time for the tumor volume to double was increased by decoys. Tumors were inoculated 5 days before castration and subsequently injected every 5 days until the duration of the experiment. Student t-test: *, p<0.05; **, p<0.01.
The time to castration-recurrence is defined here as the time for serum PSA levels to return to pre-castration levels. The longer the time for serum PSA to reach the precastration level suggests a better prognosis. Mock-injected animals took 7.7 weeks compared to decoy AR1-558 injected mice that took 10.3 weeks (p = 0.0296) to progress to castration-recurrence. Similarly, GFP injected animals took 9.1 weeks compared to decoy GFP-AR1-558 injected mice that took 15.5 weeks (p = 0.0245) to progress to castration-recurrence (Fig 1B). The time for PSA to reach pre-castration levels was almost doubled in presence of decoy AR1-558 in comparison to control tumors (mock and GFP).
The time for the tumor to double in volume also has prognostic value. The longer it takes for the tumor to double in size, the more favorable the prognosis. In examining this parameter for prognosis, mock-injected animals required 4.1 weeks to double in tumor volume compared to decoy AR1-558 injected mice that took 8.4 weeks (p = 0.0095). GFP injected animals took 7.1 weeks compared to decoy GFP-AR1-558 injected mice that took 11.5 weeks (p = 0.021) (Fig 1C). Thus, in the presence of the decoy, approximately twice the amount of time was required for the tumor volume to double in the absence of testicular androgens. Consistent with reduced tumor growth, decoy AR1-558 decreased staining of the proliferation marker Ki67 (data not shown) in xenografts as previously reported [2]. Together these data (lower nadir, delayed time for PSA to return to pre-castration levels, and increased time for tumor volume to double) suggest that decoy AR1-558 can significantly delay the time for prostate cancer to become castration recurrent.
Effects of lentivirus delivery of decoy AR1-558 on other tissues
Upon the duration of the experiment, the major organs were surgically removed for histological review to determine the effect of viral delivery of decoy on spleen, liver, lung, heart, and kidney tissues. H&E staining showed no unusual pathology implying that decoy had no effect on the morphology of these tissues (data not shown). One interpretation could be that AR1-558 decoy does not have effect on tissues that are not dependent upon functional AR. Alternatively the lentivirus may not have been delivered to these other organs. To test this, we employed an antibody to GFP and analysed whole cell lysates from these organs from hosts treated with lentivirus. Western blot analysis showed detection of GFP in xenografts harvested from the host (Fig 2A), yet levels of GFP in the organs from the same animals were below levels of detection. This suggests that intratumoral injection of lentivirus to subcutaneous tumors was not efficiently delivered or expressed in other tissues. Since the animals were castrated, we did not examine the murine prostate organ in response to decoy AR1-558. Western blot analysis using an antibody to the AR NTD (anti-AR antibody to epitope 299–315) confirmed that decoy AR1-558 was delivered to the xenograft (Fig 2B). Levels of endogenous AR were also detected by this antibody, and the levels were not altered by expression of decoy as previously reported [2].
Fig 2. Levels of expression of decoys and endogenous AR in vivo.
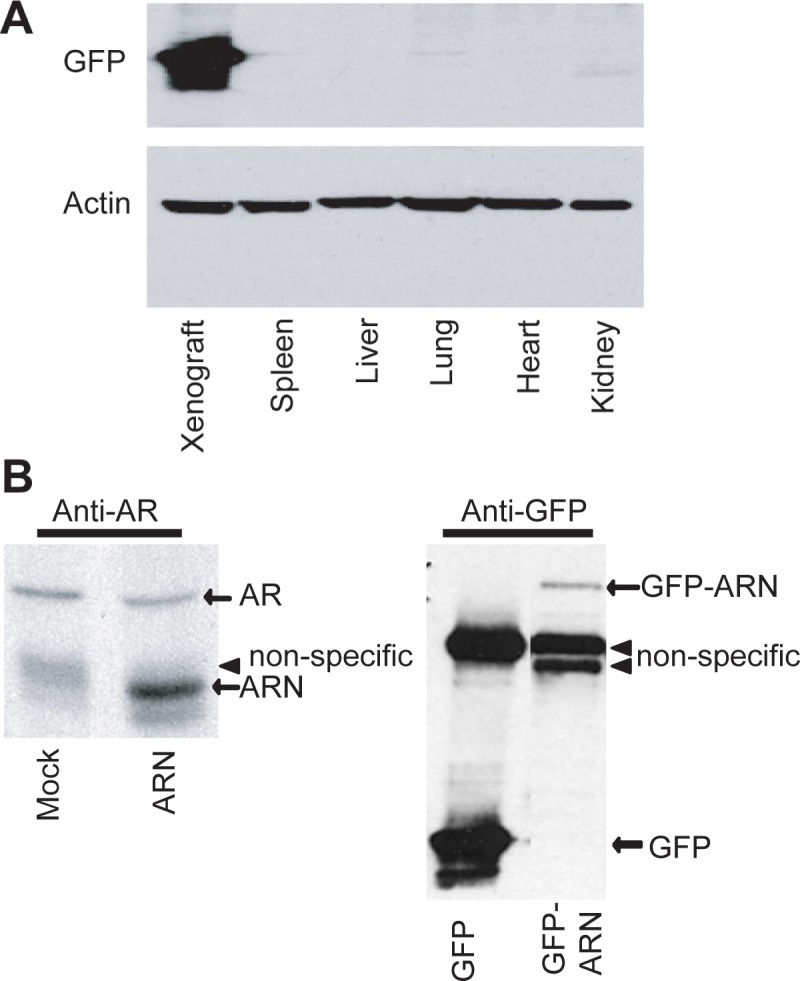
A, Western blot analysis for GFP with a representative animal showing extremely high expression of GFP delivered by lentivirus to the xenograft, yet non-detectable levels of expression in the spleen, liver, lung, heart, and kidney of the same animal. Similar levels of protein (40μg) from whole cell lysates. The membrane was stripped and re-probed for β-actin as a loading control. B, AR, AR1-558 (ARN), GFP, and GFP-AR1-558 (GFP-ARN) protein levels in harvested xenografts. A non-specific diffuse band migrates slightly slower than AR1-558 and was most apparent in the mock-treated lysates.
Decoy AR1-558 does not prevent nuclear localization of the AR
Nuclear AR protein is detected in secondary prostate cancer tumors from patients failing androgen deprivation therapy [7]. To determine if lentivirus delivery of decoys prevented nuclear localization of the AR, xenografts were stained for AR using antibodies that detect both the LBD and the NTD. Staining with an antibody to AR LBD showed predominantly nuclear staining with some cytoplasmic staining regardless of treatment (Fig 3A, column labeled C19). This antibody should only stain the AR and not decoy AR1-558. These LNCaP cells do not express detectable levels of AR splice variant protein [8]. An antibody to the NTD stains both decoy and AR and stained the nuclei of cells and was not altered by treatments (column labelled 441). LNCaP cells that stably expressed decoy AR1-558, or vector, and treated with androgen for 30 minutes induced the nuclear localization of the GFP-AR regardless of the presence of decoy AR1-558 (Fig 3B). Together these data suggest that decoy AR1-558 does not prevent nuclear localization of AR.
Fig 3. Decoy AR1-558 does not prevent nuclear localization of the AR.
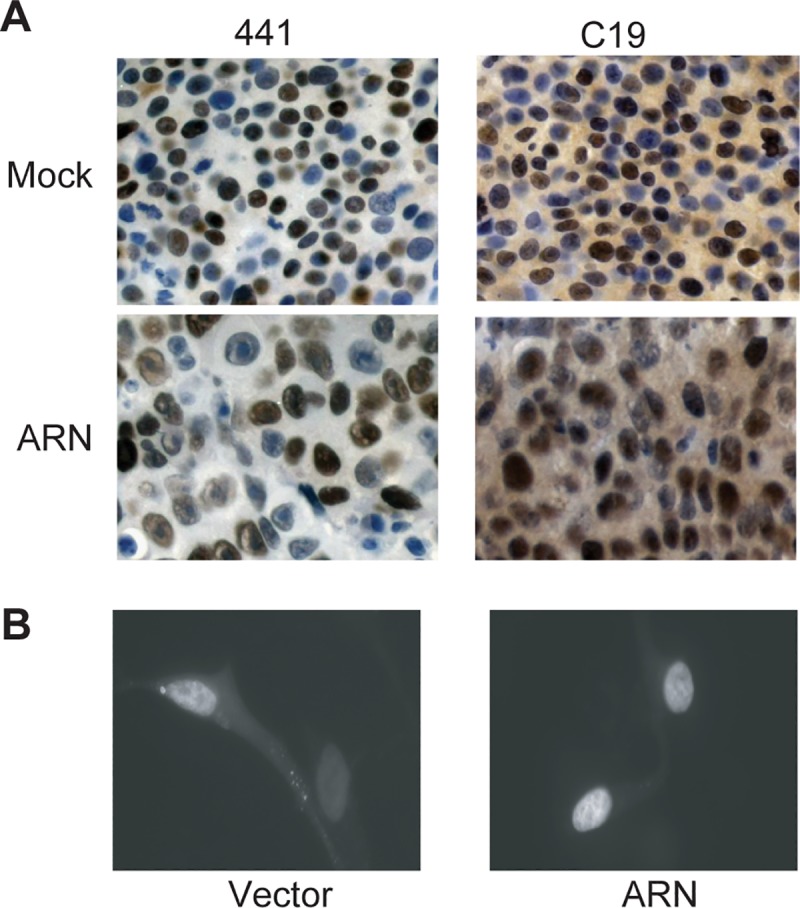
A, Xenografts were harvested at the duration of the experiments and sections were stained for AR NTD (441) or the LBD (C19). B, Fluorescent microscopy of GFP-AR in LNCaP cells stably expressing vector (left) or decoy (right) and treated with R1881 (10nM) for 30 minutes.
Decoy AR1-558 reduces the expression of androgen-regulated genes
To determine if decoy AR1-558 would alter the expression of androgen-regulated genes, qPCR was performed using total RNA from LNCaP cells that stably express decoy AR1-558, as well as from the castration-recurrent xenografts transduced with decoy AR1-558. LNCaP cells that stably express vector or decoy were treated with 10 nM R1881 for 24 hours and total RNA was harvested. qPCR was used to measure levels of expression of HMGCR, KLK2, KLK3/PSA, MAF, RHOU, and SAT1 that are normally induced by R1881 [9]. Levels of expression of these genes were significantly decreased in cells stably transfected with decoys (p<0.05 or p<0.01) in comparison to control cells expressing the vector (Fig 4A). Re-expression of some androgen-regulated genes occurs in castration-recurrent disease [10]. Therefore, we also measured the expression of the same set of genes using RNA prepared from harvested LNCaP tumors from castrated mice that had been treated lentivirus for decoys or controls (described in Fig 1). Levels of expression of these androgen-regulated genes that were decreased by decoys in vitro were also decreased in vivo in castration-recurrent tumors transduced with decoys (Fig 4B). These data suggest that decoy AR1-558 interferes with the regulation of androgen-regulated genes both in vitro in cells treated with R1881 and in vivo in castration-recurrent xenografts. Together, these in vitro and in vivo results support that decoy AR1-558 inhibits the transcriptional activity of endogenous AR.
Fig 4. Decoys block the expression of androgen-regulated genes.
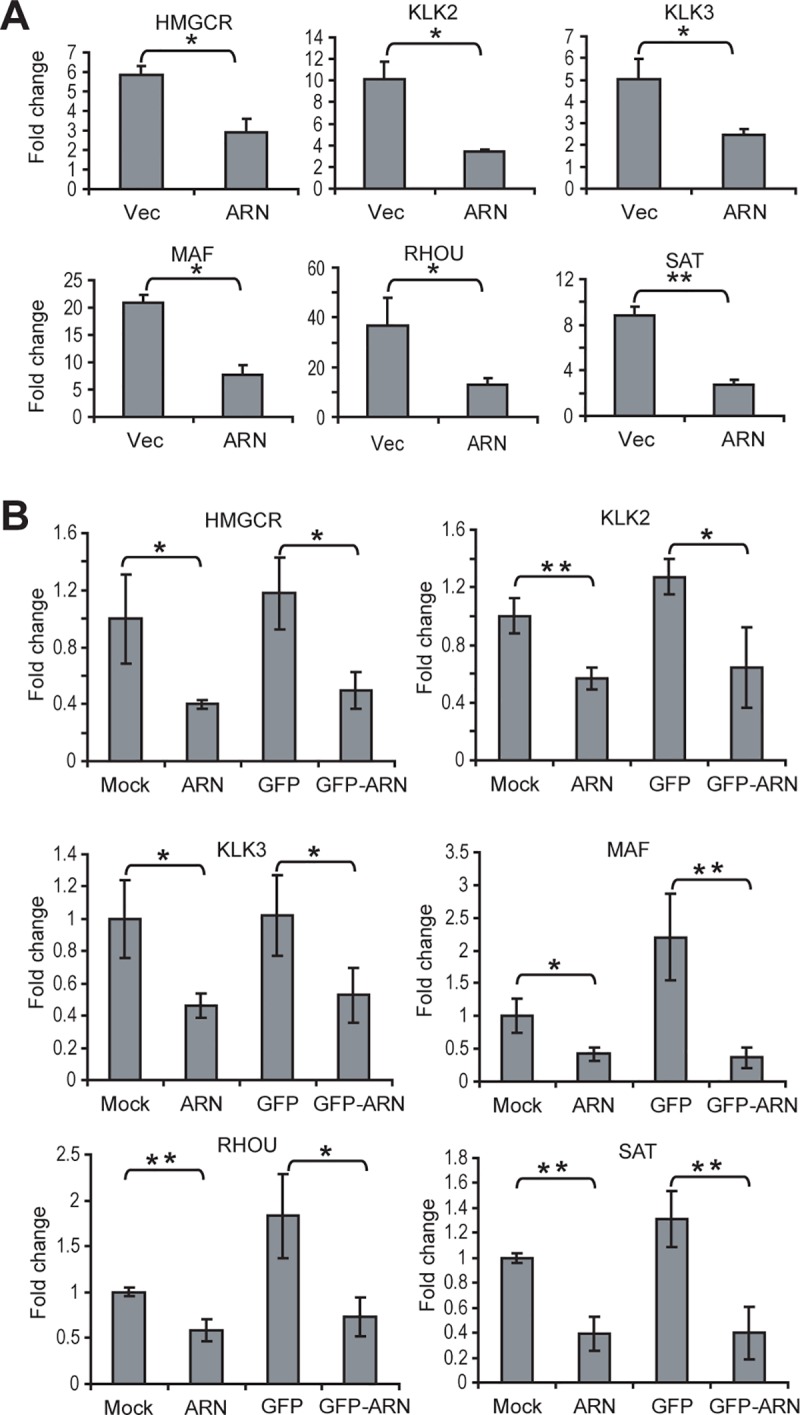
Real-time qPCR was performed using total RNA isolated from: A, LNCaP cells stably transfected with vector (Vec) or decoys (ARN) and treated for 24 hours with 10nM R1881; or B, xenografts injected with mock, AR1-558 (ARN), GFP, and GFP-AR1-558 (GFP-ARN). Transcript levels of HMGCR, KLK2, KLK3/PSA, MAF, RHOU, and SAT, were normalized to levels of GAPDH. The ratio of each transcript to GAPDH is plotted as fold-change. The bars represent the mean ± SD (n = 3). Student t-test: * p<0.05; **: p<0.01.
Decoy AR1-558 blocks AR-ARE interactions
Gene expression data generated here from in vitro and in vivo experiments provided evidence that decoy AR1-558 had a direct effect on the transcriptional activity of AR. To determine if decoy AR1-558 potentially altered the formation and stabilization of the transcriptional complex on AREs, we performed ChIP assays. Interaction of endogenous AR with the ARE in the enhancer of the PSA gene was measured in non-transfected and stably transfected (vector or decoy) cells in response to androgen. These studies revealed that the decoys significantly (p<0.05) reduced AR-ARE interaction by 50% in presence of R1881 in cells stably expressing the decoys in comparison to cells untransfected or solely expressing the vector (Fig 5). These data were consistent with reduced expression of PSA mRNA in cells expressing the decoy AR1-558 (Fig 4). Thus, decoy AR1-558 reduced interaction of endogenous AR with AREs which would result in decreased levels of transcription of androgen-regulated genes.
Fig 5. Decoys block AR-ARE interaction.
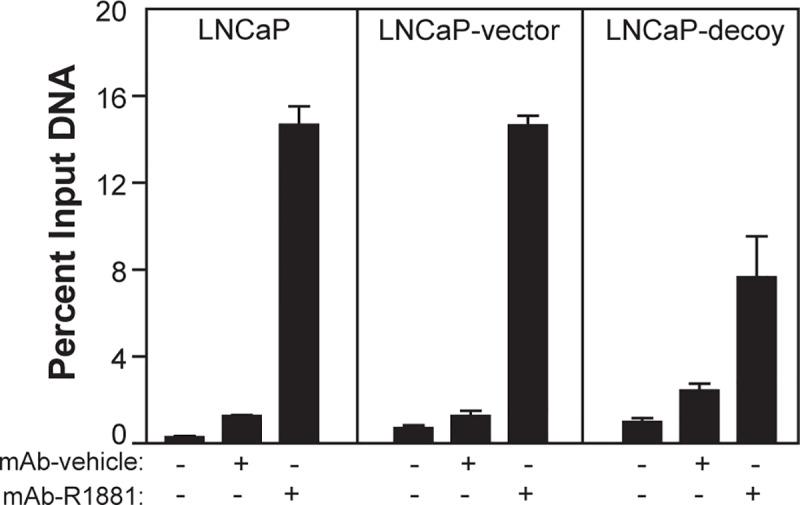
ChIP was performed in non-transfected and stably transfected (vector or decoy) cells in response to androgen. Parental LNCaP cells (untransfected) or stables expressing vector or decoy were treated ± androgens (R1881, 10 nM) for 3 hours and used in ChIP analyses with rabbit IgG (no antibody negative control) and anti-AR (mAb; C19 antibody to AR LBD). Eluted DNA fragments were purified and used for qPCR with primers designed to amplify the PSA ARE. Bars show the percentage input as the mean ± SD (n = 3). A representative result from repeated experiments is shown.
AR NTD decoys do not interact with the AR
To initiate transcription in response to androgen, dimerization of AR is needed and requires interactions between DNA-binding domains (i.e., DBD/DBD) (for a review see [11]). Heterodimerization of AR with truncated AR splice variant AR-V7 that lacks LBD and homodimerization of AR-Vs also requires DBD/DBD interactions [12]. Here the decoy lacks a DBD and is not predicted to interact with AR because of the requirement of DBD/DBD interactions. However, since 23FQNLF27 and 429WHTLF433 in AR NTD can be important in AR NTD-LBD interaction in response of AR to androgen [13], we tested if AR interacted with decoy. To do this, we immunoprecipated the AR using an antibody to the LBD of the AR in LNCaP cells that stably expressed the decoy. Cells used were either continually passaged in whole serum, or had been serum-starved prior to treatment with R1881 for 3 hr. Western blot analyses using an antibody to the NTD detected both decoy and the FL-receptor in samples prepared from whole cell lysates, supernatant, and wash (Fig 6A). Cells without decoy (stably transfected with vector) had no protein bands at the expected MW for the decoy (compare vector lanes to decoy lanes). Decoy AR1-558 was not detected in immunoprecipitated complexes with the AR (see lanes 7 and 8). This suggests that decoy AR1-558 does not interact with the AR to inhibit the receptor through a dominant negative mechanism.
Fig 6. Decoy molecules do not interact with the AR.

A, LNCaP cells stably expressing vector (V) or decoy AR1-558 (D) were incubated in serum (FBS) or with R1881 (1nM) for 3 h followed by immunoprecipation of the AR using an antibody to the LBD (Santa Cruz C19). Whole cell lysates (lanes 1 and 2), supernatant (lanes 3 and 4), wash (lanes 5 and 6), and immunoprecipitated complex-IP elution (lanes 7 and 8) were analyzed by Western blot using antibody to the AR NTD (Santa Cruz 441) to detect both AR and the decoy AR1-558. B, Decoy AR1-558 blocked ligand-independent activation of the AR by forskolin while AR1-233 and AR392-558 did not. LNCaP cells were transiently transfected with PSA(-630/+12)-luciferase reporter and expression vectors for His-tag, His-AR1-558, His-AR1-233, and His-AR392-558 and treated with forskolin (50μM) for 48 h under serum-free conditions. The percent induction of PSA-luciferase activities relative to values achieved with expression of His-tag is shown. Bars represent the mean ± SE of three separate experiments. C, Western blot analysis using an antibody to His-tag with whole cell lysates from LNCaP cells transiently transfected with expression vectors for His-tag, His-AR1-558, His-AR1-233, and His-AR392-558.
To determine if decoys that contain 23FQNLF27 or 429WHTLF433 could repress expression of PSA, we co-transfected LNCaP cells with expression vectors encoding regions with amino acids 1–233 (contains 23FQNLF27) or 392–558 (contains 429WHTLF433) of the AR NTD and PSA promoter-luciferase reporter gene construct prior to treating the cells with forskolin. Consistent with previous reports, forskolin increased PSA-luciferase activity in the absence of serum and androgen [2,14]. Decoy AR1-558 significantly reduced the induction of PSA-luciferase activity compared to control (positive control labeled “His-tag”) as shown previously [2]. Decoys encoding amino acids 1–233 or 392–558 that contain the (F/W)XXLF motifs did not inhibit induction of PSA-luciferase activity (Fig 6B). Levels of expression of these constructs were similar in cells as shown by Western blot analysis using an antibody to the his-tag motif (Fig 6C). Thus, the differences between the constructs of the AR NTD to inhibit PSA-luciferase activity was not due to reduced levels of expression. Together with the lack of detection of interaction between decoy AR1-558 and AR by co-immunoprecipitation, these data do not support a dominant negative mechanism of decoy AR1-558 to inhibit transcriptional activity of AR.
Androgens increase as well as repress gene expression through the full-length AR. Antiandrogens can de-repress the expression of some genes turned off in response to androgens. Here, in the presence of androgen, decoy AR1-558 did not de-repress the expression of the known androgen-repressed genes that were tested (Fig 7).
Fig 7. Effects of decoy on androgen-repressed genes.

Real-time qPCR was performed using total RNA isolated from LNCaP cells stably transfected with vector or decoy (ARN) and treated for 24 hours with 10nM R1881. Transcript levels of CAM2KN1, MMP16, SESN2, SLC44A1, ST7, TMEM144, UGT2B15, and UGT2B17 were normalized to levels of GAPDH. The ratio of each transcript to GAPDH is plotted as percent activity relative to vector control. The bars represent the mean ± SD (n = 3).
Discussion
Androgen deprivation therapy causes a temporary reduction in tumor burden concomitant with a decrease in serum levels of PSA [15] which is an AR-regulated gene. Unfortunately, prostate cancer will eventually begin to grow again in the absence of androgens to form castration-recurrent disease as characterized by a rising titer of serum PSA [16]. Enzalutamide and abiraterone, initially reduce serum PSA and increase survival by approximately 6 months in castration-recurrent patients [17,18]. Resistance to these drugs is associated with increasing serum levels of PSA which implies re-activation of AR transcriptional activity.
Evidence supporting the in vivo efficacy of targeting the AR NTD was shown by application of decoy AR1-558 molecules of AR NTD that inhibit the growth and hormonal progression of prostate cancer xenografts both in the presence and absence of androgen [2]. Those studies applied two models: one model used cells that stably expressed decoy AR1-558 to grow tumors with decoy; and the other model applied lentivirus delivery of decoy to established tumors in non-castrated mice. Here we investigated the effects of decoy in established tumors combined with castration. This is a model that better reflects the clinical representation of patients that undergo androgen deprivation therapy and develop castration-recurrent disease. Non-transfected LNCaP tumors were first established in NOD-SCID mice prior to castration and lentivirus delivery of the decoys. PSA nadir, time for PSA to reach pre-castration levels, and the time for the tumor to double in volume after therapy were all measured as an indication of tumor progression as reported in clinical studies [19,20]. Patients have a better prognosis when their PSA nadir reaches very low levels, or with long periods of the time required for rises in PSA or for their tumor volume to double. Our studies showed that the delivery of decoys to established tumors leads to a lower nadir, increased time for the tumor to double in volume as well as for PSA to reach the pre-castrate levels. These data concur with delayed hormonal progression in xenografts of LNCaP cells stably expressing decoy in castrated mice [2] and suggests that tumor progression is at least in part dependent on the AR NTD.
AR NTD has a high degree of intrinsic disorder and acts as a hub for protein-protein interactions. Not surprising is also the fact that the NTD is highly posttranslationally modified which can alter protein interactions and localization. Within the NTD is AF-1 which is essential for transcriptional activity. Thus targeting AR NTD for therapeutic intervention would be effective in preventing its transcriptional activity regardless of the presence or absence of ligand and LBD. Here we show that in combination with castration, lentivirus delivery of decoys to the AR NTD to established tumors delayed hormonal progression to castration-recurrence. The mechanism of action of the decoys involved decreased expression of genes normally regulated by androgen, and reduced physical interaction of the AR with AREs. The decoys did not reduce levels of endogenous AR protein, nor did decoys inhibit the activity of the AR through a dominant negative mechanism or involve amino acids 1–233 or 392–558 which contain the (F/W)XXLF motifs as well the core sequences for Tau-1 (178LKDIL182) and Tau-5 (435WHTLF439). The decoys also did not prevent nuclear localization of AR in response to ligand. Thus, overexpression of decoy AR1-558 allows the AR to be in the nucleus yet still prevents the interaction of endogenous AR with AREs to reduce transcription of genes regulated by AR.
Potentially, the mechanism by which decoy AR1-558 inhibits AR may involve changes in protein-protein interactions required for a stable and functional transcriptional complex. These interactions may involve coactivators, kinases, or enzymes altering posttranslational modifications. Currently, the AR is suggested to interact with over 169 different proteins [21], thereby complicating the identification of proteins that may be blocked by the decoys. However, decoy AR1-558 had no effect on PC3 xenografts that do not express AR, suggesting that decoy AR1-558 does not mop-up critical proteins that are generally required for survival and growth [2]. Although the AR NTD shares less than 15% homology with progesterone receptor (PR), glucocorticoid receptor (GR) and estrogen receptor (ER), these receptors do interact with some of the same proteins (e.g., SRC-1). Previous work has shown that decoy AR1-558 does not inhibit the transcriptional activity of ER or GR, suggesting it does not compete with proteins that are limiting for the activities of these transcription factors. However, PR was significantly inhibited by decoy AR1-558 [2]. These data highlight potential molecular mechanisms shared between the AR and PR. Recently, PR has been reported to be highly expressed and an independent negative prognostic factor for clinical trials for prostate cancer [22] thereby making inhibition of PR a potential benefit of an approach that blocks both AR NTD and PR.
Repression of gene expression by androgen may involve many different mechanisms and different domains of the androgen receptor. Corepressors such as SMRT can bind the AR NTD to mediate transrepression [23]. The mechanism of androgen repression of cyclin D1 gene expression involves androgen-bound AR recruited to a negative ARE and an SP1-binding site with recruitment of a repression complex that includes DAX-1 and HDAC-1 [24]. Some other repression mechanisms elucidated involve interaction of AR DNA-binding domain with Sp-1 or AR interactions with other proteins such as Runx2 protein [25], SF-1 [26], ATF-2 [27], FOXO1 [28], HOXB13 [29], and lysine-specific demethylase 1 (LSD1) [30]. Genes tested here are known to be repressed by androgen and MMP16, SLC44A1, ST7, and UGT2B15 and UGT2B17 all have AR binding sites [31,32]. However, these AREs are not necessarily negative AREs since UGTY2B15/B17 requires AR for basal expression as well [32]. If AR is knocked down, levels of UGT2B15/17 are attenuated [32] thereby highlighting the complexity of the mechanism. If decoy AR1-558 does not bind to the full-length AR, but rather competes with full-length AR for essential coactivators or other proteins that bind to the AR NTD as we suggest to inhibit transactivation, it may be that the mechanism of androgen repression does not involve AR NTD, at least not for these specific genes. Further experiments are warranted to delineate the mechanisms of how antagonists of AR NTD impact androgen-repressed genes.
In conclusion, our findings here support that the AR NTD is a feasible therapeutic target for the development of novel drugs for the treatment of prostate cancer. Recently the first AR NTD antagonist, a prodrug of EPI-002 [33,34], started Phase 1 clinical trials for prostate cancer patients that have failed abiraterone and/or enzalutamide (Clinical trials NCT02606123).
Supporting information
(PPTX)
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This research was supported by funding from the USA National Cancer Institute (2R01CA105304) to MDS. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Jenster G, van der Korput HA, Trapman J, Brinkmann AO. Identification of two transcription activation units in the N-terminal domain of the human androgen receptor. J Biol Chem. 1995; 270(13),7341–7346. [DOI] [PubMed] [Google Scholar]
- 2.Quayle SN, Mawji NR, Wang J, Sadar MD. Androgen receptor decoy molecules block the growth of prostate cancer. Proc Natl Acad Sci U S A. 2007;104(4),1331–1336. 10.1073/pnas.0606718104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Riegman PH, Vlietstra RJ, van der Korput JA, Brinkmann AO, Trapman J. The promoter of the prostate-specific antigen gene contains a functional androgen responsive element. Mol Endocrinol. 1991;5(12),1921–1930. 10.1210/mend-5-12-1921 [DOI] [PubMed] [Google Scholar]
- 4.Cleutjens KB, van Eekelen CC, van der Korput HA, Brinkmann AO, Trapman J. (1996) Two androgen response regions cooperate in steroid hormone regulated activity of the prostate-specific antigen promoter. J Biol Chem. 1996;271(11),6379–6388. [DOI] [PubMed] [Google Scholar]
- 5.Jia L, Coetzee GA. Androgen receptor-dependent PSA expression in androgen-independent prostate cancer cells does not involve androgen receptor occupancy of the PSA locus. Cancer Res. 2005;65(17),8003–8008. 10.1158/0008-5472.CAN-04-3679 [DOI] [PubMed] [Google Scholar]
- 6.Tomioka A, Tanaka N, Yoshikawa M, Miyake M, Anai S, Chihara Y,et al. Nadir PSA level and time to nadir PSA are prognostic factors in patients with metastatic prostate cancer. BMC Urol. 2014;29;14:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Kwast TH, Schalken J, Ruizeveld de Winter JA, van Vroonhoven CC, Mulder E, Boersma W, et al. Androgen receptors in endocrine-therapy-resistant human prostate cancer. Int J Cancer. 1991;48(2),189–193. [DOI] [PubMed] [Google Scholar]
- 8.Yang YC, Banuelos CA, Mawji NR, Wang J, Kato M, Haile S, et al. Targeting Androgen Receptor Activation Function-1 with EPI to Overcome Resistance Mechanisms in Castration-Resistant Prostate Cancer. Clin Cancer Res. 2016;22(17):4466–77. 10.1158/1078-0432.CCR-15-2901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang G, Jones SJ, Marra MA, Sadar MD. Identification of genes targeted by the androgen and PKA signaling pathways in prostate cancer cells. Oncogene. 2006;25(55),7311–7323. 10.1038/sj.onc.1209715 [DOI] [PubMed] [Google Scholar]
- 10.Gregory CW, Hamil KG, Kim D, Hall SH, Pretlow TG, Mohler JL, et al. Androgen receptor expression in androgen-independent prostate cancer is associated with increased expression of androgen-regulated genes. Cancer Res. 1998;58(24),5718–5724. [PubMed] [Google Scholar]
- 11.Centenera MM, Harris JM, Tilley WD, Butler LM. The contribution of different androgen receptor domains to receptor dimerization and signaling. Mol. Endocrinol. 2008;22:2373–82. 10.1210/me.2008-0017 [DOI] [PubMed] [Google Scholar]
- 12.Xu D, Zhan Y, Qi Y, Cao B, Bai S, Xu W, et al. Androgen Receptor Splice Variants Dimerize to Transactivate Target Genes. Cancer Research 2015;75(17):3663–71. 10.1158/0008-5472.CAN-15-0381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He B, Kemppainen JA, Wilson EM. FXXLF and WXXLF sequences mediate the NH2-terminal interaction with the ligand binding domain of the androgen receptor. J Biol Chem. 2000;275(30),22986–22994. 10.1074/jbc.M002807200 [DOI] [PubMed] [Google Scholar]
- 14.Sadar MD. Androgen-independent induction of prostate-specific antigen gene expression via cross-talk between the androgen receptor and protein kinase A signal transduction pathways. J Biol Chem. 1999;274(12),7777–7783. [DOI] [PubMed] [Google Scholar]
- 15.Culig Z, Klocker H, Bartsch G, Hobisch A. Androgen receptors in prostate cancer. Endocr Relat Cancer. 2002;9(3),155–170. [DOI] [PubMed] [Google Scholar]
- 16.Huber PR, Schnell Y, Hering F, Rutishauser G. Prostate specific antigen. Experimental and clinical observations. Scand J Urol Nephrol Suppl. 1987;104,33–39. [PubMed] [Google Scholar]
- 17.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367(13):1187–97. 10.1056/NEJMoa1207506 [DOI] [PubMed] [Google Scholar]
- 18.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364(21):1995–2005. 10.1056/NEJMoa1014618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morote J, Esquena S, Abascal JM, Trilla E, Cecchini L, Raventós CX, et al. Usefulness of prostate-specific antigen nadir as predictor of androgen-independent progression of metastatic prostate cancer. Int J Biol Markers. 2005;20(4):209–216. [DOI] [PubMed] [Google Scholar]
- 20.Roberts SG, Blute ML, Bergstralh EJ, Slezak JM, Zincke H. PSA doubling time as a predictor of clinical progression after biochemical failure following radical prostatectomy for prostate cancer. Mayo Clin Proc. 2001;76(6):576–81. 10.4065/76.6.576 [DOI] [PubMed] [Google Scholar]
- 21.Heemers HV, Tindall DJ. Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex. Endocr Rev. 2007;28(7):778–808. 10.1210/er.2007-0019 [DOI] [PubMed] [Google Scholar]
- 22.Grindstad T, Andersen S, Al-Saad S, Donnem T, Kiselev Y, Nordahl Melbø-Jørgensen C, et al. High progesterone receptor expression in prostate cancer is associated with clinical failure. PLoS One. 2015;10(2):e0116691 10.1371/journal.pone.0116691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dotzlaw H, Moehren U, Mink S, Cato AC, Iñiguez Lluhí JA, Baniahmad A. The amino terminus of the human AR is target for corepressor action and antihormone agonism. Mol Endocrinol. 2002;16(4):661–73. 10.1210/mend.16.4.0798 [DOI] [PubMed] [Google Scholar]
- 24.Lanzino M, Sisci D, Morelli C, Garofalo C, Catalano S, Casaburi I, et al. Inhibition of cyclin D1 expression by androgen receptor in breast cancer cells-identification of a novel androgen response element. Nucleic Acids Res. 2010;38(16):5351–65. 10.1093/nar/gkq278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baniwal SK, Khalid O, Sir D, Buchanan G, Coetzee GA, Frenkel B. Repression of Runx2 by androgen receptor (AR) in osteoblasts and prostate cancer cells: AR binds Runx2 and abrogates its recruitment to DNA. Mol Endocrinol. 2009;23(8):1203–14. 10.1210/me.2008-0470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jorgensen JS, Nilson JH. AR suppresses transcription of the LH beta subunit by interacting with steroidogenic factor-1. Mol Endocrinol. 2001;15(9):1505–16. 10.1210/mend.15.9.0691 [DOI] [PubMed] [Google Scholar]
- 27.Jorgensen JS, Nilson JH. AR suppresses transcription of the alpha glycoprotein hormone subunit gene through protein-protein interactions with cJun and activation transcription factor 2. Mol Endocrinol. 2001;15(9):1496–504. 10.1210/mend.15.9.0690 [DOI] [PubMed] [Google Scholar]
- 28.Bohrer LR, Liu P, Zhong J, Pan Y, Angstman J, Brand LJ, et al. FOXO1 binds to the TAU5 motif and inhibits constitutively active androgen receptor splice variants. Prostate. 2013;73(10):1017–27. 10.1002/pros.22649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Norris JD, Chang CY, Wittmann BM, Kunder RS, Cui H, Fan D, et al. The homeodomain protein HOXB13 regulates the cellular response to androgens. Mol. Cell 2009;36:405–416. 10.1016/j.molcel.2009.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cai C, He HH, Chen S, Coleman I, Wang H, Fang Z, et al. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell. 2011;20(4):457–71. 10.1016/j.ccr.2011.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin B, Wang J, Hong X, Yan X, Hwang D, Cho JH, et al. Integrated expression profiling and ChIP-seq analyses of the growth inhibition response program of the androgen receptor. PLoS One. 2009;4(8):e6589 10.1371/journal.pone.0006589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bao BY, Chuang BF, Wang Q, Sartor O, Balk SP, Brown M, et al. Androgen receptor mediates the expression of UDP-glucuronosyltransferase 2 B15 and B17 genes. Prostate. 2008;68(8):839–48. 10.1002/pros.20749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, Myung JK, et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell. 2010;17(6):535–46. 10.1016/j.ccr.2010.04.027 [DOI] [PubMed] [Google Scholar]
- 34.Myung JK, Banuelos CA, Fernandez JG, Mawji NR, Wang J, Tien AH, et al. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J Clin Invest. 2013;123(7):2948–60. 10.1172/JCI66398 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PPTX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.