

SUMMARY
Ca2+ dynamics and oxidative signaling are fundamental mechanisms for mitochondrial bioenergetics and cell function. The MCU complex is the major pathway by which these signals are integrated in mitochondria. Whether and how these coactive elements interact with MCU has not been established. As an approach towards understanding the regulation of MCU channel by oxidative milieu, we adapted inflammatory and hypoxia models. We identified the conserved cysteine 97 to be the only reactive thiol in human MCU that undergoes S-glutathionylation. Furthermore, biochemical, structural and superresolution imaging analysis revealed that MCU oxidation promotes MCU higher-order oligomer formation. Both oxidation and mutation of MCU Cys-97 exhibited persistent MCU channel activity with higher [Ca2+]m uptake rate, elevated mROS and enhanced [Ca2+]m overload-induced cell death. In contrast, these effects were largely independent of MCU interaction with its regulators. These findings reveal a distinct functional role for Cys-97 in ROS sensing and regulation of MCU activity.
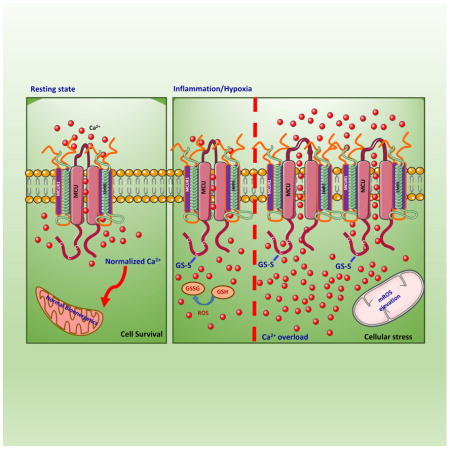
Graphical Abstract
INTRODUCTION
Oxidative stress and mitochondrial calcium ([Ca2+]m) signaling are the two functional entities that have been established often to go hand-in-hand in order to maintain a normal cellular physiology (Brookes et al., 2004; Hamanaka and Chandel, 2010; Kiselyov and Muallem, 2016). Paradoxically, a cell that predominantly relies on oxidative phosphorylation for its energy source always encounters the risk of heightened oxidative stress (Murphy, 2012). This is largely due to the production of superoxide resulting from miscreant electrons prematurely reacting with molecular oxygen. The electron transport chain H+ pumping of mitochondria is involved in regulating the major functions of the organelle that includes ATP production, generation of reactive oxygen species, the regulation of calcium uptake, and the activation of cell death (Duchen, 2000; Glancy and Balaban, 2012; Gunter et al., 1994; Nicholls, 2005; O’Rourke, 2007; Orrenius et al., 2003; Santo-Domingo and Demaurex, 2012). The Ca2+ taken up by the mitochondria participates in stimulation of bioenergetics through activation of Ca2+-sensitive dehydrogenases to promote ATP synthesis (Denton and McCormack, 1990; Glancy and Balaban, 2012; Hajnoczky et al., 1995). Although [Ca2+]m signaling has been established to be crucial for both physiological and pathological processes (Drago et al., 2011; Rizzuto et al., 2012), the molecular identity of the machinery was recently elucidated by genomic approaches and several proteins have been implicated to participate in [Ca2+]m uptake, including the MCU and its regulators MICU1, MCUR1, EMRE, MCUb, and MICU2 (Baughman et al., 2011; Chaudhuri et al., 2016; Csordas et al., 2013; De Stefani et al., 2011; Kamer and Mootha, 2014; Mallilankaraman et al., 2012a; Mallilankaraman et al., 2012b; Patron et al., 2014; Perocchi et al., 2010; Plovanich et al., 2013; Raffaello et al., 2013; Sancak et al., 2013). Increasing evidence from a number of molecular, biochemical, and structural studies indicate that MCU is the pore component of the uniporter complex (Kamer and Mootha, 2015; Kirichok et al., 2004; Lee et al., 2016; Lee et al., 2015; Oxenoid et al., 2016(Vais et al., 2016). Mitochondria function as a sink for Ca2+ during cellular activation, and thus generate copious amounts of ROS that are important mediators of many physiological and pathological processes (Duchen, 2000). Oxidative stress also stimulates [Ca2+]m overload followed by PTP opening (Kroemer et al., 2007; Pan et al., 2013; Shanmughapriya et al., 2015a), however, it is unknown whether intracellular ROS affects [Ca2+]m homeostasis by targeting MCU channels or its regulators.
Posttranslational modification (PTM) fine-tunes both physical expansion and functional activity of proteins. In cellular system, oxidation is one of the most frequently occurring PTMs used as a powerful functional on/off switch mechanisms. Cysteine (Cys) is a highly malleable amino acid susceptible to many types of oxidative PTMs (Brewer et al., 2015). The sulfur atom of Cys provides a considerable range of chemical reactivity to the major perceived oxidative stress and offers structural flexibility in the proteome. Cys is incorporated into proteins as thiol (RSH) form and thiols can be oxidized to sulfenic acids (RSO−) intermediate to form disulfides (RSSR) and higher oxidation states (e.g., RSO2−). Thus, the thiol moiety of Cys undergoes a range of chemical modifications enabling biological switching of structure and reactivity. These modifications include sulfenylation, disulfide formation, S-glutathionylation, S-nitrosylation, persulfidation, metalation, and other modifications (Go et al., 2015). Emerging evidence suggests that Cys Ox-PTMs function as redox-switches that regulate the cell’s response to oxidative stimuli and may work in concert with other PTMs (such as phosphorylation, acetylation, ubiquitination, among others) to determine the ultimate biological outcome and cell phenotype. Previous studies demonstrated that the evolutionarily conserved Cys residue in ER Ca2+ sensor STIM1 functions as a negative regulator of STIM1/Orai1 store operated Ca2+ entry and endothelial functional preservation (Gandhirajan et al., 2013; Hawkins et al., 2010; Soboloff et al., 2012). Currently there is a lack of knowledge on the PTMs of the core uniporter complex protein, MCU and thereby channel regulation. Here we sought to investigate whether redox dependent alterations in MCU potentiates persistent channel activity and affects cellular bioenergetics and cell survival.
Here, we demonstrate that of all the cysteines that exist in human MCU, Cys-97 is a reactive thiol and undergoes S-glutathionylation. To study the effects of ROS in MCU oxidation and channel activation, we utilized LPS-mediated inflammation and hypoxia/reoxygenation (H/R) models of oxidative stress and found that mitochondrial-derived oxidants markedly induced MCU oxidation and augmented MCU channel current and persistent [Ca2+]m uptake, whereas the [Ca2+]c dynamics remained unaffected. We also found that either oxidation of MCU or mutation of Cys97 to MCUC97A or MCUC97M resulted in a similar MCU clustering, higher order oligomer formation and [Ca2+]m uptake phenotype. Importantly, the oxidation of Cys-97 relieves MCU from MICU1/MICU2 gatekeeping regulation that results in basal [Ca2+]m accumulation. These results establish that MCU oxidation induces persistent [Ca2+]m uptake and perturbs mitochondrial bioenergetics and cell function.
RESULTS
Inflammatory and Hypoxia-Derived Oxidants Modulate MCU Activity Through Oxidation
Mitochondria shape cellular Ca2+ dynamics by sequestering [Ca2+]c. Following GPCR or tyrosine-kinase agonist stimulation, the increase in [Ca2+]c is sensed by mitochondria and facilitates MCU-mediated [Ca2+]c clearance (Berridge et al., 2003; Clapham, 2007). Over a range covered by submicromolar [Ca2+]c, MCU plays only a nominal role but becomes more relevant when [Ca2+]c is elevated >2 μM (Antony et al., 2016; Csordas et al., 2013; Liu et al., 2016; Mallilankaraman et al., 2012b; Nicholls, 1978; Tsai et al., 2016). Thus [Ca2+]c transients shape [Ca2+]m dynamics and vice versa. Apart from [Ca2+]c controlling MCU activity, we hypothesize that oxidative stress can also independently regulate MCU-mediated [Ca2+]m uptake.
To understand the regulatory role of oxidants in modulating MCU activity, HPMVECs were treated with 10 μg/ml LPS for 5 h. Following LPS treatment, cells were imaged for simultaneous measurement of [Ca2+]c and [Ca2+]m dynamics. HPMVECs were stimulated with GPCR agonist, thrombin (1 mU/ml) and change in [Ca2+]c and [Ca2+]m dynamics were measured. Both control and LPS treated HPMVECs on stimulation with thrombin mobilized robust [Ca2+]c (Figure 1A and 1B) that was indistinguishable between control and LPS treated cells. However, strikingly, LPS treatment resulted in a sustained elevation of [Ca2+]m uptake after thrombin stimulation and remained elevated above baseline (Figure 1B and 1C). Additionally, to assess changes in [Ca2+]m dynamics by oxidants, HPMVECs and HeLa cells were transfected with genetically encoded mitochondrial Ca2+ sensor, GCaMP2-mt. 48 hours post-transfection, HPMVECs and HeLa cells were treated with LPS (5 hours) and H2O2 (100 μM; for 30 min), respectively. The changes in [Ca2+]m dynamics were measured after stimulation with thrombin or histamine. Cells treated with LPS/H2O2 resulted in sustained [Ca2+]m accumulation phenotype (Figure 1D and 1E; Figure S1A and S1B) with no observable differences in [Ca2+]c dynamics (Figure 1F and 1G; Figure S1C and S1D).
Figure 1. Hypoxia and Inflammatory-Mediated Oxidative Stress Alters Sensitivity of MCU and [Ca2+]m Uptake.

(A and B) Mean traces for [Ca2+]c (Fluo-4) and [Ca2+]m (rhod-2) responses in HPMVECs treated with (B) or without (A) LPS (10 μg/ml) for 5 h. Cells were stimulated with thrombin (1 mU/ml) and changes in [Ca2+]c and [Ca2+]m fluorescence were measured. (C) Quantification of peak rhod-2 fluorescence. Bar represents Mean ± SEM; *** P <0.001; n = 3. (D) Mean traces for [Ca2+]m (GCaMP2-mt) fluorescence measured in control, LPS, LPS + MnSOD, and LPS+MnSOD + PRDX3 treated HPMVECs. (E) Quantification of normalized GCaMP2-mt fluorescence at peak and 600 s. Bar represents Mean ± SEM; * P <0.05; n = 4–7. (F) Mean traces of [Ca2+]c R-Geco fluorescence in HPMVECs. (G) Quantification of normalized R-GECO1 fluorescence at peak. Bar represents Mean ± SEM; * P <0.05; n = 4–7. (H) Representative traces of mitochondrial membrane potential (ΔΨm) in control (VE-Cre) and MCUΔEC MPMVECs treated with or without LPS. (I) Quantification of the ΔΨm before CCCP addition. Bar represents Mean ± SEM; *** P <0.001; n = 4–7. (J) Representative [Ca2+]out traces before and after CCCP (3 μM) in VE-Cre and MCUΔEC MPMVECs treated with or without LPS. (K) Quantification of resting matrix [Ca2+]m after the addition of CCCP. Data represents Mean ± SEM; **P <0.01; ns = not significant; n = 4. (L) Representative images showing MitoSox Red fluorescence. (M) Bar graph indicates MitoSox Red fluorescence in HPMVECs. Data indicate Mean ± SEM; *** P <0.001. n=4–6. (N) Quantification of MitoSox Red fluorescence. Data represents Mean ± SEM; ***P <0.01; ns = not significant; n = 4. (O) Oxidative stress-induced modification of MCU complex components. Lysates were prepared, incubated with mPEG5 and Western blotted with FLAG antibody (n = 3). (P) HPMVECs expressing MCU (Ad MCU) were exposed to menadione (10 μM), LPS (10 μg/ml) or BSO (200 μM). Lysates were prepared, incubated with mPEG5 and Western blotted with FLAG antibody (n = 3). (Q) Cartoon depicting the oxidation of MCU alters MCU-mediated [Ca2+]m uptake during oxidative stress.
It is predicted that the initial [Ca2+]m uptake by LPS stimulation could enhance the production of NADH that promotes electron leak, which results in mitochondrial membrane polarization and mitochondrial ROS (mROS) production. To test whether the MCU-mediated Ca2+ uptake is an effective signal for mROS production, we measured the mitochondrial membrane polarization in freshly isolated mouse microvascular pulmonary endothelial cells (MPMVECs) from control (VE-Cre) and endothelial-specific MCU KO mice (MCUΔEC) (Tomar et al., 2016). LPS stimulation promoted the mitochondrial hyperpolarization in VE-Cre MPMVEC’s but not MCUΔEC (Figure 1H and I) which is consistent with MCU-mediated [Ca2+]m accumulation in VE-Cre MPMVECs (Figure 1J and 1K). Together, these data indicate that the initial MCU-mediated [Ca2+]m uptake results in mROS production and the positive feedback role of mROS could oxidatively modify the MCU complex for persistent [Ca2+]m overload (Figure 1Q).
To further investigate whether the excessive production of mROS was responsible for the sustained [Ca2+]m mobilization, we primed HPMVECs with LPS for three hours. LPS challenged HPMVECs had elevated mROS as quantified by mitoSOX fluorescence and mitochondria-targeted H2O2-sensitive HyPer-Mito ROS sensor (Figure 1L and 1M; Figure S1 E). Additionally, LPS stimulation did not increase mROS production in MCUΔEC further substantiating the role for MCU-mediated [Ca2+]m uptake in mROS production (Figure 1N). Freshly isolated adult ventricular myocytes were subjected to hypoxia (2 h) followed by re-oxygenation (30 min). Similar to LPS response, cardiomyocytes exposed to H/R exhibited increased mitoSOX fluorescence indicative of mROS production (Figure S1F). Consistent with increased mROS levels, the rate of [Ca2+]m uptake was increased in Neonatal Rat Ventricular Myocytes (NRVMs) exposed to H/R (Figure S1G–S1J). Next we asked whether targeted scavenging of mROS by expressing mitochondrial antioxidants MnSOD (Ad-MnSOD virus) and peroxiredoxin 3 (PRDX3), could possibly revert the sustained [Ca2+]m mobilization phenotype. HPMVECs transfected with GCaMP2-mt were transduced with either Ad-MnSOD or in combination with PRDX3. 48 hours post-infection/transfection, cells were treated with LPS for 5 hours and changes in [Ca2+]m uptake was measured after thrombin stimulation. Since oxidative insults-mediated intracellular ROS production is contributed by mitochondria, ectopic expression of antioxidants alleviated the sustained [Ca2+]m uptake phenotype (Figure 1D–1F; Figure S1A and 1B), indicating a strong link between oxidative signaling and MCU-mediated [Ca2+]m uptake (Figure 1Q).
Having demonstrated that mROS levels can amplify MCU-mediated [Ca2+]m uptake, we next investigated whether uniporter complex components are sensitive to ROS. To investigate cysteine modifications of MCU complex, we used a gel-shift assay using methoxypolyethylene glycol linked maleimide (mPEG5) that measures the redox changes in proteins in situ (Leichert and Jakob, 2004). FLAG-tagged MCU, MCUb, MICU1, MCUR1 and EMRE expressing HEK293T cells were exposed to thiol oxidizing agent, menadione for 10 min and mPEG5 conjugation was performed (Figure 1O; Figure S1K). Only a robust mPEG5 conjugated MCU was resolved by Western blotting following menadione treatment suggesting at least one freely available cysteine for oxidation. Although MCUb sequence alignment predicted a conserved cysteine, we observed a nominal mPEG5 conjugated MCUb (Figure S1K blue asterisk). Similar to menadione-treated HEK293T cells, HPMVECs pretreated with LPS (10 μg/ml; for 5 hr) or glutathione depleting agent buthionine sulfoximine (24 hr) which halts GSH synthesis also resulted in MCU oxidation as indicated by MCU-mPEG formation (Figure 1P). Additionally, NRVMs either subjected to H/R or stimulation by β-adrenergic agonist, isoproterenol also elicited considerable levels of MCU oxidation (Figure S1L and S1M). Together, these data indicate that MCU is a redox sensitive channel, which could augment [Ca2+]m accumulation during oxidative stress conditions (Figure 1Q).
Cysteine Residues of MCU Are Redox Sensors and MCU Cysteine 97 Regulates IMCU That Controls Mitochondrial Matrix Ca2+ Accumulation
To determine the cysteines targeted by oxidants, computational analysis of MCU protein sequence was performed. The sequence analysis identified three potentially reactive cysteine residues at positions 67, 97, and 191 (human) of which C97 is highly conserved (Figure S2A). To evaluate the importance of cysteine residues in MCU channel activity and [Ca2+]m uptake, we substituted an alanine residue: MCUC67A, MCUC97A, MCUC191A, and MCUCF (CF-Cysteine Free) (Figure 2A) and examined the effect of these mutations on [Ca2+]m uptake and IMCU following expression in HeLa, HEK293T, and HPMVEC cells. We hypothesized that the modifications of cysteine residues in MCU could modulate the MCU channel activity and thereby control MCU-mediated [Ca2+]m uptake. To assess this, HeLa cells were co-transfected with MCU-mRFP, MCUC67A-mRFP, MCUC97A-mRFP, MCUC191A-mRFP, and MCUCF-mRFP along with GCaMP2-mt (Figure S2B). After baseline recording of [Ca2+]m fluorescence, GPCR agonist histamine was added. Cells transfected with MCU, MCUC67A, and MCUC191A mobilized robust [Ca2+]m upon histamine stimulation and returned to baseline representing modest [Ca2+]m dynamics (Figure S2F and S2G). In contrast, cells transfected with MCUC97A and MCUCF resulted in a phenotypic shift with elevated [Ca2+]m mobilization and fluorescence remained above the baseline indicating increased MCU-mediated [Ca2+]m uptake (Figure 2B and 2C) with no significant changes in [Ca2+]c dynamics (Figure 2D; Figure S2H and S2I) and mitochondrial membrane potential (Figure S2E) in any MCU mutants.
Figure 2. Oxidative Stress Modulates MCU Channel Activity.

(A) Schematic of full length MCU depicting its functional domains and mutations in cysteine residues (C67A, C97A, C191A, and CF). (B) Mean traces of [Ca2+]m (GCaMP2-mt) fluorescence in HeLa cells expressing MCU, MCUC97A, and MCUCF. (C) Quantification of normalized GCaMP2-mt fluorescence at peak and 600 s. Data represents Mean ± SEM; ***P <0.001; n = 4–6. (D) Mean traces of [Ca2+]c (R-Geco) fluorescence measured in HeLa cells expressing MCU, MCUC97A, and MCUCF. Insert is the peak quantification of R-Geco fluorescence after histamine stimulation. Data represents Mean ± SEM; ***P <0.001; n = 4–6. (E) Representative [Ca2+]out traces before and after CCCP (3 μM) addition in HEK293T cells stably expressing control vector, MCU, MCUC67A, MCUC97A, MCUC191A, and MCUCF. (F) Quantification of resting matrix [Ca2+]m after the addition of CCCP. Data represents Mean ± SEM; **P <0.01; ns = not significant; n = 4. (G) Mean traces of [Ca2+]m (GCaMP2-mt) fluorescence in HeLa cells expressing MCU and MCUC97M. (H) Quantification of normalized GCaMP2-mt fluorescence at peak and 600 s. Data represents Mean ± SEM; ***P <0.001; n = 4–6. (I) Quantification of resting matrix [Ca2+]m after the addition of CCCP as in Figure 2E. Data represents Mean ± SEM; **P <0.01; ns = not significant; n = 4. (J) Representative traces of [Ca2+]out in control and MCU mutant HEK293T cells. (K) Quantification of the rate of [Ca2+]m uptake as a function of decrease in [Ca2+]out after 10 μM Ca2+ pulse. Data represents Mean ± SEM; **P <0.01; ns = not significant; n = 4. (L) Quantification of the rate of [Ca2+]m efflux. Data represents Mean ± SEM; **P <0.01; ns = not significant; n = 4. (M) Representative [Ca2+]out traces in HEK293T cells stably expressing control vector, MCU, MCUC97A, and MCUCF. (N) Quantification of the rate of [Ca2+]m uptake as a function of decrease in [Ca2+]out. Data represents Mean ± SEM; * P <0.05, **P <0.01, ***P <0.001; n = 4. (O) Representative [Ca2+]out traces in HEK293T cells stably expressing control and MCU mutants. (P) Quantification of total matrix [Ca2+]m released following 20 μM Ca2+ pulse. Data represents Mean ± SEM; **P <0.01; n = 4. (Q) IMCU current in mitoplasts derived from HEK293T control and MCU mutant cells. The inset is a representative Western blot probed with an antibody specific for MCU (R) IMCU densities (pA/PF) in mitoplasts derived from control vector, MCU, MCUC97A, and MCUCF. Data represents Mean ± SEM; ***P <0.001; n = 5–9.
Because the Cys-97 mutant caused sustained [Ca2+]m uptake upon GPCR agonist stimulation, we next analyzed the MCU-mediated matrix [Ca2+]m accumulation. We tested the basal matrix [Ca2+]m in HEK 293T MCU, MCUC67A, MCUC97A, MCUC191A, and MCUCF cells. The basal mitochondrial matrix Ca2+ was higher in HEK 293T cells expressing MCUC97A and MCUCF (Figure 2E and 2F). Increased accumulation of basal mitochondrial matrix Ca2+ was also observed in HeLa and HPMVEC cells stably expressing MCUC97A, and MCUCF (Figure S2J–S2M).
Our results showing that the substitution of redox-reactive cysteine to an alanine mimicked the effect of oxidative conditions on MCU functions were surprising, as replacing a cysteine with alanine residue is not expected to generate an oxidomimetic mutant. In the replacement to alanine, we maintain a hydrophobic nature at the Cys97 position but prevent any new/non-native steric effects due to the truncated side chain of alanine. Nevertheless, to validate the change in phenotype is not a consequence of the specific amino acid type substitution (i.e. Ala incorporation at position 97), we substituted a methionine residue for cysteine (MCUC97M). To assess the effect of methionine substitution, HeLa cells were co-transfected with MCU-mRFP and MCUC97M-mRFP along with GCaMP2-mt (Figure S2B). HeLa cells expressing MCUC97M mutant exhibited a similar phenotype to MCUC97A (Figure 2G and 2H), suggesting that Ala and Met cause a comparable phenotype, possibly to be due to structural perturbations at Cysteine 97. Consistent with MCUC97, the MCUC97M mutant accumulated increased matrix [Ca2+ ]m (Figure 2I).
Next, we asked whether sustained [Ca2+]m fluorescence observed in MCU cysteine mutants was due to any changes in [Ca2+]m extrusion by the mitochondrial Na+/Ca2+ exchanger. To exclude this possibility, the rate of MCU-mediated [Ca2+]m uptake, and NCLX-mediated [Ca2+]m efflux was measured in HEK 293T cells (Figure 2J). Consistent with the sustained increase in [Ca2+]m fluorescence, the rate of MCU-mediated [Ca2+]m uptake was increased in both MCUC97A and MCUC97M mutants (Figure 2K) with little change in [Ca2+]m efflux rate (Figure 2L), suggesting oxidative modification of only the mitochondrial calcium influx machinery. Additionally, the extent of [Ca2+]m uptake also depends on the Ca2+ filling state of the ER. To further verify the increased [Ca2+]m uptake observed in the MCU mutants was not due to changes in the ER-Ca2+ content, we measured [Ca2+]er in MCU, MCUC97A, and MCUC97M cells (Figure S2C and S2D). Convincingly, the cysteine mutants did not alter the ER Ca2+ content.
Next we asked whether MCU cysteine mutants facilitate MCU-mediated [Ca2+]m uptake even at higher extramitochondrial [Ca2+] to mimic oxidative stress conditions. Cells expressing MCUC97A and MCUCF showed both increased [Ca2+]m uptake rate (Figure 2M and 2N) and total accumulated [Ca2+]m under active condition (Figure 2O and 2P). These data demonstrate a remarkable functional sensitivity conferred by the cysteine residue (MCUC97A) whereby enhancing MCU activity and [Ca2+]m accumulation.
Since MCUC97A constitutively triggered MCU-mediated [Ca2+]m uptake, we next examined whether the cysteine mutation in MCU can regulate IMCU. To measure IMCU, we utilized mitoplast patch clamp (Chaudhuri et al., 2013; Hoffman et al., 2013; Kirichok et al., 2004). Compared with vector control and MCU, the IMCU of mitoplast from cells expressing MCUC97A, and MCUCF was significantly higher which correlated with increased MCU-mediated [Ca2+]m uptake in these cells (Figure 2Q and 2R). In addition, the increased IMCU cannot be accounted for any difference in MCU expression (Figure 2Q; inset). We next tested whether the enhanced MCU activity was due to changes in the interactions of MCU with other uniporter complex components including MICU1, MCUR1, EMRE, and MCUb. We found cysteine residue mutations did not abolish the homo- or heteromeric interactions (Figure S3A–S3E). Taken together, these data strongly support that mutation in cysteine residues of MCU exhibit increased IMCU, and persistent channel activity irrespective of the interaction between MCU and other uniporter complex components.
Mitochondrial-derived Oxidants Promote S-Glutathionylation of MCU at Cysteine 97
Having observed oxidative stress signals to promote oxidation of MCU and increase its channel activity, we investigated whether selective cysteine residues reside in the MCU might become modified by ROS. To further understand how redox signaling through site-specific modifications permit an oxidant signal to be decoded into a biological response, we applied site-specific PEGylation approach to identify the reactive cysteines in MCU. We first exposed the Flag-tagged wild-type MCU expressing HEK293T cells to menadione for various time points followed by PEGylation with mPEG5 (Figure 3A). The mature N-terminal domain of MCU has three cysteines and are sensitive to oxidative stress in vivo, which elicited mPEGylation of MCU in a time dependent manner (Figure 3B). Pretreatment of cells with a reducing agent (DTT) prevented the menadione-induced gel-shift of MCU (Figure 3B). We next explored the MCU sequence alignment for cysteine conservation among eukaryotic species and found that Cys-97 (human) is well conserved among higher to lower eukaryotes, it is in spatial proximity to MCU regulating acidic patch (MRAP) region (Lee et al., 2016) (Figure 3C). To test whether Cys-97 is susceptible to oxidative modification, HEK293T cells stably expressing human MCU and MCUC97A were subjected to oxidative stress. As expected, wild-type MCU underwent substantial oxidation following exposure to menadione (Figure 3D). Under the same oxidation condition, MCUC97A was not affected by the oxidants (Figure 3D) revealing the potential site for oxidative modification. Having detected Cys-97 undergoes oxidant-induced modification, intact HEK293T cells stably expressing MCU and MCU mutants were treated with menadione followed by mPEGylation (Figure 3E). Notably, a single shifted mPEGylated band was observed in MCU, MCUC67A, and MCUC191A but not with MCUC97A, and MCUCF (Figure 3E). To further reinforce Cys-97 as the only cysteine susceptible to oxidation, HEK293T cells were treated with menadione followed by mPEGylation (Figure 3F). MCUC97M mutant was not oxidized with menadione strengthening our conclusion that only the cysteine residue at position 97 undergoes oxidative modifications which correlates with our functional effects of MCUC97A and cysteine free mutant on MCU channel activity and MCU-mediated [Ca2+]m uptake.
Figure 3. MCU Senses Mitochondrial Matrix ROS and MCU Cys-97 Undergoes S-glutathionylation.

(A) Scheme illustrating identification of reactive cysteine in MCU. (B) Menadione-induced modification of MCU. Modified MCU was identified by Western blot analysis and probed with MCU antibody. (C) Alignment of conserved cysteine residue in MCU. (D) Lysates from HEK293T cells stably expressing MCU or MCUC97A treated with menadione were incubated with mPEG5. (E) Representative Western blots for the identification of reactive cysteines in MCU. n=3. (F) Lysates from HEK293T cells stably expressing MCU, MCUC97A or MCUC97M treated with menadione. (G) Cartoon illustrating the identification of MCU S-glutathionylation. (H) HEK293T cells stably expressing MCU or MCUC97A were exposed to menadione and lysates were subjected to Western blotting for the identification of S-glutathionylation (Right). Flag-tagged MCU was probed for control expression (left). (I and J) A comparison of 1H-15N-HSQC spectra of the oxidized NTD-MCU protein before (magenta crosspeaks) and after (blue crosspeaks) 10 mM DTT addition shows numerous amide 1H(15N) chemical shift perturbations (green circles) indicative of a conformational change. The inset shows a Coomassie-stained SDS-PAGE gel of the NTD-MCU protein with and without the addition of DTT. (K) Structure of the human NTD-MCU and relative location of the Cys-97 residue. The Cys-97 residue is located on the β3 strand and forms backbone hydrogen bonds (yellow dashed lines) with Val88 of the β2 strand to stabilize the β-grasp-like fold.
It has been established that oxidative modifications are either reversible or irreversible in the cellular systems. Since MCU oxidation is reversible and menadione is known to oxidize the reduced glutathione in the cellular system, we next assessed whether Cys-97 undergoes S-glutathionylation upon oxidative stress using an antibody that recognizes this modification. We used MCUC97A mutant as a control, and this mutant protein remained non-oxidized under the same oxidation condition that elicited the wild-type MCU oxidative modification (Figure 3G and 3H). To preserve the S-glutathionlylation, these protein samples were separated under a non-reducing condition. This result confirms the hypothesis that Cys-97 is susceptible to S-glutathionylation.
Having demonstrated that Cys-97 is susceptible to S-glutathionylation, we sought to determine whether this modification induces structural changes within the human MCU N-terminal domain (NTD) where only Cys-97 resides (Lee et al., 2016). We prepared uniformly 15N-labeled recombinant MCU (residues 72-189) and subjected the isolated MCU-NTD to an S-glutathionylation protocol. Both monomer and dimer bands on Coomassie blue SDS-PAGE showed a retarded migration relative to the reduced sample after our in vitro S-glutathionylation reaction, consistent with S-glutathionylation of the domain (Figure 3I, inset). The 1H-15N-heteronuclear single quantum coherence spectra of the protein exhibited a wide peak dispersal in the 1H(15N) dimension, consistent with a well folded β-grasp domain. Remarkably, several amide 1H(15N)-specific chemical shift perturbations were observed after the addition of DTT indicating a conformational change within the MCU-NTD associated with the reversible modification (Figure 3I and 3J). We next assessed the structure of the human MCU-NTD domain and relative location of the Cys-97 residue. The Cys-97 residue is located on the β3 strand and forms backbone hydrogen bonds (yellow dashed lines) with Val88 of the β2 strand to stabilize the β-grasp-like fold (Figure 3K). The Cys-97 thiol group is surface exposed (orange surface and orange dashed ellipse) and primed for an oxidative PTMs. Thus, our in vitro data confirmed the susceptibility of the MCU N-terminal domain to S-glutathionylation, demonstrated robust conformational changes within the β-grasp fold and showed that the structural changes associated with this oxidative modification are reversible.
Based on Figure 1 and 2 experimental findings, one possibility is that oxidation of Cys-97 or mutation increases the likelihood of regulating MCU oligomerization. To test the effect of oxidation on MCU oligomerization, we performed size exclusion chromatography (FPLC) (Tomar et al., 2016). Our FPLC analysis revealed that MCU was eluted from monomeric to higher order oligomeric complex. While MCUC97A and MCUC97M mutants were separated as only higher order MCU oligomers (Figure 4A and 4B). We again used these conditions and exposed cells to menadione to induce oxidative stress. Analysis of the menadione-treated MCU revealed the MCU higher order oligomer formation similar to MCUC97A and MCUC97M mutants with or without treatment (Figure 4C and 4D). Collectively, these results suggest that oxidative or mutational perturbation of Cys97 increased the stability of MCU complex and promotes channel activity.
Figure 4. Oxidation of MCU or MCU Cys-97 Mutagenesis Promotes Higher Order MCU Complex Assembly.

(A and C) Lysates from HEK293T cells stably expressing MCUWT (top), MCUC97A (Middle), and MCUC97M (bottom) with (C) and without menadione (A) were subjected to FPLC analysis. FPLC fractions were subjected to Western blotting and probed with FLAG antibody (n=3). (B and D) Quantification of elution profiles of MCUWT, MCUC97A, and MCUC97M with and without menadione treatment from A and C expressed as normalized intensities.
The Conserved Redox Sensitive MCU Cysteine 97 Regulates MCU Complex Assembly at The Inner Mitochondrial Membrane for Ca2+ Uptake
Because mitochondrial diameter is typically 250–500 nm that is close to the resolution limit of light microscopy, we applied multiple imaging modalities to reveal the sub-mitochondrial MCU distribution in oxidative conditions. To gain insights into the mitochondrial morphology, confocal microscopy was utilized and live cell images were acquired. Confocal images of GCaMP2-mt and wild-type MCU-mRFP positive cells revealed proper mitochondrial targeting with indistinguishable signal distribution (Figure 5A). Strikingly, MCU mutants (MCUC97A and MCUCF) positive cells co-localized with GCaMP2-mt and appeared as unique globular like structures (Figure 5B and 5C).
Figure 5. Disruption of Cys-97 Enables MCU Complex Redistribution at The Inner Mitochondrial Membrane.

(A–C) Confocal micrographs of HeLa cells co-transfected with matrix localized GCaMP2-mt (green) and, either MCU WT-mRFP (A), MCUC97A-mRFP (B), or MCUCF-mRFP (C) (red). The blue oval in A–C marks the merged images of the respective magnified cells. (n = 3–5).
(D and E) HeLa cells were co-transfected with the mitochondrial matrix marker COX8-mRFP (red) and either MCU WT-GFP (D) or MCUC97A-GFP (E) (green). Samples were imaged using SR-SIM. The blue box in D and E marks the magnified area (right panels). (n = 4)
(F and G) Co-localization scatter plots (F) of MCU WT and MCUC97A (green) show partial co-localization with COX8 (red). Pearson, Manders, and overlap coefficients between MCU WT and COX8-RFP or MCUC97A and COX8-RFP.
(H) MCU and MCUC97A were tagged with a photo-switchable protein, mEOS3.2. HeLa cells were transfected with either construct, fixed, and imaged using PALM. The blue box marks the magnified area (right panels). (I) Nanoclustering of MCU WT-mEOS3.2, MCUC97A-mEOS3.2, and MCU WT-mEOS3.2 treated with menadione was conducted as previously described (Baumgart et al., 2016). The normalized density of (ρ/ρ0) molecules was plotted against the relative area covered by the clusters (η). The red line fitted to the graph denotes 100% randomly distributed molecules, while true clustering has a higher density (ρ/ρ0) of molecules along a higher percentage of the area covered by the clusters (η) (n = 4–9). For each condition, we randomly quantified 5–10 cells for cluster analysis. (J) Model depicting MCU clustering and matrix remodeling upon oxidative stress.
To visualize the mitochondrial distribution and distinctive mitochondrial morphology exhibited by MCU and MCUC97A, we performed super-resolution structured illumination microscopy (SIM) imaging to identify precisely the MCU localization. As predicted, COX8-mRFP is clearly localized in the matrix while wild-type MCU properly targeted to mitochondrial inner membrane (Figure 5D). Remarkably, MCUC97A mutant again resulted in globular like morphology but targeted to the inner mitochondrial membrane (Figure 5E). The MCU-positive structures did not co-localize with matrix COX8-mRFP indicating that SIM images not only resolves the localization but also distinguishes mitochondrial compartmentalization. Furthermore, the co-localization analysis revealed that MCU and MCUC97A are indistinguishable with little changes on Pearson’s correlation parameters (Figure 5F and 5G). In order to study the detailed assembly and regulation mechanisms of MCU complex structures in the mitochondrion, we employed Photo-activated localization microscopy (PALM). HeLa cells were transfected with MCU and MCUC97A tagged with photo switchable protein mEos3.2 (Baumgart et al., 2016). Similar to the above described imaging data, PALM acquisition images showed random distribution of MCU whereas MCUC97A appeared to assemble as clusters in the globular like inner mitochondrial membrane (Figure 5H). To further verify whether the distribution of oxidized MCU phenocopies the MCUC97A mutant, we performed PALM imaging in HeLa cells transfected with MCU-mEos3.2 plasmid and treated with menadione for various time points to induce oxidative stress (Figure S4). Similar to MCUC97A, menadione treatment increased the clustering of MCU WT (Figure 5H and I). Since we adapted the PALM approach for visualization of MCU clusters, we next quantified the density of clustering within a given region of inner mitochondrial membrane. MCUC97A mEos3.2 showed high density of cluster events when compared to wild-type MCU indicating that MCUC97A mEos3.2 protein is efficiently assembled on the inner mitochondrial membrane strongly supporting the notion of MCU higher order oligomerization by oxidation (Figure 5I and 5J). Collectively, this distribution is fully in line with a MCUC97A-mediated higher MCU channel activity.
MCU Oxidation Drives Mitochondrial ROS Elevation, Perturbs Cellular Bioenergetics and Sensitizes Cells to Death
Although ROS-mediated cytotoxicity is a well-recognized phenomenon in inflammatory and ischemic conditions, the constitutive MCU-mediated [Ca2+]m uptake results in [Ca2+]m overload and is generally associated with excessive mROS in cells lacking MCU gatekeeping mechanism (Mallilankaraman et al., 2012b). Unexpectedly, here we found that sustained MCU activity and mitochondrial matrix Ca2+ overload occurs (Figure 2E and F) in spite of an intact high-affinity Ca2+ sensing mechanism of MICU1. Next we asked whether constitutive elevation of [Ca2+]m results in [Ca2+]m-dependent mROS overproduction. As expected basal mROS levels were elevated in HPMVECs stably expressing MCUC97A, and MCUCF compared to control (Figure 6A and 6B). Ectopic expression of MnSOD in MCU mutant cells resulted in decreased MitoSOX, indicating mROS overproduction (Figure 6A–6C). To determine the effect of increased [Ca2+]m accumulation, on cellular bioenergetics, ATP levels and OCR were measured in HPMVECs. In line with matrix [Ca2+]m overload and increased mROS levels, ATP levels, basal and maximal OCR were significantly reduced in HPMVECs expressing MCUC97A, and MCUCF (Figure 6D–6G).
Figure 6. Mutation of MCU Cys-97 perturbs endothelial cell bioenergetics, function and cell death.

(A) Representative images showing MitoSox Red fluorescence in HPMVECs. (B) Bar graph represents quantification of fluorescence. Data indicate Mean ± SEM; *** P <0.001; n=3–7. (C) Representative Western blot for the expression of MnSOD and PRDX3. (D) Measurement of OCR in HPMVECs. After basal OCR, oligomycin (A), FCCP (B), and rotenone + Antimycin A (C) were added as indicated. (E and F) Bar represents mean basal OCR and maximal OCR. Data indicate Mean ± SEM; ** P <0.01; n=4. (G) Bar represents cellular ATP levels in HPMVECs. Data indicate Mean ± SEM; ** P <0.01; n=4.
(H) Bar represents cellular basal NADH levels. Data indicate Mean ± SEM; ** P <0.01; n=3. (I) Quantification of endothelial cell migration. Data indicate Mean ± SEM; ** P <0.01; n=3–7. (J) Representative blot for the expression of ICAM-1 in HPMVECs. (K) Quantification of ICAM-1 expression. Data indicate Mean ± SEM; ** P <0.01; n=3. (L–N) Quantification of relative mRNA abundance of IL-1β (L), IL-6 (M), and TNF-α (N) in HPMVECs with or without LPS stimulation. Data indicate Mean ± SEM; *** P <0.001; n=3. (O) Quantification of PI-positive cells by FACS analysis. Data represents Mean ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001; (n = 3).
To investigate whether quenching mROS can alleviate the bioenergetic crisis, mitochondrial antioxidant enzymes (AdMnSOD, peroxiredoxin 3; PRDX3, and a combination of MnSOD + PRDX3) were transfected in HPMVECs stably expressing control and MCU mutants (Figure 6C). Expression of antioxidants markedly restored basal OCR and cellular ATP levels in MCUC97A and MCUCF cells (Figure 6D–6G), suggesting the chronic accumulation of mitochondrial matrix Ca2+ promotes excessive mROS that inhibit mitochondrial OXPHOS. Additionally, because [Ca2+]m accumulation regulates a range of mitochondrial enzymes that participate in NADH generation, we also assessed the basal steady-state levels of NADH in HPMVECs. The basal NADH levels are significantly lower in MCUC97A and MCUCF mutant cells (Figure 6H) suggesting perturbed cellular bioenergetics by oxidation of MCU and increased channel activity.
Physiological mROS is implicated in many cellular processes, including gene expression, cell growth, proliferation, and differentiation, and conversely chronic accumulation of mROS promotes cell death (Hamanaka and Chandel, 2010). Because HPMVECs expressing MCUC97A, and MCUCF have elevated mROS levels and bioenergetic crisis, we next examined the endothelial migration, a key function of angiogenesis. To determine migration, we performed scratch assay (Mallilankaraman, 2012). Endothelial migratory capacity was attenuated in HPMVECs expressing MCUC97A, and MCUCF (Figure 6I). We next investigated whether MCU-mediated signaling primes endothelial cell activation. We assessed the proinflammatory cytokine and ICAM-1 expression in control and MCU mutant cells. Persistent elevation of [Ca2+]m and mROS possibly upregulates ICAM-1, TNF-α, IL1-β, and IL-6 protein and mRNA abundance in MCU mutant cells that was further enhanced by LPS challenge (Figure 6J–6N). Additionally, H2O2 induced cell death was augmented in HPMVECs expressing MCUC97A, and MCUCF (Figure 6O). Importantly, cell death was prevented in HPMVECs expressing MCUC97A, and MCUCF overexpressing mitochondrial antioxidants (Figure 6O). Thus, our results suggest that Cys-97 in MCU is susceptible to oxidative stress conditions, increases MCU-mediated [Ca2+]m uptake, elevates the accumulation of mitochondrial matrix Ca2+, mROS levels, causes cellular bioenergetics crisis and sensitizes cells to death.
DISCUSSION
Reversible oxidation of proteins is essential for the proper regulation of biological functions in most eukaryotes (Hamanaka and Chandel, 2010). Our work provides a strong foundation for understanding how mitochondria-derived oxidants regulate mitochondrial metabolic pathways that are dependent on MCU-mediated mitochondrial matrix Ca2+ surge. Our characterization of the oxidative modification of MCU and the identification of the conserved cysteine residue that is capable of undergoing S-glutathionylation greatly influencing MCU channel activity suggests that this modification may occur in response to both acute and chronic oxidative stress. It has been established that both [Ca2+]m overload and higher mROS elevation are known to induce mitochondrial dysfunction via mPTP opening (Orrenius et al., 2003). Since the anterograde (Ca2+ entry) and retrograde (ROS production) are often interdependent phenomena, we hypothesized that oxidative stress could play a positive feedback role in modulating MCU activity. Our biochemical analysis revealed that only Cys-97 of three available cysteines was susceptible to nucleophilic reaction (Figure 3). Having recently demonstrated that these three cysteines are dispensable for MCU oligomerization (Lee et al., 2016; Oxenoid et al., 2016), Cys-97 is less likely to undergo intra- or inter molecular disulfide bond formation. Alternative oxidative modifications include sulfenic acid or S-glutathionlyation formation.
Although numerous proteins undergo sulfenylation in mammalian cells, our biochemical analysis suggested that Cys-97 was not susceptible to sulfenic acid formation (undetectable). Here our studies provide striking evidence that MCU Cys-97 becomes S-glutathionylated when exposed to oxidative stress. The conjugation of glutathione causes a conformation change within the N-terminal domain and appears to promote MCU channel activity under resting state. Since the N-terminal domain of MCU is facing the matrix, it is an opportune location to sense matrix ROS to reprogram the Ca2+-dependent mitochondrial metabolic signals. This suggests that inflammatory or H/R stresses, possibly promote mROS overproduction in the vicinity of mitochondrial matrix that perturbs mitochondrial GSH/GSSG ratio which results in S-glutathionlyation of MCU.
S-glutathionylated MCU as well as MCUC97A mutant proteins are enriched as higher order oligomers as visualized by superresolution microscopy using SIM and PALM imaging of wild-type MCU and MCUC97A mutant. Detailed imaging and analysis revealed that wild-type MCU was randomly distributed along the IMM. In contrast, the oxidized MCU and MCUC97A mutant appeared to reorganize cristae and orderly clustered (Figure 5H). In images acquired with conventional microscopy, these MCU assemblies would be indistinguishable because of limited resolution. Importantly, we noticed that dual color images of mitochondria decorated with matrix marker COX8-mRFP and MCU-GFP often displayed that MCU-GFP signal is devoid of COX8-mRFP (Figure 5D and 5E). Intriguingly, superresolution imaging cluster analysis and size exclusion chromatography MCU complex separation are consistent with higher order oligomerization and increased MCU channel activity upon post-translational oxidative modification.
Apart from the several binding partners that form the MCU protein complexes, studies have also shown that posttranslational modifications of MCU protein can also play a key role in regulating [Ca2+]m levels. Phosphorylation of MCU S57 and S92 have been proposed to be mediated by CaMKII, while PyK2 has been implicated in the phosphorylation of MCU Y158, Y289 and Y317 to modulate MCU activity (J et al., 2014; Joiner et al., 2012). However, it still remains unclear if phosphorylation of MCU can happen in vivo because of lack of direct experimental evidence. Hence, we choose to analyze the oxidative PTMs of MCU and furthermore, it is expected that mitochondria being the major source of intracellular ROS, the local concentration of ROS is high and MCU could be a likely potential target for ROS. It is important to note that though MCU complex components MICU1 (seven), MCUb (four) and MCUR1 (six) contain several cysteine residues (Figure 1O), the comprehensive mPEG conjugation analysis revealed MCU as the primary mROS sensor.
Upon oxidative stress, several ion transport proteins are either reversibly or irreversibly modified by ROS. Our previous findings demonstrated that STIM1 undergo S-glutathionylation that promote Orai channel opening for Ca2+ entry without affecting intracellular store depletion (Hawkins et al., 2010). This supports the notion that ion channels are regulated by redox state, although the functional consequences are divergent. For example, oxidation of reactive cysteines in Orai 1 and Orai 2 channels inhibits Ca2+ entry (Bogeski et al., 2010). Interestingly, other intracellular channels like ryanodine receptor, SERCA and IP3R channels undergo oxidative modification that promote activity (Adachi et al., 2004; Bogeski and Niemeyer, 2014; Porter Moore et al., 1999). Here we highlight for the first time that S-glutathionylation of MCU substantially redistributes MCU complex and resulted in MCU channel activation. Our results also suggest that either oxidation or disruption of Cys-97 does not alter the interactions of MCU with its regulators, but rather produces a conformational change within the N-terminal β-grasp fold (Figure S3 and Figure 3I–K). These findings indicate that Cys-97 is critical for producing changes in MCU oligomerization and channel activity. It is important to note that one of the mitochondrial intermembrane space protein MIA40 undergoes oxidative folding and serves protein import and accumulation in the IMS (Tokatlidis, 2005), yet it is not known whether it senses oxidative stress. Given the unique role for MCU as a rapid mitochondrial matrix ROS sensor, our data reveal that oxidative PTM controls protein architecture and MCU channel activity.
In summary, upon activation by endotoxin or H/R, cells produce ROS-related species that are sensed by MCU and enhances channel activity. The results revealed here highlight both the oxidative stress-induced mitochondrial Ca2+ dynamics and mitochondrial MCU complex distribution that modulate mitochondrial Ca2+ homeostasis and cellular bioenergetics. Although ROS modulates MCU activity in cells, the molecular basis and functional significance of this redox regulation in vivo warrants future research. The identification of the redox-sensing role for MCU establishes a new frame of reference to ascertain how oxidative stress perturbs mitochondrial ion homeostasis. Further understanding of the mechanisms involved may contribute insight into the etiology of metabolic disorders.
EXPERIMENTAL MODEL DETAILS
HeLa, HEK-293, and COS7 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C and 5% CO2. Endothelial cells were grown in ECGS supplementation condition. Ventricular cardiomyocytes from one- to two-day-old rat hearts (NRVMs) were prepared as previously described (Brinks et al., 2010; Vagnozzi et al., 2013). NRVMs were cultured in Ham’s F-10 supplemented with 5% fetal bovine serum (FBS) and penicillin/streptomycin (100 U/ml) at 37°C in a 95% air/5% CO2 humidified atmosphere for 4 days. Mouse microvascular pulmonary endothelial cells (MPMVECs) were obtained from C57BL/6 endothelial-specific MCUKO mice (MCUΔEC), and control (VE-Cre) mice as described earlier.
METHOD DETAILS
Plasmids and Recombinant proteins
Plasmids pCMV6-Entry- MCU, MICU1, EMRE, MCU, and COX8A-C-mRFP were purchased from Origene Technologies. They were then subcloned in the pCMV6-AC-HA and pCMV6-AC-GFP vectors (Origene), and pCMV/Bsd (blasticidin) (Thermo Fisher Scientific) as required. The mutant MCU constructs (MCUC97A, MCUC67A, MCUC191A, MCUC97M, and MCUCF) were custom synthesized as gBlock gene fragments from IDT Inc. and subcloned into appropriate vectors for further use. The mEOS3.2MCU (MCUWT, MCUC97A, and MCUCF) constructs for PALM were engineered by inserting mEOS3.2 (monomeric variant of green-to-red photoswitchable fluorescent protein EosFP, for reduced aggregation relative to mEos2 (from Michael Davidson Lab)) between the MluI and XhoI sites of pCMV6-AC-DDK vector and then the MCUWT, MCUC97A, and MCUCF were inserted into the SgfI and MluI sites. All plasmids were confirmed by sequencing before use.
Generation of stable cells expressing MCUWT, MCUC67A, MCUC97A, MCUC97M, MCUC191A and MCUCF
HEK293T, HeLa, and HPMVEC cells were transfected with MCUWT, MCUC67A, MCUC97A, MCUC191A MCUC97M and MCUCF and 48 h post-transfection, cells were selected with blasticidin (2μg/ml) for 6–10 days and expanded.
Hypoxia/Reoxygenation Exposure
Freshly isolated NRVMs (rat neonates) and adult cardiomyocytes (mice) were subjected to 2 h of hypoxia (1% O2-5% CO2) followed by 30 min of reoxygenation (21% O2-5%CO2) before any measurements were performed to study the role of ROS induced modification of MCU protein and channel activity.
Simultaneous Measurement of Cytoplasmic and Mitochondrial Ca2+ uptake
HPMVECs grown on 0.2% gelatin coated 25 mm glass coverslips were treated with LPS (1 μg/ml) for 5 h. Both LPS treated and untreated cells were loaded with 2 μM rhod-2/AM (50 min) and 5 μM Fluo-4/AM (30 min) in extracellular medium as previously described. Coverslips were mounted in an open perfusion microincubator (PDMI-2; Harvard Apparatus) at 37°C and imaged. After 1 min of baseline recording, thrombin (100 mM) was added, and confocal images were recorded every 3 s (510 Meta; Carl Zeiss, Inc.) at 488 and 561 nm excitation using a 63 x oil objective. Images were analyzed and quantified by using ImageJ (NIH).
Simultaneous Measurement of Ca2+ Uptake and ΔΨm in Permeabilized Cell System
HEK293T cells stably expressing control vector, MCUWT, MCUC97A, MCUC97M and MCUCF were washed in Ca2+ free PBS, pH 7.4. An equal amount of cells (7x106 cells) were resuspended and permeabilized with 40 μg/ml digitonin in 1.5 ml of intracellular medium (ICM) composed of 120 mM KCl, 10 mM NaCl, 1 mM KH2PO4, 20 mM Hepes-Tris, pH 7.2 and 2 μM thapsigargin to block the SERCA pump. All the measurements were performed in the presence of 5 mM succinate. The simultaneous measurement of ΔΨm and extramitochondrial Ca2+ ([Ca2+]out) clearance as an indicator of [Ca2+]m uptake was achieved by loading the permeabilized cells with JC-1 (800 nM) and Fura2-FF (0.5 μM), respectively. Fluorescence was monitored in a multi-wavelength excitation dual-wavelength emission fluorimeter (Delta RAM, PTI). [Ca2+]out is represented as the excitation ratio (340 nm/380 nm) of Fura2-FF/FA fluorescence and ΔΨm as the ratio of the fluorescence of J-aggregate (570 nm excitation/595 nm emission) and monomer (490 nm excitation/535 nm emission) forms of JC-1. A single Ca2+ bolus (20 μM) and mitochondrial uncoupler, CCCP (2 μM), were added at the indicated time points. All the experiments were performed at 37°C with constant st irring.
Measurement of Resting Mitochondrial Ca2+
HEK293T cells stably expressing control vector, MCUWT, MCUC67A, MCUC97A, MCUC97M, MCUC191A and MCUCF were resuspended and permeabilized with digitonin (40 μg/ml) in 1.5 ml of intracellular medium composed of 120 mM KCl, 10 mM NaCl, 1 mM KH2PO4, 20 mM HEPES-tris (pH 7.2), 5 mM succinate, bath Ca2+ indicator Fura-2FF (0.5 μM), MCU blocker Ru360 (1 μM) and NCLX blocker, CGP (1 μM), and 2 μM thapsigargin to block the SERCA pump. The experiments were performed at 37°C with c onstant stirring. To assess the resting [Ca2+]m, after baseline recording of bath Ca2+ ([Ca2+]out), CCCP (2 μM) was added to release matrix mitochondrial Ca2+. Fluorescence was monitored in a multiwavelength excitation, dual-wavelength emission fluorimeter (DeltaRAM, PTI). [Ca2+]out was recorded as an excitation ratio (340 nm/380 nm) and emission at 510 nm of Fura2FF fluorescence (Mallilankaraman et al., 2012).
Mitoplast Patch-Clamp Recording
Mitoplast patch-clamp recordings were performed at 30°C as detailed previously with the following modifications. Freshly prepared mitoplasts from HeLa cells stably expressing MCUWT, MCUC97A, and MCUCF were plated on the Cell-Tak–coated coverslips and mounted on the microscope. The isolated mitoplasts were bathed in a solution containing sodium gluconate (150 mM), KCl (5.4 mM), CaCl2 (5 mM), and Hepes (10 mM) (pH 7.2). The pipette solution contained sodium gluconate (150 mM), NaCl (5 mM), sucrose (135 mM), Hepes (10 mM), and EGTA (1.5 mM) (pH 7.2). After formation of GΩ seals (20 to 35 MΩ), the mitoplasts were ruptured with a 200- to 400-mV pulse for 2 to 6 ms. Mitoplast capacitance was measured (2.5 to 3.0 pF). After capacitance compensation, mitoplasts were held at 0 mV and IMCU was elicited with a voltage ramp (from −160 to 80 mV, 120 mV/s). Samples were discarded if the break-in took longer than 5 s after addition of 5 mM Ca2+. Currents were recorded using an Axon200B patchclamp amplifier with a Digidata 1320A acquisition board (pCLAMP 10.0 software; Axon Instruments). The bath solution (5 mM Ca2+) was chosen on the basis of previous measurements.
Co-immunoprecipitation and Western Blot Analysis
Cell extracts were prepared from transiently transfected COS-7 cells using RIPA buffer (50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.25% deoxycholic acid, 1 mM EDTA, 1% NP-40, protease inhibitor cocktail (Complete: Roche and 1 mM PMSF). To study the interaction of MICU1 with MCUWT, MCUC97A, and MCUCF, HA tagged MICU1 was co-transfected with Flag tagged MCUWT, MCUC97A, and MCUCF plasmids. Following immunoprecipitation with HA antibody (Sigma-aldrich, USA), total cell lysates and immunoprecipitated materials were subjected to Western blot analysis. 10% of cell lysates were probed with Flag (Sigma-Aldrich) and HA antibodies to serve as inputs, and similarly, immunoprecipitated samples were probed with their corresponding antibodies to assess protein binding.
We also looked for interaction between MCUWT, MCUC97A, and MCUCF with MCU, EMRE, MCUR1, and MCUb. To study the interaction of MCU complex components, either MCU-HA, EMRE-HA, MCUR1-V5, or MCUb-HA was co-transfected with Flag-tagged MCUWT, MCUC97A, and MCUCF plasmids. Co-immunoprecipitation and western blotting was performed as described earlier.
Size Exclusion Chromatographic Analysis of MCU Complex
Gel filtration was performed by fast protein liquid chromatography (ÄKTA Pure FPLC; GE Healthcare), and the Superdex 200 10/300 column was equilibrated with PBS. Column calibration was carried out with a gel filtration protein standards kit (Bio-Rad). Cleared lysates from HEK293T cells stably expressing MCU-FLAG, and MCUC97A –FLAG with and without menadione treatment were directly loaded onto a Superdex 200 FPLC column at a flow rate of 0.5 ml/min. 40 fractions were collected, and aliquots containing 150 μl of the 1 ml fractions were used to assay complexes.
Superresolution Structured Illumination Microscopy
HeLa cells were co-transfected with MCU-GFP and Cox8-mRFP. Forty-eight hours post transfection, transfected HeLa cells were fixed with 4% paraformaldehyde in PHEM buffer (60 mM PIPES, 25 mM HEPES, 10 mM EGTA, 2mM MgCl2 at pH 6.9) at 4°C for 20 min. The fixed cells were washed thrice with 1x PHEM buffer and incubated for 5 min in diluted gold fiducials (100 nm 5:1 in 1X PHEM buffer), washed and stored at 4°C until imaging. For SR-SIM, images made of 15 different Z-planes separated by 111 nm over a range of 1.53 μm were acquired. 5 different rotation angles and five different phase steps were acquired and then reconstructed using Zen 2011 software (Carl Zeiss). Multicolor 120 nm beads were used to align the GFP and RFP channels in X, Y, and Z using affine alignment. Images were captured using an Andor iXon DU885 EMCCD camera. The camera acquisition parameters for GFP and RFP were 100 ms exposure and an EM-CCD gain of 10. The laser excitation was 488 nm laser line at 12% power for GFP and 561 nm laser line at 8% for RFP. A band pass 495–590 filter was used for GFP and a long-pass 570 for RFP.
Photoactivated Localization Microscopy (PALM)
HeLa cells were transfected with mEOS3.2MCU plasmids. Forty-eight hours post transfection, transfected HeLa cells were washed with 2 ml of 4% paraformaldehyde fixative. 2 ml of 4% paraformaldehyde fixative was added, cells were incubated at 4°C for 20 minutes. Following the incubation step, cells were washed with 2 ml of 1X PHEM buffer for 10 min (3 times) and replaced with diluted gold fiducials (100 nm were gold fiducials were diluted 5:1 ratio in 1x PHEM buffer; 4 ml PHEM buffer 1 ml gold fiducials) and incubated for 5 min. As a final step, cells were again washed with 1x PHEM buffer and stored at 4°C until ready to image. Samples were first checked for mEos3.2 expression using a 488 nm laser line and bandpass 495–590 filter. The mEos3.2 proteins were switched green to red using a 405 nm laser line. The red fluorescence was excited with the 561 nm laser line and collected with a long pass 570 filter. Approximately 65,000–80,000 images were collected with an Andor iXon DU897 EM-CCD camera set to 100 ms exposure and an EMCCD gain of 30. The 100 nm gold fiducials were used to autofocus every 2,000 frames. Drift correction was perform and images were generated in Zen 2012 software.
Nanocluster Analysis
The nanocluster analysis was performed as described in Baumgart et al. (2016). Images were processed using the FIJI distribution of ImageJ and the ThunderSTORM plug-in (Ovesny et al, 2014). Images were filtered using the wavelet filter (B-Spline; order =3, scale = 2). Approximate localization of molecules was conducted using local maximum and sub-pixel localization was determined using an integrated Gausssian PSF method with a fitting radius of 3 pixels, weighted least squares and initial sigma of 1.6 pixels. Drift was removed using the drift correction feature of ThunderSTORM. Comma separated (CSV) files were exported and then divided into smaller 10,000 frame CSV files. The files were in MATLAB R2016a (Mathworks) and processed using the nanoclustering code provided by Baumgart, et al (2016). Individual mitochondria were selected as regions of interest for either wild-type MCU or the mutant MCUC97A. A threshold value of 3 was used to plot the normalized density of (ρ/ρ0) molecules against the relative area covered by the clusters (η).
Measurement of Mitochondrial Superoxide
Mitochondrial superoxide was measured by using the mitochondrial oxygen free radical indicator MitoSOX Red (molecular probes; Invitrogen) as described previously (Mukhopadhyay et al., 2007). Briefly, cells grown on 0.2% gelatin coated glass coverslips were loaded with 5 μM MitoSOX Red for 30 min, and coverslips were mounted in an open perfusion microincubator (PDMI-2; Harvard Apparatus) at 37°C and imaged. Con focal (510 Meta; Carl Zeiss, Inc.) images were obtained at 561 nm excitation by using a 63 x oil objective. Images were analyzed, and the mean MitoSOX Red fluorescence was quantified by using Image J software (NIH).
ATP Measurement
Total ATP abundance was assessed using CellTiter-Glo® luminescent assay and the luminescence was measured using a plate reader (Infinite M1000 PRO, Tecan).
Mitochondrial Oxygen Consumption Rate
Intact HPMVECs stably expressing MCUWT, MCUC97A, and MCUCF were subjected to oxygen consumption rate (OCR) measurement at 37°C in an XF 96 extracellular flux analyzer (Seahorse Bioscience). HPMVECs (3 x 105) were sequentially challenged with oligomycin, FCCP, and rotenone plus antimycin A (Doonan et al., 2014).
NADH Measurement
Intact HPMVECs stably expressing MCUWT, MCUC97A, and MCUCF (5 × 106 cells) were suspended in Hank’s balanced salt solution (Sigma). Autofluorescence of NAD(P)H was monitored at 350/460 nm (excitation/emission) using a multiwavelength excitation, dual-wavelength emission fluorimeter (DeltaRAM, Photon Technology International) (Shanmughapriya et al., 2015). The experiments were performed at 37°C.
Endothelial Cell Migration Assay
HPMVECs stably expressing MCUWT, MCUC97A, and MCUCF were seeded at a density of 0.5×105 cells/well in 6-well plates overnight for confluent monolayer. A uniform 1.8 mm scratch running the entire length of the well was created using a sterile 200 μl tip. The wells were washed thrice with PBS to remove the cell debris and then bathed in 2 ml complete endothelial medium. After 24 h the cells were washed and fixed with CAMCO Quick Stain® II as per manufacturer’s instructions. The wells were photographed at multiple locations using phase contrast microscopy with a 4 x objective. Migration was quantified using Image J software (NIH); results are expressed as number of cells migrated (Gandhirajan et al., 2013).
Profiling of inflammatory cytokines
HPMVECs stably expressing vector control, MCUWT, MCUC97A, and MCUCF were treated with LPS for 5 h. After LPS stimulation, RNA was extracted using RNAeasy kit (QIAGEN) as per manufacturer’s instruction. The extracted RNA was reverse transcribed to cDNA. The synthesized cDNA was used for qRT-PCR using a Taqman or SYBR Green chemistry. The following primers were used. IL-1βF 5′-CAAAGGCGGCCAGGATATAA-3′; IL-1β: 5′-CTAGGGATTGAGTCCACATTCAG-3′; IL-1β Probe 5′ FAM/AGAGCTGTA/ZEN/CCCAGAGAGTCCTGT/3IABkFQ/-3′ IL-6F 5′-CCAGGAGAAGATTCCAAAGATGTA-3′; IL-6R 5′-CGTCGAGGATGTACCGAATTT -3′ IL-6 Probe: 5′-FAM/CACACAGAC/ZEN/AGCCACTCACCTCTT/3IABkFQ/-3′; TNF-αF 5′-AGCCTCTTCTCCTTCCTGATCGTG-3′; TNF-αR: 5′-GGCTGATTAGAGAGAGGTCCCTGG-3′.
Flow Cytometry and PI Staining
HPMVECs stably expressing MCUWT, MCUC97A, and MCUCF were treated with H2O2 (0.8 mM) for 6 hr. Cells were stained with propidium iodide. The cells were analyzed using FACS Canto apparatus (BD Biosciences). The percentage of PI positive cells was plotted using Flow-Jo software.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data from multiple experiments were quantified and expressed as Mean ± SEM., and differences between groups were analyzed by using two-tailed paired Student’s t-test Differences in the means among multiple data sets were analyzed using 1-way ANOVA with the Kruskal-Wallis test, followed by pairwise comparison using the Dunn test. P value less than 0.05 was considered significant in all analysis. The data were computed with either Graphpad Prism version 7.0 or SigmaPlot 11.0 Software.
DATA AND SOFTWARE AVAILABILITY
The unprocessed image files used to prepare the figures in this manuscript are in Mendeley Data and are available at http://dx.doi.org/10.17632/k6cj6vy5zm.1
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal ANTI-FLAG M2-peroxidase (HRP) antibody produced in mouse | Sigma-Aldrich | A8592-1MG |
| Anti-MCU antibody produced in rabbit | Sigma-Aldrich | HPA016480-100UL |
| Tom20 Antibody (FL-145) | Santa Cruz | Sc-11415 |
| Anti-Glutathione antibody [D8] | Abcam | Ab19534 |
| Purified Mouse Anti-MnSOD clone 19/MnSOD | BD Transduction Laboratories | 611580 |
| β-actin antibody (C4) | Santa Cruz | sc-47778 |
| HA Tag Monoclonal antibody (2-2.2.14) | ThermoFisher Scientific | 26183 |
| Clone OT14C5, Anti-DDK (FLAG) monoclonal antibody | Origene | TA50011-100 |
| ICAM-1 antibody (15.2) | Santa Cruz | sc-107 |
| V5 Tag Monoclonal Antibody | ThermoFisher Scientific | R960-25 |
| Biological Samples | ||
| Human Pulmonary Microvascular Endothelial Cells clone ST1.6R | Krump-Konvalinkova et al., 2001 | |
| Mouse Pulmonary Microvascular Endothelial Cells (VE- Cre, and endothelial-specific MUC KO) | Tomar et al., 2016 | |
| HeLa cells | ATCC | ATCC# CCL-2 |
| HEK 293T cells | ATCC | ATCC# CRL-1573 |
| Adenovirus MnSOD | University of Iowa Gene Transfer Vector Core |
Ad5CMVSOD2 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lipopolysaccharides from Escherichia coli 0111:B4 | Sigma-Aldrich | L5293-2ML |
| Rhod-2, AM, cell permeant | ThermoFisher Scientific | R1245MP |
| Menadione | Sigma-Aldrich | M57405 |
| DL-Buthionine-(S, R)-sulfoximine | Sigma-Aldrich | B2640 |
| mPEG-5 | Sigma-Aldrich | 63187-1G |
| Ru360 | Calbiochem | 557440 |
| CGP-37157 | Sigma-Aldrich | C8874-25MG |
| Blasticidin S HCl, powder | ThermoFisher Scientific | R210-01 |
| Digitonin (~50% TLC) | Sigma-aldrich | D5628 |
| Fura 2FF-pentapotassium salt | ThermoFisher Scientific | |
| Sodium succinate dibasic hexahydrate | Sigma-aldrich | S2378 |
| DTT | ||
| L-Glutathione oxidized | Sigma-aldrich | G4376 |
| L-Glutathione reduced | Sigma-aldrich | G4251 |
| Fluo-4, AM, Cell permeant | ThermoFisher Scientific | F14201 |
| Protease Inhibitor cocktail Tablets (complete) | Roche | 11873580001 |
| Seahorse XF Cell Mito Stress Test kit | Seahorse | 103015 |
| Cell Titer-Glo Luminescent Cell Viability Assay | Promega | G7571 |
| Propidium iodide | ThermoFisher Scientific | P3566 |
| Hydrogen peroxide solution 30% w/w in H2O | Sigma-Aldrich | H1009 |
| Histamine dihydrochloride | Sigma-Aldrich | H7250-10 mg |
| Calcium chloride | Sigma-Aldrich | |
| (−)-Isoproterenol hydrochloride | Sigma-Aldrich | I6504 |
| Thapsigargin | Sigma-Aldrich | T9033 |
| JC-1 Dye (Mitochondrial Membrane potential probe) | ThermoFisher Scientific | T3168 |
| Carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone | Sigma-Aldrich | C2920 |
| MitoSox Red Mitochondrial Superoxide Indicator, for live cell imaging | ThermoFisher Scientific | M36008 |
| FBS | HyClone | SH3039603 |
| Antibiotic Antimycotic | ThermoFisher Scientific | 15240062 |
| Endothelial Growth Supplement | Millipore | 02-102 |
| Deposited Data | ||
| Unprocessed Image Data | This paper | http://dx.doi.org/10.17632/k6cj6vy5zm.1 |
| Experimental Models: Cell Lines | ||
| HEK293 T cells stably expressing MCU mutants | This paper | |
| HeLa cells stably expressing MCU mutants | This paper | |
| HPMVEC cells stably expressing MCU mutants | This paper | |
| Experimental Models: Organisms/Strains | ||
| C57BL/6 MCUΔEC | Tomar et al., 2016 | |
| Recombinant DNA | ||
| MCU-Flag, MICU1-Flag, MCUb-Flag, MCUR1-Flag, EMRE-Flag (pCMV6-AC-DDK backbone) | Origene | |
| MCU-GFP (pCMV6-AC-GFP) | Origene | |
| MCU-HA, MICU1-HA, MCUb-HA, EMRE-HA (pCMV6- AC-HA) | Origene | |
| Cox8-mRFP (pCMV6-AC-RFP) | Origene | |
| mitoGCaMP2 (Plasmid) | ||
| R-GECO1 (Plasmid) | ||
| pCMV/Bsd | ThermoFisher Scientific | |
| mEOs3.2 plasmid | Michael Davidson/Addgene | Plasmid #54550 |
| Peroxiredoxin 3 (plasmid) | Origene | |
| Custom synthesized gBlock gene fragments (MCUC67A, MCUC97A, MCUC191A, MCUC97M, MCUCF) | IDT | |
| pET-28a MCU72-189 | Lee et al., 2016 | |
| Sequence-Based Reagents | ||
| IL-1βF 5′-CAAAGGCGGCCAGGATATAA-3′; IL-1βR: 5′-CTAGGGATTGAGTCCACATTCAG-3′; IL-1β Probe 5′FAM/AGAGCTGTA/ZEN/CCCAGAGAGTCCTGT/3IABk FQ/-3′ IL-6F 5′-CCAGGAGAAGATTCCAAAGATGTA-3′ IL-6R 5′-CGTCGAGGATGTACCGAATTT -3′ IL-6 Probe: 5′-FAM/CACACAGAC/ZEN/AGCCACTCACCTCTT/3IABk FQ/-3′; TNF-αF 5′-AGCCTCTTCTCCTTCCTGATCGTG-3′; TNF-αR: 5′-GGCTGATTAGAGAGAGGTCCCTGG-3′. |
This paper | |
| Software and Algorithms | ||
| NMRPipe/NMRDraw | Delalglio et al., 1995 | |
| PyMOL | The PyMOL Molecular Graphics System, Version 1.7.2.1 Schrödinger, LLC | |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the corresponding author, Dr. Muniswamy Madesh (mandeshm@temple.edu).
Supplementary Material
Acknowledgments
This research was funded by the National Institutes of Health (R01GM109882, R01HL086699, R01HL119306, and 1S10RR027327 to M.M., P01 DA037830, PI: K. Khalili and RO1DK103558 to SKJ/GH). Z.D. is supported by China Scholarship Council (No.201403170252). S.L. is supported by R01GM109882 supplement. F.J. is supported by FONDECYT postdoctoral fellowship #3140458. The Natural Sciences and Engineering Research Council of Canada (05239) and the Canadian Foundation for Innovation/Ontario Research Fund to P.B.S. The authors declare no financial interest.
Footnotes
AUTHOR CONTRIBUTIONS
Z.D., S.S., D.T., N.N., F.J., A.T. P.P., S.L.B., M.R., R.S., A.S., E.C., S.R., and M.M. performed and analyzed experiments involving biochemical, mitochondrial bioenergetics, electrophysiology, molecular and cellular experiments. D.T., S.S. and S.K.J. performed mPEG experiments. X.Z., and J.Y.C. performed electrophysiology experiments. S.L. and J.C. performed super resolution imaging and analysis. N.S. and P.B.S. performed MCU protein production and NMR studies. Z.D., S.S., D.T. S.R., and M.M. conceived, designed, analyzed and interpreted experiments and data. S.S., S.R. and M.M. wrote the manuscript with contributions from J.C., P.B.S., and S.K.J. All authors commented on the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schoneich C, Cohen RA. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med. 2004;10:1200–1207. doi: 10.1038/nm1119. [DOI] [PubMed] [Google Scholar]
- Antony AN, Paillard M, Moffat C, Juskeviciute E, Correnti J, Bolon B, Rubin E, Csordas G, Seifert EL, Hoek JB, et al. MICU1 regulation of mitochondrial Ca(2+) uptake dictates survival and tissue regeneration. Nature communications. 2016;7:10955. doi: 10.1038/ncomms10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgart F, Arnold AM, Leskovar K, Staszek K, Folser M, Weghuber J, Stockinger H, Schutz GJ. Varying label density allows artifact-free analysis of membrane-protein nanoclusters. Nat Methods. 2016;13:661–664. doi: 10.1038/nmeth.3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- Bogeski I, Kummerow C, Al-Ansary D, Schwarz EC, Koehler R, Kozai D, Takahashi N, Peinelt C, Griesemer D, Bozem M, et al. Differential redox regulation of ORAI ion channels: a mechanism to tune cellular calcium signaling. Sci Signal. 2010;3:ra24. doi: 10.1126/scisignal.2000672. [DOI] [PubMed] [Google Scholar]
- Bogeski I, Niemeyer BA. Redox regulation of ion channels. Antioxid Redox Signal. 2014;21:859–862. doi: 10.1089/ars.2014.6019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer TF, Garcia FJ, Onak CS, Carroll KS, Chang CJ. Chemical approaches to discovery and study of sources and targets of hydrogen peroxide redox signaling through NADPH oxidase proteins. Annual review of biochemistry. 2015;84:765–790. doi: 10.1146/annurev-biochem-060614-034018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. American journal of physiology. Cell physiology. 2004;287:C817–833. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- Chaudhuri D, Artiga DJ, Abiria SA, Clapham DE. Mitochondrial calcium uniporter regulator 1 (MCUR1) regulates the calcium threshold for the mitochondrial permeability transition. Proc Natl Acad Sci U S A. 2016;113:E1872–1880. doi: 10.1073/pnas.1602264113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri D, Sancak Y, Mootha VK, Clapham DE. MCU encodes the pore conducting mitochondrial calcium currents. eLife. 2013;2:e00704. doi: 10.7554/eLife.00704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE. Calcium signaling. Cell. 2007;131:1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys J. 2004;86:3993–4003. doi: 10.1529/biophysj.103.038422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordas G, Golenar T, Seifert EL, Kamer KJ, Sancak Y, Perocchi F, Moffat C, Weaver D, de la Fuente Perez S, Bogorad R, et al. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca(2)(+) uniporter. Cell Metab. 2013;17:976–987. doi: 10.1016/j.cmet.2013.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton RM, McCormack JG. Ca2+ as a second messenger within mitochondria of the heart and other tissues. Annu Rev Physiol. 1990;52:451–466. doi: 10.1146/annurev.ph.52.030190.002315. [DOI] [PubMed] [Google Scholar]
- Drago I, Pizzo P, Pozzan T. After half a century mitochondrial calcium in- and efflux machineries reveal themselves. EMBO J. 2011;30:4119–4125. doi: 10.1038/emboj.2011.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR. Mitochondria and calcium: from cell signalling to cell death. J Physiol. 2000;529(Pt 1):57–68. doi: 10.1111/j.1469-7793.2000.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, Shoelson SE, Pawson T, Forman-Kay JD, Kay LE. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- Gandhirajan RK, Meng S, Chandramoorthy HC, Mallilankaraman K, Mancarella S, Gao H, Razmpour R, Yang XF, Houser SR, Chen J, et al. Blockade of NOX2 and STIM1 signaling limits lipopolysaccharide-induced vascular inflammation. J Clin Invest. 2013;123:887–902. doi: 10.1172/JCI65647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glancy B, Balaban RS. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry. 2012;51:2959–2973. doi: 10.1021/bi2018909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go YM, Chandler JD, Jones DP. The cysteine proteome. Free radical biology & medicine. 2015;84:227–245. doi: 10.1016/j.freeradbiomed.2015.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter TE, Gunter KK, Sheu SS, Gavin CE. Mitochondrial calcium transport: physiological and pathological relevance. Am J Physiol. 1994;267:C313–339. doi: 10.1152/ajpcell.1994.267.2.C313. [DOI] [PubMed] [Google Scholar]
- Hajnoczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995;82:415–424. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci. 2010;35:505–513. doi: 10.1016/j.tibs.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins BJ, Irrinki KM, Mallilankaraman K, Lien YC, Wang Y, Bhanumathy CD, Subbiah R, Ritchie MF, Soboloff J, Baba Y, et al. S-glutathionylation activates STIM1 and alters mitochondrial homeostasis. J Cell Biol. 2010;190:391–405. doi: 10.1083/jcb.201004152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman NE, Chandramoorthy HC, Shamugapriya S, Zhang X, Rajan S, Mallilankaraman K, Gandhirajan RK, Vagnozzi RJ, Ferrer LM, Sreekrishnanilayam K, et al. MICU1 motifs define mitochondrial calcium uniporter binding and activity. Cell reports. 2013;5:1576–1588. doi: 10.1016/j.celrep.2013.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOU, Jhun BS, Xu S, Hurst S, Raffaello A, Liu X, Yi B, Zhang H, Gross P, Mishra J, et al. Adrenergic signaling regulates mitochondrial Ca2+ uptake through Pyk2-dependent tyrosine phosphorylation of the mitochondrial Ca2+ uniporter. Antioxid Redox Signal. 2014;21:863–879. doi: 10.1089/ars.2013.5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joiner ML, Koval OM, Li J, He BJ, Allamargot C, Gao Z, Luczak ED, Hall DD, Fink BD, Chen B, et al. CaMKII determines mitochondrial stress responses in heart. Nature. 2012;491:269–273. doi: 10.1038/nature11444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamer KJ, Mootha VK. MICU1 and MICU2 play nonredundant roles in the regulation of the mitochondrial calcium uniporter. EMBO reports. 2014 doi: 10.1002/embr.201337946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamer KJ, Mootha VK. The molecular era of the mitochondrial calcium uniporter. Nat Rev Mol Cell Biol. 2015;16:545–553. doi: 10.1038/nrm4039. [DOI] [PubMed] [Google Scholar]
- Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360–364. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- Kiselyov K, Muallem S. ROS and intracellular ion channels. Cell Calcium. 2016;60:108–114. doi: 10.1016/j.ceca.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- Lee SK, Shanmughapriya S, Mok MC, Dong Z, Tomar D, Carvalho E, Rajan S, Junop MS, Madesh M, Stathopulos PB. Structural Insights into Mitochondrial Calcium Uniporter Regulation by Divalent Cations. Cell Chem Biol. 2016;23:1157–1169. doi: 10.1016/j.chembiol.2016.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Min CK, Kim TG, Song HK, Lim Y, Kim D, Shin K, Kang M, Kang JY, Youn HS, et al. Structure and function of the N-terminal domain of the human mitochondrial calcium uniporter. EMBO reports. 2015;16:1318–1333. doi: 10.15252/embr.201540436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leichert LI, Jakob U. Protein thiol modifications visualized in vivo. PLoS Biol. 2004;2:e333. doi: 10.1371/journal.pbio.0020333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JC, Liu J, Holmstrom KM, Menazza S, Parks RJ, Fergusson MM, Yu ZX, Springer DA, Halsey C, Liu C, et al. MICU1 Serves as a Molecular Gatekeeper to Prevent In Vivo Mitochondrial Calcium Overload. Cell reports. 2016;16:1561–1573. doi: 10.1016/j.celrep.2016.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madesh M, Hawkins BJ, Milovanova T, Bhanumathy CD, Joseph SK, Ramachandrarao SP, Sharma K, Kurosaki T, Fisher AB. Selective role for superoxide in InsP3 receptor-mediated mitochondrial dysfunction and endothelial apoptosis. J Cell Biol. 2005;170:1079–1090. doi: 10.1083/jcb.200505022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallilankaraman K. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell. 2012;151:630–644. doi: 10.1016/j.cell.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallilankaraman K, Cardenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenar T, Csordas G, Madireddi P, Yang J, Muller M, et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol. 2012a;14:1336–1343. doi: 10.1038/ncb2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallilankaraman K, Doonan P, Cardenas C, Chandramoorthy HC, Muller M, Miller R, Hoffman NE, Gandhirajan RK, Molgo J, Birnbaum MJ, et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell. 2012b;151:630–644. doi: 10.1016/j.cell.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG. The regulation of extramitochondrial free calcium ion concentration by rat liver mitochondria. Biochem J. 1978;176:463–474. doi: 10.1042/bj1760463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG. Mitochondria and calcium signaling. Cell Calcium. 2005;38:311–317. doi: 10.1016/j.ceca.2005.06.011. [DOI] [PubMed] [Google Scholar]
- O’Rourke B. Mitochondrial ion channels. Annu Rev Physiol. 2007;69:19–49. doi: 10.1146/annurev.physiol.69.031905.163804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- Oxenoid K, Dong Y, Cao C, Cui T, Sancak Y, Markhard AL, Grabarek Z, Kong L, Liu Z, Ouyang B, et al. Architecture of the mitochondrial calcium uniporter. Nature. 2016;533:269–273. doi: 10.1038/nature17656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15:1464–1472. doi: 10.1038/ncb2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, Granatiero V, Szabo I, De Stefani D, Rizzuto R. MICU1 and MICU2 Finely Tune the Mitochondrial Ca Uniporter by Exerting Opposite Effects on MCU Activity. Mol Cell. 2014 doi: 10.1016/j.molcel.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, Mootha VK. MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature. 2010;467:291–296. doi: 10.1038/nature09358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, Girgis HS, Kuchimanchi S, De Groot J, Speciner L, et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One. 2013;8:e55785. doi: 10.1371/journal.pone.0055785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter Moore C, Zhang JZ, Hamilton SL. A role for cysteine 3635 of RYR1 in redox modulation and calmodulin binding. J Biol Chem. 1999;274:36831–36834. doi: 10.1074/jbc.274.52.36831. [DOI] [PubMed] [Google Scholar]
- Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, Checchetto V, Moro S, Szabo I, Rizzuto R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013;32:2362–2376. doi: 10.1038/emboj.2013.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol. 2012;13:566–578. doi: 10.1038/nrm3412. [DOI] [PubMed] [Google Scholar]
- Sancak Y, Markhard AL, Kitami T, Kovacs-Bogdan E, Kamer KJ, Udeshi ND, Carr SA, Chaudhuri D, Clapham DE, Li AA, et al. EMRE is an Essential Component of the Mitochondrial Calcium Uniporter Complex. Science. 2013 doi: 10.1126/science.1242993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santo-Domingo J, Demaurex N. Perspectives on: SGP symposium on mitochondrial physiology and medicine: the renaissance of mitochondrial pH. J Gen Physiol. 2012;139:415–423. doi: 10.1085/jgp.201110767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanmughapriya S, Rajan S, Hoffman NE, Higgins AM, Tomar D, Nemani N, Hines KJ, Smith DJ, Eguchi A, Vallem S, et al. SPG7 Is an Essential and Conserved Component of the Mitochondrial Permeability Transition Pore. Mol Cell. 2015a;60:47–62. doi: 10.1016/j.molcel.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanmughapriya S, Rajan S, Hoffman NE, Zhang X, Guo S, Kolesar JE, Hines KJ, Ragheb J, Jog NR, Caricchio R, et al. Ca2+ signals regulate mitochondrial metabolism by stimulating CREB-mediated expression of the mitochondrial Ca2+ uniporter gene MCU. Sci Signal. 2015b;8:ra23. doi: 10.1126/scisignal.2005673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soboloff J, Rothberg BS, Madesh M, Gill DL. STIM proteins: dynamic calcium signal transducers. Nat Rev Mol Cell Biol. 2012;13:549–565. doi: 10.1038/nrm3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokatlidis K. A disulfide relay system in mitochondria. Cell. 2005;121:965–967. doi: 10.1016/j.cell.2005.06.019. [DOI] [PubMed] [Google Scholar]
- Tomar D, Dong Z, Shanmughapriya S, Koch DA, Thomas T, Hoffman NE, Timbalia SA, Goldman SJ, Breves SL, Corbally DP, et al. MCUR1 Is a Scaffold Factor for the MCU Complex Function and Promotes Mitochondrial Bioenergetics. Cell reports. 2016;15:1673–1685. doi: 10.1016/j.celrep.2016.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MF, Phillips CB, Ranaghan M, Tsai CW, Wu Y, Willliams C, Miller C. Dual functions of a small regulatory subunit in the mitochondrial calcium uniporter complex. eLife. 2016:5. doi: 10.7554/eLife.15545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vais H, Mallilankaraman K, Mak DO, Hoff H, Payne R, Tanis JE, Foskett JK. EMRE Is a Matrix Ca(2+) Sensor that Governs Gatekeeping of the Mitochondrial Ca(2+) Uniporter. Cell reports. 2016;14:403–410. doi: 10.1016/j.celrep.2015.12.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.