

Abstract
Caffeic acid O-methyltransferase (COMT) catalyzes preferentially the methylation of 5-hydroxyconiferaldehyde to sinapaldehyde in monolignol biosynthesis. Here, we have compared HPLC profiles of the methanol-soluble phenolics fraction of xylem tissue from COMT-deficient and control poplars (Populus spp.), using statistical analysis of the peak heights. COMT down-regulation results in significant concentration differences for 25 of the 91 analyzed peaks. Eight peaks were exclusively detected in COMT-deficient poplar, of which four could be purified for further identification using mass spectrometry/mass spectrometry, nuclear magnetic resonance, and spiking of synthesized reference compounds. These new compounds were derived from 5-hydroxyconiferyl alcohol or 5-hydroxyconiferaldehyde and were characterized by benzodioxane moieties, a structural type that is also increased in the lignins of COMT-deficient plants. One of these four benzodioxanes amounted to the most abundant oligolignol in the HPLC profile. Furthermore, all of the differentially accumulating oligolignols involving sinapyl units were either reduced in abundance or undetectable. The concentration levels of all identified oligolignols were in agreement with the relative supply of monolignols and with their chemical coupling propensities, which supports the random coupling hypothesis. Chiral HPLC analysis of the most abundant benzodioxane dimer revealed the presence of both enantiomers in equal amounts, indicating that they were formed by radical coupling reactions under simple chemical control rather than guided by dirigent proteins.
Lignins are aromatic heteropolymers that are predominantly deposited in the walls of secondary thickened cells. In angiosperms, lignins are principally derived from coniferyl and sinapyl alcohols. When these hydroxycinnamyl alcohols are polymerized into lignin, the resulting units in the polymer are designated guaiacyl and syringyl (G and S), respectively. In addition to these two main monolignols, other monomers, such as p-coumaryl alcohol, hydroxycinnamaldehydes, and hydroxycinnamate esters, may be incorporated into the polymer as well (Boerjan et al., 2003; Lu et al., 2004; Ralph et al., 2004b).
The biosynthesis of the monolignols starts with the deamination of Phe and proceeds through the general phenylpropanoid pathway (Fig. 1). The first step of the monolignol-specific pathway is the conversion of feruloyl-CoA to coniferaldehyde, which is catalyzed by cinnamoyl-CoA reductase (CCR). For the biosynthesis of coniferyl alcohol, coniferaldehyde is reduced by cinnamyl alcohol dehydrogenase (CAD). The biosynthesis of sinapyl alcohol involves a further hydroxylation of the aromatic C5 position of coniferaldehyde by ferulic acid 5-hydroxylase (F5H, alternatively called coniferaldehyde 5-hydroxylase [Cald5H]; Humphreys et al., 1999; Osakabe et al., 1999), followed by the methylation of this hydroxyl group by caffeic acid O-methyltransferase (COMT, alternatively called 5-hydroxyconiferaldehyde O-methyltransferase [AldOMT]; Li et al., 2000). The subsequent reduction by CAD or sinapyl alcohol dehydrogenase (SAD) leads to sinapyl alcohol (Li et al., 2001; Boerjan et al., 2003).
Figure 1.

Monolignol biosynthetic pathway. PAL, Phenylalanine ammonia-lyase; C4H, cinnamate 4-hydroxylase; 4CL, 4-coumarate:CoA ligase; HCT, p-hydroxycinnamoyl-CoA:shikimate/quinate p-hydroxycinnamoyltransferase; C3H, p-coumaroyl-shikimic acid/quinic acid 3-hydroxylase; SAD; sinapyl alcohol dehydrogenase; H, p-hydroxyphenyl; G, guaiacyl; 5H, 5-hydroxyguaiacyl; S, syringyl; ??, direct conversion not convincingly demonstrated; ?, conversion demonstrated; 4CL??; some species have 4CL activity toward sinapic acid; CCR? and F5H?, substrate not tested; others, enzymatic activity shown in vitro. Bold arrows indicate the presumed flux throughout the metabolic grid (for further discussion, see Boerjan et al., 2003).
Transgenic plants altered in the expression of monolignol biosynthesis genes have been very instructive in unraveling the monolignol biosynthetic pathway. Most of them have been analyzed for their lignin amount, composition, and structure (Baucher et al., 2003; Boerjan et al., 2003), but only a few for potential alterations in soluble phenolics (Meyermans et al., 2000; Chabannes et al., 2001; Guo et al., 2001; Ranocha et al., 2002; Chen et al., 2003; Goujon et al., 2003). The analysis of soluble phenolics is important to understand the in vivo function of the different enzymes, the flux through the monolignol pathway, and the complexity of monolignol coupling and lignin deposition (Morreel et al., 2004).
Previously, we have analyzed the methanol-soluble phenolic fraction of poplar xylem, a tissue that is heavily lignified (Morreel et al., 2004). By liquid chromatography (LC)-mass spectrometry (MS), we elucidated the structures of 38 phenolic compounds, corresponding to a family of dimers, trimers, and tetramers of monolignols and their corresponding aldehydes, linked by 8–O–4, 8–5, and 8–8 bonds. In poplar down-regulated for caffeoyl-CoA O-methyltransferase (CCoAOMT) that produces less lignin (Meyermans et al., 2000; Zhong et al., 2000), the synthesis of these oligolignols is reduced and compensated for by the production of glucosides of vanillic, caffeic, and sinapic acids (Meyermans et al., 2000; Morreel et al., 2004). All oligolignol structures were in agreement with a combinatorial coupling of monolignols according to chemical principles.
Here, we analyzed statistically the levels of 91 peaks after HPLC profiling of the methanol-soluble phenolic fraction of COMT-deficient poplar. Previous research has shown that COMT deficiency leads to reduced incorporation of S units and that of 5-hydroxyconiferyl alcohol into the lignin polymer (Van Doorsselaere et al., 1995; Lapierre et al., 1999; Jouanin et al., 2000; Ralph et al., 2001a, 2001b; Marita et al., 2003a, 2003b). We found that the abundance of several oligolignols is significantly altered in transgenic compared to wild-type plants, and that their relative abundance is in agreement with the relative supply of monolignols and with the composition of lignin. Importantly, four new structures, abundantly present in the xylem extracts of COMT-deficient plants and below detection limit in wild-type plants, were identified by LC-mass spectrometry/mass spectrometry (MS/MS), NMR, and/or chemical synthesis. All four compounds were benzodioxane structures formally derived from 5-hydroxyconiferyl alcohol or 5-hydroxyconiferaldehyde, both substrates for COMT. Furthermore, chiral HPLC of the most abundant benzodioxane dimer demonstrated that the two enantiomers were present in equal amounts, suggesting that these dimers are formed via radical coupling reactions under chemical control without involvement of dirigent proteins.
RESULTS
LC-MS/MS Analysis of Hydroxycinnamic Acids and Hydroxycinnamaldehydes
To investigate whether COMT down-regulation affected the concentration of monolignol biosynthesis precursors, the in vivo concentrations of the different hydroxycinnamic acid and hydroxycinnamaldehyde intermediates in phenylpropanoid biosynthesis were compared statistically in COMT-deficient (transgenic lines ASB 2B and ASB 10B) and control poplars. LC-atmospheric pressure chemical ionization (APCI)-MS/MS was performed on extracts of the stem xylem from 144 individuals. Concentrations of p-coumaric acid, caffeic acid, 5-hydroxyferulic acid, caffealdehyde, and 5-hydroxyconiferaldehyde were below the detection limit (approximately 0.5 pmol mg−1 dry weight) in all analyzed plants, whereas that of p-coumaraldehyde was close to it. Ferulic acid, sinapic acid, coniferaldehyde, and sinapaldehyde were detectable in all poplar lines, with concentrations (mean ± se of the mean) of 147 ± 70, 197 ± 30, 28.4 ± 4, and 89.5 ± 14 pmol mg−1 dry weight in the control poplars, and 69.5 ± 14, 182 ± 40, 30.8 ± 6, and 21.4 ± 4 pmol mg−1 dry weight in the COMT-deficient lines, respectively. Analysis of this data set by the nested ANOVA Model A (see “Materials and Methods”) revealed a significantly lower concentration of sinapaldehyde in the COMT-deficient poplars, in agreement with the predominant role played by COMT in the methylation of 5-hydroxyconiferaldehyde to sinapaldehyde (Humphreys et al., 1999; Li et al., 2000). COMT also catalyzes the methylation of 5-hydroxyferulic acid to sinapic acid in vitro (Van Doorsselaere et al., 1993), albeit not very efficiently, but the concentration of sinapic acid was not significantly affected by COMT down-regulation. Still, the inability to detect a reduction in the amount of sinapic acid could be due to a variable MS response obtained for the compounds from run to run, to the quantification procedure, or to biological variation among the samples. These sources of variation can be eliminated by calculating the logarithmically transformed ratios (Birks and Kanowski, 1993) of the abundance of two phenylpropanoids present in one and the same LC-MS run. Thus, the nested ANOVA Model A was used to analyze the six possible log ratios between the four phenylpropanoids (mean ratios are shown in Table I). Also with this method, the concentration of sinapic acid relative to that of ferulic acid was not different in the COMT-deficient poplars, whereas that of sinapaldehyde was significantly lower relative to the other three phenylpropanoids.
Table I.
Mean ratios of the concentration of phenylpropanoid pathway intermediates
| Ratio
|
Mean Ratio ± s.e.m.
|
||||
|---|---|---|---|---|---|
| Control
|
COMT
|
||||
| WT | 35S 17B | 35S 21B | ASB 2B | ASB 10B | |
| FA/SA | 0.28 ± 0.06 | 0.35 ± 0.05 | 0.26 ± 0.06 | 0.42 ± 0.10 | 0.29 ± 0.06 |
| FA/Cal | 1.11 ± 0.30 | 1.40 ± 0.38 | 1.21 ± 0.64 | 1.49 ± 0.54 | 0.82 ± 0.23 |
| Cal/SA | 0.27 ± 0.06 | 0.30 ± 0.08 | 0.30 ± 0.07 | 0.50 ± 0.27 | 0.37 ± 0.07 |
| FA/Sal | 0.18 ± 0.05 | 0.30 ± 0.07 | 0.24 ± 0.11 | 1.18 ± 0.25* | 0.72 ± 0.21* |
| SA/Sal | 0.67 ± 0.11 | 0.90 ± 0.18 | 0.80 ± 0.19 | 3.47 ± 1.11* | 2.66 ± 0.56* |
| Cal/Sal | 0.16 ± 0.01 | 0.23 ± 0.03 | 0.21 ± 0.03 | 0.96 ± 0.18* | 0.91 ± 0.18* |
The mean ratios of the LC-MS selected ion currents obtained for ferulic acid (FA), sinapic acid (SA), coniferaldehyde (Cal), and sinapaldehyde (Sal) are given for the control (wild-type and transgenic lines 35S 17B and 35S 21B) and COMT-deficient poplars (transgenic lines ASB 2B and ASB 10B). Significantly changed ratios, obtained by applying the nested ANOVA Model A, between both groups of poplar lines are indicated with an asterisk. s.e.m., se of the mean.
Phenolic Profiling of COMT-Deficient Transgenic Poplars
To investigate whether COMT down-regulation affected the concentration of phenolic metabolites other than the phenylpropanoid intermediates, the individual chromatogram peaks were quantified by standardizing peak height to dry weight. The total amount of soluble phenolics, approximated by the sum of the heights of all peaks in the chromatogram divided by the dry weight, did not significantly differ between wild-type and COMT-deficient poplars based on the analysis of 144 individuals. An overlay of the chromatograms of a representative wild-type and COMT-deficient line is shown in Figure 2. Of a total of 91 peaks analyzed, 8 peaks were specifically associated with the COMT-deficient lines and 4 had disappeared in the COMT-deficient poplars. For the remaining 79 peaks, a weighted nested ANOVA or one-way ANOVA, combined with the appropriate post hoc test (see “Materials and Methods”), showed significantly increased concentrations for three peaks in the COMT-deficient poplars, whereas those of 10 peaks were lower in the transgenic poplars. Thus, 25 of the 91 peaks had a significantly altered concentration, of which six had been identified previously as coupling products of monolignols and their aldehydes (Morreel et al., 2004; see below). Four of the 19 unidentified peaks, notably compounds 1a, 1b, 2, and 3a that are not detected in the wild-type poplars, were present in sufficient amounts in the transgenic lines, allowing their isolation for further characterization. The remaining 15 unknown peaks were of too low abundance for isolation and subsequent MS/MS and/or NMR analysis. Notably, 14 of these 15 unknowns showed UV/visible (Vis) absorption spectra reminiscent of oligolignols.
Figure 2.

HPLC analysis. Overlay of the HPLC chromatograms of the xylem extract of a wild-type (black) and a COMT down-regulated line (ASB 2B, red), showing the position of the different oligolignols. Oligolignols that are significantly different in abundance between wild-type and COMT-deficient poplars are in blue.
The mean concentrations of the oligolignol peaks whose structures could be characterized, including both differential and nondifferential peaks, are presented in Table II. Compounds 1a, 1b, 2, and 3a were found only in the COMT-deficient poplars. The most prominent change was seen for compound 1a, which was at 666 to 683 pmol mg−1 dry weight, or at least 260-fold increased in COMT-deficient poplars, taking the detection limit (2.6 pmol mg−1) into account for the control group. The concentration of compounds 1b, 2, and 3a were at least 19-, 29-, and 71-fold higher than the detection limit, respectively. G(8–5)G′ (balanophonin) was also present in the control lines, but its level was 6.8-fold higher in COMT-deficient than in wild-type poplar. By contrast, the levels of the oligomers G(t8–O–4)S(8–5)G (threo-buddlenol B), G(e8–O–4)S(8–5)G (erythro-buddlenol B), G(e8–O–4)S(8–5)G′ (erythro-buddlenol A), and G(8–O–4)S(8–8)S(8–O–4)G (hedyotisol) were reduced by 60% to 90%, whereas SP(8–8)S was undetectable in the COMT-deficient poplars. If present at all, the concentration of this product was at least 9-fold lower than that of the control lines based on the detection limit.
Table II.
Concentration of oligolignols identified in COMT-deficient or control poplars
| Product
|
Mean pmol mg−1 Dry Weight ± s.e.m.
|
Fold Change
|
||||
|---|---|---|---|---|---|---|
| Control
|
COMT
|
|||||
| WT | 35S 17B | 35S 21B | ASB 2B | ASB 10B | ||
| G(8–O–4)5H 1a | ND | ND | ND | 683 ± 70* | 666 ± 120* | 260 |
| G(8–O–4)5H 1b | ND | ND | ND | 51.4 ± 5* | 49.6 ± 7* | 19 |
| G(8–5)G′(8–O–4)5H 2 | ND | ND | ND | 68.0 ± 5* | 84.7 ± 20* | 29 |
| G(8–O–4)5H′ 3a | ND | ND | ND | 180 ± 20* | 191 ± 40* | 71 |
| G(t8–O–4)G | 69.5 ± 31 | 38.8 ± 29 | 94.3 ± 40 | 111 ± 30 | 86.8 ± 32 | |
| G(8–5)G | 105 ± 40 | 95.5 ± 28 | 98.1 ± 24 | 134 ± 40 | 111 ± 30 | |
| G(8–8)G | 53.3 ± 8 | 46.5 ± 3 | 42.2 ± 5 | 68.7 ± 8 | 77.3 ± 3 | |
| S(8–8)S | coel. | coel. | coel. | 17.8 ± 5 | 14.2 ± 7 | |
| S(8–5)G | 41.3 ± 6 | 31.2 ± 9 | 39.3 ± 5 | 12.0 ± 1 | 26.6 ± 3 | |
| G(8–5)V′ | ND | ND | ND | ND | ND | |
| G(8–5)G′ | 13.5 ± 7 | 2.52 ± 0.4 | 4.64 ± 1.2 | 50.0 ± 9* | 43.9 ± 10* | 6.8 |
| G(t8–O–4)G(t8–O–4)G | 20.4 ± 4 | 15.6 ± 7 | 23.3 ± 5 | 14.1 ± 3 | 9.22 ± 3.0 | |
| S(t8–O–4)S(8–5)G | ND | ND | ND | ND | ND | |
| G(t8–O–4)S(8–5)G | 151 ± 30 | 153 ± 30 | 152 ± 20 | 61.8 ± 9* | 57.9 ± 10* | 0.40 |
| G(e8–O–4)S(8–5)G | 35.7 ± 4 | 37.7 ± 7 | 34.9 ± 6 | 3.35 ± 0.9* | 4.06 ± 1.2* | 0.10 |
| S(t8–O–4)S(8–5)G′ | coel. | coel. | coel. | coel. | coel. | |
| SP(8–8)S | 20.3 ± 6 | 27.2 ± 5 | 24.4 ± 6 | ND* | ND* | 0.11 |
| G(t8–O–4)S(8–5)G′ | 107 ± 10 | 125 ± 20 | 96.0 ± 13 | 68.6 ± 5 | 126 ± 10 | |
| G(e8–O–4)S(8–5)G′ | 85.8 ± 7 | 92.9 ± 25 | 82.1 ± 13 | 14.8 ± 4* | 18.7 ± 8* | 0.19 |
| G(8–O–4)S(8–8)S(8–O–4)G | 66.4 ± 6 | 65.0 ± 11 | 67.5 ± 6 | 19.1 ± 2* | 26.6 ± 4* | 0.34 |
Mean concentrations, expressed as pmol mg−1 dry weight, are shown for the control poplar lines (wild-type and transgenic lines 35S 17B and 35S 21B) and poplars down-regulated for COMT (transgenic lines ASB 2B and ASB 10B). Those oligolignols for which the concentration could be determined in control (Morreel et al., 2004) and/or COMT-deficient poplar are presented. The fold change of the product abundance in COMT-deficient poplars relative to the control lines is indicated. When the peak was not detected, the relative increase was calculated using a detection limit of 2.6 pmol mg−1 dry weight. Significantly different concentrations between control and COMT-deficient poplars are marked by an asterisk. coel., Concentration not determined because of coelution of two or more compounds; ND, not detected; s.e.m., se of the mean.
Characterization of Compounds 1a, 1b, 2, 3a, and 3b
Compounds 1a and 1b
Compound 1a, eluting at 16.7 min, had a UV/Vis spectrum (λmax at 226.5 and 274.7 nm) similar to that of (8–5)-dehydrodiconiferyl alcohol, G(8–5)G, and identical to that of compound 1b, eluting at 16.5 min immediately in front of compound 1a. Both compounds have a molecular mass of 374 g mol−1 as determined by MS/MS (Fig. 3). The isolated compound 1a (containing 10% 1b; see “Materials and Methods”) was characterized by NMR. The aromatic region of the 1H-NMR spectrum showed eight signals: four in a pattern typical for G units, namely a broad singlet for the phenolic proton at 7.68 ppm, a doublet with a small J value (1.9 Hz) at 7.11 ppm, a doublet with a J value of 8.2 Hz at 6.88 ppm, a double doublet at 6.97 ppm, two signals indicative of a 5-oxy-substituted coniferyl moiety (two mutually coupled small doublets at 6.70 and 6.60 ppm), and two trans-olefinic (J = 15.9 Hz) signals at 6.50 and 6.27 ppm. Furthermore, a doublet with a J = 8 Hz at 4.99 ppm, a two-proton multiplet at 4.20 ppm typical for an allylic CH2OH group, and a one-proton multiplet at 4.04 ppm coupled with the previously mentioned 4.99-ppm signal and also with the multiplets at 3.77 and 3.50 ppm; at approximately 3.86 ppm, two singlets typical for two methoxy groups were found. Assignments were confirmed by two-dimensional correlation spectroscopy spectra and also by several one-dimensional total correlation spectroscopy experiments with selective excitation of the signals at 6.48, 4.98, or 4.05 ppm, using an array of mixing times ranging from 90 to 250 ms. The 1H-NMR data together with the mass spectra led to the assignment of a structure arising from cross-coupling of coniferyl alcohol and 5-hydroxyconiferyl alcohol and characterized by a benzodioxane ring. The trans-configuration of the two groups on the benzodioxane ring (7R,8R or 7S,8S) was assigned by the large coupling constant (J = 8.0 Hz) between the pseudoaxial protons H-7 and H-8 at 4.98 and 4.04 ppm, respectively.
Figure 3.

Spectra, proposed or authenticated structures, and chiral HPLC data. APCI-MS/MS spectra of compounds 1, 2, and 3 were obtained in the negative-ion mode. The presented UV/Vis spectra for each of these products were taken between 200 and 450 nm. Based on MS/MS and UV/Vis spectra, molecular structures were proposed and subsequently synthesized for authentication for products 1 and 3. Two pairs of enantiomers were detected for both the natural product (N) and the synthesized (S) compounds 1a and 1b by chiral HPLC analysis. G, Guaiacyl; 5H, 5-hydroxyguaiacyl; G′, unit derived from coniferaldehyde; 5H′, unit derived from 5-hydroxyconiferaldehyde.
To authenticate this compound, the proposed structure was synthesized (Fig. 4). Both synthetic isomers eluted at the same time and in the same ratio (10:1, w/w) as the natural compounds 1a (trans-isomer) and 1b (cis-isomer); 1H-NMR spectra matched completely. Both isomers together will be designated compound 1 hereafter: [G(8–O–4)5H, 4-[3-hydroxymethyl-7-(E)-(3-hydroxypropenyl)-5-methoxy-2,3-dihydro-benzo[1,4] dioxin-2-yl]-2-methoxyphenol (named nocomtol)].
Figure 4.

Synthesis of benzodioxane compound 1. Compound a, Coniferyl alcohol. Compound b, Ethyl 5-hydroxyferulate. Compound c, 3-[3-(4-Hydroxy-4-methoxyphenyl)-2-hydroxymethyl-8-methoxy-2,3-dihydro-benzo[1,4] dioxin-6-yl]-acrylic acid ethyl ester.
Compound 2
Compound 2 elutes at 19.4 min and has a similar UV/Vis spectrum (λmax at 210.1 and 273.5 nm) as (8–5)-dehydrodiconiferyl alcohol [G(8–5)G]. The molecular mass of 550 g mol−1, indicated by LC-MS/MS (Fig. 3), suggests a trimer of monolignols.
The molecular structure was deduced from the following observations. (1) In the MS/MS spectra, ions at m/z 531 (M-H2O)− and 519 (M-CH2O)− imply the presence of a –CH2OH group. (2) Because the magnitude of the ion abundance at m/z 501 (M-H2O-CH2O)− was clearly lower than for both former ions, a conventional β-aryl ether (8–O–4) unit did not seem to be involved in the MS/MS fragmentation. The detection of daughter ions corresponding to neutral losses of 18 g mol−1 (H2O), 30 g mol−1 (CH2O), and 48 g mol−1 (H2O + CH2O) have been associated with a threo-β-aryl ether unit, whenever the latter ion is prominent and with an erythro configuration, when all three ions are equally abundant (Morreel et al., 2004). (3) The major peaks in the MS/MS spectra did not correspond with neutral losses associated with the fragmentation of resinol or phenylcoumaran structures (Morreel et al., 2004), not even when a shift of 14 or 30 g mol−1 was considered to take into account the incorporation of units other than coniferyl alcohol. Its MS/MS spectrum was clearly different from those of G(8–5)G [(8–5)-dehydrodiconiferyl alcohol] and G(8–8)G (pinoresinol; Morreel et al., 2004). (4) The major peaks found here at m/z 179 and 193 point to the release of a G and a 5H unit, respectively, as also found in the MS/MS fragmentation of nocomtol. Together with the occurrence of an ion at m/z 195, these observations indicate that a terminal 5H unit is present, implying that a second G unit is attached to the benzodioxane G unit by means of a phenylcoumaran unit. The loss of 180 g mol−1 is associated with the release of the phenylcoumaran-linked G unit, whereby the remaining daughter ion at m/z 369 points to the presence of an aliphatic aldehyde group instead of an alcohol group in the dioxane structure. Hence, this structure appears to arise from the cross-coupling of coniferaldehyde (at its C8 position) with 5-hydroxyconiferyl alcohol (at its 4–O position), followed by the cross-coupling of the resultant benzodioxane dimer G′(8–O–4)5H at its C5 position with coniferyl alcohol at its favored C8 position. This trimeric structure is given in Figure 3 and shorthand annotated as G(8–5)G′(8–O–4)5H, but remains only tentatively assigned and cannot currently be authenticated. Attempts to cross-couple coniferaldehyde with 5-hydroxyconiferyl alcohol failed to produce G′(8–O–4)5H, reducing the certainty in this assignment.
Compounds 3a and 3b
Compound 3a was observed at a retention time of 19.7 min, and HPLC repurification showed equilibrium (10:1, w/w) between two compounds with retention times at 19.7 min (3a) and 21.0 min (3b). Both compounds had identical UV/Vis spectra, indicating a terminal cinnamaldehyde unit and a molecular mass of 372 g mol−1 (Fig. 3). The same neutral losses were observed in the MS/MS spectra of both compounds 3a and 3b, and 1a and 1b. UV/Vis spectra showed absorption at longer wavelengths (λmax at 204.3, 274.7, and 335.1 nm), suggesting that the compounds 3a and 3b were the corresponding aldehydes of compounds 1a and 1b. Structure 3 was also synthesized and confirmed the identification of the natural compounds 3 as trans- and cis-G(8–O–4)5H′, 3-[3-(4-hydroxy-3-methoxyphenyl)-2-hydroxymethyl-8-methoxy-2,3-dihydro-benzo[1,4]-dioxin-6-yl]acrylaldehyde, named nocomtal (Fig. 3). Both benzodioxane ring isomers were expected in analogy with compounds 1 (She et al., 1998; Sousa et al., 2002). The cis-isomer could not be directly detected in the xylem extract profiles of COMT-deficient poplars because of coelution.
Chiral HPLC Analysis
The new compounds in COMT-deficient poplars were all benzodioxanes derived from cross-coupling of a 5-hydroxyconiferyl monomer. All compounds contained asymmetric carbon atoms. If the formation of this structure were to be controlled by dirigent proteins as that of the 8–8 coupling of coniferyl alcohol in the lignan pinoresinol (Davin et al., 1997), the abundance of the two stereoisomers would deviate from the 50:50 ratio expected for racemic coupling. Hence, the major benzodioxane present in COMT-deficient poplars was analyzed on a chiral HPLC column to observe whether the isolated natural product was optically active or racemic. The HPLC separation (Fig. 3) of the natural and synthesized nocomtol showed that both trans-enantiomers, at 46 to 48 min and 54 to 57 min, were present in approximately the same 1:1 ratio. Experimentally, the integrals were 49:51 for the natural isolate and 47:53 for the synthetic nocomtol. The cis-enantiomers were detected separately at 39 to 41 min and 42 to 43 min. All peaks had the typical UV/Vis spectrum of compound 1, G(8–O–4)5H.
DISCUSSION
COMT Down-Regulation Reduces the Concentration of Sinapaldehyde But Not That of Sinapic Acid
Down-regulation of COMT in poplar results in a reduced S:G ratio in the noncondensed fraction of the lignin polymer, mainly because of a decrease in S units (Van Doorsselaere et al., 1995; Tsai et al., 1998; Lapierre et al., 1999; Jouanin et al., 2000). In vitro enzymatic assays, radiotracer experiments, and studies based on the x-ray structure of COMT have shown that COMT is predominantly active at the cinnamaldehyde and/or cinnamyl alcohol level of the monolignol biosynthetic pathway (Inoue et al., 1998, 2000; Meng and Campbell, 1998; Maury et al., 1999; Osakabe et al., 1999; Li et al., 2000; Matsui et al., 2000; Chen et al., 2001; Parvathi et al., 2001; Zubieta et al., 2002) rather than at the cinnamic acid level as was originally thought. Furthermore, the conversions catalyzed by COMT and F5H at the cinnamic acid level of the pathway would not be operational in lignifying tissue because of the inhibition by 5-hydroxyconiferaldehyde and coniferaldehyde (Humphreys et al., 1999; Osakabe et al., 1999; Li et al., 2000). Hence, when monolignol biosynthesis is active, COMT controls the C5 methylation in monolignol biosynthesis predominantly at the aldehyde level, and only a limited flux is thought to exist from caffeic acid to sinapic acid, involving COMT and F5H. In agreement, a 4.5-fold reduction in sinapaldehyde concentration was observed with LC-MS/MS of xylem extracts of COMT down-regulated poplars.
Despite the low 5% residual activity of COMT in the transgenic poplar lines ASB 2B and ASB 10B (Van Doorsselaere et al., 1995), no significant differences in the concentrations of sinapic acid were observed. These results imply either that there is an additional O-methyltransferase without enough sequence similarity with the COMT gene to be down-regulated in the antisense COMT transgenic poplars, or that an alternative biosynthetic route to sinapic acid exists in poplar.
New Benzodioxanes Accumulate in COMT Down-Regulated Poplars
HPLC analysis of the methanol-soluble phenolics present in poplar xylem revealed a significantly different concentration for 25 of the 91 analyzed peaks because of COMT down-regulation. The large number of metabolites in these profiles and the biological variation in their concentration necessitated the use of statistical methods to evaluate whether the concentration of a compound differed significantly in concentration between the control group and the COMT down-regulated plants. Significantly affected unknown peaks that were clearly resolved in the chromatogram and present as at least 1% of the total peak height were subsequently purified for further characterization.
The major difference in the metabolite profiles was the strong accumulation of compound 1a (Table II), below the detection limit in the control plants, but one of the most prominent peaks present in the chromatograms of the COMT down-regulated poplars (Fig. 2). By means of LC-MS/MS, NMR, and independent synthesis, this compound was identified as a condensation product of coniferyl alcohol and 5-hydroxyconiferyl alcohol, formed by radical coupling of 5-hydroxyconiferyl alcohol at its 4–O position with the favored C8 position of coniferyl alcohol. The cyclization (by internal trapping of the quinone methide coupling intermediate by the 5-OH) was established by the diagnostic 3J C,H correlation between H-7 and C-5′ in the gradient-selected heteronuclear multiple-bond correlation experiment. The predominance of the trans-ring-isomer (R,R or S,S) was established from the 1H-NMR 3J7–8 coupling constant. As in synthetic cross-coupling reactions, there is regioselectivity for 4–O over 5–O coupling of 5-hydroxyconiferyl alcohol to give the 8–O–4/7–O–5 benzodioxane rather than the 8–O–5/7–O–4 regioisomer.
The compound 1, G(8–O–4)5H, which has not been described before, has a 1,4-benzodioxane structure and has been called nocomtol. Compound 1b formed spontaneously from 1a at approximately the 10% level from purified as well as synthesized nocomtol 1. The cis-/trans-conversion of the benzodioxane ring is well documented (She et al., 1998; Sousa et al., 2002). In addition to nocomtol 1a and 1b, other compounds 2, 3a, and 3b were typically observed in COMT-deficient poplar samples. Compounds 3a and 3b were identified as the cinnamaldehyde analogs of nocomtol 1a and 1b, i.e. trans- and cis-G(8–O–4)5H′, and were called trans- and cis-nocomtal.
A trimeric oligolignol, containing a benzodioxane structure and an aliphatic aldehyde, was proposed for compound 2. From the UV spectrum, the compound does not contain a cinnamaldehyde end group, yet the MS/MS-derived molecular weights of the daughter ions require an aldehyde to be present. All data for this compound were consistent with a G(8–5)G′(8–O–4)5H structure, as shown in Figure 3.
All of these benzodioxanes resulted from the cross-coupling of coniferyl and 5-hydroxyconiferyl monomers. Analogous benzodioxane structures have been identified at striking levels in the lignin polymer of COMT-deficient alfalfa (Medicago sativa; Marita et al., 2003a), poplar (Jouanin et al., 2000; Ralph et al., 2001a, 2001b), Arabidopsis (Arabidopsis thaliana; Ralph et al., 2001b), and maize (Zea mays; Lapierre et al., 1988; Marita et al., 2003b) by NMR and thioacidolysis. Evidence for 5H units coupled with sinapyl alcohol can be found in trace quantities in lignin of COMT-deficient alfalfa (Marita et al., 2003a), yet this S(8–O–4)5H lignin unit is minor because of the limited synthesis of sinapyl alcohol in the COMT-deficient plants. The corresponding S(8–O–4)5H dilignols were detectable neither in HPLC profiles of wild-type and COMT-deficient poplars nor in LC-MS/MS chromatograms (Ishikawa et al., 1995) based on the corresponding m/z values (data not shown). Coupling products involving two 5H units have not been identified either, yet there is evidence in polymeric lignins for sequential 5H units in extended benzodioxane structures (Marita et al., 2003a). Such structures are more likely to be found in higher oligomers because the 5-hydroxyguaiacyl group is reactive and not found as an end group.
Cross-coupling products between coniferaldehyde and 5-hydroxyconiferaldehyde were not detected either, which may be because of their (unknown)cross-coupling propensities that may not favor such products (Kim et al., 2000). For example, although dehydrodimerization of coniferaldehyde and 8–O–4 cross-coupling of coniferaldehyde with either coniferyl or sinapyl alcohol occurs efficiently, coniferaldehyde cross-couples only efficiently with S units but not with G units in the evolving lignin polymer, both in vitro and in vivo (Kim et al., 2000). Consequently, in CAD-deficient plants that accumulate the cinnamaldehyde substrates for CAD (Kim et al., 2003), coniferaldehyde incorporates well into S-G lignins in hardwoods, such as poplar, but is poorly incorporated into softwoods, which have only G lignins. An alternative reason for the apparent lack of a G′-5H′ coupling product is that the in vivo concentrations of both cinnamaldehydes may be much lower than those of their corresponding alcohols, which would favor cross-coupling with a cinnamyl alcohol. The apparent absence of 5-hydroxyconiferaldehyde in LC-MS profiles further supports the latter hypothesis.
No dimers involving a 5-hydroxyconiferyl unit linked via its C8 position to a G, G′, S, S′, or 5H′ unit were detected by HPLC or LC-MS/MS analysis, suggesting that 5-hydroxyconiferyl units are very reactive and preferentially couple via their 4–O rather than their C8 position. The seeming lack of dimers involving 5-hydroxyconiferyl units other than compounds G(8–O–4)5H 1 and G(8–O–4)5H′ 3 suggests that the remaining seven peaks, with increased abundance in COMT down-regulated lines but still unidentified because of their low concentration, are higher-order oligomers containing 5-hydroxyconiferyl units.
Hydroxycinnamaldehydes Are Monomers
Previously elucidated oligolignol structures have shown that hydroxycinnamaldehydes or hydroxybenzaldehydes can be used in oligomerization reactions (Morreel et al., 2004). For example, in xylem of wild-type poplar, a trimeric and a tetrameric oligolignol involving an S′(8–8)S moiety were implicated, for which the structure is most logically explained by the 8–8 coupling of a sinapaldehyde with a sinapyl alcohol monomer (Morreel et al., 2004). Similarly, the aliphatic aldehyde functionality in G(8–5)G′(8–O–4)5H 2 would indicate that this compound was not synthesized by the oxidation of its corresponding aliphatic alcohol (which is much more difficult to oxidize than the cinnamyl alcohol also present in the structure), but arises from the initial cross-coupling between coniferaldehyde and 5-hydroxyconiferyl alcohol radicals. Therefore, the compounds G(8–5)G′(8–O–4)5H 2 (if authenticated) and G(8–O–4)5H′ 3 further support the notion that the aromatic aldehydes are monomeric components and that COMT down-regulation provides an additional cinnamaldehyde precursor, 5-hydroxyconiferaldehyde.
The Oligolignol Composition Reflects the Relative Supply of Monolignols and Their Coupling Propensities
The remaining 21 of the 25 differentially accumulating peaks had UV/Vis spectra resembling those of oligolignols for all but one. Previously, we have identified six of these peaks (Morreel et al., 2004). G(8–5)G′ (balanophonin) accumulates about 7-fold in COMT-deficient poplars, whereas the levels of other coupling products between two G units, which were identified and quantified {G(8–8)G [pinoresinol] and G(8–5)G [(8–5)-dehydrodiconiferyl alcohol]} were not significantly altered nor did those of G(t8–O–4)G [(8–O–4)-dehydrodiconiferyl alcohol], possibly because of the very low levels observed for this compound in all xylem extracts. The fact that G(8–5)G′ increases 7-fold, whereas G-G dimers did not, further supports the predominant role of COMT at the cinnamaldehyde level of the pathway (Humphreys et al., 1999; Osakabe et al., 1999; Li et al., 2000) and provides further evidence that coniferaldehyde end groups in the oligomers and in the lignin of COMT down-regulated poplars (Tsai et al., 1998; Jouanin et al., 2000) are derived from coniferaldehyde monomers rather than from postcoupling oxidation. The concentration of the remaining five previously identified oligolignols (Morreel et al., 2004) were all lower in COMT-deficient poplars. These oligomers were all composed of at least one S unit coupled to a G and/or a G′ unit. Both the threo- and the erythro-isomers of buddlenol B, G(t8–O–4)S(8–5)G and G(e8–O–4)S(8–5)G, and of buddlenol A, G(t8–O–4)S(8–5)G′ (data not shown) and G(e8–O–4)S(8–5)G′, were affected (Table II). In addition, hedyotisol, G(8–O–4)S(8–8)S(8–O–4)G, was reduced in abundance and SP(8–8)S, i.e. the 8–8 coupling product of sinapyl p-hydroxybenzoate and sinapyl alcohol, was undetectable in the HPLC profiles of COMT-deficient poplars.
Taken together, all oligolignols that were decreased in abundance or reduced to undetectable levels involved an S unit. On the other hand, the levels of at least one of the oligomers (G(8–5)G′ [balanophonin]) that did not involve sinapyl alcohol, and novel oligomers composed of the substrates for COMT, the benzodioxanes, were increased. Similar relative changes were observed in the lignin polymer of COMT-deficient poplar (Lapierre et al., 1999; Ralph et al., 2001a, 2001b). Thus, these data indicate that the composition and the relative abundance of the oligolignols are determined by the relative supply of monolignols and their chemical coupling propensities.
Is Nocomtol 1 Incorporated into Lignin Biosynthesis?
The detection by thioacidolysis of 5H units in the lignins of COMT down-regulated plants (Atanassova et al., 1995; Van Doorsselaere et al., 1995; Vignols et al., 1995; Tsai et al., 1998; Jouanin et al., 2000; Guo et al., 2001; Piquemal et al., 2002) demonstrated that down-regulation of COMT results in the incorporation of 5-hydroxyconiferyl alcohol as a monolignol into the lignin polymer. Benzodioxane structures, involving G and 5H units, have been revealed as major structures in these COMT-deficient lignins (Ralph et al., 2001a, 2001b) and concluded to be formed during lignin polymerization itself, i.e. when a coniferyl alcohol radical couples endwise at its favored C8 position with the 4–O position of a 5H unit in the growing polymer. Obviously, such structures resemble the structure of G(8–O–4)5H (nocomtol 1), which is the most abundant dimer in the xylem extracts of COMT down-regulated poplars. Could the benzodioxane structures in lignin result in a significant way from the incorporation of G(8–O–4)5H dimers into lignins rather than exclusively from the incorporation of 5-hydroxyconiferyl alcohol? The concatenation of preformed dimers in the formation of lignin is unlikely because analysis of the lignin structure supports a polymerization process in which monolignols are added to the growing polymer (Hatfield and Vermerris, 2001; Boerjan et al., 2003; Ralph et al., 2004b); dimers can only add to lignin via the phenolic moiety, which would result in a high proportion of terminal alcohol residues in lignin because the unsaturated side chain would be blocked from any further reactions (Hatfield and Vermerris, 2001). Although the lignins analyzed by NMR are only a fraction of the total lignin in the cell wall, NMR analysis of the lignin structure of COMT down-regulated plants showed that the amount of cinnamyl alcohol end groups only slightly increases in COMT down-regulated poplars—5% of the common structures measured in COMT-deficient poplars versus 2% in wild types (Ralph et al., 2001a) and 4% versus 3% in COMT-deficient alfalfa (Marita et al., 2003a). Hence, the G(8–O–4)5H dimer probably does not contribute to a large extent to lignification. In addition, the massive amounts of G(8–O–4)5H in xylem extracts of COMT down-regulated transgenic poplar suggest that the dimer is not efficiently incorporated into the polymer.
Chiral HPLC Provides No Evidence for the Involvement of Dirigent Proteins in Benzodioxane Formation
Some monolignol dehydrodimers, such as (8–5)-dehydrodiconiferyl alcohol [G(8–5)G] and pinoresinol [G(8–8)G], may exist either as nearly racemic mixtures or in optically pure forms. In the latter case, the dimer is considered a lignan (Dixon, 2004; Umezawa, 2004). Lignans are nonstructural compounds derived from monolignols primarily linked via 8–8, 8–5, or 8–O–4 bonds (Lewis and Davin, 1999). In Forsythia sp., the stereoselective coupling affording pinoresinol is controlled by so-called dirigent proteins (Davin et al., 1997). Dirigent proteins lack any catalytic activity by themselves but serve to align the phenoxy radical intermediates generated by an oxidase. Dirigent protein-like genes have been identified in a variety of plant species (Lewis and Davin, 1999; Umezawa, 2004). It has been suggested that monolignol coupling in lignification is rigorously controlled and that all linkage types would be specified by different dirigent isoforms (Lewis, 1999). Until now, however, no dirigent proteins have been shown to direct the regio- and stereoselective synthesis of dilignols with linkage types other than 8–8.
To observe to what extent the new benzodioxane oligolignols were optically pure, chiral HPLC analysis was conducted on natural as well as synthesized nocomtol 1 G(8–O–4)5H. For the isolated and the synthesized compounds, the two enantiomers were found in approximately equal amounts. This result indicates that the biosynthesis of the benzodioxanes is not under stereoselective control in poplar and that the precursors are simply coupled chemically. Therefore, our data do not support the presence of a specific dirigent protein for benzodioxane formation in poplar xylem. Because benzodioxane dimers are not normally present in the cell wall, the lack of a specific dirigent protein may not be surprising, but, for the same reason, the benzodioxane dimers are the ideal model to demonstrate that a combinatorial chemical coupling can occur in the cell wall.
CONCLUSION
We have shown that down-regulation of COMT in poplar results in a reduced abundance of sinapaldehyde, and our data further support the finding that COMT does not play a role in sinapic acid synthesis in xylem tissue. The abundance of oligolignols derived from sinapyl alcohol decreases and a new set of oligolignols accumulates that derives from the cross-coupling of 5-hydroxyconiferaldehyde and 5-hydroxyconiferyl alcohol with other available units. Because the relative abundance of the oligolignols is in agreement with the relative supply of monolignols and with their chemical coupling propensities, and because, at least for nocomtol G(8–O–4)5H, both enantiomers were present in equal amounts, our data strongly support the recently challenged (Lewis, 1999) combinatorial coupling hypothesis for monolignols (Boerjan et al., 2003; Ralph et al., 2004b).
MATERIALS AND METHODS
Growth Conditions and Plant Material
Wild-type poplar (Populus tremula × P. alba clone INRA no. 717-1B4), poplar transformed with a PCaMV35S-GUS construct (lines 35S 17B and 35S 21B; Nilsson et al., 1996; Chen et al., 2000), transgenic poplar down-regulated for COMT (lines ASB 2B and ASB 10B; Van Doorsselaere et al., 1995; Lapierre et al., 1999), and a set of 13 transgenic poplar lines down-regulated for various enzymes involved in phenylpropanoid biosynthesis (CCoAOMT, CCR, CAD, phenylcoumaran benzylic ether reductase, and both COMT and CCR) were included to increase the statistical power of the experiment. After the propagation of the poplar lines in vitro on Murashige and Skoog medium (Duchefa, Haarlem, The Netherlands), rooted plantlets were transferred to the greenhouse (21°C, 60% humidity, 16-h-light/8-h-dark regime, 40–60 μmol m−2 s−1 photosynthetic photon flux) and grown for 3 months according to a completely randomized design (Neter et al., 1996), reaching a height of approximately 1.5 m. Approximately seven ramets were grown for each poplar line.
LC-MS/MS Analysis of Cinnamic Acids and Cinnamaldehydes
For each individual, xylem tissue was harvested from a 10-cm-long, debarked stem cut 15 cm above ground by scraping with a scalpel (approximately 300 mg). After grinding in liquid nitrogen, the tissue was extracted with 15 mL of methanol. The debris was subsequently removed, and the solute freeze-dried and weighed (approximately 70 mg). An aliquot (1.5 mL) of the methanol phase was lyophilized and extracted with cyclohexane/water acidified with 0.1% trifluoroacetic acid (TFA; 1:1, v/v) as described previously (Meyermans et al., 2000). Individual samples were injected by means of a SpectraSystem AS1000 autosampler (Thermo Separation Products, Riviera Beach, FL), cooled at 4°C, onto a reversed phase Luna C18(2) column (150 × 2.1 mm, 3 μm; Phenomenex, Torrance, CA). A gradient separation (SpectraSystem P1000XR HPLC pump; Thermo Separation Products) was run from 1% aqueous triethylammonium acetate (TEAA; solvent A, pH 5) to methanol-acetonitrile (25:75, v/v; 1% TEAA; solvent B) using the following conditions: flow 0.3 mL min−1; time 0 min, 0% B; time 25 min, 40% B; time 35 min, 100% B.
A SpectraSystem UV6000LP detector (Thermo Separation Products) measured UV/Vis absorption between 200 and 450 nm with a scan rate of 2 Hz. APCI, operated in the negative-ion mode, was used as an ion source to couple HPLC with an LCQ Classic (ThermoQuest, San Jose, CA) MS instrument (vaporizer temperature 450°C, capillary temperature 175°C, source current 5 μA, sheath gas flow set at 27, aux gas flow set at 2). Using selected ion monitoring, a clear MS signal corresponding to the different cinnamic acid (p-coumaric, caffeic, ferulic, 5-hydroxyferulic, and sinapic acids) and cinnamaldehyde (p-coumaraldehyde, caffealdehyde, coniferaldehyde, 5-hydroxyconiferaldehyde, and sinapaldehyde) standards was observed. An unstable MS signal was obtained for p-coumaryl, coniferyl, and sinapyl alcohols because these compounds fragmented during the ionization process. To verify whether the cinnamic acid and cinnamaldehyde standards were chemically stable during the extraction procedure, 20 μL of methanol:water (10:90, v/v) solution containing 0.25 μg mL−1 of each of the cinnamic acid and cinnamaldehyde standards was added to a xylem sample. After grinding and cleaning the sample as described above, all standards were detected in the LC-MS chromatogram of the xylem extract in the same concentration range as when the standard solution was immediately analyzed on LC-MS. The phenylpropanoids were quantified with the total ion current signal, obtained by selected ion monitoring scans of the cinnamic acids and cinnamaldehydes, divided by the total ion current signal of the internal standard 3,4,5-trimethoxycinnamic acid.
HPLC Profiling
A 1-mL aliquot of the prepared methanol phase of each sample (see above) was treated and separated on HPLC as described previously (Meyermans et al., 2000; Morreel et al., 2004).
Statistical Analyses
Differences in the mean abundance of soluble phenolics between the poplar lines were analyzed statistically according to the following nested ANOVA:
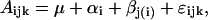 |
(A) |
where Aijk is abundance of the compound in individual k belonging to the jth poplar line of the ith group; μ is overall mean abundance of the compound, αi is deviation from the overall mean due to the effect of the ith group; βj(i) is specific effect of the jth poplar line nested within group i; and ɛijk is residual deviation of the compound abundance in individual k of the jth poplar line within the ith group. The residual deviations are independent and normally distributed with mean 0 and variance σ2.
Model A detects differences between groups of similarly affected poplar lines and simultaneously between the poplar lines nested in each group. As such, this model is more powerful to reveal significant differences than the one-way ANOVA Model B, which detects only differences between the different poplar lines. A large set of transgenic poplar lines was included in the statistical analysis to augment the statistical power (see above). Besides the control group (wild type, 35S 17B, and 35S 21B) and the group composed of the COMT-deficient poplar lines (ASB 2B and ASB 10B), five additional groups composed of lines deficient in CCoAOMT, CCR, CAD, and phenylcoumaran benzylic ether reductase, and poplars deficient for both CCR and COMT were included in the nested ANOVA model. Following the overall nested ANOVA analysis, specific differences between the control and the COMT-deficient lines were detected by a lsd multiple comparison test. Whenever necessary, homoscedasticity was obtained by Box-Cox transformations.
If insufficient homoscedasticity could be achieved by transforming the original data to be able to apply Model A, the one-way ANOVA Model B was chosen instead.
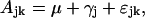 |
(B) |
where Ajk is abundance of the compound in individual k of the jth poplar line; μ is overall mean of the compound abundance; γj is deviation from the overall mean due to the effect of the jth poplar line; and ɛjk is residual deviation of the compound abundance in individual k of the jth poplar line. The residual deviations are independent and normally distributed with mean 0 and variance σ2.
In this case, a significant difference between the control lines and transgenic poplar lines down-regulated for the same gene was considered when the two following requirements were met: (1) the lsd post hoc test (type I error = 0.05) was not significant between any of the control poplar lines, and (2) the lsd post hoc test (type I error = 0.05) was significant and the sign of the difference in mean value was the same when each of the transgenic lines down-regulated for the same gene was compared with each of the control poplar lines.
Because of the large number of statistical tests, Bonferroni corrections were applied to obtain an experiment-wise significance threshold of 0.05. All statistical analyses were performed by using the software program SPSS 9.0 (SPSS, Chicago). Here, only the results obtained for the down-regulation of COMT are presented.
Characterization of Components
Sufficient material for spectral identifications was isolated by preparative HPLC on a Luna C18(2) column (250 × 10 mm, 10 μm; Phenomenex) using a flow and temperature of 5 mL min−1 and 40°C, respectively, with a gradient from 0.1% aqueous TFA (solvent A, pH 2) to methanol-acetonitrile (25:75, v/v; 0.1% TFA; solvent B) with the same instrument and under the same time and gradient conditions as described for the analytical HPLC analysis of soluble phenolics (Meyermans et al., 2000). Compounds of interest were subjected to NMR analysis and/or LC-MS/MS.
1H- and 13C-NMR spectra were recorded on a Unity 500 spectrometer (Varian, Palo Alto, CA), operating at 499.193 MHz for 1H and at 125.534 MHz for 13C, with an inverse 5-mm broad-band probe with π/2 pulses of 5.5 and 18.5 μs, respectively, and equipped with pulsed magnetic field gradient coils. The standard Varian software Vnmr version 6.1b was used throughout. The measurements were performed at 27°C in deuteroacetone solution and with tetramethylsilane as internal standard. Spectral assignments were based not only on chemical shift rules and coupling patterns, but also especially on two-dimensional correlations from routine experiments, such as COSY45, GHSQC (single-bond C,H correlations), and GHMBC experiments (multiple-bond or 3J/2J C,H correlations). The assignments especially for the carbon spectrum were based on single-bond C,H correlations (GHSQC spectra) and on multiple-bond correlations (GHMQC spectra) for the quaternary carbons. NMR data for synthetic compounds in acetone-d6, including the data deposited in the NMR database (Ralph et al., 2004a), were acquired on a 360 MHz machine (DRX-360; Bruker, Karlsruhe, Germany) with an inverse triple-axis gradient probe and the standard array of one-dimensional and two-dimensional Bruker pulse programs.
LC-MS/MS analysis was performed with a Luna C18(2) column (150 × 2.1 mm, 3 μm; Phenomenex), using a gradient from 100% solvent A (aqueous 1% acetic acid) to 65% solvent B (methanol, 1% acetic acid) in 8 min and to 100% solvent B within the next 12 min at a flow of 0.25 mL min−1 and a column temperature of 40°C. The most abundant ion in each full MS scan, in the negative-ion mode, was subsequently fragmented in the next scan using the dependent MS/MS mode. Vaporizer temperature, capillary temperature, and sheath gas flow of the APCI ion source were set at 500°C, 170°C, and 80. Source current and aux gas flow were as mentioned above.
Chemicals
p-Coumaric acid, ferulic acid, and 3,4-dihydroxy-5-methoxy-benzaldehyde were purchased from Sigma-Aldrich (St. Louis), and caffeic acid and sinapic acid from ACROS (Beerse, Belgium). 5-Hydroxyferulic acid was synthesized according to Akabori and Tsuchiya (1990), whereas p-coumaraldehyde, caffealdehyde, coniferaldehyde, sinapaldehyde, and sinapyl alcohol were synthesized according to Daubresse et al. (1994). Coniferyl alcohol (Fig. 4, compound a) was synthesized by sodium borohydride reduction of coniferaldehyde according to Lu and Ralph (1998). Nitidanin (Ishikawa et al., 1995) was kindly provided by Tsutomu Ishikawa (Chiba University, Chiba, Japan).
Synthesis of Compounds
The same shorthand naming convention for oligolignols is used as described (Morreel et al., 2004), where bold capitals are used for units formally derived from coniferyl alcohol (G), sinapyl alcohol (S), coniferaldehyde (G′), sinapaldehyde (S′), vanillin (V′), sinapyl p-hydroxybenzoate (SP), 5-hydroxyconiferyl alcohol (5H), and 5-hydroxyconiferaldehyde (5H′), and the interunit bond formed during the radical coupling reaction is specified in parentheses: (8–O–4), (8–5), or (8–8). Conformational information is mentioned when important with R and S configuration and the carbon number of the chiral center.
Compound 1 is G(8–O–4)5H, 4-[3-hydroxymethyl-7-(E)-(3-hydroxypropenyl)-5-methoxy-2,3-dihydro-benzo[1,4]dioxin-2-yl]-2-methoxyphenol (named nocomtol; see Fig. 3).
In the synthesis, 3,4-dihydroxy-5-methoxy-benzaldehyde was acetylated with acetic anhydride/pyridine to produce 3,4-diacetoxy-5-methoxy-benzaldehyde in essentially quantitative yield. Ethyl 4,5-diacetoxy-5-methoxyferulate was prepared by a Wittig-Horner reaction with triethyl phosphonoacetate (yield 90%) according to Ralph et al. (1992). Ethyl 5-hydroxyferulate (Fig. 4, compound b) was obtained in 92% yield by deacetylation with pyrrolidine for approximately 10 min at room temperature (Lu and Ralph, 1998). NMR of ethyl 5-hydroxyferulate was δH 1.27 (1H, t, J = 7.1, methyl), 3.86 (3H, s, methoxyl), 4.20 (q, J = 7.1, methylene), 6.30 (1H, d, J = 15.8, 8), 6.85 (1H, s, 6), 6.88 (1H, s, 2), 7.5 (1H, d, J = 15.8 Hz, 7); δC 14.5 (methyl), 56.4 (OMe), 60.8 (methylene), 104.2 (2), 110.3 (6), 115.9 (8), 126.4 (1), 137.3 (4), 145.8 (7), 146.2 (5), 149.0 (3), 167.4 (9).
Coniferyl alcohol (Fig. 4, compound a; 788 mg, 4.38 mmol) and ethyl 5-hydroxyferulate (Fig. 4, compound b; 870 mg, 3.65 mmol) were dissolved in acetone:benzene (1:2, v/v, 45 mL). Silver carbonate (2.52 g, 9.13 mmol) was added. The mixture was stirred for 14 h. The solid oxidant was removed by filtration through a Celite pad and washed with acetone. The filtrate and acetone washings were combined and evaporated under reduced pressure. The residue was applied to flash silica gel chromatography (1:1 chloroform:ethyl acetate as eluant) resulting in pure 3-[3-(4-hydroxy-4-methoxyphenyl)-2-hydroxymethyl-8-methoxy-2,3-dihydrobenzo[1,4] dioxin-6-yl]-acrylic acid ethyl ester as a pale-yellow oil (Fig. 4, compound c; 800 mg, 53% yield). NMR was δH 1.25 (1H, t, J = 7.1, methyl), 3.50 (1H, m, G-91), 3.83 (1H, m, G-92), 3.85 (3H, s, 5H-OMe), 3.92 (3H, s, G-OMe), 4.09 (1H, m, G-8), 4.17 (q, J = 7.1, methylene), 4.98 (1H, d, J = 8.0 Hz, G-7), 6.40 (1H, d, J = 15.9 Hz, 5H-8), 6.85 (1H, d, J = 1.5 Hz, 5H-6), 6.88 (1H, d, J = 8.1 Hz, G-5), 6.95 (1H, d, J = 1.5 Hz, 5H-2), 6.96 (1H, dd, J = 8.1, 1.8 Hz, G-6), 7.10 (1H, d, J = 1.8 Hz, G-2), 7.54 (1H, d, J = 15.9 Hz, 5H-7); δC 14.6 (methyl), 56.2 (5H-OMe), 56.3 (G-OMe), 61.6 (G-9), 61.6 (methylene), 76.9 (G-7), 79.5 (G-8), 104.7 (5H-2), 111.1 (5H-6), 111.7 (G-2), 115.7 (G-5), 117.0 (5H-8), 121.4 (G-6), 127.4 (5H-1), 128.8 (G-1), 136.4 (5H-4), 145.2 (5H-7), 145.3 (5H-5), 147.8 (G-4), 148.3 (5H-3), 150.1 (G-3), 167.2 (5H-9).
Compound c (10 mmol) was dissolved in toluene (10 mL) at 0°C and added to a stirred solution of diisobutylaluminum hydride (DIBAL-H; Aldrich, Milwaukee, WI) in toluene (5 mL) through a syringe (Fig. 4; 700 mg, 1.68 mmol). The mixture was stirred continuously for 1 h. The excess reducing agent was quenched with ethanol; the mixture was diluted with ethyl acetate (200 mL) and washed with 3% aqueous HCl solution (100 mL × 2) and saturated aqueous NH4Cl (50 mL). The ethyl acetate portion was dried over MgSO4 and evaporated under reduced pressure, resulting in compound 1 as a white foam (Fig. 4; 580 mg, 93% yield). Compound 1 is a mixture of trans- and cis-isomers 1a and 1b, (w/w; 10:1). Upon acid or base catalysis, the cis-isomer 1b is almost exclusively converted to the more thermodynamically stable trans-isomer 1a (She et al., 1998; Sousa et al., 2002). Both 1a and 1b are chiral molecules and occur as enantiomeric mixtures 1a (7R,8R/7S,8S) and 1b (7S,8R/7R,8S), as can be shown with chiral chromatography. NMR of synthesized 1 (i.e. mixture of 1a and 1b, but only data for the trans-isomer [R,R and S,S] are reported) was δH 3.50 (1H, m, G-91), 3.76 (1H, m, G-92), 3.85 (3H, s, 5H-OMe), 3.85 (3H, s, G-OMe), 4.05 (1H, m, G-8), 4.20 (2H, dd, J = 5.4, 1.6 Hz, 5H-9's), 4.96 (1H, d, J = 7.9 Hz, G-7), 6.26 (1H, dt, J = 15.9, 5.4 Hz, 5H-7), 6.47 (1H, dt, J = 15.9, 1.6 Hz, 5H-8), 6.59 (1H, d, J = 1.8 Hz, 5H-2), 6.68 (1H, d, J = 1.8 Hz, 5H-6), 6.87 (1H, d, J = 8.1 Hz, G-5), 6.95 (1H, dd, J = 8.1, 1.8 Hz, G-6), 7.10 (1H, d, J = 1.8 Hz, G-2); δC 56.2 (5H-OMe), 56.2 (G-OMe), 61.6 (G-9), 63.6 (5H-9), 76.7 (G-7), 79.3 (G-8), 103.2 (5H-2), 108.5 (5H-6), 111.7 (G-2), 115.7 (G-5), 121.4 (G-6), 129.0 (5H-8), 129.1 (G-1), 130.1 (5H-7), 130.3 (5H-1), 133.7 (5H-4), 145.2 (5H-5), 147.8 (G-4), 148.3 (G-3), 149.8 (5H-3).
Compound 3, G(8–O–4)5H′, (2E)-3-[3-(4-hydroxy-3-methoxy-phenyl)-2-(hydroxymethyl)-8-methoxy-2,3-dihydro-1,4-benzodioxin-6-yl]acrylaldehyde (which we have named nocomtal) is for the same reason as compound 1 an equilibrium mixture of 3a and 3b, the trans- and cis-isomers, in a 10:1 (w/w) ratio.
For the synthesis, G(8–O–4)5H′ 3, the cinnamaldehyde analog of 1, was prepared by 2,3-dichloro-5,6-dicyanobenzoquinone oxidation of G(8–O–4)5H (compound 1; nocomtol) as described for the synthesis of balanophonin, G(8–5)G′ (Morreel et al., 2004). The product 3 was obtained in 83% yield as a pale-yellow oil after purification through a 3-mL silica gel sep-pack eluted with 2:1 CHCl3:EtOAc. NMR of the trans-isomer (R,R and S,S): δH 3.53 (1H, m, G-9a), 3.82 (1H, m, G-9b), 3.86 (3H, s, G-OMe), 3.91 (3H, s, 5H-OMe), 4.14 (1H, ddd, J = 8.0, 3.8, 2.6 Hz, G-8), 5.01 (1H, d, J = 8.0 Hz, G-7), 6.67 (1H, dd, J = 15.8, 7.6 Hz, 5H-8), 6.88 (1H, d, J = 8.0 Hz, G-5), 6.92 (1H, dd, J = 2.0, 0.4 Hz, 5H-6), 6.97 (1H, ddd, J = 8.0, 2.0, 0.5 Hz, G-6), 7.02 (1H, d, J = 2.0 Hz, 5H-2), 7.12 (1H, d, J = 2.0 Hz, G-2), 7.53 (1H, d, J = 15.8 Hz, 5H-7), 7.59 (<1H, bs, G4-OH), 9.64 (1H, d, J = 7.6 Hz, 5H-9); δC 56.4 (G-OMe), 56.5 (5H-OMe), 61.6 (G-9), 77.0 (G-7), 79.8 (G-8), 105.3 (5H-2), 111.9 (5H-6), 112.0 (G-2), 115.8 (G-5), 121.6 (G-6), 127.5 (5H-1), 128.0 (5H-8), 128.9 (G-1), 137.4 (5H-7), 145.6 (5H-5), 148.1 (G-3), 148.5 (G-4), 150.4 (5H-3), 153.7 (5H-4), 193.8 (5H-9).
Chiral HPLC Analysis
The synthesized as well as the isolated forms of compounds 1a and 1b, trans- and cis-nocomtol (w/w; 10:1), in amounts of approximately 0.2 and 0.02 μg, respectively, were dissolved in 25 μL ethanol and isocratically separated on a polysaccharide-type chiral column (ChiralcelOD, 10 μm, 250 × 4.6 mm; Daicel Chemical Industries, Osaka) at 35°C, by using a flow of 1 mL min−1 of hexane:ethanol (85:15; v/v) by an Agilent 1100 series instrument (type HPLC-diode array detector; Agilent Technologies, Foster City, CA).
Distribution of Materials
Upon request, all novel materials described in this publication will be made available in a timely manner for noncommercial research purposes, subject to the requisite permission from any third-party owners of all or parts of the material. Obtaining any permissions will be the responsibility of the requester.
Acknowledgments
The authors thank Martine De Cock for help in preparing the manuscript and Tsutomu Ishikawa (Chiba University, Chiba, Japan) for providing nitidanin.
This work was supported by the Fund for Scientific Research-Flanders (grant no. G0040.00N), the European Commission program (grant no. QLK5–CT–2000–01493 to E.M., P.H., and W.B.), in part by the Department of Energy Biosciences program (grant no. DE–AI02–00ER15067) and the U.S. Department of Agriculture-National Research Initiatives (grant no. 2001–02176 to J.R.), and the Instituut voor de aanmoediging van Innovatie door Wetenschap en Technologie in Vlaanderen (predoctoral fellowship to K.M.).
Article, publication date, and citation information can be found at www.plantphysiol.org/cgi/doi/10.1104/pp.104.049312.
References
- Akabori S, Tsuchiya S (1990) The preparation of crown ethers containing dicinnamoyl groups and their complexing abilities. Bull Chem Soc Jpn 63: 1623–1628 [Google Scholar]
- Atanassova R, Favet N, Martz F, Chabbert B, Tollier MT, Monties B, Fritig B, Legrand M (1995) Altered lignin composition in transgenic tobacco expressing O-methyltransferase sequences in sense and antisense orientation. Plant J 8: 465–477 [Google Scholar]
- Baucher M, Halpin C, Petit-Conil M, Boerjan W (2003) Lignin: genetic engineering and impact on pulping. Crit Rev Biochem Mol Biol 38: 305–350 [DOI] [PubMed] [Google Scholar]
- Birks JS, Kanowski PJ (1993) Analysis of resin compositional data. Silvae Genet 42: 340–350 [Google Scholar]
- Boerjan W, Ralph J, Baucher M (2003) Lignin biosynthesis. Annu Rev Plant Biol 54: 519–546 [DOI] [PubMed] [Google Scholar]
- Chabannes M, Barakate A, Lapierre C, Marita JM, Ralph J, Pean M, Danoun S, Halpin C, Grima-Pettenati J, Boudet AM (2001) Strong decrease in lignin content without significant alteration of plant development is induced by simultaneous down-regulation of cinnamoyl CoA reductase (CCR) and cinnamyl alcohol dehydrogenase (CAD) in tobacco plants. Plant J 28: 257–270 [DOI] [PubMed] [Google Scholar]
- Chen C, Meyermans H, Burggraeve B, De Rycke RM, Inoue K, De Vleesschauwer V, Steenackers M, Van Montagu MC, Engler GJ, Boerjan WA (2000) Cell-specific and conditional expression of caffeoyl-CoA O-methyltransferase in poplar. Plant Physiol 123: 853–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Duran AL, Blount JW, Sumner LW, Dixon RA (2003) Profiling phenolic metabolites in transgenic alfalfa modified in lignin biosynthesis. Phytochemistry 64: 1013–1021 [DOI] [PubMed] [Google Scholar]
- Chen F, Kota P, Blount JW, Dixon RA (2001) Chemical syntheses of caffeoyl and 5-OH coniferyl aldehydes and alcohols and determination of lignin O-methyltransferase activities in dicot and monocot species. Phytochemistry 58: 1035–1042 [DOI] [PubMed] [Google Scholar]
- Daubresse N, Francesch C, Mhamdi F, Rolando C (1994) A mild synthesis of coumaryl, coniferyl, sinapyl aldehydes and alcohols. Synthesis 1994: 369–371 [Google Scholar]
- Davin LB, Wang H-B, Crowell AL, Bedgar DL, Martin DM, Sarkanen S, Lewis NG (1997) Stereoselective bimolecular phenoxy radical coupling by an auxiliary (dirigent) protein without an active center. Science 275: 362–366 [DOI] [PubMed] [Google Scholar]
- Dixon RA (2004) Phytoestrogens. Annu Rev Plant Biol 55: 225–261 [DOI] [PubMed] [Google Scholar]
- Goujon T, Sibout R, Pollet B, Maba B, Nussaume L, Bechtold N, Lu F, Ralph J, Mila I, Barrière Y, et al (2003) A new Arabidopsis thaliana mutant deficient in the expression of O-methyltransferase impacts lignins and sinapoyl esters. Plant Mol Biol 51: 973–989 [DOI] [PubMed] [Google Scholar]
- Guo D, Chen F, Inoue K, Blount JW, Dixon RA (2001) Downregulation of caffeic acid 3-O-methyltransferase and caffeoyl CoA 3-O-methyltransferase in transgenic alfalfa: impacts on lignin structure and implications for the biosynthesis of G and S lignin. Plant Cell 13: 73–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatfield R, Vermerris W (2001) Lignin formation in plants. The dilemma of linkage specificity. Plant Physiol 126: 1351–1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys JM, Hemm MR, Chapple C (1999) New routes for lignin biosynthesis defined by biochemical characterization of recombinant ferulate 5-hydroxylase, a multifunctional cytochrome P450-dependent monooxygenase. Proc Natl Acad Sci USA 96: 10045–10050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Parvathi K, Dixon RA (2000) Substrate preferences of caffeic acid/5-hydroxyferulic acid 3/5-O-methyltransferases in developing stems of alfalfa (Medicago sativa). Arch Biochem Biophys 375: 175–182 [DOI] [PubMed] [Google Scholar]
- Inoue K, Sewalt VJH, Ballance GM, Ni W, Stürzer C, Dixon RA (1998) Developmental expression and substrate specificities of alfalfa caffeic acid 3-O-methyltransferase and caffeoyl coenzyme A 3-O-methyltransferase in relation to lignification. Plant Physiol 117: 761–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Seki M, Nishigaya K, Miura Y, Seki H, Chen I-S, Ishii H (1995) Studies on the chemical constituents of Xanthoxylum nitidum (Roxb.) D. C. (Fagara nitida Roxb.). III. The chemical constituents of wood. Chem Pharm Bull 43: 2014–2018 [Google Scholar]
- Jouanin L, Goujon T, de Nadaï V, Martin M-T, Mila I, Vallet C, Pollet B, Yoshinaga A, Chabbert B, Petit-Conil M, et al (2000) Lignification in transgenic poplars with extremely reduced caffeic acid O-methyltransferase activity. Plant Physiol 123: 1363–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Ralph J, Lu F, Ralph SA, Boudet A-M, MacKay JJ, Sederoff RR, Ito T, Kawai S, Ohashi H, et al (2003) NMR analysis of lignins in CAD-deficient plants. Part 1. Incorporation of hydroxycinnamaldehydes and hydroxybenzaldehydes into lignin. Org Biomol Chem 1: 268–281 [DOI] [PubMed] [Google Scholar]
- Kim H, Ralph J, Yahiaoui N, Pean M, Boudet A-M (2000) Cross-coupling of hydroxycinnamyl aldehydes into lignins. Org Lett 2: 2197–2200 [DOI] [PubMed] [Google Scholar]
- Lapierre C, Tollier MT, Monties B (1988) A new type of constitutive unit in lignins from the corn bm3 mutant. C R Acad Sci Ser III Sci Vie 307: 723–728 [Google Scholar]
- Lapierre C, Pollet B, Petit-Conil M, Toval G, Romero J, Pilate G, Leplé J-C, Boerjan W, Ferret V, De Nadai V, et al (1999) Structural alterations of lignins in transgenic poplars with depressed cinnamyl alcohol dehydrogenase or caffeic acid O-methyltransferase activity have opposite impact on the efficiency of industrial Kraft pulping. Plant Physiol 119: 153–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis NG (1999) A 20th century roller coaster ride: a short account of lignification. Curr Opin Plant Biol 2: 153–162 [DOI] [PubMed] [Google Scholar]
- Lewis NG, Davin LB (1999) Lignans: biosynthesis and function. In U Sankawa, ed, Comprehensive Natural Products Chemistry, Vol 1: Polyketides and Other Secondary Metabolites Including Fatty Acids and Their Derivatives. Elsevier Science, Amsterdam, pp 639–712
- Li L, Cheng XF, Leshkevich J, Umezawa T, Harding SA, Chiang VL (2001) The last step of syringyl monolignol biosynthesis in angiosperms is regulated by a novel gene encoding sinapyl alcohol dehydrogenase. Plant Cell 13: 1567–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Popko JL, Umezawa T, Chiang VL (2000) 5-Hydroxyconiferyl aldehyde modulates enzymatic methylation for syringyl monolignol formation, a new view of monolignol biosynthesis in angiosperms. J Biol Chem 275: 6537–6545 [DOI] [PubMed] [Google Scholar]
- Lu F, Ralph J (1998) Facile synthesis of 4-hydroxycinnamyl p-coumarates. J Agric Food Chem 46: 2911–2913 [Google Scholar]
- Lu F, Ralph J, Morreel K, Messens E, Boerjan W (2004) Preparation and relevance of a cross-coupling product between sinapyl alcohol and sinapyl p-hydroxybenzoate. Org Biomol Chem 2: 2888–2890 [DOI] [PubMed] [Google Scholar]
- Marita JM, Ralph J, Hatfield RD, Guo D, Chen F, Dixon RA (2003. a) Structural and compositional modifications in lignin of transgenic alfalfa down-regulated in caffeic acid 3-O-methyltransferase and caffeoyl coenzyme A 3-O-methyltransferase. Phytochemistry 62: 53–65 [DOI] [PubMed] [Google Scholar]
- Marita JM, Vermerris W, Ralph J, Hatfield RD (2003. b) Variations in the cell wall composition of maize brown midrib mutants. J Agric Food Chem 51: 1313–1321 [DOI] [PubMed] [Google Scholar]
- Matsui N, Chen F, Yasuda S, Fukushima K (2000) Conversion of guaiacyl to syringyl moieties on the cinnamyl alcohol pathway during the biosynthesis of lignin in angiosperms. Planta 210: 831–835 [DOI] [PubMed] [Google Scholar]
- Maury S, Geoffroy P, Legrand M (1999) Tobacco O-methyltransferases involved in phenylpropanoid metabolism. The different caffeoyl-coenzyme A/5-hydroxyferuloyl-coenzyme A 3/5-O-methyltransferase and caffeic acid/5-hydroxyferulic acid 3/5-O-methyltransferase classes have distinct substrate specificities and expression patterns. Plant Physiol 121: 215–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng H, Campbell WH (1998) Substrate profiles and expression of caffeoyl coenzyme A and caffeic acid O-methyltransferases in secondary xylem of aspen during seasonal development. Plant Mol Biol 38: 513–520 [DOI] [PubMed] [Google Scholar]
- Meyermans H, Morreel K, Lapierre C, Pollet B, De Bruyn A, Busson R, Herdewijn P, Devreese B, Van Beeumen J, Marita JM, et al (2000) Modification in lignin and accumulation of phenolic glucosides in poplar xylem upon down-regulation of caffeoyl-coenzyme A O-methyltransferase, an enzyme involved in lignin biosynthesis. J Biol Chem 275: 36899–36909 [DOI] [PubMed] [Google Scholar]
- Morreel K, Ralph J, Kim H, Lu F, Goeminne G, Ralph SA, Messens E, Boerjan W (2004) Profiling of oligolignols reveals monolignol coupling conditions in lignifying poplar xylem. Plant Physiol 136: 3537–3549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neter J, Kutner MH, Nachtsheim CJ, Wasserman W (1996) Applied Linear Statistical Models, Ed 4. WCB/McGraw-Hill, Boston
- Nilsson O, Little CHA, Sandberg G, Olsson O (1996) Expression of two heterologous promoters, Agrobacterium rhizogenes rolC and cauliflower mosaic virus 35S, in the stem of transgenic hybrid aspen plants during the annual cycle of growth and dormancy. Plant Mol Biol 31: 887–895 [DOI] [PubMed] [Google Scholar]
- Osakabe K, Tsao CC, Li L, Popko JL, Umezawa T, Carraway DT, Smeltzer RH, Joshi CP, Chiang VL (1999) Coniferyl aldehyde 5-hydroxylation and methylation direct syringyl lignin biosynthesis in angiosperms. Proc Natl Acad Sci USA 96: 8955–8960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvathi K, Chen F, Guo D, Blount DW, Dixon RA (2001) Substrate preferences of O-methyltransferases in alfalfa suggest new pathways for 3-O-methylation of monolignols. Plant J 25: 193–202 [DOI] [PubMed] [Google Scholar]
- Piquemal J, Chamayou S, Nadaud I, Beckert M, Barrière Y, Mila I, Lapierre C, Rigau J, Puigdomenech P, Jauneau A, et al (2002) Down-regulation of caffeic acid O-methyltransferase in maize revisited using a transgenic approach. Plant Physiol 130: 1675–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralph J, Helm RF, Quideau S, Hatfield RD (1992) Lignin—feruloyl ester cross-links in grasses. Part 1. Incorporation of feruloyl esters into coniferyl alcohol dehydrogenation polymers. J Chem Soc Perkin Trans I 1: 2961–2969 [Google Scholar]
- Ralph SA, Landucci LL, Ralph J (2004. a) NMR database of lignin and cell wall model compounds. U.S. Dairy Forage Research Center, U.S. Department of Agriculture-Agricultural Research Service. http://www.dfrc.ars.usda.gov/software.html (November 15, 2004)
- Ralph J, Lapierre C, Lu F, Marita JM, Pilate G, Van Doorsselaere J, Boerjan W, Jouanin L (2001. a) NMR evidence for benzodioxane structures resulting from incorporation of 5-hydroxyconiferyl alcohol into lignins of O-methyltransferase-deficient poplars. J Agric Food Chem 49: 86–91 [DOI] [PubMed] [Google Scholar]
- Ralph J, Lapierre C, Marita JM, Kim H, Lu F, Hatfield RD, Ralph S, Chapple C, Franke R, Hemm MR, et al (2001. b) Elucidation of new structures in lignins of CAD- and COMT-deficient plants by NMR. Phytochemistry 57: 993–1003 [DOI] [PubMed] [Google Scholar]
- Ralph J, Lundquist K, Brunow G, Lu F, Kim H, Schatz PF, Marita JM, Hatfield RD, Ralph SA, Christensen JH, et al (2004. b) Lignins: natural polymers from oxidative coupling of 4-hydroxyphenylpropanoids. Phytochem Rev 3: 29–60 [Google Scholar]
- Ranocha P, Chabannes M, Chamayou S, Danoun S, Jauneau A, Boudet A-M, Goffner D (2002) Laccase down-regulation causes alterations in phenolic metabolism and cell wall structure in poplar. Plant Physiol 129: 145–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- She XG, Qi SH, Gu WX, Pan XF (1998) Study of the total synthesis of neolignans(I): synthesis of the methyl ethers of americanin A and americanol A. J Chem Res S 1998: 436–437 [Google Scholar]
- Sousa EP, Silva AMS, Pinto MMM, Pedro MM, Cerqueira FAM, Nascimento MSJ (2002) Isomeric kielcorins and dihydroxyxanthones: synthesis, structure elucidation, and inhibitory activities of growth of human cancer cell lines and on the proliferation of human lymphocytes in vitro. Helv Chim Acta 85: 2862–2876 [Google Scholar]
- Tsai C-J, Popko JL, Mielke MR, Hu W-J, Podila GK, Chiang VL (1998) Suppression of O-methyltransferase gene by homologous sense transgene in quaking aspen causes red-brown wood phenotypes. Plant Physiol 117: 101–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umezawa T (2004) Diversity of lignan biosynthesis. Phytochemistry Rev (in press)
- Van Doorsselaere J, Baucher M, Chognot E, Chabbert B, Tollier M-T, Petit-Conil M, Leplé J-C, Pilate G, Cornu D, Monties B, et al (1995) A novel lignin in poplar trees with a reduced caffeic acid/5-hydroxyferulic acid O-methyltransferase activity. Plant J 8: 855–864 [Google Scholar]
- Van Doorsselaere J, Dumas B, Baucher M, Fritig B, Legrand M, Van Montagu M, Inzé D (1993) One-step purification and characterization of a lignin-specific O-methyltransferase from poplar. Gene 133: 213–217 [DOI] [PubMed] [Google Scholar]
- Vignols F, Rigau J, Torres MA, Capellades M, Puigdomènech P (1995) The brown midrib3 (bm3) mutation in maize occurs in the gene encoding caffeic acid O-methyltransferase. Plant Cell 7: 407–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong R, Morrison WH III, Himmelsbach DS, Poole FL II, Ye Z-H (2000) Essential role of caffeoyl coenzyme A O-methyltransferase in lignin biosynthesis in woody poplar plants. Plant Physiol 124: 563–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubieta C, Kota P, Ferrer J-L, Dixon RA, Noel JP (2002) Structural basis for the modulation of lignin monomer methylation by caffeic acid/5-hydroxyferulic acid 3/5-O-methyltransferase. Plant Cell 14: 1265–1277 [DOI] [PMC free article] [PubMed] [Google Scholar]