

Abstract
Recent work has indicated that nitric oxide (NO) and its synthesis are important elements of signal cascades in plant pathogen defense and are a prerequisite for drought and abscisic acid responses in Arabidopsis (Arabidopsis thaliana) and Vicia faba guard cells. Nonetheless, its mechanism(s) of action has not been well defined. NO regulates inward-rectifying K+ channels of Vicia guard cells through its action on Ca2+ release from intercellular Ca2+ stores, but alternative pathways are indicated for its action on the outward-rectifying K+ channels (IK,out), which are Ca2+ insensitive. We report here that NO affects IK,out when NO is elevated above approximately 10 to 20 nm. NO action on IK,out was consistent with oxidative stress and was suppressed by several reducing agents, the most effective being British anti-Lewisite (2,3-dimercapto-1-propanol). The effect of NO on the K+ channel was mimicked by phenylarsine oxide, an oxidizing agent that cross-links vicinal thiols. Neither intracellular pH buffering nor the phosphotyrosine kinase antagonist genistein affected NO action on IK,out, indicating that changes in cytosolic pH and tyrosine phosphorylation are unlikely to contribute to NO or phenylarsine oxide action in this instance. Instead, our results strongly suggest that NO directly modifies the K+ channel or a closely associated regulatory protein, probably by nitrosylation of cysteine sulfhydryl groups.
The gas nitric oxide (NO) is a highly reactive, membrane-permeant free radical that is a natural constituent of all living cells and serves as a signaling molecule. In animals, NO acts indirectly through guanylate cyclase to activate cGMP-dependent cellular responses and affects the gating of Ca2+-dependent K+ channels, Ca2+ and Na+ channels (Bolotina et al., 1994; Tang et al., 2001; Renganathan et al., 2002). Although actions of NO in plants have been known for almost one-half century (Bhatia and Sybenga, 1965), only recently has the gas been recognized to play a physiological role in events as diverse as photomorphogenesis (Zhang et al., 2003), cell growth (Lamattina et al., 2003; Pagnussat et al., 2003), abiotic stress (Garcia-Mata and Lamattina, 2003), and plant pathogen defense (Delledonne et al., 1998; Durner et al., 1998). In plants, NO occurs as a by-product of metabolism from NO2 through photoconversion by carotenoids, reaction with nitrate reductases and with Gly decarboxylase (Rockel et al., 2002; Garcia-Mata and Lamattina, 2003); it is formed nonenzymatically from 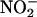 in the apoplast (Bethke et al., 2004); and it is also released by specific NO synthases (Chandok et al., 2003; Guo et al., 2003).
in the apoplast (Bethke et al., 2004); and it is also released by specific NO synthases (Chandok et al., 2003; Guo et al., 2003).
NO affects guard cells and their control of gas exchange and transpirational water loss through the stomata of the leaf epidermis. The gas enhances plant tolerance to drought (Garcia-Mata and Lamattina, 2003) and contributes to stomatal closure evoked by the water-stress hormone abscisic acid (ABA). NO scavengers suppress ABA action in closing stomata, NO donors promote closure in the absence of ABA (Garcia-Mata and Lamattina, 2002; Neill et al., 2002), and, in Arabidopsis (Arabidopsis thaliana) deficient in NO synthesis, stomata fail to close in ABA (Desikan et al., 2002). ABA closes stomata by facilitating osmotic solute loss to reduce guard cell turgor. Among its actions, ABA raises cytosolic-free [Ca2+] ([Ca2+]i) and cytosolic pH (pHi) signals that, in turn, inactivate inward-rectifying K+ channels to prevent K+ uptake, and activate outward-rectifying K+ channels (IK,out) and Cl− (anion) channels at the plasma membrane for solute efflux (Blatt, 2000a; Hetherington, 2001; Schroeder et al., 2001).
Our recent work (Garcia-Mata et al., 2003) demonstrated that NO acts on inward-rectifying K+ channels and anion channels by activating ryanodine-sensitive Ca2+ channels of intercellular Ca2+ stores to elevate [Ca2+]i in Vicia guard cells. At these very low levels, NO had no influence on IK,out, consistent with the Ca2+ insensitivity of these K+ channels. However, NO might be expected to have additional effects on stomatal behavior at higher concentrations. Oxidative stress in plants is known to suppress stomatal closure (see Willmer and Fricker, 1996) and, in some circumstances, can suppress IK,out (Kohler et al., 2003) and promote stomatal opening (Black and Black, 1979). Indeed, NO can bond covalently with the SH residues of Cys to form S-nitrosothiols, and this simple reaction is the basis of many regulatory cascades (Stamler et al., 2001; Ahern et al., 2002), including vascular homeostasis and endotoxic shock in animals (Liu et al., 2004). In subsequent experiments, we observed a reversible decrease in IK,out with moderate elevation to submicromolar NO and have since explored the effects of NO on the IK,out in vivo. Our key observations are that a NO-mediated block of IK,out is suppressed by reducing reagents, especially British anti-Lewisite (BAL; 2,3-dimercapto-1-propanol), and is mimicked by the oxidizing reagent phenylarsine oxide (PAO) that targets viscinal SH residues. These findings indicate that the IK,out can become locked down under nitrosative stress, and they lead us to propose that NO action on IK,out is mediated by direct S-nitrosylation of Cys residues closely associated with the ion channel.
RESULTS
NO Inactivates IK,out
Although our previous work indicated that IK,out is not appreciably sensitive to NO application at rates up to 10 nm/min, we found that exposures to higher levels of NO resulted in a reduction in the amplitude of this K+ current. Figure 1 (inset) shows current traces and steady-state current-voltage (I-V) curves from one Vicia guard cell recorded before (○), during (▪), and after (•) exposure to 50 nm/min NO. Clamp voltage steps from −100 mV to voltages between −90 and +50 mV yielded current typical of IK,out, showing a time-dependent activation. NO exposure of 2 min reduced the current to less than 10% of the control at all clamp voltages. Washing NO from the bath for 10 min led to a gradual recovery and, in this cell, an overshoot of the current relative to the control that we ascribe to secondary effects of NO (see “Discussion”). Similar results were obtained in 15 other experiments with this level of NO (see also Fig. 2). In every case, exposures to NO suppressed IK,out within 2 to 5 min of the treatments, and washout with fresh solution minus NO led to complete recovery over periods of 5 to 10 min (recovery half-time [t1/2], 6 ± 2 min). We noted a very sharp dependence in IK,out to the level of NO exposure. Figure 2 summarizes data from 117 separate experiments with NO exposure levels between 1 nm/min and 500 nm/min. Plotted as the mean of the current complement (=relative inactivation [block]), these data were well fitted to the Hill equation (Hill, 1910) with an apparent Hill coefficient of 4.6 ± 0.4 and Ki of 19.5 ± 0.5 nm/min. Thus, the effect of NO showed a remarkably high degree of cooperativity and was essentially complete at NO levels of 30 nm/min and above.
Figure 1.

NO inactivates IK,out. Steady-state current-voltage curves derived from voltage clamp recordings (insets) from an intact Vicia guard cell before (○), during exposure to 50 nm/min NO (▪), and 8 min after washing in buffer without NO (•). Curves are corrected for instantaneous current recorded at each voltage. Inset, Corresponding current traces cross-referenced by symbol with the voltage protocol shown above (conditioning voltage, −100 mV; test voltages, −90 to +50 mV; tailing voltage, −100 mV). Scale, Horizontal, 2 s; vertical, 1 nA.
Figure 2.

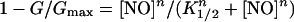 |
(2) |
NO did not appear to affect K+ channel gating, as evident from two observations. First, no indication could be found for a change in gating kinetics. Using NO levels that gave partial suppression of IK,out, we found no significant difference in half-times for current activation compared with current activation kinetics before NO treatments from the same guard cells. Activation half-times at +50 mV before treatments were 291 ± 23 ms and during exposures to 20 nm/min NO were 315 ± 38 ms (n = 11). Second, fitting the steady-state I-V curves from these same experiments to a Boltzmann function showed no appreciable change in the voltage sensitivity for gating. Visually satisfactory and statistically best fittings were obtained with joint fittings (±NO) to the equation
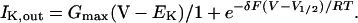 |
(1) |
in which only the maximum conductance (Gmax) varied between curves. Here R, T, and F have their usual meanings. The remaining parameters, the gating charge coefficient (δ), K+ equilibrium voltage (EK), and the voltage giving half-maximal activation (V1/2), were held in common between data sets. Joint fittings in which V1/2 also varied between curves gave statistically equivalent results and yielded values for V1/2 of 11 ± 4 mV before, and 16 ± 5 mV during NO treatments. Thus, the voltage dependence for activation was unaffected by the presence of NO.
NO Action Is Not Mediated by pHi
In guard cells, IK,out rises steeply with pHi above 7.3, and driving pHi to values near 7.0 virtually eliminates the current (Grabov and Blatt, 1997). Indeed, control of the IK,out by ABA is mediated through a rise in pHi (Blatt and Armstrong, 1993). Because oxidative and related stresses are known to affect pHi, (Jackson, 1991; Low and Merida, 1996), we suppressed changes in pHi by passive loading to buffer the cytosol near pHi 7.5 to test its possible role in NO-mediated inactivation of the channel. Guard cells were impaled with microelectrodes containing 200 mm K+-HEPES, pH 7.5, conditions that previously were found to prevent ABA-mediated increases in pHi and its enhancement of IK,out (Blatt and Armstrong, 1993). Figure 3 shows the results from one guard cell exposed to NO after 15-min buffer loading from the microelectrode. As before, NO treatment led to a rapid block of IK,out, which recovered subsequently on extended washing of NO from the bath. Despite the pH buffer, we noted an increase in the leak current (see legends, Figs. 3 and 4) consistent with the effect of NO in promoting Cl− channel activity through its action on [Ca2+]i (Garcia-Mata et al., 2003). Similar results were obtained in five independent experiments, yielding values for Gmax (see Eq. 1) of 0.73 ± 0.08 and 0.04 ± 0.03 before and during NO treatment, respectively. In no case was there any indication that buffering pHi suppressed the NO-evoked decrease in IK,out. Although we cannot categorically rule out an effect on pHi from these experiments, the data do suggest that changes in pHi are not the primary mechanism leading to the inactivation of IK,out by NO.
Figure 3.

Buffering pHi does not suppress NO-evoked inactivation of IK,out. Steady-state current-voltage curves derived from voltage clamp recordings (inset) from an intact Vicia guard cell before (○), during 3-min exposure to 50 nm NO (▪), and 8 min after washing in buffer without NO (▴). Curves are corrected for instantaneous current recorded at each voltage. The guard cell was first loaded with 200 mm K+-HEPES, pH 7.5, from the microelectrode for 10 min. Inset, Corresponding current traces cross-referenced by symbol. Voltage protocol as in Figure 1. Scale, Horizontal, 2 s; vertical, 1 nA. Note the inward (anion) current in the final clamp steps of each cycle during and, to a lesser extent, after NO treatment.
Figure 4.

The vicinal dicysteine reducing agent BAL restores IK,out and protects against subsequent NO treatment. Steady-state current-voltage curves derived from voltage clamp recordings (inset) from an intact Vicia guard cell before (○) and during 2-min exposure to 50 nm/min NO (□). The guard cell was then washed with 0.3 mm BAL for 2 min (▾). After washing with buffer solution alone (BAL) for 6 min, the cell was again challenged with 50 nm NO, this time for 8 min (▪), but without any appreciable effect on IK,out. Voltage protocol as in Figure 1. Scale, Horizontal, 2 s; vertical, 1 nA.
Reducing Reagents Protect IK,out from NO Inactivation
In addition to its action on Ca2+ signaling, NO can modify the thiol group of Cys residues directly by reversible S-nitrosylation (Stamler et al., 2001; Ahern et al., 2002). As a test for possible redox-mediated inactivation of IK,out by NO, we applied the reductants dithiothreitol (DTT) and BAL. Both DTT and BAL are membrane permeant and capable of reducing Cys thiol adducts. In control experiments, neither reagent affected IK,out at concentrations as high as 1 mm (DTT treatment did lead to a small, but not very significant, increase relative to the control; see Fig. 5). However, after NO exposures, both DTT and BAL dramatically accelerated the recovery of IK,out, reversing the effect of NO within the period required for full solution exchange in the chamber (BAL recovery t1/2, 21 ± 6 s; DTT recovery t1/2, 27 ± 8 s; see also Fig. 5).
Figure 5.

Reducing agents restore IK,out and protect against subsequent NO treatment. Summary of steady-state IK,out determined at +50 mV before and after exposure to 50 nm/min NO and/or 0.3 mm BAL and DTT. Data are means ± se of 5 to 15 independent experiments in each case. Bars on left are data from single treatments only or none (control); bars on right are data from guard cells exposed first to 50 nm/min NO and the gas then washed out (1) with buffer alone for 10 min (=washout); (2) with buffer alone for 10 min before a second exposure to 50 nm/min NO (=washout→NO); (3) with 0.3 mm BAL for 2 min and then with buffer alone for 6 to 8 min before a second exposure to 50 nm/min NO (=BAL→NO); and (4) with 0.3 mm DTT for 2 min and then with buffer alone for 6 to 8 min before a second exposure to 50 nm min−1 NO (=DTT→NO). Measurements were taken in buffer only (no shading), BAL (light shading), DTT (dark shading), and NO (diagonal lines).
Remarkably, we found that prior treatments with BAL also protected IK,out from subsequent exposures to NO, even over 10-min periods of continuous superfusion. Figure 4 shows currents and steady-state I-V curves obtained from one guard cell exposed to 50 nm/min NO before and after BAL treatment. Following the control voltage clamp recording (○), the guard cell was challenged with NO for 2 min (□) before washing with 0.3 mm BAL for 3 min (▴). After washing with buffer solution alone for 6 min, the cell was again challenged with 50 nm/min NO, this time for 8 min (▪), but without any appreciable effect on IK,out. Virtually identical results were obtained in five other experiments with BAL (Fig. 5). By contrast, repeated treatments with NO invariably led to a suppression of IK,out that was quantitatively comparable to the effect of the first exposure (Fig. 5). Similar experiments with DTT showed a limited, but statistically significant, suppression of NO action on IK,out (Fig. 5) We cannot discount the possibility that a residue of BAL, especially, might remain in the lipid phase of the membrane to suppress NO action. It is more difficult, however, to explain the absence of any NO action, even after 10-min continuous superfusion with NO. In all events, the ability for both reductants to reverse NO-evoked inactivation of IK,out suggests that NO targets SH residues of Cys associated with the K+ channel, either on the cytosolic side of the membrane or within the lipid bilayer.
Membrane-Permeant SH Oxidants Inactivate IK,out
On the assumption that NO modifies one or more free Cys SH groups, we used several redox reagents with known chemistry to explore a role for Cys thiols in NO action. A summary of these results is shown in Figure 6. N-ethylmaleimide (NEM) is a highly reactive SH reagent that covalently and irreversibly alkylates free Cys thiols; like NEM, iodoacetamide (IodAA) alkylates free Cys thiols; and 5,5′-dithiobis-2-nitrozoic acid (DTNB) is an oxidizing reagent that acts by formation of a disulfide bond between itself and free Cys SH groups to give dithiobenzoate complexes with proteins. NEM is moderately membrane permeant, but neither IodAA nor DTNB penetrates membranes readily (Broillet and Firestein, 1996; Broillet, 2000). We found that exposure to 1 mm NEM led to rapid and complete inactivation of IK,out and the current could not be recovered, even after extensive washing with fresh solution (minus NEM) for periods of up to 20 min. By contrast, exposures to 0.2 mm DTNB and 1 mm IodAA had little effect on IK,out, although the guard cells were continuously superfused with these solutions for periods of 6 to 12 min.
Figure 6.

Membrane-impermeant SH-modifying agents IodAA and DTNB are ineffective in suppressing IK,out. Summary of steady-state IK,out determined at +50 mV before (control) and after exposure to 50 nm/min NO, to 1 mm of the membrane-permeant N-ethylmaleimide (NEM), to 1 mm IodAA, and to 0.2 mm DTNB. Data are means ± se of 5 to 15 independent experiments in each case.
PAO Mimics NO Suppression of IK,out
The organic arsenical PAO is a redox reagent that preferentially reacts with closely spaced thiol groups and forms stable ring complexes. In mammalian tissues, PAO targets protein Tyr phosphatases (Carballo et al., 1999) through its ability to oxidatively bridge critical Cys, and it also affects other protein-signaling intermediates, including human syntaxin 1A (Arien et al., 2003) and Rho GTPases (Gerhard et al., 2003). In guard cells, PAO has been reported to counter the effects of ABA, dark, and H2O2 in closing stomata and to promote stomatal opening, at least in part through its effect on tonoplast ion transport (MacRobbie, 2002), and to affect ABA-mediated gene expression (Heimovaaradijkstra et al., 1996).
Because the actions of PAO on stomatal aperture are also consistent with a suppression of IK,out, we challenged guard cells with PAO during voltage clamp experiments. Figure 7 shows measurements from one guard cell exposed to 10 μm PAO. Like NO, exposure to PAO treatment led to a rapid block of IK,out. Unlike NO, the effect was not reversed on washing PAO from the bath. However, IK,out activity was restored fully within 3 min of washing with BAL (Fig. 7; see also Fig. 8A), consistent with the ability of BAL to reduce arsenical bridged dicysteines, and recovered partially with DTT treatments (see Fig. 8A). Similar results were obtained in 11 other experiments with BAL and four experiments with DTT. PAO has been suggested to act through a protein Tyr phosphatase pathway in its action on guard cells (MacRobbie, 2002). Therefore, we also tested genestein, a protein Tyr phosphatase antagonist, for its ability to suppress NO action on the K+ channel. Our reasoning was that, if PAO-sensitive Tyr dephosphorylation was also essential for control of IK,out by NO, then antagonism of the corresponding kinase might counteract the effects of NO. However, genestein treatments showed no influence on NO-evoked block of IK,out (Fig. 8B).
Figure 7.

Dicysteine cross-linker PAO inactivates IK,out. Steady-state current-voltage curves derived from voltage clamp recordings (inset) from an intact Vicia guard cell before (○), during 5-min exposure to 10 μm PAO (▾), and after 3 min washing in buffer with 0.3 mm BAL (▴). Curves are corrected for instantaneous current recorded at each voltage. Inset, Corresponding current traces cross-referenced by symbol. Voltage protocol as in Figure 1. Scale, Horizontal, 2 s; vertical, 1 nA.
Figure 8.

Reducing agents restore IK,out and protect against subsequent PAO treatment, but the kinase antagonist genestein is ineffective. A, Summary of steady-state IK,out determined at +50 mV before and after exposure to 10 μm PAO and/or 0.3 mm BAL and DTT. Data are means ± se of 5 to 12 independent experiments in each case. Bars on left are data from single treatments only or none (control); bars on right are data from guard cells exposed first to 10 μm PAO and the oxidizing reagent then washed out (1) with buffer alone for 10 min (=washout); (2) with 0.3 mm BAL for 2 min and then with buffer alone for 6 to 8 min before a second exposure to PAO (=PAO→BAL); and (3) with 0.3 mm DTT for 2 min and then with buffer alone for 6 to 8 min before a second exposure to PAO (=PAO→DTT). Measurements taken in buffer only (no shading), BAL (light shading), DTT (dark shading), PAO (diagonal lines). B, Summary of steady-state IK,out determined at +50 mV before and after exposure to 10 μm genestein and to 50 nm min−1 NO with and without concurrent treatment with 10 μm genestein.
As with NO, we found a steep dependence of IK,out on PAO concentration (Fig. 9). Plotted as the mean of the current complement (=relative inactivation), these data were well fitted to the Hill equation (Hill, 1910), with an apparent Hill coefficient of 2.3 ± 0.5 and Ki of 3.2 ± 0.4 μm. Thus, the effect of PAO showed a degree of cooperativity precisely one-half that of NO and was fully reversed by BAL, consistent with the ability of PAO and BAL to target vicinal Cys pairs.
Figure 9.

PAO inactivation of IK,out is cooperative. IK,out steady-state currents derived as in Figure 1 at +50 mV before and during PAO treatments. Data are given as means ± se of the conductance complement (= relative inactivation) from 47 experiments (>5 experiments per data point) and are plotted as a function of PAO concentration. The solid curve is the best fit to the Hill equation (Eq. 2) yielding a cooperativity coefficient of 2.3 ± 0.5 and an apparent K1/2 of 3.2 ± 0.4 μm with a maximum block of 91% ± 6%.
DISCUSSION
NO is now widely recognized to contribute to cellular signaling in plants, especially in response to environmental stress and pathogen attack. It has been widely implicated in Ca2+-dependent responses (Delledonne et al., 1998; Durner et al., 1998; Klessig et al., 2000; Desikan et al., 2002; Garcia-Mata and Lamattina, 2003) and, in guard cells, NO was recently demonstrated to promote Ca2+ release from endomembrane stores thereby potentiating evoked Ca2+-induced Ca2+ release and elevation of [Ca2+]i (Garcia-Mata et al., 2003). Nonetheless, other actions of NO can be anticipated that are unlinked to Ca2+. As a free radical, NO is highly reactive in redox exchange reactions and, thus, is capable of targeting a wide variety of proteins. In animals, NO reacts preferentially with exposed Cys thiols to form S-nitrosyl adducts that affect the function of a number of cellular functions, including ion transport (Stamler et al., 1992, 2001; Broillet and Firestein, 1996; Broillet, 2000; Ahern et al., 2002; Sun et al., 2003). With the exception of its influence on mitochondrial respiration (Chiandussi et al., 2002; Zottini et al., 2002), however, in plants S-nitrosylation of proteins has attracted much less attention. Our results now implicate NO-mediated S-nitrosylation in regulation of the IK,out of Vicia stomatal guard cells and indicate target site(s) for NO action that are on or closely associated with the channel protein at the inner face of the membrane.
A key observation favoring this hypothesis is that NO-mediated inactivation of IK,out was reversible and its recovery accelerated by treatments with membrane-permeant reducing reagents (Figs. 4 and 5). S-Nitrosylated thiols are readily targeted by reductants such as DTT. Thus, the fact that IK,out inactivation was reversed rapidly by DTT, as well as BAL, argues for this simplest of oxidative modifications by NO. The hypothesis finds additional support in the parallel actions of other oxidizing reagents on IK,out (Figs. 6–9). Both the alkylating reagent NEM and the SH cross-linking arsenical PAO mimicked the action of NO. Significantly, the membrane-impermeant Cys-modifying reagents IodAA and DTNB had little or no effect on the K+ current, leading us to propose that the primary sites for oxidative modification are not accessible at the external face of the membrane. Similar lines of evidence have been drawn for direct activation of olfactory cyclic nucleotide-gated channels (Broillet and Firestein, 1996; Broillet, 2000), and inactivation of Ca2+ channels (Summers et al., 1999; Sun et al., 2001) by NO.
Equally important, we found that buffering pHi was ineffective in countering the inactivation of IK,out by NO (Fig. 3). IK,out in guard cells is insensitive to changes in [Ca2+]i, but its activity is strongly dependent on pHi (for review, see Assmann and Shimazaki, 1999; Blatt, 2000b; Hetherington, 2001; Schroeder et al., 2001; Webb et al., 2001). Activation of IK,out at alkaline pHi shows cooperativity consistent with two H+-binding sites with an apparent pKa near 7.4 (Grabov and Blatt, 1997). Thus, the inactivation of IK,out might be understood if NO transiently reduced pHi to values near 7.0. As the simplest test for an effect of NO-evoked changes in pHi, we buffered the cytosol by diffusional loading from microelectrodes filled with 200 mm HEPES. This approach was shown to raise pH buffer capacity 4-fold, sufficient to prevent ABA-evoked increases in pHi and IK,out (Blatt and Armstrong, 1993). That NO treatments inactivated IK,out under these conditions therefore argues strongly against any role for pHi as an intermediate.
It is significant that S-nitrosylation of Cys thiols is reversible in vivo. NO is thought to undergo oxidation to form nitrous anhydride (N2O3), which, in turn, can donate NO either to a Cys thiol of a protein or to a small organic thiol such as glutathione. Thus, protein thiol groups can be either primary or secondary targets for S-nitrosylation, and denitrosylation may occur through a similar exchange process (Sun et al., 2001; Ahern et al., 2002). In fact, the mechanisms for denitrosylation in many cases have yet to be fully understood (Stamler et al., 2001), but there is evidence that specific enzymes govern levels of S-nitrosylation in vivo (Liu et al., 2001), and peroxidase and transaminase activities are implicated in denitrosylation (Abu-Soud and Hazen, 2000; Lai et al., 2001; Mannick and Schonhoff, 2004). S-Nitrosylation does confer precisely regulated posttranslational modification and probably affects the function of many proteins in much the same way as protein (de)phosphorylation (Stamler et al., 1997, 2001; Hess et al., 2001; Ahern et al., 2002). One emerging pattern is of covalent linkage to Cys in an acid-base pocket that supports (de)nitrosylation reactions similar to hemoglobin (Stamler et al., 1997; Hess et al., 2001) that confers specificity. Indeed, the action of NO on ryanodine-sensitive Ca2+ channels in skeletal muscle results from addition/removal of NO at a single Cys moiety out of the >40 Cys residues in the channel protein (Sun et al., 2001).
By contrast with NO, inactivation of IK,out by PAO was not reversible on washout within the time frame of these experiments but, like NO, its action was readily reversed by treatments with the dithiol BAL (Figs. 7 and 8). PAO preferentially reacts with closely spaced (vicinal) thiol groups in proteins to form stable, sulfur-metal ring complexes, and its targets include protein Tyr phosphatases (Carballo et al., 1999), vesicle trafficking, and associated proteins (Arien et al., 2003; Gerhard et al., 2003), as well as inducible NO synthase (Oda et al., 2000). A feature of PAO complexes is their lability to reduction by BAL. Thus, the efficacy of this reducing agent in rescuing IK,out suggests the presence of critical, paired Cys thiols that are exposed to oxidation by both NO and PAO within the cell. In support of this argument, we found that inactivation of IK,out by PAO was best fitted with a binding function and cooperativity index (Hill coefficient) precisely one-half that for NO-mediated inactivation, as if PAO oxidized paired Cys overlapping with the targets of NO action. However, these results do not identify the protein target(s) in either case.
How might NO action be mediated, then? The inactivation of IK,out is sensitive to NO over a remarkably narrow range of concentrations. We found that raising NO generation to levels only marginally above 20 nm/min gave almost complete inactivation, consistent with at least a 4-fold cooperativity for the effect. No appreciable change was evident in the relaxation kinetics for the current, nor in its voltage sensitivity in the presence of NO. These are characteristics most easily explained with a reduction in the number of functional channels at the membrane. Voltage-gated ion channels, presumably including IK,out, operate as tetramers with each subunit contributing to both gating and permeation (Doyle et al., 1998; Yellen, 2002; Jiang et al., 2003; Very and Sentenac, 2003). So one interpretation is that NO targets a single pair of Cys within the K+ channel protein that are critical for channel activation, and oxidizing these Cys thiols in two of the four subunits is sufficient to block channel opening. However, at present, other interpretations that do not entail a direct action on the K+ channel are equally plausible. We note that NO washout (Fig. 1; see also Fig. 5) occasionally led to an overshoot in IK,out, consistent with additional secondary actions on other regulatory factors. In this context, it is of interest, for example, that the Arabidopsis ABI1 protein phosphatase is similarly sensitive to micromolar PAO (Ki, 3 μm), although in vitro its inactivation shows no cooperativity (Meinhard and Grill, 2001). The dominant-negative mutant of this protein phosphatase also suppresses the activity of IK,out in tobacco guard cells (Armstrong et al., 1995).
An equally important question is of the physiological function for NO sensitivity of IK,out. At present, our knowledge of the roles for NO in plants is expanding rapidly, so a definitive answer is not possible at this time. However, we can offer two conjectures. (1) On the assumption that NO action is not specifically targeted to IK,out, its inactivation may represent a broad-range response to oxidative stress. The K+ channel is known to be sensitive to concentrations of H2O2 only marginally higher than the steady-state levels of NO achieved in these experiments and, like NO, H2O2 inactivates the current (Kohler et al., 2003). This effect is consistent with previous reports that oxidative stress in plants can suppress stomatal closure (see Willmer and Fricker, 1996) and even promote stomatal opening (Black and Black, 1979). (2) On the assumption that NO action is targeted to IK,out, its inactivation may reflect an imbalance between nitrosylation and denitrosylation (Liu et al., 2001) superimposed by the exogenous addition of NO in these experiments. From this explanation, it would follow that the K+ channel activity is finely tuned to the balance of S-nitrosylation in vivo. Indeed, these two explanations are not mutually exclusive, but a resolution of this question is not yet possible.
MATERIALS AND METHODS
Plant Material and Electrophysiology
Epidermal peels were prepared from Vicia faba grown under a 16-h light/8-h dark and 21°C/14°C cycle (Blatt and Armstrong, 1993). All operations were carried out on a Zeiss Axiovert microscope (Zeiss, Jena, Germany) with 63× LWD DIC optics. Epidermal peels were fixed in the experimental chamber with an optically clear, pressure-sensitive adhesive (50/50 medical adhesive; Dow Corning, Brussels) and were bathed in 5 mm Ca2+-MES, pH 6.1 [MES titrated to its pKa with Ca(OH)2], with 10 mm KCl. Measurements were carried out in continuously flowing solution at 20 chamber volumes/min (Grabov and Blatt, 1998).
Microelectrodes
Recordings were obtained with two-barrelled microelectrodes coated with paraffin wax to reduce electrode capacitance (Blatt and Armstrong, 1993). Current-passing and voltage-recording barrels were filled with 200 mm K+ acetate, pH 7.5, to minimize salt leakage and salt loading artifacts associated with the Cl− anion without imposing a pH load. In some experiments, the electrodes were filled with 200 mm K+-HEPES, pH 7.5, to suppress any changes in pHi. Connections to amplifier headstages were via 1 m KCl Ag|AgCl half-cells, and a matching half-cell and 1 m KCl-agar bridge served as the reference (bath) electrode.
NO Release
NO was generated in solution from S-nitroso-N-acetyl-penacillamine (SNAP), which spontaneously releases NO in a pseudo first-order reaction with a half-time in solution of approximately 5 h (Hou et al., 1999). NO generation was assayed by the Griess reaction (Zhang et al., 2003) in perfusion buffer and indicated that 10 μm SNAP releases 2.5 to 3.0 μm NO over 2 h, equivalent to approximately 2 nm NO min−2 μm−1 SNAP in standing solution (not shown). Because the K+ channel measurements were carried out in continuously flowing solution, this figure represents a lower estimate of the rate of NO generation.
Electrical and Numerical Analysis
Mechanical and electrical design has been described previously (Blatt and Armstrong, 1993). Voltage clamp control, data acquisition, and analysis were carried out using Henry II software (Y-Science, Glasgow, UK; available for academic use by download at http://www.gla.ac.uk/ibls/BMB/mrb/lppbh.htm). Currents were normally filtered with a low-pass Butterworth filter (cutoff frequency, 1 kHz) and sampled at 2 kHz. IK,out was determined and activation half-times were taken from two-step voltage clamp protcols after subtracting instantaneous currents at each voltage. Where appropriate, analyses were carried out by nonlinear, least-squares fittings using a Marquardt-Levenberg alorithm (Marquardt, 1963). Results are reported as means ± se and taken to be significant at P < 0.05.
Chemicals and Solutions
SNAP was dissolved in 1:1 ethanol:water, and PAO was dissolved in DMSO before >1,000-fold dilution for use. Ethanol and DMSO alone at this concentration had no effect (Grabov and Blatt, 1998; Hamilton et al., 2000). All other compounds were used directly. All reagents were from Sigma (Poole, UK) or Calbiochem (Darmstadt, Germany).
Acknowledgments
We thank Dr. I. Johansson for comments on the manuscript.
This work was supported by the Biotechnology and Biological Science Research Council (grants P09640, C10234, and P09561 to M.R.B.).
Article, publication date, and citation information can be found at www.plantphysiol.org/cgi/doi/10.1104/pp.104.050344.
References
- Abu-Soud HM, Hazen SL (2000) Nitric oxide is a physiological substrate for mammalian peroxidases. J Biol Chem 275: 37524–37532 [DOI] [PubMed] [Google Scholar]
- Ahern GP, Klyachko VA, Jackson MB (2002) cGMP and S-nitrosylation: two routes for modulation of neuronal excitability by NO. Trends Neurosci 25: 510–517 [DOI] [PubMed] [Google Scholar]
- Arien H, Wiser O, Arkin IT, Leonov H, Atlas D (2003) Syntaxin 1A modulates the voltage-gated L-type calcium channel (Ca(v)1.2) in a cooperative manner. J Biol Chem 278: 29231–29239 [DOI] [PubMed] [Google Scholar]
- Armstrong F, Leung J, Grabov A, Brearley J, Giraudat J, Blatt MR (1995) Sensitivity to abscisic acid of guard cell K+ channels is suppressed by abi1-1, a mutant Arabidopsis gene encoding a putative protein phosphatase. Proc Natl Acad Sci USA 92: 9520–9524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assmann SM, Shimazaki K (1999) The multisensory guard cell. Stomatal responses to blue light and abscisic acid. Plant Physiol 119: 809–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bethke PC, Badger MR, Jones RL (2004) Apoplastic synthesis of nitric oxide by plant tissues. Plant Cell 16: 332–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia CR, Sybenga J (1965) Effect of nitric oxide on x-ray sensitivity of Crotalaria intermedia seeds. Mutat Res 2: 332–338 [DOI] [PubMed] [Google Scholar]
- Black CR, Black VJ (1979) The effects of low concentrations of sulphur dioxide on stomatal conductance and epidermal cell survival in field bean (Vicia faba L). J Exp Bot 30: 291–298 [Google Scholar]
- Blatt MR (2000. a) Ca2+ signaling and control of guard-cell volume in stomatal movements. Curr Opin Plant Biol 3: 196–204 [PubMed] [Google Scholar]
- Blatt MR (2000. b) Cellular signaling and volume control in stomatal movements in plants. Annu Rev Cell Dev Biol 16: 221–241 [DOI] [PubMed] [Google Scholar]
- Blatt MR, Armstrong F (1993) K+ channels of stomatal guard cells: abscisic acid-evoked control of the outward rectifier mediated by cytoplasmic pH. Planta 191: 330–341 [Google Scholar]
- Bolotina VM, Najibi S, Palacino JJ, Pagano PJ, Cohen RA (1994) Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature 368: 850–853 [DOI] [PubMed] [Google Scholar]
- Broillet MC (2000) A single intracellular cysteine residue is responsible for the activation of the olfactory cyclic nucleotide-gated channel by NO. J Biol Chem 275: 15135–15141 [DOI] [PubMed] [Google Scholar]
- Broillet MC, Firestein S (1996) Direct activation of the olfactory cyclic nucleotide-gated channel through modification of sulfhydryl groups by NO compounds. Neuron 16: 377–385 [DOI] [PubMed] [Google Scholar]
- Carballo M, Conde M, El Bekay R, Martin-Nieto J, Camacho MJ, Monteseirin J, Conde J, Bedoya FJ, Sobrino F (1999) Oxidative stress triggers STAT3 tyrosine phosphorylation and nuclear translocation in human lymphocytes. J Biol Chem 274: 17580–17586 [DOI] [PubMed] [Google Scholar]
- Chandok MR, Ytterberg AJ, van Wijk KJ, Klessig DF (2003) The pathogen-inducible nitric oxide synthase (iNOS) in plants is a variant of the P protein of the glycine decarboxylase complex. Cell 113: 469–482 [DOI] [PubMed] [Google Scholar]
- Chiandussi E, Petrussa E, Macri F, Vianello A (2002) Modulation of a plant mitochondrial K-ATP(+) channel and its involvement in cytochrome c release. J Bioenerg Biomembr 34: 177–184 [DOI] [PubMed] [Google Scholar]
- Delledonne M, Xia YJ, Dixon RA, Lamb C (1998) Nitric oxide functions as a signal in plant disease resistance. Nature 394: 585–588 [DOI] [PubMed] [Google Scholar]
- Desikan R, Griffiths R, Hancock J, Neill S (2002) A new role for an old enzyme: nitrate reductase-mediated nitric oxide generation is required for abscisic acid-induced stomatal closure in Arabidopsis thaliana. Proc Natl Acad Sci USA 99: 16314–16318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle DA, Cabral JM, Pfuetzner RA, Kuo AL, Gulbis JM, Cohen SL, Chait BT, MacKinnon R (1998) The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280: 69–77 [DOI] [PubMed] [Google Scholar]
- Durner J, Wendehenne D, Klessig DF (1998) Defense gene induction in tobacco by nitric oxide, cyclic GMP, and cyclic ADP-ribose. Proc Natl Acad Sci USA 95: 10328–10333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Mata C, Gay R, Sokolovski S, Hills A, Lamattina L, Blatt MR (2003) Nitric oxide regulates K+ and Cl− channels in guard cells through a subset of abscisic acid-evoked signaling pathways. Proc Natl Acad Sci USA 100: 11116–11121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Mata C, Lamattina L (2002) Nitric oxide and abscisic acid cross talk in guard cells. Plant Physiol 128: 790–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Mata C, Lamattina L (2003) Abscisic acid, nitric oxide and stomatal closure: is nitrate reductase one of the missing links? Trends Plant Sci 8: 20–26 [DOI] [PubMed] [Google Scholar]
- Gerhard R, John H, Aktories K, Just I (2003) Thiol-modifying phenylarsine oxide inhibits guanine nucleotide binding of Rho but not of Rac GTPases. Mol Pharmacol 63: 1349–1355 [DOI] [PubMed] [Google Scholar]
- Grabov A, Blatt MR (1997) Parallel control of the inward-rectifier K+ channel by cytosolic-free Ca2+ and pH in Vicia guard cells. Planta 201: 84–95 [Google Scholar]
- Grabov A, Blatt MR (1998) Membrane voltage initiates Ca2+ waves and potentiates Ca2+ increases with abscisic acid in stomatal guard cells. Proc Natl Acad Sci USA 95: 4778–4783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo FQ, Okamoto M, Crawford NM (2003) Identification of a plant nitric oxide synthase gene involved in hormonal signaling. Science 302: 100–103 [DOI] [PubMed] [Google Scholar]
- Hamilton DWA, Hills A, Kohler B, Blatt MR (2000) Ca2+ channels at the plasma membrane of stomatal guard cells are activated by hyperpolarization and abscisic acid. Proc Natl Acad Sci USA 97: 4967–4972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimovaaradijkstra S, Nieland TJF, Vandermeulen RM, Wang M (1996) Abscisic acid-induced gene expression requires the activity of protein(s) sensitive to the protein-tyrosine-phosphatase inhibitor phenylarsine oxide. Plant Growth Regul 18: 115–123 [Google Scholar]
- Hess DT, Matsumoto A, Nudelman R, Stamler JS (2001) S-nitrosylation: spectrum and specificity. Nat Cell Biol 3: E46–E49 [DOI] [PubMed] [Google Scholar]
- Hetherington AM (2001) Guard cell signaling. Cell 107: 711–714 [DOI] [PubMed] [Google Scholar]
- Hill AV (1910) The possible effects of the aggregation of the molecules of hemoglobin on its dissociation curves. J Physiol 40: 4–7 [Google Scholar]
- Hou YC, Wang JQ, Ramirez J, Wang PG (1999) Glyco-S-nitrosothiols: sugar-SNAP, a new type of nitric oxide donor. Nitric Oxide 301: 242–249 [DOI] [PubMed] [Google Scholar]
- Jackson MB (1991) Regulation of water relationships in flooded plants by ABA from leaves, roots and xylem sap. In WJ Davies, HG Jones, eds, Abscisic Acid Physiology and Biochemistry. BIOS Scientific Publishers, Oxford, pp 217–226
- Jiang YX, Lee A, Chen JY, Ruta V, Cadene M, Chait BT, MacKinnon R (2003) X-ray structure of a voltage-dependent K+ channel. Nature 423: 33–41 [DOI] [PubMed] [Google Scholar]
- Klessig DF, Durner J, Noad R, Navarre DA, Wendehenne D, Kumar D, Zhou JM, Shah J, Zhang SQ, Kachroo P, et al (2000) Nitric oxide and salicylic acid signaling in plant defense. Proc Natl Acad Sci USA 97: 8849–8854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler B, Hills A, Blatt MR (2003) Control of guard cell ion channels by hydrogen peroxide and abscisic acid indicates their action through alternate signaling pathways. Plant Physiol 131: 385–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai TS, Hausladen A, Slaughter TF, Eu JP, Stamler JS, Greenberg CS (2001) Calcium regulates S-nitrosylation, denitrosylation, and activity of tissue transglutaminase. Biochemistry 40: 4904–4910 [DOI] [PubMed] [Google Scholar]
- Lamattina L, Garcia-Mata C, Graziano M, Pagnussat G (2003) Nitric oxide: the versatility of an extensive signal molecule. Annu Rev Plant Biol 54: 109–136 [DOI] [PubMed] [Google Scholar]
- Liu L, Yan Y, Zeng M, Zhang J, Hanes MA, Ahearn G, McMahon TJ, Dickfeld T, Marshall HE, Que LG, et al. (2004) Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell 116: 617–628 [DOI] [PubMed] [Google Scholar]
- Liu LM, Hausladen A, Zeng M, Que L, Heitman J, Stamler JS (2001) A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature 410: 490–494 [DOI] [PubMed] [Google Scholar]
- Low PS, Merida JR (1996) The oxidative burst in plant defense: function and signal transduction. Physiol Plant 96: 533–542 [Google Scholar]
- MacRobbie EAC (2002) Evidence for a role for protein tyrosine phosphatase in the control of ion release from the guard cell vacuole in stomatal closure. Proc Natl Acad Sci USA 99: 11963–11968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannick JB, Schonhoff CM (2004) NO means no and yes: regulation of cell signaling by protein nitrosylation. Free Radic Res 38: 1–7 [DOI] [PubMed] [Google Scholar]
- Marquardt D (1963) An algorithm for least-squares estimation of nonlinear parameters. J Soc Ind Appl Math 11: 431–441 [Google Scholar]
- Meinhard M, Grill E (2001) Hydrogen peroxide is a regulator of ABI1, a protein phosphatase 2C from Arabidopsis. FEBS Lett 508: 443–446 [DOI] [PubMed] [Google Scholar]
- Neill SJ, Desikan R, Clarke A, Hancock JT (2002) Nitric oxide is a novel component of abscisic acid signaling in stomatal guard cells. Plant Physiol 128: 13–16 [PMC free article] [PubMed] [Google Scholar]
- Oda M, Sakitani K, Kaibori M, Inoue T, Kamiyama Y, Okumura T (2000) Vicinal dithiol-binding agent, phenylarsine oxide, inhibits inducible nitric-oxide synthase gene expression at a step of nuclear factor-kappa B DNA binding in hepatocytes. J Biol Chem 275: 4369–4373 [DOI] [PubMed] [Google Scholar]
- Pagnussat GC, Lanteri ML, Lamattina L (2003) Nitric oxide and cyclic GMP are messengers in the indole acetic acid-induced adventitious rooting process. Plant Physiol 132: 1241–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renganathan M, Cummins TR, Waxman SG (2002) Nitric oxide blocks fast, slow, and persistent Na+ channels in C-type DRG neurons by S-nitrosylation. J Neurophysiol 87: 761–775 [DOI] [PubMed] [Google Scholar]
- Rockel P, Strube F, Rockel A, Wildt J, Kaiser WM (2002) Regulation of nitric oxide (NO) production by plant nitrate reductase in vivo and in vitro. J Exp Bot 53: 103–110 [PubMed] [Google Scholar]
- Schroeder JI, Allen GJ, Hugouvieux V, Kwak JM, Waner D (2001) Guard cell signal transduction. Annu Rev Plant Physiol Plant Mol Biol 52: 627–658 [DOI] [PubMed] [Google Scholar]
- Stamler JS, Lamas S, Fang FC (2001) Nitrosylation: the prototypic redox-based signaling mechanism. Cell 106: 675–683 [DOI] [PubMed] [Google Scholar]
- Stamler JS, Singel DJ, Loscalzo J (1992) Biochemistry of nitric oxide and its redox-activated forms. Science 258: 1898–1902 [DOI] [PubMed] [Google Scholar]
- Stamler JS, Toone EJ, Lipton SA, Sucher NJ (1997) (S)NO signals: translocation, regulation, and a consensus motif. Neuron 18: 691–696 [DOI] [PubMed] [Google Scholar]
- Summers BA, Overholt JL, Prabhakar NR (1999) Nitric oxide inhibits L-type Ca2+ current in glomus cells of the rabbit carotid body via a cGMP-independent mechanism. J Neurophysiol 81: 1449–1457 [DOI] [PubMed] [Google Scholar]
- Sun JH, Eu JP, Stamler JS, Meissner G (2001) Classes of thiols that influence the activity of the skeletal muscle calcium release channel. J Biol Chem 276: 15625–15630 [DOI] [PubMed] [Google Scholar]
- Sun JH, Xu L, Eu JP, Stamler JS, Meissner G (2003) Nitric oxide, NOC-12, and S-nitrosoglutathione modulate the skeletal muscle calcium release channel/ryanodine receptor by different mechanisms: an allosteric function for O2 in S-nitrosylation of the channel. J Biol Chem 278: 8184–8189 [DOI] [PubMed] [Google Scholar]
- Tang XD, Daggett H, Hanner M, Garcia ML, Mcmanus OB, Brot N, Weissbach H, Heinemann SH, Hoshi T (2001) Oxidative regulation of large conductance calcium-activated potassium channels. J Gen Physiol 117: 253–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Very AA, Sentenac H (2003) Molecular mechanisms and regulation of K+ transport in higher plants. Annu Rev Plant Biol 54: 575–603 [DOI] [PubMed] [Google Scholar]
- Webb AAR, Larman MG, Montgomery LT, Taylor JE, Hetherington AM (2001) The role of calcium in ABA-induced gene expression and stomatal movements. Plant J 26: 351–362 [DOI] [PubMed] [Google Scholar]
- Willmer C, Fricker MD (1996) Stomata. London, Chapman and Hall, pp 1–375
- Yellen G (2002) The voltage-gated potassium channels and their relatives. Nature 419: 35–42 [DOI] [PubMed] [Google Scholar]
- Zhang MX, An LZ, Feng HY, Chen T, Chen K, Liu YH, Tang HG, Chang JF, Wang XL (2003) The cascade mechanisms of nitric oxide as a second messenger of ultraviolet B in inhibiting mesocotyl elongations. Photochem Photobiol 77: 219–225 [DOI] [PubMed] [Google Scholar]
- Zottini M, Formentin E, Scattolin M, Carimi F, Lo Schiavo F, Terzi M (2002) Nitric oxide affects plant mitochondrial functionality in vivo. FEBS Lett 515: 75–78 [DOI] [PubMed] [Google Scholar]