

Abstract
Palliative surgery for congenital heart disease has allowed patients with previously lethal heart malformations to survive and, in most cases, to thrive. However, these procedures often place pressure and volume loads on the heart, and over time these chronic loads can cause heart failure. Current therapeutic options for initial surgery and chronic heart failure that results from failed palliation are limited, in part, by the mammalian heart’s low inherent capacity to form new cardiomyocytes (CMs). Surmounting the heart regeneration barrier would transform the treatment of congenital, as well as acquired, heart disease, and likewise would enable development of personalized, in vitro cardiac disease models. Although these remain distant goals, studies of heart development are illuminating the path forward and suggest unique opportunities for heart regeneration, particularly in fetal and neonatal periods. Here we review major lessons from heart development that inform current and future studies directed at enhancing cardiac regeneration.
Subject Terms: Congenital heart Disease, Treatment
Keywords: Cardiac development, regeneration, proliferation, cardiac progenitor cell, transcription factor
Introduction
Since Robert Gross inaugurated pediatric cardiac surgery in 1938 and since the advent of open heart surgery on cardiopulmonary bypass in the 1950s, mortality from congenital heart disease has plummeted. However, many of the more complex forms of congenital heart disease still cannot be physiologically corrected; rather, patients are surgically palliated, permitting survival but leaving significant residual cardiac pressure and volume loads. Over time, these patients are at risk for heart failure and some ultimately require heart transplantation.
A major biological factor that constrains treatment options for congenital heart disease patients, as well as adults with acquired heart disease, is the limited ability of mature mammalian cardiomyocytes (CMs) to proliferate. The low innate regenerative capacity of the heart narrows initial treatment strategies as well as the therapeutic options for maximizing the longevity of pressure- or volume-loaded hearts. Overcoming this barrier by developing methods to engineer new myocardium or to regenerate injured myocardium would transform the management of congenital heart disease patients who are confronted primarily by declining pump function.
Not long ago, this vision of cardiac regeneration was confined to science fiction; however, expanding mechanistic understanding of heart formation and development achieved over the past decades has enabled cardiac biologists to make significant inroads into the challenge of therapeutic heart regeneration. In this Compendium article, we review the key signaling pathways and transcriptional regulators that govern cardiogenesis from gastrulation through the formation of cardiac mesoderm to the specification of CMs. We go on to review mechanisms that control CM proliferation and maturation. We highlight recent studies that exemplify redeployment of developmental signaling pathways to further the goals of cardiac regeneration and discuss further challenges that must be overcome for achieving success in myocardial regeneration. This review is focused on myocardial regeneration. Due to space limitations, we do not address valve and pulmonary vascular disease, additional important problems faced by some palliated congenital heart disease patients that will need to be corrected to make regenerative approaches viable for the full spectrum of congenital heart disease.
Development of the cardiac mesoderm
Mesodermal Commitment from Epiblast
During gastrulation, the developing embryo reorganizes itself into three primary germ layers: the ectoderm, mesoderm, and endoderm. CMs are derived from the mesoderm that forms during early development. Besides the heart, the mesoderm also gives rise to skeletal muscle, bones, blood and other tissues1. It has been recognized that molecular signaling and cell-to-cell interactions between germ layers play significant roles in mesoderm induction and cardiac differentiation.
At embryonic day 5 (E5) in the mouse, the embryo exhibits an elongated cylinder shape that is divided into two portions—the proximal and distal regions. A combination of signaling events leads to initial patterning and separation of the embryo into embryonic and extraembryonic layers. Along the distal region of the extraembryonic visceral endoderm (EVE), NODAL is expressed and forms a concentration gradient along the proximal-distal axis1, 2. Through a positive feedback loop, NODAL activates its expression throughout the epiblast and induces an organizing center in the distal EVE that later becomes known as the anterior visceral endoderm3. As the embryo further develops, the anterior visceral endoderm migrates towards the anterior of the embryo and expresses NODAL antagonists such as Cerberus-like (CER1) and LEFTY1 proteins4. These antagonists prevent NODAL signaling from taking place in the anterior but not the posterior of the epiblast (Figure 1). Along with NODAL5, fibroblast growth factor (FGF)6, bone morphogenic protein (BMP)7–9, and wingless-type MMTV integration site family member 3 (WNT3A)10 are expressed in the posterior of the embryo11, 12. Together, NODAL, FGF, BMP, and WNT signaling induce early mesoderm that is marked by the primitive streak (PS)/pan-mesoderm marker, Brachyury.
Figure 1. Specification of mesodermal precursors.
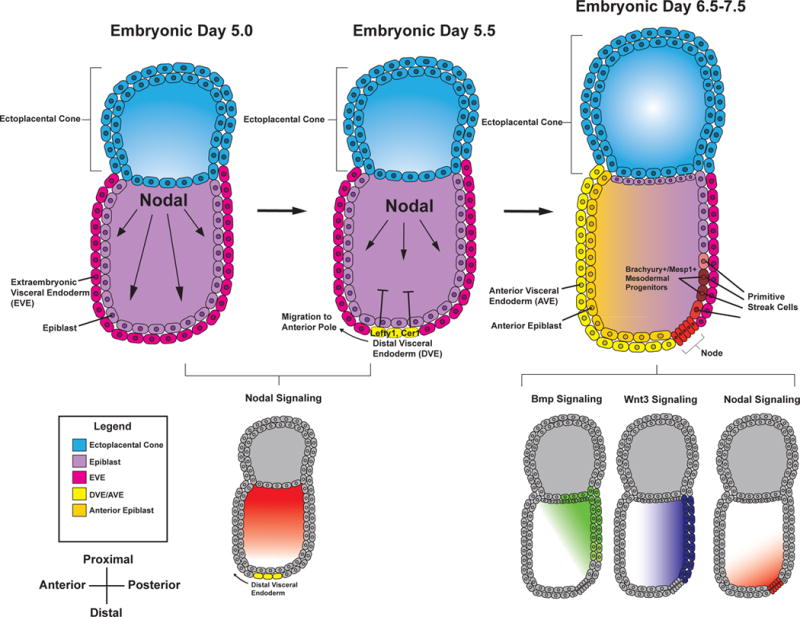
Schematic representing the signaling events leading to mesodermal specification during early development. NODAL is first expressed proximally at E5.0. Through an autoregulatory loop, NODAL activates its own expression throughout the epiblast (shown in light purple) and goes on to induce the expression of NODAL antagonists, LEFTY1 and CER1 in the distal visceral endoderm at E5.5 (DVE). The DVE migrates anteriorly where it specifies the anterior portion of the embryo as shown in the yellow hues at E6.5-7.5. The anterior visceral endoderm (AVE, yellow) limits NODAL signaling to the posterior of the embryo. Along with WNT3 and BMP signaling, NODAL specifies early primitive streak progenitors to the mesoderm fate.
The PS marks the site where epiblast cells begin to ingress into the embryo and differentiate into the three embryonic germ layers. Several NODAL signaling gradients pattern epiblast cells along the streak. High concentrations of NODAL at the anterior PS specify definitive endoderm precursors, while lower levels of NODAL in concert with BMP and WNT signaling specify mesodermal precursors in the intermediate and posterior PS1, 13. Cardiac mesoderm precursors form anterior and lateral to the PS as a result of this morphogen patterning2. The T-box transcription factor eomesodermin (EOMES), expressed at the anterior PS, is important for both definitive endoderm and cardiac mesoderm development14. EOMES+ cells activate the basic-helix-loop-helix transcription factor, mesoderm posterior-1 (MESP1)14, which subsequently regulates the activation of several cardiac and mesodermal genes and plays a vital role in allowing mesodermal precursor cells to migrate through the PS15. While MESP1 is broadly expressed throughout mesoderm precursors16, a subpopulation of MESP1+ cells anterior to the PS become specified to cardiac lineages as they ingress through the PS17 (Figure 1).
Migration from Primitive Streak to Cardiogenic Mesoderm
With the patterning of the primitive streak and the activation of MESP1, mesoderm cells begin to migrate away from the PS. A subpopulation of these MESP1+ cells marches anteriorly and laterally away from the mid- and posterior aspect of the mesoderm to become the anterior lateral plate mesoderm17. The migration of these early cardiac precursors away from the WNT/β-catenin rich PS, mediated by WNT chemorepulsion18, marks a significant step in the commitment of early mesoderm cells towards a cardiac fate. At E7.5 in the mouse/week 3 of human development, these migratory cells make two pools of cardiac progenitors known as the first and second heart fields (FHF and SHF)19.
Canonical WNT signaling, mediated by β-catenin, plays a crucial role in the differentiation of these cardiac progenitors. First, as noted above, WNT3A from the posterior of the embryo induces early mesoderm. Subsequently, WNT signaling from the overlying ectoderm inhibits cardiac induction in the region posterior to the cardiac crescent, limiting the cardiogenic region to the anterior lateral plate mesoderm20, 21. Canonical WNT signaling thus has a biphasic effect on cardiac differentiation by enhancing mesoderm formation followed by repression of cardiac fates in mesoderm-specified cell populations21–24. This biphasic role of WNT is evolutionarily conserved. In chick embryos, canonical WNT inhibition by Dickkopf WNT signaling pathway inhibitor 1 (DKK1) and Crescent secreted from the anterior endoderm is necessary for cardiac differentiation, while ectopic expression of canonical WNT ligands WNT3A and WNT8C induces erythroid cell formation in cardiac precursor regions21. Other chemotactic and inductive signals are likely involved in mesoderm migration and differentiation into the cardiac fate; however, their identities remain to be determined.
This understanding of the major signaling pathways that govern mesoderm formation has permitted the development of robust protocols to differentiate pluripotent stem cells into cardiomyocytes (CMs)25, 26. These protocols rely on activation of canonical WNT signaling to induce mesoderm formation, followed by WNT inhibition to stimulate CM differentiation. These protocols are remarkably efficient, often yielding 100 CMs per input pluripotent stem cells, and at purities of over 90%25, 26. Efficient pluripotent stem cell to CM differentiation protocols are fundamental to using pluripotent cell differentiation systems to study cardiac differentiation27, model human heart disease28, and produce CMs as replacement therapy for heart failure29.
Cardiomyogenesis In The First and Second Heart Fields
Signals regulating commitment to cardiac progenitor cells from mesodermal precursors
While the early signals regulating mesoderm induction have been well characterized, the molecular signaling and genetic regulation driving MESP1+ mesoderm precursors to FHF and SHF cardiac progenitors have only been uncovered in recent years. The FHF gives rise to the left ventricular free wall, part of the septum, and a portion of the atria, while the second heart field gives rise to the right ventricle, a portion of the septum, the outflow tracts, and a portion of the atria30. Identifying the signals and molecular markers that regulate the segregation of early mesodermal progenitors to either of these fields is an important step towards understanding cardiac development in vivo as well as developing novel differentiation protocols that can give rise to specific subpopulations within the heart.
During mesodermal cell migration, the earliest cells to reach the anterolateral plate form the FHF while later cells form the SHF17. Recent work from the Mummery lab has shown that modulating FGF and BMP signaling in vitro can direct pluripotent stem cell-derived cardiac progenitors into either FHF- or SHF-like cells, suggesting that differential signaling underlies the differentiation of precursor cells into SHF or FHF lineages31. At E7.5 in mouse development, FHF cells are spatially organized in a crescent-shaped structure, the cardiac crescent. SHF cells lie more medial and dorsal to these FHF cells (Figure 2A). These FHF progenitors receive BMP232, FGF833, and non-canonical WNT34 signals from the underlying endoderm to promote their differentiation. Meanwhile, the SHF progenitors receive FGFs35, sonic hedgehog36, and canonical WNT/β-catenin37 signals to promote their proliferation and multilineage differentiation (Figure 2B). While the signaling pathways described above are important for the multipotency of their respective heart fields, the question remains as to how the MESP1+ mesodermal progenitors are appropriately allocated into FHF or SHF populations, and what factors regulate the size of each heart field.
Figure 2. Regulation of cardiac progenitor proliferation and differentiation.
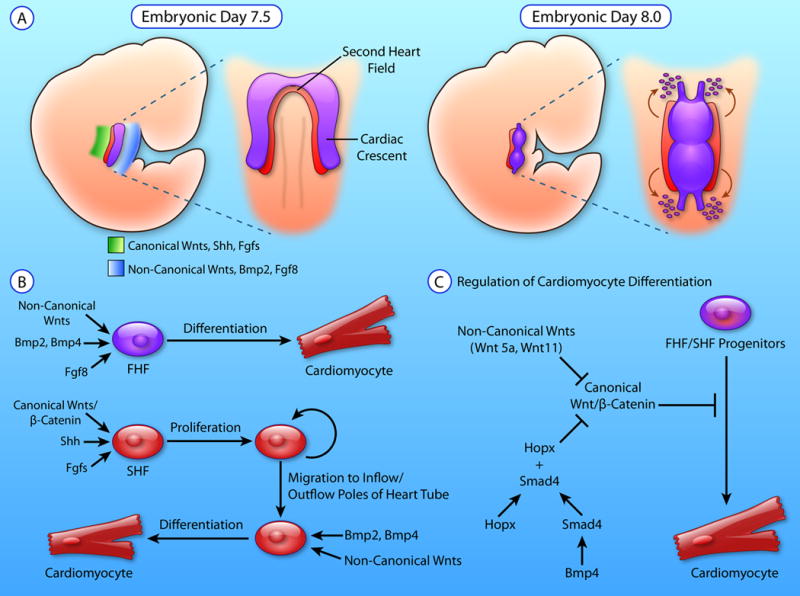
A. Schematic showing the anterolateral position of first heart field (FHF) progenitors and dorsomedial position of second heart field (SHF) progenitors at E7.5. Canonical WNTs, Sonic Hedgehog (SHH), and fibroblast growth factors (FGFs) are expressed dorsally in the region encompassed by the SHF, while non-canonical WNTs, BMP2, and FGF8 are expressed ventrally, where the FHF is present. FHF progenitors make up the cardiac crescent and differentiate prior to the SHF in order to form the developing heart tube at E8.0. SHF maintain their proliferative state and elongate the heart tube by migrating and differentiating at the inflow and outflow poles of the heart. B. Non-canonical WNTs, BMP2/4, and FGF8 signaling drives FHF progenitors to differentiates towards the myocyte lineage. Meanwhile, Canonical WNT/β-catenin, SHH, and FGFs maintain SHF progenitor proliferation. SHF progenitor migration to the outflow and inflow poles of the heart tube exposes them to BMP2/4 and non-canonical WNTs which drives SHF progenitors to exit their proliferative state and differentiate. C) Canonical WNT/ β-catenin signaling inhibits the differentiation of cardiac progenitors to the myocytes. BMP signaling activates SMAD4 which binds to the transcription factor HOPX to directly inhibit Canonical WNT/β-catenin. Moreover, non-canonical WNTs such as WNT5a and WNT11 also inhibit Canonical WNT/β-catenin to drive cardiac progenitor differentiation. (Illustration credit: Ben Smith)
Despite the incomplete understanding of FHF and SHF progenitor cell regulation from their mesodermal precursors, there has been enthusiasm for the generation of renewable cardiac progenitor cells from the application of these signaling pathways for cardiac regeneration. Aside from deriving FHF/SHF cardiac progenitor cells from pluripotent stem cells,31 investigators have recently generated cardiac progenitor cells by direct reprogramming of fibroblasts using cardiac transcription factors and small molecules that activate WNT3A, JAK/STAT, BMP, and TGF-β signaling to regulate the formation and renewal of induced, expandable cardiac progenitor cells.38, 39 Remarkably, when transplanted in vivo into an infarcted heart, these cells differentiated into CMs and improved cardiac function. Further studies will be necessary to reproduce and validate the findings from these studies. Nevertheless, these studies illustrate the potential importance of harnessing early developmental signaling to generate cardiac progenitor cells for therapeutic benefit.
Signaling Pathways Regulating Cardiomyogenesis from First and Second Heart Field Progenitors
The regulation of cardiomyogenesis from multipotent cardiovascular progenitors remains a major focus of ongoing research due to a combination of interest in understanding the mechanisms of cardiac disease as well as an ever-growing interest in generating CM subtypes (e.g. atrial, ventricular, nodal) for therapeutic purposes. BMP2 and BMP4, secreted by underlying endoderm, induce cardiomyogenesis of the overlying lateral plate mesoderm, and exogenous BMP2 and BMP4 induced ectopic cardiac differentiation in chick embryos.32 SHF progenitors switch from a proliferative state toward cardiac differentiation in the setting of increased BMP expression as they migrate to the outflow tract.40, 41 Notably, inhibition of BMP signaling via Noggin resulted in complete lack of cardiac differentiation in chick embryos, highlighting the critical importance of this pathway in heart development.32 The central role of BMP signaling in promoting cardiomyogenesis is in part mediated by the induction of Gata4, Mef2c, Srf and Nkx2-5 expression42, 43. Within SHF, BMP is also required for up-regulation of T-Box2 (Tbx2) and T-Box3 (Tbx3), which are required for maintaining the slow conduction velocity and reduced proliferation of myocardium within the outflow tract, atrioventricular canal, and sinus horns44, 45. Compared to FHF cells, SHF cells have delayed commitment to the CM lineage. Canonical WNT/β-catenin signaling is critical for maintaining SHF progenitors in a proliferative state while inhibiting differentiation towards terminal lineages.46–48 In developing mouse hearts, as SHF progenitors migrate into the developing outflow tract, canonical Wnt signaling is significantly downregulated, coinciding with activation of CM specific gene expression.41, 49
Non-canonical WNT signaling, through calcium-dependent pathways involving protein kinase C and calmodulin-dependent kinase, or through so-called planar cell polarity pathways involving Rho-associated kinase 2 (ROCK2) and Jun amino-terminal kinase, is crucial for normal CM specification.50 Two non-canonical WNT ligands, WNT5A and WNT11, are essential for heart development across multiple species. WNT11 was necessary for expression of cardiac genes and induced ectopic heart formation in both chick and frog embryos34, 51. Developing mouse embryos lacking both Wnt5a and Wnt11 showed a dramatic reduction in SHF progenitors. These roles of Wnt5a and Wnt11 are thought to be due in part to the suppressive effect of non-canonical WNT signaling on the canonical WNT/β-catenin pathway.52
Recent work has identified HOPX, a homeodomain-containing transcriptional repressor, as a key link between canonical WNT/β-catenin and BMP signaling pathways during heart development.41 HOPX expression initiates in FHF and SHF derivatives that are exclusively committed to the CM lineage. Notably, Hopx expression in FHF occurs earlier than its expression in the SHF, consistent FHF preceding SHF differentiation. Using an embryoid body directed differentiation system, it was further shown that Hopx promotes CM differentiation via inhibition of WNT signaling (Figure 2C). This inhibition is mediated by direct interaction between HOPX and SMAD4, a transcription factor that is essential for transducing BMP signals. These data suggest that as SHF cells migrate into the outflow tract, they are exposed to increased BMP4 signaling, which cooperates with newly expressed HOPX to diminish canonical WNT/β-catenin signaling and thereby promote CM differentiation.41
Transcriptional Events During Cardiomyogenic Commitment
The signaling pathways described above act in a coordinated fashion with a complex network of cardiac transcription factors to regulate cardiomyogenesis. The cardiac genetic program is initiated during the MESP1+ pre-cardiac mesoderm stage15. As MESP1+ pre-cardiac progenitors march toward the anterolateral plate mesoderm, a subunit of the SWI/SNF chromatin remodeling complex, SMARCD3, is expressed after MESP1 is downregulated and before upregulation of cardiac progenitor markers NKX2-5 and ISL homeobox 1 (ISL1)53. SMARCD3 allows the zinc-finger transcription factor GATA4 to bind the enhancer regions of several transcription factors that initiate the cardiac gene program54 including GATA4, NKX2-5, ISL1, T-box 5 (TBX5), and myocyte enhancer factor 2c (MEF2C)53, 55. Along with GATA4, yin yang 1 (YY1) has been shown to bind directly to a cardiac enhancer region of NKX2-5 and plays a vital role in its early cardiac transcriptional activation56. The central roles of GATA4 and NKX2-5 in early cardiac development are highlighted by the observations that GATA4/6 double mutants lack hearts entirely, and NKX2-5 is the most frequently mutated gene in congenital heart disease patients57, 58.
FHF progenitors are the earliest cells to express NKX2-5 and constitute the earliest wave of developing cardiac progenitors. Early FHF progenitors quickly activate TBX5, which interacts with GATA4 and NKX2-5 to drive cardiac muscle development and specification of the left ventricle through induction of many cardiac genes, including natriuretic peptide A (Nppa) and the gap junction protein connexin 40 (Gja5)59, 60. TBX5 misexpression can lead to the improper positioning of the interventricular septum or the complete absence of the septum60. GATA4 and NKX2-5 repress the hemangiogenic gene program, through suppression of the hematopoietic transcription factor GATA1, and upregulate cardiac specific genes including Hand1, Mef2c, myosin light chain-2v (Myl2, also known as Mlc2v), and other genes necessary for cardiomyocyte structure and function61–63. Study of FHF progenitors has been complicated by their transience and by a paucity of well-established markers. One of the few markers for FHF progenitors is hyperpolarization-activated cyclic nucleotide gated potassium channel 4 (HCN4)64, 65, which is initially expressed in the cardiac crescent and gradually becomes confined to the cardiac conduction system.
SHF progenitors persist after the formation of the heart tube66. These cells, located at the tube’s arterial and venous poles, migrate and differentiate into CMs, endothelial cells, and smooth muscle cells67, 68, thereby contributing to the growth of the heart tube, and formation of the inflow and outflow tracts. SHF progenitor cells are marked by sustained expression of ISL166, 67, although the FHF also transiently expresses this transcription factor69. ISL1 knockout mice exhibit severe defects in the looping of the heart, and the development of the right ventricle and outflow tract. ISL1 activates FGF and BMP genes, which play important roles in cardiac progenitor proliferation and differentiation as discussed above66. Moreover, ISL1 cooperates with GATA4 to activate Mef2c, which activates the expression of the basic-helix-loop-helix transcription factor HAND270–72. HAND2 is an essential transcription factor that regulates right ventricular development, as seen by Hand2 knockouts that display varying degrees of right ventricular hypoplasia73. Moreover, Hand2 is required for the survival and proliferation of SHF progenitors73. As SHF progenitors differentiate, Nkx2-5 is activated and directly represses ISL1 to limit progenitor proliferation and promote progenitor differentiation74. Interestingly, a recent study has shown that the direct repression of Isl1 by NKX2-5 is required for the proper development of ventricular cardiomyocytes75. In contrast, this same study showed that overexpression of ISL1 in mouse ESCs enhanced specification of cardiac progenitors and led to increased nodal type cardiomyocytes relative to ventricular subtypes, thus demonstrating complex regulatory feedback between Isl1 and Nkx2-5 in cardiac progenitor specification and differentiation. The role of these transcription factors in regulating cardiac progenitor proliferation and differentiation continues to be a point of investigation. Understanding the complex interactions that regulate cardiac progenitor proliferation and differentiation is vital for understanding the etiology of congenital heart defects and for applications of cardiac regeneration to congenital and acquired heart disease.
Role of Micro RNAs in Cardiac Differentiation
Micro-RNAs (miRNAs) are single-stranded, non-coding RNA molecules that negatively affect gene expression at the post-transcriptional level, either by guiding mRNA degradation or by preventing protein translation.76, 77 DICER is an RNase that is critical for cytoplasmic processing of pre-miRNAs into mature miRNAs, which will subsequently be incorporated into the RNA-induced silencing complex. Selective knock-out of Dicer in murine cardiac precursors using Nkx2-5Cre resulted in embryonic lethality due to dilated cardiomyopathy, ventricular hypoplasia, and heart failure by E12.578. However, when Dicer was knocked out in CMs via myosin heavy chain 6 (Myh6)-Cre, which initiates recombination later in heart development, the phenotype was milder, as mice survived to birth but died soon after from dilated cardiomyopathy and heart failure.79 These studies demonstrate the critical importance of miRNAs in cardiac differentiation and morphogenesis.
Several specific miRNAs have been shown to play key roles in cardiac development. The miR-1 and miR-133 families are co-transcribed as miR-1-1/miR-133a-2 and miR-1-2/miR-133a-1, and are expressed in the developing heart as well as skeletal muscle.76, 80 In the developing heart, miR-1 and miR-133 family members are regulated by serum response factor (SRF) and MEF2 transcription factors, which are intricately involved in myocyte differentiation.81, 82 Homozygous deletion of miR-1-2 resulted in a wide range of severe cardiac defects that lead to embryonic or perinatal lethality, including ventricular septal defect, heart failure, and dysrhythmias.78 Notably, driving over-expression of miR-1 from the myosin heavy chain 7 (Myh7) promoter, active in fetal CMs, resulted in thinning of the ventricular wall and heart failure.78 Intriguingly, double miR-1-1 and miR-1-2 knockout caused predominantly postnatal lethality prior to weaning as a result of severe cardiac dysfunction.83 While the milder phenotype in these double knockout mice compare with miR-1-2 single knockout requires further clarification, it appears that miR-1 plays a key role in modulating the relative proliferation and differentiation of cardiac precursors. Similar to miR-1-2 deletion, mice lacking both miR-133a-1 and miR-133a-2 developed severe heart failure due to ventricular septal defects and dilated cardiomyopathy.84 Interestingly, these mutants also showed ectopic expression of smooth muscle genes, pointing to a role for miR-133a in mediating the lineage decision between cardiac and smooth muscle. Deletion of individual miR-1-1/miR-133a-2 or miR-1-2/miR-133a-1 gene clusters did not affect survival to birth, cardiac morphogenesis, or function, suggesting functional redundancy85. However, double knock-out of both miR-1-1/miR-133a-2 and miR-1-2/miR-133a-1 caused embryonic lethality at E11.5 with marked thinning of the compact ventricular myocardium and impaired cardiomyocyte maturation and proliferation. This effect is, in part, mediated through loss of suppression of the transcription factor Myocardin, leading to persistent expression of smooth muscle genes and incomplete cardiomyocyte maturation.
Fibroblast to CM reprogramming
One exciting application of our knowledge of the factors that govern CM specification and differentiation is direct reprogramming of non-myocytes such as fibroblasts to CM-like cells (induced CMs or iCMs). This can be achieved by transduction of non-myocytes with a cocktail of transcriptional regulators that promote myocardial transdifferentiation. While this strategy is still in its infancy, results have been promising. Multiple groups have reported direct reprograming via transduction of fibroblasts with retrovirus expressing transcription factor cocktails.86–89 The most commonly used cocktails have used GATA4, MEF2C, and TBX587, with some reports indicating that HAND2 enhances reprogramming90. The combination of MESP1 and ETS2 has also been reported to successfully reprogram human fibroblasts to cardiac progenitors91. An additional recent study utilizing a cocktail of four miRNAs also reported direct reprogramming of cardiac fibroblasts to CMs92. A range of phenotypes were observed in iCMs, with some cells demonstrating contractile and calcium handling characteristics of CMs. However, the robustness of reprogramming in vitro, and the extent to which in vitro iCMs express complex physiological phenotypes characteristic of bona fide CMs requires further improvement93. The efficiency of non-myocyte to iCM reprogramming, and the extent to which iCMs can be induced to attain functional properties of mature bona fide CMs (see sections on CM maturation, below) will determine whether this exciting concept ultimately will bear fruit.
Regulation of CM proliferation
CM proliferation during embryonic heart development
After cardiac progenitor cells differentiate into CMs, new CMs are generated through division of existing CMs, and this expansion of CM number primarily accounts for embryonic heart growth. Early events of CM proliferation have been studied through lineage tracing experiments and clonal analyses. One of the earliest CM clonal analyses was performed in chicken embryos94, using a low dose of retrovirus to genetically label a small fraction of CMs and cardiac progenitor cells. Later on, these labeled cells were observed to form clusters and the number of cells per cluster increased during heart development. This result suggested a coherent expansion of CMs through their local proliferation. More precise labeling and tracing experiments done in mouse embryos showed that coherent CM colonies derived from single CMs could be found at E8.5, when heart tube looping initiates95. This clonal expansion of CMs during heart growth was further demonstrated in zebrafish. In elegant studies that took advantage of the ease with which the developing zebrafish heart can be visualized, stochastic CM labeling by combinations of multiple fluorescent proteins permitted detailed clonal analysis of heart growth96. Proliferation and expansion of as little as ~55 CMs during early development was sufficient to generate all of the CMs of the ventricular myocardium96.
A number of signaling pathways precisely control CM proliferation to regulate heart growth and morphogenesis. Among the best characterized is the Hippo/YAP signaling pathway, which controls the size of several organs during development97, 98. Elevated cell density signals through incompletely understood upstream pathways to activate Hippo pathway kinases (MST1/2 and LATS1/2), which put the brakes on CM proliferation by phosphorylating and inactivating the transcriptional co-activators YAP and TAZ (formally known as WWTR1), partially redundant drivers of cell proliferation99, 100. Hippo/YAP regulation of CM proliferation and heart growth has been well characterized using genetic mouse models to manipulate pathway components. Inactivation of Salvador (Sav1), encoding a scaffold protein required for Hippo kinase activity, in CMs increased CM proliferation, resulting in overgrowth of the fetal ventricular walls and trabeculae101. Forced expression of constitutively active YAP in CMs also strongly stimulated their proliferation, while YAP/TAZ loss-of-function in CMs reduced their proliferation, causing embryonic lethal cardiac hypoplasia and hypotrabeculation102–104. YAP functions by interacting with transcription factor TEAD1105, 106, which activates downstream mitogenic pathways, including the PI3K-AKT pathway107, 108. YAP could also interact with β-catenin and directly modify WNT signaling to upregulate CM proliferation101. Thus the Hippo/YAP pathway is an essential regulator of CM proliferation that matches cardiac growth to physiological needs of the fetus.
Proper spatiotemporal control of CM proliferation is required for normal cardiac morphogenesis. CMs in the compact myocardium proliferate more rapidly than trabecular CMs. Differences in Hippo/YAP signaling may contribute to this regional difference in CM proliferation, since relieving Hippo inhibition or increasing YAP activity abrogates the difference and causes dramatic trabecular myocardial hyperplasia101, 103. Gradients of mitogenic factors may also contribute to more rapid proliferation of CMs in the compact myocardium. The epicardium, an epithelial sheet that covers the outer surface of the heart, is essential for growth of the compact myocardium, as disrupting epicardium perturbs the proliferation of underlying myocardium109. Recent studies showed that epicardium secretes IGF2, which activates IGF1R and subsequently ERK in CMs to induce proliferation110, 111. Follistatin-like 1 (FSTL1) is another epicardially-secreted cardiac mitogen112, although the importance of FSTL1 for cardiac development remains to be determined. Gradients of environmental stimuli, such as oxygen or blood flow, also regulate regional CM proliferation rates. Nuclear localization of hypoxia inducible factor 1 alpha (HIF1A), which is governed by oxygen tension, was enriched in CMs within the outer compact myocardium and interventricular septum, where it was required for robust CM proliferation113.
Another heart morphogenic process that is tightly linked to CM proliferation is trabeculation (Figure 3). Trabeculae are ridge-shaped myocardial protrusions that are derived from the sub-endocardial myocardium. Although the key mechanistic events are still being elucidated, in mouse the process of trabeculation is thought to be initiated when a subset of sub-endocardial CMs alter their polarity from parallel to perpendicular relative to the chamber wall114. These CMs then proliferate and expand to form the trabeculae115, 116, 117. Rodent models show that CM proliferation during trabeculation is tightly controlled by communication between myocardium and trabecular endocardium, which involves a complex signaling pathway composed of NOTCH1, BMP10, ephrin B2 (EFNB2), HAND2, and Neuregulin-1 (NRG1)115–118. Zebrafish based studies of trabecular development support crucial roles for Notch and Neuregulin signaling, however, the expression profiles and presumed functions of individual pathway components appears to be markedly different from rodent models96, 119, 120;Han, 2016 #139}. Furthermore, trabeculation in zebrafish initiates through depolarization and delamination of a subset of CMs rather than altered polar orientation as is thought to occur in mouse121.
Figure 3. Model of the trabeculation process.
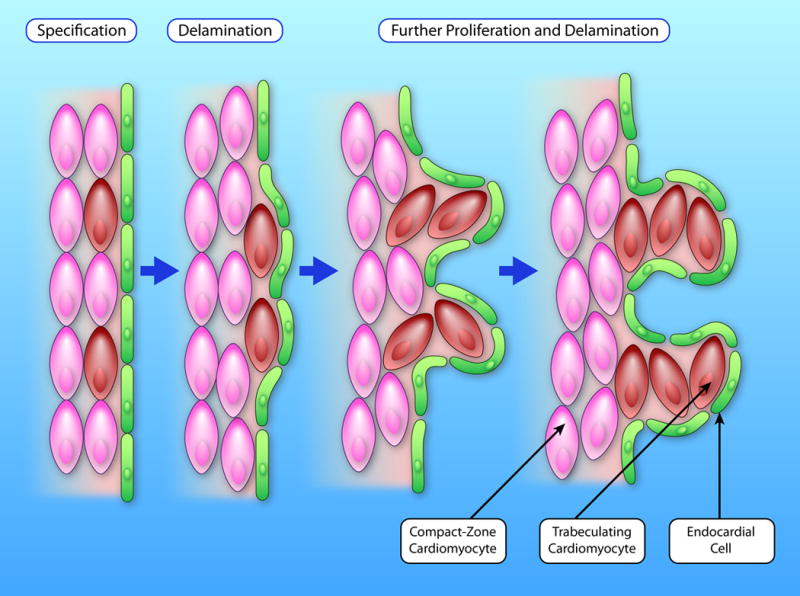
During trabeulation, a small fraction of CMs in the compact myocardium (pink) are first specified as trabeculating CMs (brown). These cells delaminate from the compact myocardium and migrate inward to form the first trabecular CMs. CMs in both compacted and trabecular myocardium further proliferate. This proliferation, together with CM migration and rearrangement, results in protrusion and expansion of the trabecular myocardium. (Illustration Credit: Ben Smith)
CM cell cycle exit during postnatal development
While CM proliferation is responsible for fetal heart growth, shortly after birth mammalian CMs largely exit the cell cycle. In adult hearts, there is measurable albeit very limited CM turnover122–125. By taking advantage of the spike in atmospheric C14 that accompanied above ground nuclear bomb tests, Bergmann et al. showed that human CM turnover rate is about 1% per year at 20 years-of-age and gradually decreases with advancing age122, 125. This estimate is similar to the 0.76% annual turnover rate measured in adult mouse heart by stable isotope labeling coupled with imaging mass spectrometry and genetic fate mapping123. CM proliferation extends into the first postnatal week in mice126, 127. In humans, this postnatal proliferative window may last for several years128, suggesting a potential window in infancy for therapeutic regenerative approaches in congenital heart disease. Several factors that trigger and enforce adult CM cell cycle exit have been identified. Mitogenic signaling pathways that drive fetal CM proliferation are attenuated postnatally. For example, expression of ERBB2 quickly decreases in CMs after birth, which reduces the mitogenic potency of NRG1129. The mitogenic activity of YAP is likewise restrained by multiple mechanisms, including activity of Hippo pathway kinases101, downregulation of YAP and TEAD1, and TEAD1 sequestration by the protein VGLL4105. In addition to loss of pro-mitotic signals, several mechanisms actively inhibit expression and function of the cell cycle machinery in adult CMs. Transcriptional regulators such as MEIS homeobox 1 (MEIS1)130 and retinoblastoma protein (RB)131, 132, epigenetic modifiers such as polycomb repressive complex 2 (PRC2)133, and microRNAs such as the miR-15 family134 actively repress core cell-cycle activators and/or activate cell-cycle inhibitors. P38 MAP kinase signaling also inhibits CM proliferation135, 136. Increased oxidative stress in post-mitotic CMs has been implicated in causing DNA damage response-mediated cell-cycle arrest137. Downregulation of telomerase activity has been shown to block proliferation in a p21-dependent manner138, 139. Disassembly of centrosomes, a hub of mitogenic signaling140 as well as the organizing center for mitotic spindle assembly141, 142, in postnatal CMs may add another layer of negative regulation of CM proliferation143.
Heart regeneration through CM proliferation
Heart injury such as myocardial infarction causes massive CM death. In response, the rate of CM proliferation increases in the peri-infarct region by about 5-fold above its very low basal rate123. However, this level of innate cardiac regeneration is far too limited to effectively replace the lost CMs. As a result, the remaining CMs become overburdened and experience chronically elevated biomechanical stress that induces further CM loss, precipitating a vicious cycle that ultimately leads to heart failure and pathological heart remodeling144, 145. Interrupting or reversing this process requires replacing or regenerating the lost CMs.
To gain insights into how heart regeneration could be therapeutically enhanced, cardiac biologists have studied models competent in effective heart regeneration. Several species including fish and amphibians retain heart regenerative capacity throughout life144, 146–148. A key finding from the zebrafish system is that CMs lost in heart injury models are replaced by proliferation of pre-existing CMs, rather than by generation of CMs from non-myocytes149, 150. Enhanced CM proliferation rate is observed in zebrafish hearts following several different types of injuries such as resection and genetic ablation149–151, indicating a robust injury response mechanism is present in zebrafish that promotes CM proliferation. Regenerating CMs had unique morphological and molecular signatures consistent with sarcomere disassembly, which has been described as CM “dedifferentiation” (Figure 4). Sarcomere disassembly has been inferred to be a pre-requisite for productive mitosis and cytokinesis149, 150. Thus CM proliferation provides the basis of heart regeneration in zebrafish.
Figure 4. Model of cardiomyocyte regeneration.
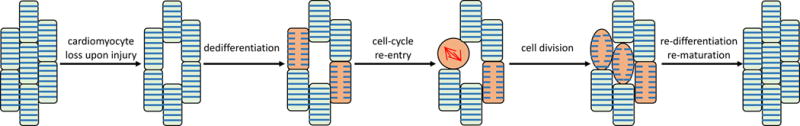
During heart regeneration, myocardial injury and CM loss induce the dedifferentiation of a fraction of CMs (pink). These CMs reenter the cell cycle, produce new CMs, and replenish the lost CMs. CMs that are generated from cell division re-differentiate to a fully mature state to improve heart contraction.
CM proliferation also compensates for the damage or loss of CMs in fetal and neonatal mice. At fetal stages, CM-specific mosaic knockout of Hccs, an X-linked gene that is essential for mitochondrial function, damages ~50% CMs in E10.5 heterozygous females due to random X chromosome inactivation. However, this perturbation does not cause embryonic lethality because the healthy CMs proliferate to compensate for the injury. Consequently, only ~10% CMs are Hccs deficient at birth and over 85% of these animals develop and survive normally152. Similarly, we used diphtheria toxin A to ablate various fractions of CMs in embryonic hearts. We found that embryos tolerated loss of up to 60% of CMs and maintain normal heart development by upregulating the proliferation of remaining unablated CMs153.
Murine cardiac regeneration capacity remains robust into the first postnatal week of life, as hearts injured by apical resection on postnatal day 1 efficiently replenish the lost CMs, resulting in hearts with minimal residual scar and with normal heart morphology and function by 1 month of life127, 134. Although multiple groups have reproduced this result154, confirmation has not been uniform155, possibly because the extent of scarring and regeneration depends on surgical technique and the extent of myocardial resection.156 Interestingly, macrophages are essential for robust neonatal cardiac regeneration157. Lineage tracing showed that the proliferation of preexisting CMs also underlies murine heart regeneration123, 127. CM “dedifferentiation”, characterized by sarcomere disassembly, has also been implicated in mammalian CM proliferation129, 134, 158–160 (Figure 4), although it has not yet been observed directly during in vivo mammalian CM proliferation. Thus induction of CM proliferation is a conserved mechanism of heart regeneration from zebrafish to mammals.
Enhancing cardiac regeneration by driving CM proliferation
The induction of CM proliferation as the major mechanism for heart regeneration in lower vertebrates and fetal and neonatal mice provides a rational foundation for current efforts to augment CM proliferation in mature mammalian hearts to achieve therapeutic cardiac regeneration after heart injury. Developmental CM cell cycle exit is accompanied by the silencing of fetal pathways that drive proliferation; thus reactivating and augmenting these fetal pathways is an attractive strategy to stimulate the adult CM proliferation. Although many strategies are currently being pursued, due to space constraints here we will focus on NRG1 and Hippo/YAP. We refer readers to other excellent reviews that cover other efforts to stimulate CM proliferation161–164.
Several studies have shown that activating NRG1/ERBB signaling boosts proliferation of adult CMs and improves heart repair upon injury. In zebrafish, overexpressing NRG1 in CMs was sufficient to stimulate CM proliferation, resulting in accelerated myocardial expansion after injury165, 166. NRG1 treatment stimulated proliferation of in vitro cultured neonatal and adult murine CMs159, 167, as well as CMs from myocardium removed from pediatric patients at the time of heart surgery167. NRG1 delivery to mice, or over-expression of a constitutive active form of ErbB2, improved heart structure and function after myocardium infarction129, 159. In patients with stable chronic heart failure, NRG1 infusion has been shown to be safe and to have beneficial haemodynamic effects168. Although several studies have suggested that NRG1’s therapeutic effect is due in part to stimulation of adult CM proliferation, other studies have demonstrated that NRG1’s CM mitogenic activity quickly diminishes after birth129, 167. Indeed, one group could not detect a mitogenic effect of NRG1 on adult CMs and assigned its salutary effects to other mechanisms169.
Because YAP has robust CM mitogenic activity during fetal heart development, elevating YAP activity in CMs is another potential approach to stimulate CM proliferation and heart regeneration. Forced expression of constitutively active YAP in CMs stimulated CM proliferation in both neonatal and adult hearts102, 104. In the neonatal murine heart regeneration model, abolishing YAP activity disrupted the regenerative capacity of the neonatal murine heart, whereas YAP overexpression enhanced it163. A study that used adeno-associated virus, a promising gene therapy vector, to selectively overexpress activated YAP in adult CMs, provided proof-of-concept that this treatment can improve myocardial function and survival after myocardial infarction102. Importantly, YAP activation did not have deleterious effects on the heart. Thus YAP activation has therapeutic potential to stimulate heart repair and regeneration.
It is worth noting that NRG1 and YAP both play additional biological functions other than regulating CM proliferation, including CM survival170, migration119, 171 and calcium handling172. NRG1 also acts on non-myocytes to promote neo-vascularization after heart injury165. YAP regulates CM survival107, 173 and transcriptional responses to mechanical stress174, 175. In CMs, YAP promotes actin cytoskeleton remodeling and the formation of CM protrusions at heart injury sites176. Recently, YAP was also implicated in controlling CM oxidative stress177. Thus the beneficial effects of NRG1 and YAP activation in injured hearts are likely to be multifactorial.
Although stimulating CM proliferation is a promising strategy to boost myocardial regeneration in adult hearts, multiple obstacles need to be surmounted before this strategy can be used in clinical applications. First, induction of CM proliferation remains inefficient in adult hearts, likely due to multiple layers of negative regulation that block mature CM cell cycle activity. Thus, a critical task in future research is to precisely define the signals that trigger and maintain cell-cycle withdrawal in adult CMs and design methods to remove or circumvent these obstacles. A second challenge is the potential for increased cancer risk due to cell cycle activation in non-CMs. For example, both NRG1 and Yap signals have oncogenic potentials and are known to be involved in cancers178, 179. Precise targeting of mitogenic stimuli to CMs is necessary to minimize oncogenic risk.
The “dedifferentiation” of regenerating CMs has been observed in both zebrafish and mouse. This process, marked by sarcomere disassembly, occurs during regeneration in response to heart injury127, 134, 149 as well as during NRG1-stimulated myocardial regeneration129, 165. These data imply the reversing CM differentiation state might stimulate CM proliferation, a strategy which could potentially be more efficient and less risky than direct manipulation of cell cycle regulators. However, the merits of this strategy are unknown and depend upon better understanding the “dedifferentiation” process. Although sarcomere disassembly has been suggested as a prerequisite for completion of CM mitosis158, 180, it is unclear whether the “dedifferentiation” observed during regeneration is a true reversal of differentiation state or simply a change of cytoskeletal architecture of CMs undergoing mitosis. Second, whether dedifferentiation will necessarily enhance proliferation is also unclear. Although correlation between dedifferentiation and the increase of CM proliferation has been well documented127, 129, 134, 149, 165, the causal relationship between these two processes has not been established.
How newly regenerated CMs re-establish the morphology of myocardium damaged by injury is also an open question. During heart regeneration in adult zebrafish and fetal and neonatal mice, regenerated heart walls largely retain the morphology of the original structures96, 127, 153. Given that CM proliferation plays an essential role in morphogenesis during development, it is hoped that the proliferation of regenerative CMs follows similar rules to rebuild the adult myocardium. Understanding these rules and their underlying molecular mechanisms will be important to properly enhance heart regeneration by CM proliferation, or to properly sculpt myocardium regenerated by other means (e.g. by delivery of stem cell-derived CMs).
CM maturation
Hallmarks of CM maturation
The dynamic process of heart development includes dramatic alterations in CM metabolism, form, and function, particularly in the neonatal period (Figure 5; Table 1). Fetal CMs actively proliferate, and the resulting increase in CM number largely accounts for fetal heart growth. These cells are adapted to the hypoxic intrauterine environment, as they rely primarily on glycolytic metabolism. At birth, CMs undergo dramatic changes linked to greater demand for left ventricular pump function and greater availability of oxygen. Postnatal CMs become highly specialized for efficient contraction: they shift to oxidative phosphorylation as their primary energy source, increase in volume by 30-40-fold between birth and adulthood, and develop accompanying ultrastructural specializations and changes in gene expression that enable efficient and coordinated CM contraction. Perhaps intimately linked with specialization for efficient contraction, postnatal CMs largely exit the cell cycle and become predominantly polyploid (a single polyploid nucleus in humans, or two diploid nuclei in rodents).
Figure 5. Representative cellular events during CM maturation.
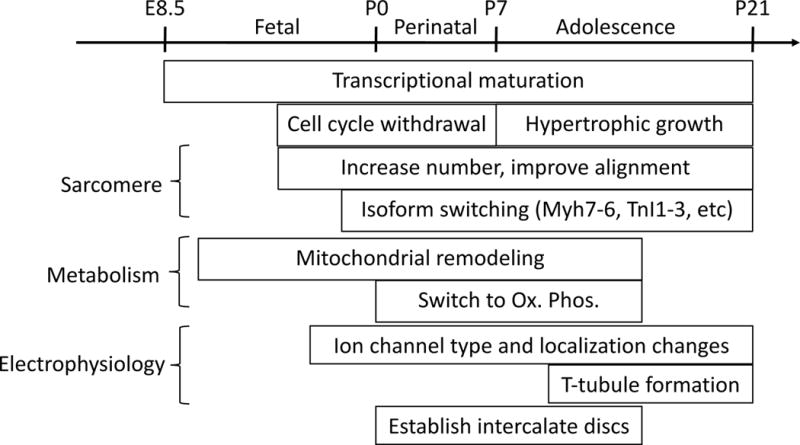
During CM maturation, transcriptional changes occur throughout both embryonic and postnatal development. CMs stop hyperplastic growth at the perinatal stage and transition to hypertrophic growth, in which sarcomere number increases and sarcomere alignment improves. Altered expression of critical sarcomere isoforms modifies the contractile properties of CMs. Mitochondria increase in number and size and become well organized with respect to sarcomeres. After birth, CMs switch from deriving most of their energy from glycolysis to depending upon oxidative phosphorylation. The maturation of electrophysiologic properties is characterized by proper expression and localization of ion channels characteristic of adult CMs. T-tubules form at a late stage of postnatal development, perhaps to permit rapid AP penetration with rapidly enlarging CMs. Maturation is also characterized by the postnatal establishment of intercalated discs and proper formation of cell-cell contacts.
Table 1.
Hallmarks of CM Maturation.
| Phenotype | Immature (late fetal) | Mature (Adult) | PSC-CMs | Regulators | References |
|---|---|---|---|---|---|
| Cell Size | small | large | small | GATA4/6 | 185, 210 |
| Cell Shape | In situ: rod-shaped (4:1::L:W); cultured: stellate | In situ and dissociated: rod-shaped (7:1::L:W) | irregular/round | ECM composition and patterning, mechanical load, ERK1/2 signaling | 184, 186, 190, 224 |
| Sarcomere Alignment | organized | superbly organized | disorganized | cell shape, mechanical load, substrate stiffness | 190, 216, 217 |
| Sarcomere Components | TNNI1, MYH7, TTN-N2BA, EH-MYOM1,TNNT2 exon 5 inclusion | TNNI3, MYH6, TTN-N2B, TNNT2 exon 5 exclusion | variable | Thyroid hormone; maturation related transcriptional programs are currently not well understood | 191–195, 221 |
| Metabolism | glycolysis | oxidative phosphorylation | Glycolysis | PPAR signaling, greater oxygen availability after birth | 200, 205, 208 |
| Mitochondria | low number, small and round | high number, large and ovoid, close proximity to myofibers | ovoid, clustered around nucleus and cell periphery | AMPK, PPAR signaling, Mitofusins 1 and 2 | 201–203 |
| Proliferative Capacity | high | very low | can proliferate for ~1month after contraction begins | NRG1/ERBB2/4, YAP/TEAD, IGF2/IGF1R/PI3K/ERK, BMP10, WNT/β-catenin, NOTCH | 101, 103, 110, 115, 116, 159 |
| Nucleus | mononucleated | multinucleated or polyploid | mononucleated | Proliferation regulators above | 101, 103, 110, 115, 116, 159 |
| T-Tubules | none | extensive network | none | JPH2, BIN1 | 198, 199 |
While much has been learned about the fetal specification of CMs, much less is known about how CM maturation is coordinately regulated. Understanding CM maturation is critical to advance the field of cardiac regeneration, since any new source of CMs must be sufficiently mature to effectively contribute to cardiac function and to seamlessly electrically couple with pre-existing myocardium. Indeed, a recent landmark study demonstrated that while significant numbers of human pluripotent stem cell derived CMs (PSC-CMs) could be engrafted in infarcted primate hearts, all treated hearts exhibited ventricular arrhythmias that were attributed to insufficient maturity of donor CMs29. Immaturity of stem-cell derived CMs is also a major barrier to their use for in vitro modeling of human heart disease.181–183 Thus discovering ways to induce CM maturation has become a major goal in the field. Progress will require developing a detailed understanding of the normal processes that drive developmental CM maturation. Here we review key aspects of postnatal CM maturation, survey current strategies to mature PSC-CMs, and recommend directions for future study.
Structural remodeling during postnatal physiological maturation is extensive and affects virtually all aspects of CM cytoarchitecture. However, three prominent hallmarks stand out: an increase in CM size leading to formation of large rod shaped cells with high length:width aspect ratio, a higher myofibrillar density with increased sarcomere prominence, and formation of T-tubules. The molecular mechanisms that control these morphological and structural changes are incompletely understood. The length and diameter of CMs appear to be regulated by distinct types of mechanical loads, with CM elongation occurring through addition of new sarcomere units at the cell poles in response to diastolic strain, and CM girth increasing through parallel addition of filaments in response to systolic strain.184, 185 In the disease state this process is mediated by ERK1/2 signaling, which likely also plays important roles in maturation-related remodeling.186 These mechanisms are influenced by substrate stiffness, which together with additional cues make the ECM an important determinant of CM shape.187–189
Myofibrillar density and organization directly correlate with CM maturation status, with fully mature CMs having superbly organized sarcomeres. Sarcomere organization is intimately linked to cell shape, as patterning CMs with micro-contact printing to induce the formation of different cell geometries directly impacts sarcomere alignment, with rectangular shape and higher length to width ratios driving increased organization.190 This increase in sarcomeric organization and prominence corresponds to alterations in the expression ratio of many crucial sarcomere protein isoforms. In mouse, this transition includes shifts in gene transcription from fetal to adult isoforms (e.g. troponin I1 (TNNI1) to troponin I3 (TNNI3); Myh7 to Myh6) and altered gene splicing (e.g. titin (TTN)-N2BA to TTN-N2B; myomesin 1 embryonic isoform (EH-MYOM1) to mature isoform; troponin T2 (TNNT2) exon 5 inclusion to exclusion).191–195 T-tubules, invaginations of the plasma membrane that allow membrane depolarization to quickly penetrate to the CM interior, are a hallmark of mature CMs. Development of the murine T-tubule network initiates at approximately two weeks after birth, and similar to sarcomere development gradually becomes better defined and organized as the CMs mature to adulthood.196, 197 Currently, besides junctophilin 2 and Bin 1 (Table 1), few proteins involved in T-tubule formation have been identified, and the process is poorly understood.198, 199
In addition to structural remodeling during maturation, neonatal CMs undergo dramatic alterations in metabolism, shifting from predominantly non-oxidative to oxidative metabolism in the neonatal period.200 This metabolic transition coincides with an increase in mitochondrial density, as well as a mitochondrial shift in shape from small and round to large and ovoid.201, 202 Positional differences are also observed, with mitochondria of mature CMs being located in closer proximity with myofibrils and the sarcoplasmic reticulum.203 While distinguishing cause and effect relationships from associations has been difficult, there is mounting evidence that metabolic remodeling is a key driver of cytoskeletal and sarcomeric maturation.204–207
Many aspects of the structural and metabolic remodeling that takes place during CM maturation have been appreciated for some time. However, understanding the regulatory networks driving these changes has been more challenging. External signals influencing maturation include neurohormonal factors and mechanical stimuli related to hemodynamic load, force generation, and ECM composition. These external signals activate internal cell signaling pathways, which in turn activate transcriptional regulators to orchestrate global changes in gene expression. One recent study that analyzed hundreds of micro-array datasets identified stage specific gene regulatory networks,205 and future studies will determine if major nodes within these networks act as regulators of maturation. One strong theme of the analysis was the presence of multiple networks related to metabolism, with PPAR signaling pathways constituting central components. Indeed, PPAR signaling acts as an activator of fatty acid oxidation, and pathway activation increases as maturation proceeds.208 While this study succeeded in describing how the CM gene expression profile changes during maturation, proposed transcriptional regulators have not been functionally validated in vivo. This is a very difficult task, as global CM specific overexpression or ablation of important transcriptional regulators inevitably results in cardiac stress or failure, which confounds efforts to gauge the effect on maturation. Indeed, due to this complication many maturation investigations have been limited to the study of neonatal rat CMs or PSC-CMs in an artificial culture environment. While this line of study has yielded some notable advances, such as the identification of Let-7 miRNA family members as positive regulators of maturation,209 the field continues to be hampered by insufficient knowledge of maturational regulation in vivo. Our recent study of the postnatal roles of transcription factors GATA4 and GATA6 offers clues as to how regulation of CM maturation can be dissected in vivo.210 Mosaic gene knockout, achieved by administering a low dose of Cre-expressing adeno-associated virus serotype 9 that only transduced a fraction of CMs, enabled the study of mutant CMs in functionally healthy hearts. In this context, GATA4/6 double knockout CMs were dramatically smaller and less mature than control CMs, indicating that GATA4 and GATA6 are crucial cell autonomous regulators of postnatal cardiomyocyte growth and maturation. Combining this mosaic gene manipulation strategy with single cell RNA-seq and emerging technologies such as somatic CRISPR/Cas9 mutagenesis211, 212 offers an experimental strategy to dissect the regulatory networks that govern CM maturation.
Enhancing maturation of CMs differentiated from non-myocytes or pluripotent stem cells
Despite limitations in our understanding of the mechanisms driving CM maturation, many approaches to improving PSC-CM maturity have capitalized on knowledge of developmental paradigms. Approaches such as three dimensional tissue engineering, mechanical loading, modulation of substrate stiffness, and electric stimulation have all had varying degrees of success by more closely mimicking the in vivo environment.213–217 Indeed, just as tension-sensing mechanisms are currently generating considerable interest as determinants of how the heart remodels in response to disease,218 similar mechano-sensing signaling pathways have been shown to play important roles in maturation related remodeling.219, 220 In addition, treatment with hormonal factors has also been shown to modulate CM maturation, with thyroid hormone being a major stimulant of fetal CM maturation.221 Finally, long term culture of PSC-CMs has been shown to induce more complete maturation.222 While expression profile analyses of CMs cultured for up to one month show arrest at or before a stage equivalent to the late fetal period,205 reportedly culture for approximately one year results in PSC-CMs with expression profiles similar to adult CMs.209 Although these results are impressive, the inconvenience and cost of the long term culture approach makes it unsuitable as an experimental model system or as a regenerative therapeutic approach. However, combinations of the above strategies are currently being examined, and better understanding of the mechanisms that govern normal CM maturation promises to enable further progress.
Interestingly, CMs differentiated from non-myocytes appear to undergo enhanced maturation within the native milieu of the beating heart. For example, when human PSC-CMs were injected into a non-human primate model of myocardial infarction29. The injected PSC-CMs engrafted and over three months they were observed to have increased myofibril alignment and sarcomere registration. The engrafted PSC-CMs attained a diameter comparable to the size of normal adult money cardiomyocytes. On the other hand, transient ventricular arrhythmias were common early in the engraftment process and spontaneously resolved. The pro-arrhythmic effect may have resulted from the immaturity and heterogeneous electrical coupling of the engrafting PSC-CMs. Generalizing from this experience, pro-arrhythmia may be a consequence of many regenerative strategies that yield immature CMs, and cardiac regeneration experiments need to be designed to detect these adverse events. A second example is the maturation of iCMs reprogrammed from fibroblasts by viral delivery of cardiac transcription factors (see non-CM to CM reprogramming, above). iCMs created by reprogramming in vitro often are highly immature in size, morphology, and functional parameters such as calcium handling93. When reprogramming was performed in an in vivo context, a significant fraction iCMs properly localized structural maturation markers and had mature electrophysiological and calcium handling phenotypes87, 90. Indeed, while the overall number of in vivo reprogrammed CMs is still limited, recent reprogramming studies showed a surprisingly potent salutary effect on heart function and myocardial outcome in an experimental murine MI model87, 90. It is possible that in vivo reprogramming has additional benefits beyond cell autonomous gains in contractile ability. Translating this exciting work towards clinical applications will require further improvements in reprogramming efficiency and iCM maturation, development of reprogramming platforms that do not require viral integration, evaluation of this strategy’s efficacy in large animal models, and improving efficiency of reprogramming human fibroblasts, which have been more refractory to reprogramming compared to murine cells88, 223. The enhanced maturity achieved by in vivo CM differentiation likely reflects limitations of current in vitro culture conditions and the overall importance of the cell’s environment on CM differentiation and maturation.
Conclusions
By following the lessons of normal development, cardiac biologists have made rapid inroads towards generating cardiomyocytes to make human disease models and to enhance myocardial outcomes in heart injury models. The conceptual focus of most work in therapeutic cardiac regeneration has been ischemic heart disease. However, myocardial failure in congenital heart disease typically results from chronic volume and pressure loads produced by palliated circulations. Greater appreciation of the need for therapeutic cardiac regeneration in congenital heart disease and its specific biology will allow advances to be applied to this rapidly growing patient population. The spectrum of therapeutic options for congenital heart disease could expand through creative uses of regenerative and stem cell biology to engineer heart tissue. Although this review has focused of myocardial regeneration, parallel efforts in engineering valves and vessels are also needed to address the panoply of difficulties faced by patients with palliated congenital heart disease. It is our hope that the ongoing efforts of investigators in developmental and regenerative biology will one day lead to groundbreaking advances in the diagnosis, prevention, and treatment of congenital and acquired heart disease in children and adults.
Supplementary Material
Acknowledgments
Funding Sources
WTP was supported by grants from NIH (HL098166 and HL116461) and by a charitable contribution from Dr. and Mrs. Edwin A. Boger. SMW was supported by the NIH Office of Director’s Pioneer Award LM012179, NIH/NHLBI U01 0099776, the Cardiovascular Institute, the Division of Cardiology, Department of Medicine, the Institute for Stem Cell Biology and Regenerative Medicine, and an endowed faculty scholar award from the Child Health Research Institute/Lucile Packard Foundation for Children at Stanford (S.M.W). SLP was supported by the Department of Pediatrics at Stanford, and FXG was supported by the Stanford Medical Scientist Training Program NIH T32 GM007365. NVD was supported by F32HL13423501.
Non-standard abbreviations and acronyms
- CM
Cardiomyocyte
- E
Embryonic day (days of gestation)
- P
Postnatal day (days after birth)
- FHF
First heart field
- SHF
Second heart field
- BMP
Bone morphogenic protein
- WNT
wingless-type MMTV integration site family member
- FGF
Fibroblast growth factor
- miRNA
Micro-RNA
- PSC-CM
pluripotent stem cell-derived cardiomyocyte
Footnotes
Author Contributions
Co-first authors are listed in alphabetical order. FXG and SLP were primarily responsible for the cardiomyocyte specification and differentiation sections, respectively. YG and NVD were primarily responsible for the cardiomyocyte proliferation and maturation sections, respectively. SMW and WTP contributed equally to organize and edit the manuscript.
Conflict of Interest
None
References
- 1.Arnold SJ, Robertson EJ. Making a commitment: cell lineage allocation and axis patterning in the early mouse embryo. Nature Reviews Molecular Cell Biology. 2009;10:91–103. doi: 10.1038/nrm2618. [DOI] [PubMed] [Google Scholar]
- 2.Brennan J, Lu CC, Norris DP, Rodriguez TA. Nodal signalling in the epiblast patterns the early mouse embryo. Nodal signalling in the epiblast patterns the early mouse embryo. 2001 doi: 10.1038/35082103. [DOI] [PubMed] [Google Scholar]
- 3.Waldrip WR, Bikoff EK, Hoodless PA, Wrana JL, Robertson EJ. Smad2 signaling in extraembryonic tissues determines anterior-posterior polarity of the early mouse embryo. Cell. 1998;92:797–808. doi: 10.1016/s0092-8674(00)81407-5. [DOI] [PubMed] [Google Scholar]
- 4.Perea-Gomez A, Vella F, Shawlot W, Oulad-Abdelghani M, Chazaud C, Meno C, Pfister V, Chen L, Robertson E, Hamada H, Behringer RR, Ang S-L. Nodal Antagonists in the Anterior Visceral Endoderm Prevent the Formation of Multiple Primitive Streaks. Developmental Cell. 2002;3:745–756. doi: 10.1016/s1534-5807(02)00321-0. [DOI] [PubMed] [Google Scholar]
- 5.Conlon FL, Lyons KM, Takaesu N, Barth KS, Kispert A, Herrmann B, Robertson EJ. A primary requirement for nodal in the formation and maintenance of the primitive streak in the mouse. Development (Cambridge, England) 1994;120:1919–1928. doi: 10.1242/dev.120.7.1919. [DOI] [PubMed] [Google Scholar]
- 6.Ciruna B, Rossant J. FGF signaling regulates mesoderm cell fate specification and morphogenetic movement at the primitive streak. Developmental cell. 2001;1:37–49. doi: 10.1016/s1534-5807(01)00017-x. [DOI] [PubMed] [Google Scholar]
- 7.Mishina Y, Suzuki A, Ueno N, Behringer RR. Bmpr encodes a type I bone morphogenetic protein receptor that is essential for gastrulation during mouse embryogenesis. Genes & development. 1995;9:3027–3037. doi: 10.1101/gad.9.24.3027. [DOI] [PubMed] [Google Scholar]
- 8.Winnier G, Blessing M, Labosky PA. Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes & …. 1995 doi: 10.1101/gad.9.17.2105. [DOI] [PubMed] [Google Scholar]
- 9.Ben-Haim N, Lu C, Guzman-Ayala M, Pescatore L, Mesnard D, Bischofberger M, Naef F, Robertson EJ, Constam DB. The nodal precursor acting via activin receptors induces mesoderm by maintaining a source of its convertases and BMP4. Developmental cell. 2006;11:313–323. doi: 10.1016/j.devcel.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 10.Liu P, Wakamiya M, Shea MJ, Albrecht U, Behringer RR, Bradley A. Requirement for Wnt3 in vertebrate axis formation. Nature Genetics. 1999;22:361–365. doi: 10.1038/11932. [DOI] [PubMed] [Google Scholar]
- 11.Rivera-Pérez JA, Magnuson T. Primitive streak formation in mice is preceded by localized activation of Brachyury and Wnt3. Developmental biology. 2005;288:363–371. doi: 10.1016/j.ydbio.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 12.Arnold SJ, Stappert J, Bauer A, Kispert A. Brachyury is a target gene of the Wnt/β-catenin signaling pathway. Mechanisms of …. 2000 doi: 10.1016/s0925-4773(99)00309-3. [DOI] [PubMed] [Google Scholar]
- 13.Robertson EJ. Dose-dependent Nodal/Smad signals pattern the early mouse embryo. Seminars in cell & developmental biology. 2014;32:73–79. doi: 10.1016/j.semcdb.2014.03.028. [DOI] [PubMed] [Google Scholar]
- 14.Costello I, Pimeisl I-MM, Dräger S, Bikoff EK, Robertson EJ, Arnold SJ. The T-box transcription factor Eomesodermin acts upstream of Mesp1 to specify cardiac mesoderm during mouse gastrulation. Nature cell biology. 2011;13:1084–1091. doi: 10.1038/ncb2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bondue A, Lapouge G, Paulissen C, Semeraro C, Iacovino M, Kyba M, Blanpain C. Mesp1 acts as a master regulator of multipotent cardiovascular progenitor specification. Cell stem cell. 2008;3:69–84. doi: 10.1016/j.stem.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 16.Chan S, Shi X, Toyama A, Arpke RW, Dandapat A, Iacovino M, Kang J, Le G, Hagen HR, Garry DJ, Kyba M. Mesp1 Patterns Mesoderm into Cardiac, Hematopoietic, or Skeletal Myogenic Progenitors in a Context-Dependent Manner. Cell Stem Cell. 2013;12:587–601. doi: 10.1016/j.stem.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lescroart F, Chabab S, Lin X, Rulands S, Paulissen C, Rodolosse A, Auer H, Achouri Y, Dubois C, Bondue A, Simons BD, Blanpain C. Early lineage restriction in temporally distinct populations of Mesp1 progenitors during mammalian heart development. Nature cell biology. 2014;16:829–840. doi: 10.1038/ncb3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yue Q, Wagstaff L, Yang X, Weijer C. Wnt3a-mediated chemorepulsion controls movement patterns of cardiac progenitors and requires RhoA function. Development. 2008 doi: 10.1242/dev.015321. [DOI] [PubMed] [Google Scholar]
- 19.Später D, Hansson EM, Zangi L, Chien KR. How to make a cardiomyocyte. Development. 2014;141:4418–4431. doi: 10.1242/dev.091538. [DOI] [PubMed] [Google Scholar]
- 20.Brade T, Pane LS, Moretti A. Embryonic heart progenitors and cardiogenesis. Cold Spring …. 2013 doi: 10.1101/cshperspect.a013847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marvin MJ, Di Rocco G, Gardiner A, Bush SM, Lassar AB. Inhibition of Wnt activity induces heart formation from posterior mesoderm. Genes Dev. 2001;15:316–27. doi: 10.1101/gad.855501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schneider VA, Mercola M. Wnt antagonism initiates cardiogenesis in Xenopus laevis. Genes Dev. 2001;15:304–15. doi: 10.1101/gad.855601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ueno S, Weidinger G, Osugi T, Kohn AD, Golob JL, Pabon L, Reinecke H, Moon RT, Murry CE. Biphasic role for Wnt/beta-catenin signaling in cardiac specification in zebrafish and embryonic stem cells. Proc Natl Acad Sci U S A. 2007;104:9685–90. doi: 10.1073/pnas.0702859104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paige SL, Osugi T, Afanasiev OK, Pabon L, Reinecke H, Murry CE. Endogenous Wnt/beta-catenin signaling is required for cardiac differentiation in human embryonic stem cells. PLoS One. 2010;5:e11134. doi: 10.1371/journal.pone.0011134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD, Lan F, Diecke S, Huber B, Mordwinkin NM, Plews JR, Abilez OJ, Cui B, Gold JD, Wu JC. Chemically defined generation of human cardiomyocytes. Nat Methods. 2014;11:855–60. doi: 10.1038/nmeth.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lian X, Bao X, Zilberter M, Westman M, Fisahn A, Hsiao C, Hazeltine LB, Dunn KK, Kamp TJ, Palecek SP. Chemically defined, albumin-free human cardiomyocyte generation. Nat Methods. 2015;12:595–6. doi: 10.1038/nmeth.3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wamstad JA, Alexander JM, Truty RM, Shrikumar A, Li F, Eilertson KE, Ding H, Wylie JN, Pico AR, Capra JA, Erwin G, Kattman SJ, Keller GM, Srivastava D, Levine SS, Pollard KS, Holloway AK, Boyer LA, Bruneau BG. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell. 2012;151:206–20. doi: 10.1016/j.cell.2012.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsa E, Burridge PW, Wu JC. Human stem cells for modeling heart disease and for drug discovery. Sci Transl Med. 2014;6:239ps6. doi: 10.1126/scitranslmed.3008921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chong JJ, Yang X, Don CW, Minami E, Liu YW, Weyers JJ, Mahoney WM, Van Biber B, Cook SM, Palpant NJ, Gantz JA, Fugate JA, Muskheli V, Gough GM, Vogel KW, Astley CA, Hotchkiss CE, Baldessari A, Pabon L, Reinecke H, Gill EA, Nelson V, Kiem HP, Laflamme MA, Murry CE. Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature. 2014;510:273–7. doi: 10.1038/nature13233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vincent SDD, Buckingham ME. How to make a heart: the origin and regulation of cardiac progenitor cells. Current topics in developmental biology. 2010;90:1–41. doi: 10.1016/S0070-2153(10)90001-X. [DOI] [PubMed] [Google Scholar]
- 31.Birket MJ, Ribeiro MC, Verkerk AO, Ward D, Leitoguinho A, den Hartogh SC, Orlova VV, Devalla HD, Schwach V, Bellin M, Passier R, Mummery CL. Expansion and patterning of cardiovascular progenitors derived from human pluripotent stem cells. Nature Biotechnology. 2015;33:970–979. doi: 10.1038/nbt.3271. [DOI] [PubMed] [Google Scholar]
- 32.Schultheiss TM, Burch JB, Lassar AB. A role for bone morphogenetic proteins in the induction of cardiac myogenesis. A role for bone morphogenetic proteins in the induction of cardiac myogenesis. 1997 doi: 10.1101/gad.11.4.451. [DOI] [PubMed] [Google Scholar]
- 33.Reifers F, Walsh EC, Léger S, Stainier DY. Induction and differentiation of the zebrafish heart requires fibroblast growth factor 8 (fgf8/acerebellar) Development. 2000 doi: 10.1242/dev.127.2.225. [DOI] [PubMed] [Google Scholar]
- 34.Pandur P, Läsche M, Eisenberg LM, Kühl M. Wnt-11 activation of a non-canonical Wnt signalling pathway is required for cardiogenesis. Nature. 2002;418:636–641. doi: 10.1038/nature00921. [DOI] [PubMed] [Google Scholar]
- 35.Ilagan R, Abu-Issa R, Brown D, Yang YP, Jiao K. Fgf8 is required for anterior heart field development. Fgf8 is required for anterior heart field development. 2006 doi: 10.1242/dev.02408. [DOI] [PubMed] [Google Scholar]
- 36.Dyer LA, Kirby ML. Sonic hedgehog maintains proliferation in secondary heart field progenitors and is required for normal arterial pole formation. Sonic hedgehog maintains proliferation in secondary heart field progenitors and is required for normal arterial pole formation. 2009 doi: 10.1016/j.ydbio.2009.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cohen ED, Wang Z, Lepore JJ, Lu MM, Taketo MM, Epstein DJ, Morrisey EE. Wnt/beta-catenin signaling promotes expansion of Isl-1-positive cardiac progenitor cells through regulation of FGF signaling. The Journal of clinical investigation. 2007;117:1794–1804. doi: 10.1172/JCI31731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lalit PA, Salick MR, Nelson DO, Squirrell JM, Shafer CM, Patel NG, Saeed I, Schmuck EG, Markandeya YS, Wong R, Lea MR, Eliceiri KW, Hacker TA, Crone WC, Kyba M, Garry DJ, Stewart R, Thomson JA, Downs KM, Lyons GE, Kamp TJ. Lineage Reprogramming of Fibroblasts into Proliferative Induced Cardiac Progenitor Cells by Defined Factors. Cell Stem Cell. 2016;18:354–67. doi: 10.1016/j.stem.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y, Cao N, Huang Y, Spencer CI, Fu JD, Yu C, Liu K, Nie B, Xu T, Li K, Xu S, Bruneau BG, Srivastava D, Ding S. Expandable Cardiovascular Progenitor Cells Reprogrammed from Fibroblasts. Cell Stem Cell. 2016;18:368–81. doi: 10.1016/j.stem.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hutson MR, Zeng XL, Kim AJ, Antoon E, Harward S, Kirby ML. Arterial pole progenitors interpret opposing FGF/BMP signals to proliferate or differentiate. Development. 2010;137:3001–11. doi: 10.1242/dev.051565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jain R, Li D, Gupta M, Manderfield LJ, Ifkovits JL, Wang Q, Liu F, Liu Y, Poleshko A, Padmanabhan A, Raum JC, Li L, Morrisey EE, Lu MM, Won KJ, Epstein JA, HEART DEVELOPMENT Integration of Bmp and Wnt signaling by Hopx specifies commitment of cardiomyoblasts. Science. 2015;348:aaa6071. doi: 10.1126/science.aaa6071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klaus A, Müller M, Schulz H, Saga Y, Martin JF, Birchmeier W. Wnt/β-catenin and Bmp signals control distinct sets of transcription factors in cardiac progenitor cells. Proc Natl Acad Sci U S A. 2012;109:10921–6. doi: 10.1073/pnas.1121236109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lien CL, McAnally J, Richardson JA, Olson EN. Cardiac-specific activity of an Nkx2-5 enhancer requires an evolutionarily conserved Smad binding site. Dev Biol. 2002;244:257–66. doi: 10.1006/dbio.2002.0603. [DOI] [PubMed] [Google Scholar]
- 44.Paige SL, Plonowska K, Xu A, Wu SM. Molecular regulation of cardiomyocyte differentiation. Circulation research. 2015;116:341–353. doi: 10.1161/CIRCRESAHA.116.302752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang L, Cai CL, Lin L, Qyang Y, Chung C, Monteiro RM, Mummery CL, Fishman GI, Cogen A, Evans S. Isl1Cre reveals a common Bmp pathway in heart and limb development. Development. 2006;133:1575–85. doi: 10.1242/dev.02322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kwon C, Arnold J, Hsiao EC, Taketo MM, Conklin BR, Srivastava D. Canonical Wnt signaling is a positive regulator of mammalian cardiac progenitors. Proc Natl Acad Sci U S A. 2007;104:10894–9. doi: 10.1073/pnas.0704044104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kwon C, Qian L, Cheng P, Nigam V, Arnold J, Srivastava D. A regulatory pathway involving Notch1/beta-catenin/Isl1 determines cardiac progenitor cell fate. Nat Cell Biol. 2009;11:951–7. doi: 10.1038/ncb1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Klaus A, Saga Y, Taketo MM, Tzahor E, Birchmeier W. Distinct roles of Wnt/beta-catenin and Bmp signaling during early cardiogenesis. Proc Natl Acad Sci U S A. 2007;104:18531–6. doi: 10.1073/pnas.0703113104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van den Berg G, Abu-Issa R, de Boer BA, Hutson MR, de Boer PA, Soufan AT, Ruijter JM, Kirby ML, van den Hoff MJ, Moorman AF. A caudal proliferating growth center contributes to both poles of the forming heart tube. Circ Res. 2009;104:179–88. doi: 10.1161/CIRCRESAHA.108.185843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ruiz-Villalba A, Hoppler S, van den Hoff MJ. Wnt signaling in the heart fields: Variations on a common theme. Dev Dyn. 2016;245:294–306. doi: 10.1002/dvdy.24372. [DOI] [PubMed] [Google Scholar]
- 51.Eisenberg CA, Eisenberg LM. WNT11 promotes cardiac tissue formation of early mesoderm. Dev Dyn. 1999;216:45–58. doi: 10.1002/(SICI)1097-0177(199909)216:1<45::AID-DVDY7>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 52.Cohen ED, Miller MF, Wang Z, Moon RT, Morrisey EE. Wnt5a and Wnt11 are essential for second heart field progenitor development. Development. 2012;139:1931–40. doi: 10.1242/dev.069377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Devine PW, Wythe JD, George M, Koshiba-Takeuchi K, Bruneau BG. Early patterning and specification of cardiac progenitors in gastrulating mesoderm. eLife. 2014;3 doi: 10.7554/eLife.03848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takeuchi JK, Bruneau BG. Directed transdifferentiation of mouse mesoderm to heart tissue by defined factors. Nature. 2009;459:708–11. doi: 10.1038/nature08039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vliet P, Wu SM, Zaffran S, Pucéat M. Early cardiac development: a view from stem cells to embryos. Cardiovascular research. 2012;96:352–362. doi: 10.1093/cvr/cvs270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gregoire S, Karra R, Passer D, Deutsch M-AA, Krane M, Feistritzer R, Sturzu A, Domian I, Saga Y, Wu SM. Essential and unexpected role of Yin Yang 1 to promote mesodermal cardiac differentiation. Circulation research. 2013;112:900–910. doi: 10.1161/CIRCRESAHA.113.259259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhao R, Watt AJ, Battle MA, Li J, Bondow BJ, Duncan SA. Loss of both GATA4 and GATA6 blocks cardiac myocyte differentiation and results in acardia in mice. Developmental biology. 2008;317:614–619. doi: 10.1016/j.ydbio.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McElhinney DB, Geiger E, Blinder J, Benson DW, Goldmuntz E. NKX2.5 mutations in patients with congenital heart disease. J Am Coll Cardiol. 2003;42:1650–5. doi: 10.1016/j.jacc.2003.05.004. [DOI] [PubMed] [Google Scholar]
- 59.Bruneau BG, Nemer G, Schmitt JP, Charron F, Robitaille L, Caron S, Conner DA, Gessler M, Nemer M, Seidman CE, Seidman JG. A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell. 2001;106:709–21. doi: 10.1016/s0092-8674(01)00493-7. [DOI] [PubMed] [Google Scholar]
- 60.Takeuchi JK, Ohgi M, Koshiba-Takeuchi K. Tbx5 specifies the left/right ventricles and ventricular septum position during cardiogenesis. 2003 doi: 10.1242/dev.00797. [DOI] [PubMed] [Google Scholar]
- 61.Tanaka M, Chen Z, Bartunkova S, Yamasaki N. The cardiac homeobox gene Csx/Nkx2. 5 lies genetically upstream of multiple genes essential for heart development. 1999 doi: 10.1242/dev.126.6.1269. [DOI] [PubMed] [Google Scholar]
- 62.Olson EN. Gene regulatory networks in the evolution and development of the heart. Science (New York, NY) 2006;313:1922–1927. doi: 10.1126/science.1132292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Caprioli A, Koyano-Nakagawa N, Iacovino M, Shi X. Nkx2-5 represses Gata1 gene expression and modulates the cellular fate of cardiac progenitors during embryogenesis. Circulation. 2011 doi: 10.1161/CIRCULATIONAHA.110.008185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Später D, Abramczuk MK, Buac K, Zangi L, Stachel MW, Clarke J, Sahara M, Ludwig A, Chien KR. A HCN4+ cardiomyogenic progenitor derived from the first heart field and human pluripotent stem cells. Nature cell biology. 2013;15:1098–1106. doi: 10.1038/ncb2824. [DOI] [PubMed] [Google Scholar]
- 65.Liang X, Wang G, Lin L, Lowe J, Zhang Q, Bu L, Chen Y, Chen J, Sun Y, Evans SM. HCN4 dynamically marks the first heart field and conduction system precursors. Circ Res. 2013;113:399–407. doi: 10.1161/CIRCRESAHA.113.301588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cai C-LL, Liang X, Shi Y, Chu P-HH, Pfaff SL, Chen J, Evans S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Developmental cell. 2003;5:877–889. doi: 10.1016/s1534-5807(03)00363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moretti A, Caron L, Nakano A, Lam JT, Bernshausen A, Chen Y, Qyang Y, Bu L, Sasaki M, Martin-Puig S, Sun Y, Evans SM, Laugwitz K-LL, Chien KR. Multipotent embryonic isl1+ progenitor cells lead to cardiac, smooth muscle, and endothelial cell diversification. Cell. 2006;127:1151–1165. doi: 10.1016/j.cell.2006.10.029. [DOI] [PubMed] [Google Scholar]
- 68.Bu L, Jiang X, Martin-Puig S, Caron L, Zhu S, Shao Y, Roberts DJ, Huang PL, Domian IJ, Chien KR. Human ISL1 heart progenitors generate diverse multipotent cardiovascular cell lineages. Nature. 2009;460:113–7. doi: 10.1038/nature08191. [DOI] [PubMed] [Google Scholar]
- 69.Ma Q, Zhou B, Pu WT. Reassessment of Isl1 and Nkx2-5 cardiac fate maps using a Gata4-based reporter of Cre activity. Dev Biol. 2008;323:98–104. doi: 10.1016/j.ydbio.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zeisberg EM, Ma Q, Juraszek AL, Moses K, Schwartz RJ, Izumo S, Pu WT. Morphogenesis of the right ventricle requires myocardial expression of Gata4. J Clin Invest. 2005;115:1522–31. doi: 10.1172/JCI23769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lin Q, Schwarz J, Bucana C, Olson EN. Control of mouse cardiac morphogenesis and myogenesis by transcription factor MEF2C. Science. 1997;276:1404–7. doi: 10.1126/science.276.5317.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dodou E, Verzi MP, Anderson JP, Xu SM. Mef2c is a direct transcriptional target of ISL1 and GATA factors in the anterior heart field during mouse embryonic development. Development. 2004 doi: 10.1242/dev.01256. [DOI] [PubMed] [Google Scholar]
- 73.Tsuchihashi T, Maeda J, Shin CH, Ivey KN, Black BL, Olson EN, Yamagishi H, Srivastava D. Hand2 function in second heart field progenitors is essential for cardiogenesis. Dev Biol. 2011;351:62–9. doi: 10.1016/j.ydbio.2010.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Prall OW, Menon MK, Solloway MJ, Watanabe Y, Zaffran S, Bajolle F, Biben C, McBride JJ, Robertson BR, Chaulet H, Stennard FA, Wise N, Schaft D, Wolstein O, Furtado MB, Shiratori H, Chien KR, Hamada H, Black BL, Saga Y, Robertson EJ, Buckingham ME, Harvey RP. An Nkx2-5/Bmp2/Smad1 negative feedback loop controls heart progenitor specification and proliferation. Cell. 2007;128:947–59. doi: 10.1016/j.cell.2007.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dorn T, Goedel A, Lam JT, Haas J, Tian Q, Herrmann F, Bundschu K, Dobreva G, Schiemann M, Dirschinger R, Guo Y, Kuhl SJ, Sinnecker D, Lipp P, Laugwitz KL, Kuhl M, Moretti A. Direct nkx2-5 transcriptional repression of isl1 controls cardiomyocyte subtype identity. Stem Cells. 2015;33:1113–29. doi: 10.1002/stem.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Katz MG, Fargnoli AS, Kendle AP, Hajjar RJ, Bridges CR. The role of microRNAs in cardiac development and regenerative capacity. Am J Physiol Heart Circ Physiol. 2016;310:H528–41. doi: 10.1152/ajpheart.00181.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Takaya T, Nishi H, Horie T, Ono K, Hasegawa K. Roles of microRNAs and myocardial cell differentiation. Prog Mol Biol Transl Sci. 2012;111:139–52. doi: 10.1016/B978-0-12-398459-3.00006-X. [DOI] [PubMed] [Google Scholar]
- 78.Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, Tsuchihashi T, McManus MT, Schwartz RJ, Srivastava D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell. 2007;129:303–17. doi: 10.1016/j.cell.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 79.Chen JF, Murchison EP, Tang R, Callis TE, Tatsuguchi M, Deng Z, Rojas M, Hammond SM, Schneider MD, Selzman CH, Meissner G, Patterson C, Hannon GJ, Wang DZ. Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proc Natl Acad Sci U S A. 2008;105:2111–6. doi: 10.1073/pnas.0710228105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, Conlon FL, Wang DZ. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet. 2006;38:228–33. doi: 10.1038/ng1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhao Y, Samal E, Srivastava D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature. 2005;436:214–20. doi: 10.1038/nature03817. [DOI] [PubMed] [Google Scholar]
- 82.Liu N, Williams AH, Kim Y, McAnally J, Bezprozvannaya S, Sutherland LB, Richardson JA, Bassel-Duby R, Olson EN. An intragenic MEF2-dependent enhancer directs muscle-specific expression of microRNAs 1 and 133. Proc Natl Acad Sci U S A. 2007;104:20844–9. doi: 10.1073/pnas.0710558105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Heidersbach A, Saxby C, Carver-Moore K, Huang Y, Ang YS, de Jong PJ, Ivey KN, Srivastava D. microRNA-1 regulates sarcomere formation and suppresses smooth muscle gene expression in the mammalian heart. Elife. 2013;2:e01323. doi: 10.7554/eLife.01323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liu N, Bezprozvannaya S, Williams AH, Qi X, Richardson JA, Bassel-Duby R, Olson EN. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008;22:3242–54. doi: 10.1101/gad.1738708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wystub K, Besser J, Bachmann A, Boettger T, Braun T. miR-1/133a clusters cooperatively specify the cardiomyogenic lineage by adjustment of myocardin levels during embryonic heart development. PLoS Genet. 2013;9:e1003793. doi: 10.1371/journal.pgen.1003793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Inagawa K, Miyamoto K, Yamakawa H, Muraoka N, Sadahiro T, Umei T, Wada R, Katsumata Y, Kaneda R, Nakade K. Induction of cardiomyocyte-like cells in infarct hearts by gene transfer of Gata4, Mef2c, and Tbx5. Circulation research. 2012;111:1147–1156. doi: 10.1161/CIRCRESAHA.112.271148. [DOI] [PubMed] [Google Scholar]
- 87.Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L, Conway SJ, Fu J-d, Srivastava D. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature. 2012;485:593–598. doi: 10.1038/nature11044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fu J-D, Stone NR, Liu L, Spencer CI, Qian L, Hayashi Y, Delgado-Olguin P, Ding S, Bruneau BG, Srivastava D. Direct reprogramming of human fibroblasts toward a cardiomyocyte-like state. Stem cell reports. 2013;1:235–247. doi: 10.1016/j.stemcr.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Song K, Nam Y-J, Luo X, Qi X, Tan W, Huang GN, Acharya A, Smith CL, Tallquist MD, Neilson EG. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature. 2012;485:599–604. doi: 10.1038/nature11139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Song K, Nam YJ, Luo X, Qi X, Tan W, Huang GN, Acharya A, Smith CL, Tallquist MD, Neilson EG, Hill JA, Bassel-Duby R, Olson EN. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature. 2012;485:599–604. doi: 10.1038/nature11139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Islas JF, Liu Y, Weng KC, Robertson MJ, Zhang S, Prejusa A, Harger J, Tikhomirova D, Chopra M, Iyer D, Mercola M, Oshima RG, Willerson JT, Potaman VN, Schwartz RJ. Transcription factors ETS2 and MESP1 transdifferentiate human dermal fibroblasts into cardiac progenitors. Proc Natl Acad Sci U S A. 2012;109:13016–21. doi: 10.1073/pnas.1120299109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jayawardena TM, Finch EA, Zhang L, Zhang H, Hodgkinson CP, Pratt RE, Rosenberg PB, Mirotsou M, Dzau VJ. MicroRNA induced cardiac reprogramming in vivo evidence for mature cardiac myocytes and improved cardiac function. Circulation research. 2015;116:418–424. doi: 10.1161/CIRCRESAHA.116.304510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chen JX, Krane M, Deutsch MA, Wang L, Rav-Acha M, Gregoire S, Engels MC, Rajarajan K, Karra R, Abel ED, Wu JC, Milan D, Wu SM. Inefficient reprogramming of fibroblasts into cardiomyocytes using Gata4, Mef2c, and Tbx5. Circ Res. 2012;111:50–5. doi: 10.1161/CIRCRESAHA.112.270264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mikawa T, Borisov A, Brown AM, Fischman DA. Clonal analysis of cardiac morphogenesis in the chicken embryo using a replication-defective retrovirus: I. Formation of the ventricular myocardium. Dev Dyn. 1992;193:11–23. doi: 10.1002/aja.1001930104. [DOI] [PubMed] [Google Scholar]
- 95.Meilhac SM, Kelly RG, Rocancourt D, Eloy-Trinquet S, Nicolas JF, Buckingham ME. A retrospective clonal analysis of the myocardium reveals two phases of clonal growth in the developing mouse heart. Development. 2003;130:3877–89. doi: 10.1242/dev.00580. [DOI] [PubMed] [Google Scholar]
- 96.Gupta V, Poss KD. Clonally dominant cardiomyocytes direct heart morphogenesis. Nature. 2012;484:479–84. doi: 10.1038/nature11045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yu FX, Zhao B, Guan KL. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell. 2015;163:811–28. doi: 10.1016/j.cell.2015.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhao B, Tumaneng K, Guan KL. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat Cell Biol. 2011;13:877–83. doi: 10.1038/ncb2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Xiao Y, Leach J, Wang J, Martin JF. Hippo/Yap Signaling in Cardiac Development and Regeneration. Curr Treat Options Cardiovasc Med. 2016;18:38. doi: 10.1007/s11936-016-0461-y. [DOI] [PubMed] [Google Scholar]
- 100.Lin Z, Pu WT. Harnessing Hippo in the heart: Hippo/Yap signaling and applications to heart regeneration and rejuvenation. Stem Cell Res. 2014;13:571–81. doi: 10.1016/j.scr.2014.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL, Martin JF. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science. 2011;332:458–61. doi: 10.1126/science.1199010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lin Z, von Gise A, Zhou P, Gu F, Ma Q, Jiang J, Yau AL, Buck JN, Gouin KA, van Gorp PR, Zhou B, Chen J, Seidman JG, Wang DZ, Pu WT. Cardiac-specific YAP activation improves cardiac function and survival in an experimental murine MI model. Circ Res. 2014;115:354–63. doi: 10.1161/CIRCRESAHA.115.303632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.von Gise A, Lin Z, Schlegelmilch K, Honor LB, Pan GM, Buck JN, Ma Q, Ishiwata T, Zhou B, Camargo FD. YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proceedings of the National Academy of Sciences. 2012;109:2394–2399. doi: 10.1073/pnas.1116136109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, Porrello ER, Mahmoud AI, Tan W, Shelton JM, Richardson JA, Sadek HA, Bassel-Duby R, Olson EN. Hippo pathway effector Yap promotes cardiac regeneration. Proc Natl Acad Sci U S A. 2013;110:13839–44. doi: 10.1073/pnas.1313192110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lin Z, Guo H, Cao Y, Zohrabian S, Zhou P, Ma Q, VanDusen N, Guo Y, Zhang J, Stevens SM, Liang F, Quan Q, van Gorp PR, Li A, Dos Remedios C, He A, Bezzerides VJ, Pu WT. Acetylation of VGLL4 Regulates Hippo-YAP Signaling and Postnatal Cardiac Growth. Dev Cell. 2016 doi: 10.1016/j.devcel.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.von Gise A, Lin Z, Schlegelmilch K, Honor LB, Pan GM, Buck JN, Ma Q, Ishiwata T, Zhou B, Camargo FD, Pu WT. YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc Natl Acad Sci U S A. 2012;109:2394–9. doi: 10.1073/pnas.1116136109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lin Z, Zhou P, von Gise A, Gu F, Ma Q, Chen J, Guo H, van Gorp PR, Wang DZ, Pu WT. Pi3kcb links Hippo-YAP and PI3K-AKT signaling pathways to promote cardiomyocyte proliferation and survival. Circ Res. 2015;116:35–45. doi: 10.1161/CIRCRESAHA.115.304457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xin M, Kim Y, Sutherland LB, Qi X, McAnally J, Schwartz RJ, Richardson JA, Bassel-Duby R, Olson EN. Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci Signal. 2011;4:ra70. doi: 10.1126/scisignal.2002278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pennisi DJ, Ballard VL, Mikawa T. Epicardium is required for the full rate of myocyte proliferation and levels of expression of myocyte mitogenic factors FGF2 and its receptor, FGFR-1, but not for transmural myocardial patterning in the embryonic chick heart. Dev Dyn. 2003;228:161–72. doi: 10.1002/dvdy.10360. [DOI] [PubMed] [Google Scholar]
- 110.Li P, Cavallero S, Gu Y, Chen TH, Hughes J, Hassan AB, Bruning JC, Pashmforoush M, Sucov HM. IGF signaling directs ventricular cardiomyocyte proliferation during embryonic heart development. Development. 2011;138:1795–805. doi: 10.1242/dev.054338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Huang Y, Harrison MR, Osorio A, Kim J, Baugh A, Duan C, Sucov HM, Lien CL. Igf Signaling is Required for Cardiomyocyte Proliferation during Zebrafish Heart Development and Regeneration. PLoS One. 2013;8:e67266. doi: 10.1371/journal.pone.0067266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wei K, Serpooshan V, Hurtado C, Diez-Cunado M, Zhao M, Maruyama S, Zhu W, Fajardo G, Noseda M, Nakamura K, Tian X, Liu Q, Wang A, Matsuura Y, Bushway P, Cai W, Savchenko A, Mahmoudi M, Schneider MD, van den Hoff MJ, Butte MJ, Yang PC, Walsh K, Zhou B, Bernstein D, Mercola M, Ruiz-Lozano P. Epicardial FSTL1 reconstitution regenerates the adult mammalian heart. Nature. 2015;525:479–85. doi: 10.1038/nature15372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Guimaraes-Camboa N, Stowe J, Aneas I, Sakabe N, Cattaneo P, Henderson L, Kilberg MS, Johnson RS, Chen J, McCulloch AD, Nobrega MA, Evans SM, Zambon AC. HIF1alpha Represses Cell Stress Pathways to Allow Proliferation of Hypoxic Fetal Cardiomyocytes. Dev Cell. 2015;33:507–21. doi: 10.1016/j.devcel.2015.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Passer D, van de Vrugt A, Atmanli A, Domian IJ. Atypical Protein Kinase C-Dependent Polarized Cell Division Is Required for Myocardial Trabeculation. Cell Rep. 2016;14:1662–72. doi: 10.1016/j.celrep.2016.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Grego-Bessa J, Luna-Zurita L, del Monte G, Bolos V, Melgar P, Arandilla A, Garratt AN, Zang H, Mukouyama YS, Chen H, Shou W, Ballestar E, Esteller M, Rojas A, Perez-Pomares JM, de la Pompa JL. Notch signaling is essential for ventricular chamber development. Dev Cell. 2007;12:415–29. doi: 10.1016/j.devcel.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chen H, Shi S, Acosta L, Li W, Lu J, Bao S, Chen Z, Yang Z, Schneider MD, Chien KR. BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development. 2004;131:2219–2231. doi: 10.1242/dev.01094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chen H, Zhang W, Sun X, Yoshimoto M, Chen Z, Zhu W, Liu J, Shen Y, Yong W, Li D, Zhang J, Lin Y, Li B, VanDusen NJ, Snider P, Schwartz RJ, Conway SJ, Field LJ, Yoder MC, Firulli AB, Carlesso N, Towbin JA, Shou W. Fkbp1a controls ventricular myocardium trabeculation and compaction by regulating endocardial Notch1 activity. Development. 2013;140:1946–57. doi: 10.1242/dev.089920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.VanDusen NJ, Casanovas J, Vincentz JW, Firulli BA, Osterwalder M, Lopez-Rios J, Zeller R, Zhou B, Grego-Bessa J, De La Pompa JL, Shou W, Firulli AB. Hand2 is an essential regulator for two Notch-dependent functions within the embryonic endocardium. Cell Rep. 2014;9:2071–83. doi: 10.1016/j.celrep.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Liu J, Bressan M, Hassel D, Huisken J, Staudt D, Kikuchi K, Poss KD, Mikawa T, Stainier DY. A dual role for ErbB2 signaling in cardiac trabeculation. Development. 2010;137:3867–75. doi: 10.1242/dev.053736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Staudt DW, Liu J, Thorn KS, Stuurman N, Liebling M, Stainier DY. High-resolution imaging of cardiomyocyte behavior reveals two distinct steps in ventricular trabeculation. Development. 2014;141:585–93. doi: 10.1242/dev.098632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jimenez-Amilburu V, Rasouli SJ, Staudt DW, Nakajima H, Chiba A, Mochizuki N, Stainier DY. In Vivo Visualization of Cardiomyocyte Apicobasal Polarity Reveals Epithelial to Mesenchymal-like Transition during Cardiac Trabeculation. Cell Rep. 2016;17:2687–2699. doi: 10.1016/j.celrep.2016.11.023. [DOI] [PubMed] [Google Scholar]
- 122.Bergmann O, Zdunek S, Felker A, Salehpour M, Alkass K, Bernard S, Sjostrom SL, Szewczykowska M, Jackowska T, Dos Remedios C, Malm T, Andra M, Jashari R, Nyengaard JR, Possnert G, Jovinge S, Druid H, Frisen J. Dynamics of Cell Generation and Turnover in the Human Heart. Cell. 2015;161:1566–75. doi: 10.1016/j.cell.2015.05.026. [DOI] [PubMed] [Google Scholar]
- 123.Senyo SE, Steinhauser ML, Pizzimenti CL, Yang VK, Cai L, Wang M, Wu TD, Guerquin-Kern JL, Lechene CP, Lee RT. Mammalian heart renewal by pre-existing cardiomyocytes. Nature. 2013;493:433–6. doi: 10.1038/nature11682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ali SR, Hippenmeyer S, Saadat LV, Luo L, Weissman IL, Ardehali R. Existing cardiomyocytes generate cardiomyocytes at a low rate after birth in mice. Proc Natl Acad Sci U S A. 2014;111:8850–5. doi: 10.1073/pnas.1408233111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, Frisen J. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Soonpaa MH, Kim KK, Pajak L, Franklin M, Field LJ. Cardiomyocyte DNA synthesis and binucleation during murine development. Am J Physiol. 1996;271:H2183–9. doi: 10.1152/ajpheart.1996.271.5.H2183. [DOI] [PubMed] [Google Scholar]
- 127.Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, Sadek HA. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331:1078–80. doi: 10.1126/science.1200708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mollova M, Bersell K, Walsh S, Savla J, Das LT, Park SY, Silberstein LE, Dos Remedios CG, Graham D, Colan S, Kuhn B. Cardiomyocyte proliferation contributes to heart growth in young humans. Proc Natl Acad Sci U S A. 2013;110:1446–51. doi: 10.1073/pnas.1214608110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.D'Uva G, Aharonov A, Lauriola M, Kain D, Yahalom-Ronen Y, Carvalho S, Weisinger K, Bassat E, Rajchman D, Yifa O, Lysenko M, Konfino T, Hegesh J, Brenner O, Neeman M, Yarden Y, Leor J, Sarig R, Harvey RP, Tzahor E. ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat Cell Biol. 2015;17:627–38. doi: 10.1038/ncb3149. [DOI] [PubMed] [Google Scholar]
- 130.Mahmoud AI, Kocabas F, Muralidhar SA, Kimura W, Koura AS, Thet S, Porrello ER, Sadek HA. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature. 2013;497:249–53. doi: 10.1038/nature12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sdek P, Zhao P, Wang Y, Huang CJ, Ko CY, Butler PC, Weiss JN, Maclellan WR. Rb and p130 control cell cycle gene silencing to maintain the postmitotic phenotype in cardiac myocytes. J Cell Biol. 2011;194:407–23. doi: 10.1083/jcb.201012049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.MacLellan WR, Garcia A, Oh H, Frenkel P, Jordan MC, Roos KP, Schneider MD. Overlapping roles of pocket proteins in the myocardium are unmasked by germ line deletion of p130 plus heart-specific deletion of Rb. Mol Cell Biol. 2005;25:2486–97. doi: 10.1128/MCB.25.6.2486-2497.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.He A, Ma Q, Cao J, von Gise A, Zhou P, Xie H, Zhang B, Hsing M, Christodoulou DC, Cahan P, Daley GQ, Kong SW, Orkin SH, Seidman CE, Seidman JG, Pu WT. Polycomb repressive complex 2 regulates normal development of the mouse heart. Circ Res. 2012;110:406–15. doi: 10.1161/CIRCRESAHA.111.252205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Porrello ER, Mahmoud AI, Simpson E, Johnson BA, Grinsfelder D, Canseco D, Mammen PP, Rothermel BA, Olson EN, Sadek HA. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc Natl Acad Sci U S A. 2013;110:187–92. doi: 10.1073/pnas.1208863110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Engel FB, Schebesta M, Duong MT, Lu G, Ren S, Madwed JB, Jiang H, Wang Y, Keating MT. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev. 2005;19:1175–87. doi: 10.1101/gad.1306705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Engel FB, Hsieh PC, Lee RT, Keating MT. FGF1/p38 MAP kinase inhibitor therapy induces cardiomyocyte mitosis, reduces scarring, and rescues function after myocardial infarction. Proc Natl Acad Sci U S A. 2006;103:15546–51. doi: 10.1073/pnas.0607382103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Puente BN, Kimura W, Muralidhar SA, Moon J, Amatruda JF, Phelps KL, Grinsfelder D, Rothermel BA, Chen R, Garcia JA, Santos CX, Thet S, Mori E, Kinter MT, Rindler PM, Zacchigna S, Mukherjee S, Chen DJ, Mahmoud AI, Giacca M, Rabinovitch PS, Aroumougame A, Shah AM, Szweda LI, Sadek HA. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell. 2014;157:565–79. doi: 10.1016/j.cell.2014.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Aix E, Gutierrez-Gutierrez O, Sanchez-Ferrer C, Aguado T, Flores I. Postnatal telomere dysfunction induces cardiomyocyte cell-cycle arrest through p21 activation. J Cell Biol. 2016;213:571–83. doi: 10.1083/jcb.201510091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bar C, Bernardes de Jesus B, Serrano R, Tejera A, Ayuso E, Jimenez V, Formentini I, Bobadilla M, Mizrahi J, de Martino A, Gomez G, Pisano D, Mulero F, Wollert KC, Bosch F, Blasco MA. Telomerase expression confers cardioprotection in the adult mouse heart after acute myocardial infarction. Nat Commun. 2014;5:5863. doi: 10.1038/ncomms6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Doxsey S, Zimmerman W, Mikule K. Centrosome control of the cell cycle. Trends Cell Biol. 2005;15:303–11. doi: 10.1016/j.tcb.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 141.Hinchcliffe EH. Centrosomes and the art of mitotic spindle maintenance. Int Rev Cell Mol Biol. 2014;313:179–217. doi: 10.1016/B978-0-12-800177-6.00006-2. [DOI] [PubMed] [Google Scholar]
- 142.Guo Y, Zheng Y. Lamins position the nuclear pores and centrosomes by modulating dynein. Mol Biol Cell. 2015;26:3379–89. doi: 10.1091/mbc.E15-07-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zebrowski DC, Vergarajauregui S, Wu CC, Piatkowski T, Becker R, Leone M, Hirth S, Ricciardi F, Falk N, Giessl A, Just S, Braun T, Weidinger G, Engel FB. Developmental alterations in centrosome integrity contribute to the post-mitotic state of mammalian cardiomyocytes. Elife. 2015;4 doi: 10.7554/eLife.05563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.French BA, Kramer CM. Mechanisms of Post-Infarct Left Ventricular Remodeling. Drug Discov Today Dis Mech. 2007;4:185–196. doi: 10.1016/j.ddmec.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Konstam MA, Kramer DG, Patel AR, Maron MS, Udelson JE. Left ventricular remodeling in heart failure: current concepts in clinical significance and assessment. JACC Cardiovasc Imaging. 2011;4:98–108. doi: 10.1016/j.jcmg.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 146.Gemberling M, Bailey TJ, Hyde DR, Poss KD. The zebrafish as a model for complex tissue regeneration. Trends Genet. 2013;29:611–20. doi: 10.1016/j.tig.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Major RJ, Poss KD. Zebrafish Heart Regeneration as a Model for Cardiac Tissue Repair. Drug Discov Today Dis Models. 2007;4:219–225. doi: 10.1016/j.ddmod.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ausoni S, Sartore S. From fish to amphibians to mammals: in search of novel strategies to optimize cardiac regeneration. J Cell Biol. 2009;184:357–64. doi: 10.1083/jcb.200810094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Jopling C, Sleep E, Raya M, Marti M, Raya A, Izpisua Belmonte JC. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature. 2010;464:606–9. doi: 10.1038/nature08899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kikuchi K, Holdway JE, Werdich AA, Anderson RM, Fang Y, Egnaczyk GF, Evans T, Macrae CA, Stainier DY, Poss KD. Primary contribution to zebrafish heart regeneration by gata4(+) cardiomyocytes. Nature. 2010;464:601–5. doi: 10.1038/nature08804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wang J, Panakova D, Kikuchi K, Holdway JE, Gemberling M, Burris JS, Singh SP, Dickson AL, Lin YF, Sabeh MK, Werdich AA, Yelon D, Macrae CA, Poss KD. The regenerative capacity of zebrafish reverses cardiac failure caused by genetic cardiomyocyte depletion. Development. 2011;138:3421–30. doi: 10.1242/dev.068601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Drenckhahn JD, Schwarz QP, Gray S, Laskowski A, Kiriazis H, Ming Z, Harvey RP, Du XJ, Thorburn DR, Cox TC. Compensatory growth of healthy cardiac cells in the presence of diseased cells restores tissue homeostasis during heart development. Dev Cell. 2008;15:521–33. doi: 10.1016/j.devcel.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 153.Sturzu AC, Rajarajan K, Passer D, Plonowska K, Riley A, Tan TC, Sharma A, Xu AF, Engels MC, Feistritzer R, Li G, Selig MK, Geissler R, Robertson KD, Scherrer-Crosbie M, Domian IJ, Wu SM. Fetal Mammalian Heart Generates a Robust Compensatory Response to Cell Loss. Circulation. 2015;132:109–21. doi: 10.1161/CIRCULATIONAHA.114.011490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sadek HA, Martin JF, Takeuchi JK, Leor J, Nie Y, Giacca M, Lee RT. Multi-investigator letter on reproducibility of neonatal heart regeneration following apical resection. Stem Cell Reports. 2014;3:1. doi: 10.1016/j.stemcr.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Andersen DC, Ganesalingam S, Jensen CH, Sheikh SP. Do neonatal mouse hearts regenerate following heart apex resection? Stem Cell Reports. 2014;2:406–13. doi: 10.1016/j.stemcr.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bryant DM, O'Meara CC, Ho NN, Gannon J, Cai L, Lee RT. A systematic analysis of neonatal mouse heart regeneration after apical resection. J Mol Cell Cardiol. 2015;79:315–8. doi: 10.1016/j.yjmcc.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Aurora AB, Porrello ER, Tan W, Mahmoud AI, Hill JA, Bassel-Duby R, Sadek HA, Olson EN. Macrophages are required for neonatal heart regeneration. J Clin Invest. 2014;124:1382–92. doi: 10.1172/JCI72181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ahuja P, Perriard E, Perriard JC, Ehler E. Sequential myofibrillar breakdown accompanies mitotic division of mammalian cardiomyocytes. J Cell Sci. 2004;117:3295–306. doi: 10.1242/jcs.01159. [DOI] [PubMed] [Google Scholar]
- 159.Bersell K, Arab S, Haring B, Kuhn B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell. 2009;138:257–70. doi: 10.1016/j.cell.2009.04.060. [DOI] [PubMed] [Google Scholar]
- 160.O’Meara CC, Wamstad JA, Gladstone RA, Fomovsky GM, Butty VL, Shrikumar A, Gannon JB, Boyer LA, Lee RT. Transcriptional reversion of cardiac myocyte fate during mammalian cardiac regeneration. Circ Res. 2015;116:804–15. doi: 10.1161/CIRCRESAHA.116.304269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Engel FB. Cardiomyocyte proliferation: a platform for mammalian cardiac repair. Cell Cycle. 2005;4:1360–3. doi: 10.4161/cc.4.10.2081. [DOI] [PubMed] [Google Scholar]
- 162.Giacca M, Zacchigna S. Harnessing the microRNA pathway for cardiac regeneration. J Mol Cell Cardiol. 2015;89:68–74. doi: 10.1016/j.yjmcc.2015.09.017. [DOI] [PubMed] [Google Scholar]
- 163.Xin M, Olson EN, Bassel-Duby R. Mending broken hearts: cardiac development as a basis for adult heart regeneration and repair. Nat Rev Mol Cell Biol. 2013;14:529–41. doi: 10.1038/nrm3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lin Z, Pu WT. Strategies for cardiac regeneration and repair. Sci Transl Med. 2014;6:239rv1. doi: 10.1126/scitranslmed.3006681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Gemberling M, Karra R, Dickson AL, Poss KD. Nrg1 is an injury-induced cardiomyocyte mitogen for the endogenous heart regeneration program in zebrafish. Elife. 2015;4 doi: 10.7554/eLife.05871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kang J, Hu J, Karra R, Dickson AL, Tornini VA, Nachtrab G, Gemberling M, Goldman JA, Black BL, Poss KD. Modulation of tissue repair by regeneration enhancer elements. Nature. 2016;532:201–6. doi: 10.1038/nature17644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Polizzotti BD, Ganapathy B, Walsh S, Choudhury S, Ammanamanchi N, Bennett DG, dos Remedios CG, Haubner BJ, Penninger JM, Kuhn B. Neuregulin stimulation of cardiomyocyte regeneration in mice and human myocardium reveals a therapeutic window. Sci Transl Med. 2015;7:281ra45. doi: 10.1126/scitranslmed.aaa5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Jabbour A, Hayward CS, Keogh AM, Kotlyar E, McCrohon JA, England JF, Amor R, Liu X, Li XY, Zhou MD, Graham RM, Macdonald PS. Parenteral administration of recombinant human neuregulin-1 to patients with stable chronic heart failure produces favourable acute and chronic haemodynamic responses. Eur J Heart Fail. 2011;13:83–92. doi: 10.1093/eurjhf/hfq152. [DOI] [PubMed] [Google Scholar]
- 169.Reuter S, Soonpaa MH, Firulli AB, Chang AN, Field LJ. Recombinant neuregulin 1 does not activate cardiomyocyte DNA synthesis in normal or infarcted adult mice. PLoS One. 2014;9:e115871. doi: 10.1371/journal.pone.0115871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zhao YY, Sawyer DR, Baliga RR, Opel DJ, Han X, Marchionni MA, Kelly RA. Neuregulins promote survival and growth of cardiac myocytes. Persistence of ErbB2 and ErbB4 expression in neonatal and adult ventricular myocytes. J Biol Chem. 1998;273:10261–9. doi: 10.1074/jbc.273.17.10261. [DOI] [PubMed] [Google Scholar]
- 171.Han P, Bloomekatz J, Ren J, Zhang R, Grinstein JD, Zhao L, Burns CG, Burns CE, Anderson RM, Chi NC. Coordinating cardiomyocyte interactions to direct ventricular chamber morphogenesis. Nature. 2016;534:700–4. doi: 10.1038/nature18310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Brero A, Ramella R, Fitou A, Dati C, Alloatti G, Gallo MP, Levi R. Neuregulin-1beta1 rapidly modulates nitric oxide synthesis and calcium handling in rat cardiomyocytes. Cardiovasc Res. 2010;88:443–52. doi: 10.1093/cvr/cvq238. [DOI] [PubMed] [Google Scholar]
- 173.Del Re DP, Yang Y, Nakano N, Cho J, Zhai P, Yamamoto T, Zhang N, Yabuta N, Nojima H, Pan D, Sadoshima J. Yes-associated protein isoform 1 (Yap1) promotes cardiomyocyte survival and growth to protect against myocardial ischemic injury. J Biol Chem. 2013;288:3977–88. doi: 10.1074/jbc.M112.436311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–83. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 175.Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, Dupont S, Piccolo S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell. 2013;154:1047–59. doi: 10.1016/j.cell.2013.07.042. [DOI] [PubMed] [Google Scholar]
- 176.Morikawa Y, Zhang M, Heallen T, Leach J, Tao G, Xiao Y, Bai Y, Li W, Willerson JT, Martin JF. Actin cytoskeletal remodeling with protrusion formation is essential for heart regeneration in Hippo-deficient mice. Sci Signal. 2015;8:ra41. doi: 10.1126/scisignal.2005781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Tao G, Kahr PC, Morikawa Y, Zhang M, Rahmani M, Heallen TR, Li L, Sun Z, Olson EN, Amendt BA, Martin JF. Pitx2 promotes heart repair by activating the antioxidant response after cardiac injury. Nature. 2016;534:119–23. doi: 10.1038/nature17959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Vermeulen Z, Segers VF, De Keulenaer GW. ErbB2 signaling at the crossing between heart failure and cancer. Basic Res Cardiol. 2016;111:60. doi: 10.1007/s00395-016-0576-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the Roots of Cancer. Cancer Cell. 2016;29:783–803. doi: 10.1016/j.ccell.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Fan X, Hughes BG, Ali MA, Cho WJ, Lopez W, Schulz R. Dynamic Alterations to alpha-Actinin Accompanying Sarcomere Disassembly and Reassembly during Cardiomyocyte Mitosis. PLoS One. 2015;10:e0129176. doi: 10.1371/journal.pone.0129176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Dambrot C, Passier R, Atsma D, Mummery CL. Cardiomyocyte differentiation of pluripotent stem cells and their use as cardiac disease models. Biochemical Journal. 2011;434:25–35. doi: 10.1042/BJ20101707. [DOI] [PubMed] [Google Scholar]
- 182.Pillekamp F, Haustein M, Khalil M, Emmelheinz M, Nazzal R, Adelmann R, Nguemo F, Rubenchyk O, Pfannkuche K, Matzkies M. Contractile properties of early human embryonic stem cell-derived cardiomyocytes: beta-adrenergic stimulation induces positive chronotropy and lusitropy but not inotropy. Stem cells and development. 2012;21:2111–2121. doi: 10.1089/scd.2011.0312. [DOI] [PubMed] [Google Scholar]
- 183.Jonsson MK, Vos MA, Mirams GR, Duker G, Sartipy P, de Boer TP, van Veen TA. Application of human stem cell-derived cardiomyocytes in safety pharmacology requires caution beyond hERG. Journal of molecular and cellular cardiology. 2012;52:998–1008. doi: 10.1016/j.yjmcc.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 184.Russell B, Curtis MW, Koshman YE, Samarel AM. Mechanical stress-induced sarcomere assembly for cardiac muscle growth in length and width. Journal of molecular and cellular cardiology. 2010;48:817–823. doi: 10.1016/j.yjmcc.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Li F, Wang X, Capasso JM, Gerdes AM. Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. Journal of molecular and cellular cardiology. 1996;28:1737–1746. doi: 10.1006/jmcc.1996.0163. [DOI] [PubMed] [Google Scholar]
- 186.Kehat I, Davis J, Tiburcy M, Accornero F, Saba-El-Leil MK, Maillet M, York AJ, Lorenz JN, Zimmermann WH, Meloche S. Extracellular signal-regulated kinases 1 and 2 regulate the balance between eccentric and concentric cardiac growth. Circulation research. 2011;108:176–183. doi: 10.1161/CIRCRESAHA.110.231514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.McCain ML, Yuan H, Pasqualini FS, Campbell PH, Parker KK. Matrix elasticity regulates the optimal cardiac myocyte shape for contractility. Am J Physiol Heart Circ Physiol. 2014;306:H1525–H1539. doi: 10.1152/ajpheart.00799.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Bhana B, Iyer RK, Chen WLK, Zhao R, Sider KL, Likhitpanichkul M, Simmons CA, Radisic M. Influence of substrate stiffness on the phenotype of heart cells. Biotechnology and Bioengineering. 2010;105:1148–1160. doi: 10.1002/bit.22647. [DOI] [PubMed] [Google Scholar]
- 189.Ribeiro AJ, Ang Y-S, Fu J-D, Rivas RN, Mohamed TM, Higgs GC, Srivastava D, Pruitt BL. Contractility of single cardiomyocytes differentiated from pluripotent stem cells depends on physiological shape and substrate stiffness. Proceedings of the National Academy of Sciences. 2015;112:12705–12710. doi: 10.1073/pnas.1508073112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Bray MA, Sheehy SP, Parker KK. Sarcomere alignment is regulated by myocyte shape. Cell motility and the cytoskeleton. 2008;65:641–651. doi: 10.1002/cm.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Schiaffino S, Gorza L, Ausoni S. Troponin isoform switching in the developing heart and its functional consequences. Trends in cardiovascular medicine. 1993;3:12–17. doi: 10.1016/1050-1738(93)90022-X. [DOI] [PubMed] [Google Scholar]
- 192.Opitz CA, Leake MC, Makarenko I, Benes V, Linke WA. Developmentally regulated switching of titin size alters myofibrillar stiffness in the perinatal heart. Circulation research. 2004;94:967–975. doi: 10.1161/01.RES.0000124301.48193.E1. [DOI] [PubMed] [Google Scholar]
- 193.Weeland CJ, van den Hoogenhof MM, Beqqali A, Creemers EE. Insights into alternative splicing of sarcomeric genes in the heart. Journal of molecular and cellular cardiology. 2015;81:107–113. doi: 10.1016/j.yjmcc.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 194.Agarkova I, Auerbach D, Ehler E, Perriard J-C. A novel marker for vertebrate embryonic heart, the EH-myomesin isoform. Journal of Biological Chemistry. 2000;275:10256–10264. doi: 10.1074/jbc.275.14.10256. [DOI] [PubMed] [Google Scholar]
- 195.Bedada FB, Chan SS, Metzger SK, Zhang L, Zhang J, Garry DJ, Kamp TJ, Kyba M, Metzger JM. Acquisition of a quantitative, stoichiometrically conserved ratiometric marker of maturation status in stem cell-derived cardiac myocytes. Stem Cell Reports. 2014;3:594–605. doi: 10.1016/j.stemcr.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Brette F, Orchard C. Resurgence of cardiac t-tubule research. Physiology. 2007;22:167–173. doi: 10.1152/physiol.00005.2007. [DOI] [PubMed] [Google Scholar]
- 197.Ibrahim M, Gorelik J, Yacoub MH, Terracciano CM. The structure and function of cardiac t-tubules in health and disease. Proceedings of the Royal Society of London B: Biological Sciences. 2011;278:2714–2723. doi: 10.1098/rspb.2011.0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Reynolds JO, Chiang DY, Wang W, Beavers DL, Dixit SS, Skapura DG, Landstrom AP, Song L-S, Ackerman MJ, Wehrens XH. Junctophilin-2 is necessary for T-tubule maturation during mouse heart development. Cardiovascular research. 2013:cvt133. doi: 10.1093/cvr/cvt133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Hong T, Yang H, Zhang S-S, Cho HC, Kalashnikova M, Sun B, Zhang H, Bhargava A, Grabe M, Olgin J. Cardiac BIN1 folds T-tubule membrane, controlling ion flux and limiting arrhythmia. Nature medicine. 2014;20:624–632. doi: 10.1038/nm.3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kreipke RE, Wang Y, Miklas JW, Mathieu J, Ruohola-Baker H. Metabolic remodeling in early development and cardiomyocyte maturation. Seminars in cell & developmental biology. 2016;52:84–92. doi: 10.1016/j.semcdb.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Papanicolaou KN, Kikuchi R, Ngoh GA, Coughlan KA, Dominguez I, Stanley WC, Walsh K. Mitofusins 1 and 2 are essential for postnatal metabolic remodeling in heart. Circulation research. 2012;111:1012–1026. doi: 10.1161/CIRCRESAHA.112.274142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Yang X, Pabon L, Murry CE. Engineering adolescence: maturation of human pluripotent stem cell-derived cardiomyocytes. Circ Res. 2014;114:511–23. doi: 10.1161/CIRCRESAHA.114.300558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Piquereau J, Novotova M, Fortin D, Garnier A, Ventura‐Clapier R, Veksler V, Joubert F. Postnatal development of mouse heart: formation of energetic microdomains. The Journal of physiology. 2010;588:2443–2454. doi: 10.1113/jphysiol.2010.189670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Chung S, Dzeja PP, Faustino RS, Perez-Terzic C, Behfar A, Terzic A. Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nature clinical practice Cardiovascular medicine. 2007;4:S60–S67. doi: 10.1038/ncpcardio0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Uosaki H, Cahan P, Lee DI, Wang S, Miyamoto M, Fernandez L, Kass DA, Kwon C. Transcriptional landscape of cardiomyocyte maturation. Cell reports. 2015;13:1705–1716. doi: 10.1016/j.celrep.2015.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Hom JR, Quintanilla RA, Hoffman DL, de Mesy Bentley KL, Molkentin JD, Sheu S-S, Porter GA. The permeability transition pore controls cardiac mitochondrial maturation and myocyte differentiation. Developmental cell. 2011;21:469–478. doi: 10.1016/j.devcel.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Kolwicz SC, Purohit S, Tian R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circulation research. 2013;113:603–616. doi: 10.1161/CIRCRESAHA.113.302095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Lopaschuk GD, Jaswal JS. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. Journal of cardiovascular pharmacology. 2010;56:130–140. doi: 10.1097/FJC.0b013e3181e74a14. [DOI] [PubMed] [Google Scholar]
- 209.Kuppusamy KT, Jones DC, Sperber H, Madan A, Fischer KA, Rodriguez ML, Pabon L, Zhu W-Z, Tulloch NL, Yang X. Let-7 family of microRNA is required for maturation and adult-like metabolism in stem cell-derived cardiomyocytes. Proceedings of the National Academy of Sciences. 2015;112:E2785–E2794. doi: 10.1073/pnas.1424042112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Prendiville T, Guo H, Lin Z, Zhou P, Stevens S, He A, VanDusen N, Chen J, Zhong L, Wang D. Novel Roles of GATA4/6 in the Postnatal Heart Identified through Temporally Controlled, Cardiomyocyte-Specific Gene Inactivation by Adeno-Associated Virus Delivery of Cre Recombinase. PloS one. 2015;10:e0128105. doi: 10.1371/journal.pone.0128105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Carroll KJ, Makarewich CA, McAnally J, Anderson DM, Zentilin L, Liu N, Giacca M, Bassel-Duby R, Olson EN. A mouse model for adult cardiac-specific gene deletion with CRISPR/Cas9. Proc Natl Acad Sci U S A. 2016;113:338–43. doi: 10.1073/pnas.1523918113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, Graham DB, Jhunjhunwala S, Heidenreich M, Xavier RJ, Langer R, Anderson DG, Hacohen N, Regev A, Feng G, Sharp PA, Zhang F. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159:440–55. doi: 10.1016/j.cell.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Zhang D, Shadrin IY, Lam J, Xian HQ, Snodgrass HR, Bursac N. Tissue-engineered cardiac patch for advanced functional maturation of human ESC-derived cardiomyocytes. Biomaterials. 2013;34:5813–20. doi: 10.1016/j.biomaterials.2013.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Nunes SS, Miklas JW, Liu J, Aschar-Sobbi R, Xiao Y, Zhang B, Jiang J, Massé S, Gagliardi M, Hsieh A. Biowire: a platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Nature methods. 2013;10:781–787. doi: 10.1038/nmeth.2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Mihic A, Li J, Miyagi Y, Gagliardi M, Li S-H, Zu J, Weisel RD, Keller G, Li R-K. The effect of cyclic stretch on maturation and 3D tissue formation of human embryonic stem cell-derived cardiomyocytes. Biomaterials. 2014;35:2798–2808. doi: 10.1016/j.biomaterials.2013.12.052. [DOI] [PubMed] [Google Scholar]
- 216.Tulloch NL, Muskheli V, Razumova MV, Korte FS, Regnier M, Hauch KD, Pabon L, Reinecke H, Murry CE. Growth of engineered human myocardium with mechanical loading and vascular coculture. Circulation research. 2011;109:47–59. doi: 10.1161/CIRCRESAHA.110.237206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Jacot JG, McCulloch AD, Omens JH. Substrate stiffness affects the functional maturation of neonatal rat ventricular myocytes. Biophysical journal. 2008;95:3479–3487. doi: 10.1529/biophysj.107.124545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Davis J, Davis LC, Correll RN, Makarewich CA, Schwanekamp JA, Moussavi-Harami F, Wang D, York AJ, Wu H, Houser SR. A tension-based model distinguishes hypertrophic versus dilated cardiomyopathy. Cell. 2016;165:1147–1159. doi: 10.1016/j.cell.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Young JL, Kretchmer K, Ondeck MG, Zambon AC, Engler AJ. Mechanosensitive kinases regulate stiffness-induced cardiomyocyte maturation. Scientific reports. 2014;4 doi: 10.1038/srep06425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Jacot JG, Martin JC, Hunt DL. Mechanobiology of cardiomyocyte development. Journal of biomechanics. 2010;43:93–98. doi: 10.1016/j.jbiomech.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Chattergoon NN, Giraud GD, Louey S, Stork P, Fowden AL, Thornburg KL. Thyroid hormone drives fetal cardiomyocyte maturation. The FASEB Journal. 2012;26:397–408. doi: 10.1096/fj.10-179895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Lundy SD, Zhu W-Z, Regnier M, Laflamme MA. Structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Stem cells and development. 2013;22:1991–2002. doi: 10.1089/scd.2012.0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Wada R, Muraoka N, Inagawa K, Yamakawa H, Miyamoto K, Sadahiro T, Umei T, Kaneda R, Suzuki T, Kamiya K, Tohyama S, Yuasa S, Kokaji K, Aeba R, Yozu R, Yamagishi H, Kitamura T, Fukuda K, Ieda M. Induction of human cardiomyocyte-like cells from fibroblasts by defined factors. Proc Natl Acad Sci U S A. 2013;110:12667–72. doi: 10.1073/pnas.1304053110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Hirschy A, Schatzmann F, Ehler E, Perriard JC. Establishment of cardiac cytoarchitecture in the developing mouse heart. Dev Biol. 2006;289:430–41. doi: 10.1016/j.ydbio.2005.10.046. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.