

Summary
Reported here are twins, both of whom have a 1q21.3 microdeletion and who exhibit key features common to previously reported cases such as microcephaly and developmental delay. However, some clinical findings and deleted genes differed from those in previously reported cases. The karyotype was normal 46, XX for both of the twins. Array comparative genomic hybridization (CGH) identified a 2.6 Mb deletion on chromosome 1q21.3 (chr1: 153,514,121–156,171,335 bp) in case 1 and a 1.6 Mb deletion on chromosome 1q21.3 (chr1: 154,748,365–156,358,923 bp) in case 2. The deleted region includes DPM3, MUC1, GBA, PKLR, RIT1, and LAMTOR2 in both siblings. To the extent known, this is the second report of a 1q21.3 microdeletion in a family with mental retardation, developmental delay, seizures, and some dysmorphic features, thus expanding the phenotypic spectrum.
Keywords: 1q21.3 microdeletion syndrome, developmental delay, mental retardation, microarray-CGH, seizures
1. Introduction
The introduction of genome-wide approaches to identify deletions and duplications throughout the human genome has facilitated the discovery of numerous novel causes of intellectual disability (ID) and epilepsy (1, 2). In a clinical work-up of undiagnosed intellectual disability, array comparative genomic hybridization (CGH) can facilitate a diagnosis in 10–30% of cases. Although there is vast amounts of data on the clinical features of and standardized management guidelines for well-known classic microdeletion syndromes, systematic characterization of newly identified patients provides a host of essential information for clinicians and patients.
Only one previous report described 1q21.3 microdeletion syndrome (3), and the deleted region in question spanned about 1.4 Mb with approximate genomic location chr1:152,511,593–153,993,103 including at least 30 genes such as CHRNB2, KCNN3, HAX1, ADAR, PKLR, EFNA1, EFNA2, and EFNA3 (NCBI genome build 36). The current case report seeks to further characterize 1q21.3 microdeletion syndrome. Described here are two siblings with a 1q21.3 microdeletion that was associated with mental retardation, microcephaly, epilepsy, and some dysmorphic features.
2. Case Report
2.1. Case 1
A baby girl was born by cesarean section at 29 weeks 5 days' gestation as a set of triplets, and the girl had a birth weight of 1,240 g (Figure 1). The girl was kept in the neonatal intensive care unit because of an intraventricular hemorrhage and she also underwent surgery for a duodenal obstruction and perforation. At 4 months of age, the girl underwent surgery for hydrocephalus and a ventriculoperitoneal shunt was placed. At 5 years of age, right focal motor convulsions started. In spite of therapy with three antiepileptics, intractable seizure resumed and the girl was hospitalized for status epilepticus. The girl is now 11 years old and she has been seizure-free for three years with three antiepileptics.
Figure 1.
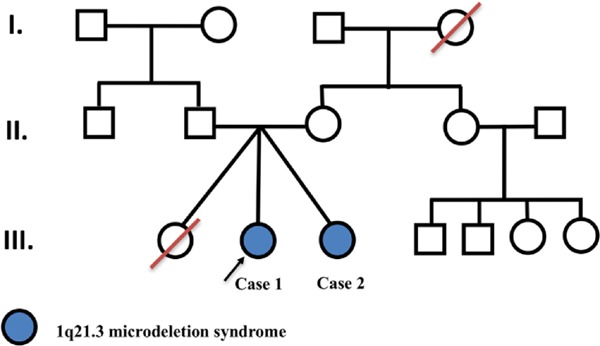
Pedigree of patients with 1q21.3 microdeletions.
A physical and neurological examination indicated growth retardation (weight and height under the 3rd percentile), pectus carinatus, scoliosis, microcephaly, spastic quadriparesis, laxity in both hands, and severe spasticity in the extremities. The girl only speaks single words, she cannot walk, and she can only sit with support.
Laboratory results revealed a normal hemogram and normal biochemical test results. Electroencephalography revealed a right frontocentral epileptiform abnormality. Brain MRI revealed periventricular leukomalacia and pontocerebellar hypoplasia (Figure 2A). Results of a karyotype analysis were normal. The girl is receiving Na-valproate, levetiracetam, and oxcarbazepine therapy.
Figure 2.
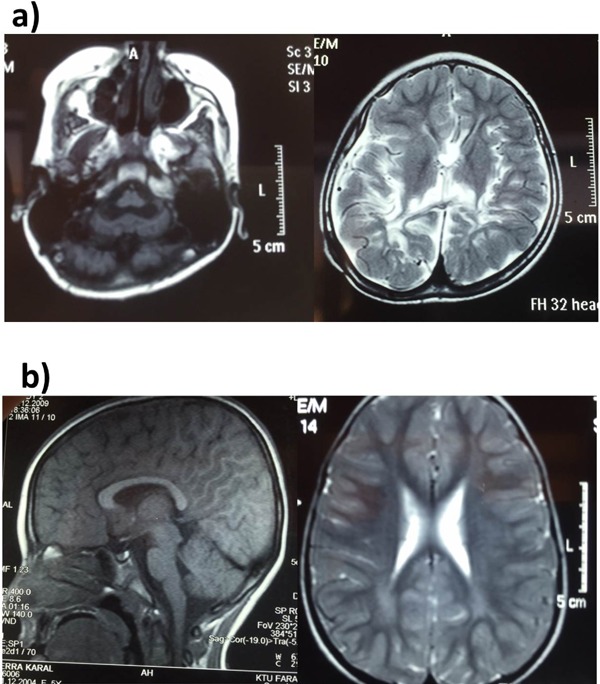
Cranial MRI findings. (a) periventricular leukomalacia and pontocerebellar hypoplasia in Case 1; (b) Periventricular hyperintensity in Case 2.
Array CGH analysis identified a 2.6 Mb deletion on chromosome 1q21.3 (chr1: 153,514,121–156,171,335 bp). The deleted region contains 68 OMIM genes. Several of these genes cause disease, including GATAD2B, TPM3, HAX1, IL6R, CHRNB2, ADAR, DPM3, MUC1, GBA, PKLR, RIT1, LAMTOR2, LMNA, and SEMA4A. Some disease-causing genes such as GATAD2B, TPM3, LMNA, and SEMA4A are present in this case but not in the case reported by Reddy et al. (Figure 3) (3). Parental array CGH analysis and FISH testing for balanced translocations were not performed because of family's decision to forego further testing. Parental testing could help to determine if the parents are carriers in terms of investigating whether the microdeletion in question is de novo or inherited. If parental testing was performed, the mutation would presumably be a de novo germline mutation because the parents are healthy and two sisters in the same family were affected with the same disorder.
Figure 3.
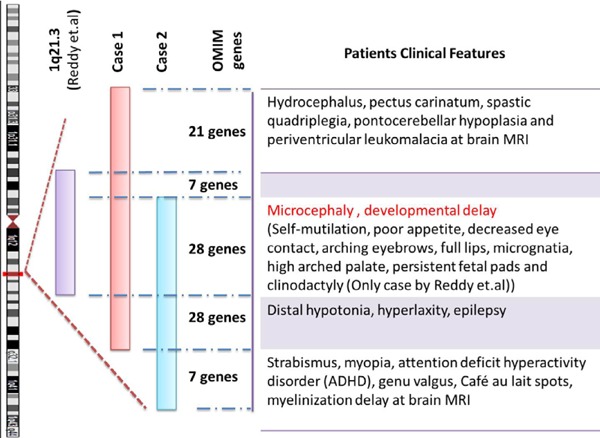
Comparison of fraternal twins to the earlier patient with a 1q21.3 microdeletion in terms of clinical-radiologic features and the deleted chromosomal region.
2.2. Case 2
The patient in this case is the sibling of the patient in case 1. After birth, the girl in case 2 was kept in the neonatal intensive care unit because of respiratory distress syndrome, hyperbilirubinemia, neonatal convulsions, and cholelithiasis. The girl could walk and speak at 2 years of age. At 6 years of age, the girl was hospitalized at another facility because of status epilepticus, hyperglycemia, and encephalitis. At that time, EEG and cranial MRI were normal, and Na-valproate therapy was started. The girl can now walk without support and she only receives risperidone therapy for attention deficit hyperactivity disorder.
On physical and neurological examination, the girl's weight was 25 kg (25%) and her height was 132 cm (50%). The girl has microcephaly, a systolic murmur, distal laxity, genu valgus, and sacral dimples. The girl can walk with a long gait.
Laboratory results revealed a normal hemogram and normal biochemical test results except for low levels of vitamin D. EEG was normal, and cranial MRI revealed bilateral periventricular hyperintensity commensurate with leukomalacia, and delayed myelination (Figure 2B). Spinal MRI was normal. The girl is now seizure-free and she can attend school. A karyotype analysis yielded normal results.
Array CGH analysis identified a 1.6 Mb deletion on chromosome 1q21.3 (chr1:154748365–156358923 bp). The deleted region in question contains 47 OMIM genes. Several of these genes cause disease, including DPM3, MUC1, GBA, PKLR, RIT1, LAMTOR2, LMNA, and SEMA4A. Some disease-causing genes such as LAMTOR2, LMNA, and SEMA4A are present in this case but not in the case reported by Reddy et al. (3). However, genes like HAX1, IL6R, CHRNB2, and ADAR were deleted in case 1 and the previously reported case of 1q21.3 microdeletion syndrome but those genes were not deleted in case 2 (Figure 3).
The third of the triplets was a girl who died 12 hours after birth because of respiratory distress. There is no other information available concerning that child.
3. Discussion
Several novel chromosome aberration syndromes have already been characterized, and undoubtedly, more descriptions will follow.
Earlier reports indicated that proximal 1q (1q21–q25) deletions present with developmental delay, microcephaly, cardiac, renal and genital abnormalities, and some dysmorphic features (4, 5). A submicroscopic deletion with a location similar/identical to the one in the current patients was previously reported by Reddy et al. (3). Reddy et al. described a 2 and a half-year-old girl with moderate mental retardation, microcephaly, arching eyebrows, low-set ears, long eyelashes, persistent fetal pads, and clinodactyly. They detected a 1.4 Mb deleted region with approximate genomic location at chr1: 152,511,593–153,993,103 (NCBI genome build 36). The deleted region in that case mostly coincided with the deleted region noted in the current patients (Figure 4). Major clinical features such as microcephaly and developmental delay were common to the current patients and the previously reported case. Furthermore, the patient in case 1 has additional clinical features like hydrocephalus, pectus carinatum, spastic quadriplegia, pontocerebellar hypoplasia, and periventricular leukomalacia on brain MRI and the patient in case 2 has additional clinical features like strabismus, myopia, genu valgus, attention deficit hyperactivity disorder, and a myelination delay on brain MRI. These results conform with the case previously reported by Reddy et al. and expand the phenotypic spectrum of 1q21.3 syndromes.
Figure 4.

Results of array CGH analysis in patients (Deleted regions). CGH, comparative genomic hybridization.
The occurrence of rare disease-causing microdeletions in the genome is not random and is possibly affected by neighboring genes in the genome (6). The high sequence similarity over large stretches of DNA makes low copy repeats (LCRs) susceptible to germline non-allelic homologous recombination (NAHR) events that cause deletion or duplication. More than 30 genomic disorders originate from NAHR-mediated events occurring in the germ line (7). Copy number variations with the same length and clustered breakpoints for a group of patients with the same disorder originate from NAHR-mediated microdeletions (6). Since the observed deletions range from 1.4 kb to 2.6 Mb in length and the breakpoint locations differ in the 2 cases reported here, a common mechanism of deletion, such as NAHR-mediated by LCRs, can be ruled out in 1q21.31 microdeletion syndrome. However, non-recurrent rearrangements vary in length, have scattered breakpoints, and the majority of these microdeletions can result from microhomology-mediated mechanisms like microhomology-mediated end-joining (MMEJ), fork stalling and template switching (FoSTeS), microhomology-mediated break-induced replication (MMBIR), serial replication slippage (SRS), or break-induced SRS (BISRS). Hence, the only option to explore the underlying mechanisms in 1q21.31 microdeletion syndrome is to characterize it at the level of base pairs and to examine the sequences found at breakpoints.
Some variations in the genomic architecture in a parent might stimulate the development of these rare germline deletions via a non-recurrent CNV, increasing the susceptibility to DNA breakage or promoting replication fork stalling. This means that recurrence of a microdeletion in different sizes would be apparent in the same family.
Although there is no parental consanguinity, the current patients could have some other recessive disorder. Array CGH is known to be unable to exclude a recessive disease in instances of a small DNA substitution or rearrangement that does not result in any loss or gain of DNA. At this point, whole-exome sequencing (WES) would provide more comprehensive information, but WES was not possible in the current cases.
Epilepsy is one of the features of this syndrome. Muhle et al. described a boy with absence seizures whose parents both had childhood absence epilepsy. A 192-kb duplication in 1q21.3, encompassing the genes IL6R, SHE, TDRD10, UBE2Q1, CHRNB2, and ADAR, was identified in the proband and his father. Both CHRNB2 and ADAR are genes potentially responsible for seizure disorders. All of the duplicated genes in that case were also deleted in the patient in Case 1. The duplication was not identified in 191 patients with independent idiopathic generalized epilepsy or in 1,157 population controls (8).
The current report has several limitations. One is that parental testing was not possible. Another is that WES was not performed. WES can be used to investigate whether there are other mutations that array CGH has failed to detect, such as small deletions and single nucleotide polymorphisms. A third limitation is that this report did not verify breakpoints in both of the current patients. Breakpoints could be mapped using methods such as quantitative PCR (qPCR), long-range PCR, and Sanger sequencing. Hence, this is an initial report, and interpretations in this report may change over time as additional cases are assembled.
In conclusion, the current report found that the 1q21.3 deletion might be a genetic risk factor in this family, contributing to mental retardation, microcephaly, epilepsy, and some dysmorphic features. This is the second report of a deletion of the 1q21.3 region and further cases are required to clarify which of the genes in the deleted interval contribute to the phenotype of the children and their long-term outcomes.
Acknowledgements
The authors wish to thank the patients and their family for their participation in this study.
References
- 1. Mefford HC, Eichler EE. Duplication hotspots, rare genomic disorders, and common disease. Curr Opin Genet Dev. 2009; 19:196-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Slavotinek AM. Novel microdeletion syndromes detected by chromosome microarrays. Hum Genet. 2008; 124:1-17. [DOI] [PubMed] [Google Scholar]
- 3. Reddy S, Dolzhanskaya N, Krogh J, Velinov M. A novel 1.4 Mb de novo microdeletion of chromosome 1q21.3 in a child with microcephaly, dysmorphic features and mental retardation. Eur J Med Genet. 2009; 52:443-445. [DOI] [PubMed] [Google Scholar]
- 4. Lo LJ, Noordhoff MS, Huang CS, Chen KT, Chen YR. Proximal deletion of the long arm of chromosome 1: [del(1) (q23–q25)]. Cleft Palate Craniofac J. 1993; 30:586-589. [DOI] [PubMed] [Google Scholar]
- 5. Pallotta R, Dalpra L, Miozzo M, Ehresmann T, Fusilli P. A patient defines the interstitial 1q deletion syndrome characterized by antithrombin III deficiency. Am J Med Genet. 2001; 104:282-286. [DOI] [PubMed] [Google Scholar]
- 6. Verdin H, D'Haene B, Beysen D, Novikova Y, Menten B, Sante T, Lapunzina P, Nevado J, Carvalho CM, Lupski JR, De Baere E. Microhomology-mediated mechanisms underlie non-recurrent disease-causing microdeletions of the FOXL2 gene or its regulatory domain. PLoS Genet. 2013; 9:e1003358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sasaki M, Lange J, Keeney S. Genome destabilization by homologous recombination in the germ line. Nat Rev Mol Cell Biol. 2010; 11:182-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Muhle H, Steinich I, von Spiczak S, et al. A duplication in 1q21.3 in a family with early onset and childhood absence epilepsy. Epilepsia. 2010; 51:2453-2456. [DOI] [PubMed] [Google Scholar]