

Abstract
For many infections transmitting to humans from reservoirs in nature, disease dispersal patterns over space and time are largely unknown. Here, a reversed genomics approach helped us understand disease dispersal and yielded insight into evolution and biological properties of Francisella tularensis, the bacterium causing tularemia. We whole-genome sequenced 67 strains and characterized by single-nucleotide polymorphism assays 138 strains, collected from individuals infected 1947-2012 across Western Europe. We used the data for phylogenetic, population genetic and geographical network analyses. All strains (n=205) belonged to a monophyletic population of recent ancestry not found outside Western Europe. Most strains (n=195) throughout the study area were assigned to a star-like phylogenetic pattern indicating that colonization of Western Europe occurred via clonal expansion. In the East of the study area, strains were more diverse, consistent with a founder population spreading from east to west. The relationship of genetic and geographic distance within the F. tularensis population was complex and indicated multiple long-distance dispersal events. Mutation rate estimates based on year of isolation indicated null rates; in outbreak hotspots only, there was a rate of 0.4 mutations/genome/year. Patterns of nucleotide substitution showed marked AT mutational bias suggestive of genetic drift. These results demonstrate that tularemia has moved from east to west in Europe and that F. tularensis has a biology characterized by long-range geographical dispersal events and mostly slow, but variable, replication rates. The results indicate that mutation-driven evolution, a resting survival phase, genetic drift and long-distance geographical dispersal events have interacted to generate genetic diversity within this species.
Keywords: epidemiology, disease transmission, human, population genetics, Francisella tularensis, genetic variation
Data Summary
This study uses whole-genome sequence data of F. tularensis samples and connected metadata, which are available through the information given in Table S1 (available in the online Supplementary Material) including the GenBank accession numbers. The single-nucleotide polymorphisms and the canSNPer software used for the canSNP analysis are available at https://github.com/adrlar/CanSNPer.
Impact Statement
In this work, we used genome data to understand biological properties and geographical spread of the bacterium Francisella. tularensis, which causes the disease tularemia. Humans may contract tularemia from infected mammals, ticks or mosquitoes or from environmental dust, but it is unclear where the bacterium survives between infections. By mapping the genomes of F. tularensis strains from many infected individuals across Western Europe, we found that tularemia has moved from east to west in Europe. Unexpectedly, we observed a movement pattern of big jumps across the continent. Our study advances the research field by showing that F. tularensis has a mechanism for long-distance transport. We additionally found more evidence that F. tularensis spends much of the time in a resting survival phase between infection episodes. More generally, this work demonstrates the value of analysing microbial genome data at large scales for learning more about an infectious organism´s biology and for interpreting epidemiological patterns of infectious diseases that currently are poorly understood.
Introduction
Geographical dispersal of microbes causing disease can be difficult to study by genetic approaches, mainly because dispersal of microbes may be rapid in relation to the rate of mutation and genetic diversification is often characterized by horizontal gene transfer events that can quickly obscure phylogenetic signatures of dispersal. It remains uncertain if barriers to geographical dispersal exist for microbes which could influence the genetic diversity of populations and would be analogous to those observed among plants and animals (Finlay, 2002; Nemergut et al., 2013). With the advent of large-scale genetic population approaches for microbes more knowledge is accumulating; results from basal studies in saline environments, experimental systems and soils indicate that spatial distance may contribute to microbial genetic diversity patterns (Low-Décarie et al., 2015; Ramette & Tiedje, 2007; Wang et al., 2015). These approaches may additionally provide novel insights into an organism´s biology (Vellend et al., 2014).
Francisella tularensis, a facultative intracellular bacterium causing the disease tularemia, is best known as a potential agent of bioterrorism due to its high virulence, low infectious dose and ease of spread by aerosol; it historically was stockpiled as a biological weapon (Dennis et al., 2001). Natural disease outbreaks present an opportunity to investigate microbial population diversity and geographical dispersal of F. tularensis, which is a bacterium with little genetic variation (Johansson & Petersen, 2010). Dispersal can be studied by investigating F. tularensis isolated from various geographic locations from diseased humans, other mammals, or transmitting arthropod vectors. There are two subspecies of F. tularensis important with respect to infection in mammals of which only the less aggressive F. tularensis subspecies holarctica exists in Europe (Dennis et al., 2001).
Tularemia has only recently been reported from Western Europe and it appears that a single genetic subpopulation of F. tularensis is specific to this region (Dempsey et al., 2007; Gyuranecz et al., 2012; Pilo et al., 2009; Vogler et al., 2009). Seasonal disease outbreaks of tularemia were first reported in central Europe in the late 1930s around Marchfeld, north of Vienna, and continued to appear intermittently in Austria, Czechoslovakia, Poland and Eastern Germany throughout the next decade (Jusatz, 1952b). In the early 1950s independent outbreaks were documented in Western Germany and France (Correspondent, 1947; Gelman, 1961; Jusatz, 1952b). Italy reported its first tularemia outbreak only in 1964 and Spain as recently as 1997 (Gutiérrez et al., 2003; Instituto de Salud Carlos III, 1997; Rinaldi et al., 1964).
We applied whole-genome and canonical single-nucleotide polymorphism (canSNP) analysis to a comprehensive set of F. tularensis samples from Western Europe to characterize microbial genetic diversity through the lens of the four classical processes of population genetics: selection, genetic drift, mutation and dispersal. Our findings demonstrate that tularemia has moved east to west in Europe in big jumps.
Methods
Study location and data collection.
A total of 205 F. tularensis subspecies holarctica strains were collected from countries in continental Western Europe, including: Belgium, Germany, France, Netherlands, Italy, Spain and Switzerland. These strains were isolated over 65 years (1947–2012) from infected humans, infected mammals in zoos, arthropod vectors, including ticks, and free-ranging wild animals. Thus, they represent a very diverse set of hosts and vectors over large spatial and temporal scales.
Genome sequencing.
Whole-genome sequences were used to identify SNPs in 67 F. tularensis samples from Western Europe. A set of 62 whole-genome sequences were generated for this study using Illumina sequencing platforms (Illumina) and five were retrieved from the public domain (see Table S1). The sequencing instruments used were HiSeq 2000 (SciLifeLabs, Uppsala, Sweden and Spietz laboratory, Spietz, Switzerland), GA IIx [Translational Genomics Research Institute (TGen), Flagstaff, Arizona], and MiSeq [Swedish Defence Research Agency (FOI), Umeå, Sweden; Northern Arizona University and TGen, Flagstaff, USA]. Steps of DNA preparation, library construction, and genome sequencing were done according to the manufacturer’s instructions. Library preparations were performed using TruSeq kits (Illumina), Nextera XT kits (Illumina), or KAPA library preparation kits (KAPA Biosystems). The KAPA kits were used with Illumina sequencing per a modified protocol including the incorporation of customized 8 bp tags for multiplexing (Kozarewa & Turner, 2011), with the adapters and oligos purchased from IDT (Integrated DNA Technologies).
CanSNP assays.
Data from comparisons of the whole genomes were used to construct 20 new canSNP assays for the characterization of F. tularensis strain samples following previously published guidelines (Birdsell et al., 2012). In addition, assays described in previous studies were used (Svensson et al., 2009; Vogler et al., 2009) to assign each sample to a phylogenetic subpopulation defined by canSNPs (see Tables S1, S2 and S3 for more information on samples and canSNP assays including primer concentrations and PCR conditions).
Genomic assembly and alignment.
The F. tularensis genome sequences from the study region were assembled using ABySS 1.5.2 (Simpson et al., 2009) and compared with a global database of more than 600 F. tularensis genomes maintained at the Swedish Defense Research Agency, Umeå, Sweden. All F. tularensis genomes in our database that were found to differ by less than 10 SNPs from any genome in the study region, and nine additional public genome sequences, were used to generate a global diversity tree (see Fig. 1a). Genome alignments were generated using a stepwise procedure: (1) each sequence was aligned with the F. tularensis strain FSC200 genome (NC 009749) to generate a nucleotide position reference. (2) All genomes were merged into a single alignment that was visually reviewed for misalignments around gaps. (3) Five nucleotides upstream and five downstream of an alignment gap were excluded to remove uncertain SNPs because read alignment in these regions is error-prone.
Fig. 1.

Whole genome neighbor-joining phylogenetic trees representing relationships among F. tularensis strains. (a) shows the relationships of 67 strains from Western Europe (Branch B.11) relative to the known global diversity within F. tularensis subsp. holarctica. (b) shows detailed relationships among strains from Western Europe. Country of origin and year of isolation are indicated at the branch tips, with colors representing different phylogenetic clades.
Phylogeny and genetic diversity.
The software mega v. 5.13 (Tamura et al., 2011) was used for the calculation of genomic distances and for phylogenetic analysis of genomic data, employing the number of differences-model and the neighbor-joining algorithm. The mean nucleotide diversity (Pi) per country was calculated using mega for countries with more than five genomes. Pi for a comparison of the East and the West part of the study region was estimated using DnaSP 5.10.01 (Librado & Rozas, 2009). Using the genome-based phylogeny and the strategically selected canSNP assays representing the branches of this phylogeny, strains with canSNP data were assigned to a node of the tree. The canSNP approach was highly accurate for node assignments but did not expose potential new genetic diversity as compared with the genomes used for reconstructing the whole-genome tree (Alland et al., 2003; Pearson et al., 2004).
Phylogeographic analysis.
Each sample was assigned to a whole-genome phylogenetic clade or to a canSNP group, mapped to geographical coordinates using Google Maps, and geographical clustering was generated by Marker Clusterer (https://github.com/googlemaps/js-marker-clusterer). The genetic network analysis was manually performed by connecting locations with identical F. tularensis canSNP genotypes. The ties connecting two locations were drawn to reflect the number of shared unique genotypes. The network was manually drawn as an arc diagram.
Analysis of genetic to geographic distance.
Genetic clades of the whole-genome phylogeny containing more than five genomes were identified and used to analyze the relationship between genetic and geographic distances. A genetic distance matrix for all pairs within a clade was created using the SNP distance between strains, and a corresponding geographic distance matrix was created using the fossil package in R 2.10 (Vavrek, 2011).
Historic and contemporary endemic regions.
Data on the spatial distribution of tularemia in Europe 1926–1955 were retrieved from publications by Jusatz (Jusatz, 1952a, b; 1955; 1961), and compared with the 1947–2012 data of this study. Comparison with the historic disease distribution was made by plotting instances of more than five strains located nearby as a cluster on a map, and by showing all strains located outside the historic disease distribution.
Mutation rate analysis.
Mutation rate estimates were made using the software BEAST 1.8.1 (Drummond et al, 2012) with 100 million iterations, out of which 10 million were used as burn-in. The lognormal relaxed clock model and the GTR without site heterogeneity substitution model was selected. The full 67 genome dataset and the set of genomes from two outbreaks in Spain (Ariza-Miguel et al, 2014) were utilized in separate analyses.
Nucleotide sequence accession numbers.
Whole-genome sequence data have been deposited at in GenBank. Accession numbers of sequence data and metadata for each sample are available in Table S1.
Results
Phylogeny for F. tularensis in Western Europe
Using a whole-genome assembly approach and SNP discovery, 67 F. tularensis strains from Western Europe were found to form a tightly clustered population distinct from all other worldwide F. tularensis subsp. holarctica genome sequences selected to represent the currently known genetic diversity of the subspecies (Fig. 1a). This tight cluster was found at the end of branch B.11 of F. tularensis subsp. holarctica and was divided further into two distinct genetic clades, B.45 and B.46 – each represented by multiple strains, and also a single strain (FDC310) separate from the other strains (Fig. 1b). The B.45 and B.46 clades were separated by just 12 SNPs. There were no conflicting SNP character states in the phylogeny (i.e. no homoplasy). The absence of homoplasy among 251 SNPs in the 1 531 265 nucleotide alignment of the 67 genomes added credibility to the phylogenetic reconstruction, but despite the temporal and spatial extent of our dataset, relationships among many F. tularensis strains within the B.45 clade remained unresolved. The clade was densely populated with 60 genomes and some of them divided into several subclades originating independently from a common internal node (Fig. 1). Such star-like phylogenetic structures with relatively long terminal branches indicate a population expansion compressed in evolutionary time. There were eight additional subclades within the B.45 clade (B.48 through B.55) that also exhibited star phylogenies. Synapomorphic SNPs shared by all of the strains within these different subclades signified their common ancestry, with 6–8 synapomorphic SNPs for the B.48 and B.52 subclades and 1–3 SNPs for the B.49–51 and B.53–55 subclades. The B.46 clade contained just six genomes and generally exhibited longer branch lengths compared with B.45, as well as a more sparsely populated hierarchical tree structure, indicating that this F. tularensis population was less abundant in Western Europe and had a longer evolutionary history.
Phylogeography
Phylogeographic analyses (Fig. 2a) revealed major differences between the two main clades. Despite high sampling intensity, strains assigned to the B.46 clade (n=9) were isolated only towards the Eastern boundary of the study area with the majority of strains isolated in the Alps region of Switzerland and Italy; no B.46 strains were recorded west of the French Alps region. In contrast, strains assigned to clade B.45 (n=195) were isolated throughout Western Europe, occurring widely across the study area from east to west and from north to south. The B.45 and B.46 strains examined in this study were isolated from diverse infected hosts, with the relatively few B.46 strains being isolated from hares, humans and a lion tamarin at a zoo, indicating that under-sampling of any particular specific B.46-reservoir in Western parts of Europe is an unlikely explanation for its absence there. Several subclades within the B.45 clade were distributed throughout the study area, including subclades B.49 and B.50. However, other subclades within the B.45 clade were restricted to specific geographical locations: strains from subclades B.48 and B.52 were only isolated in Spain and strains from subclade B.54 were only isolated in the Southeast portion of the study area.
Fig. 2.

Genetic diversity and geography of F. tularensis in Western Europe. (a) shows phylogeographic patterns of 205 strains. The colors in the circles are consistent with the genetic clade colors of the phylogenetic tree in the upper-left. The distribution of the colors within a circle corresponds to the frequency of particular genetic clades. The size of the circle represents the number of strains isolated in the region. The large circle represents the total number of strains in the study. An asterisk indicates missing information about the exact geographical location (n=15). (b) shows the mean nucleotide diversity of 67 F. tularensis genomes among different countries ordered from West to East (x-axis) and South to North (y-axis).
Genetic diversity and nucleotide substitution patterns
An analysis of SNP accumulation in the 67 whole-genome sequences revealed higher genetic diversity among strains isolated in the Eastern versus the Western part of the study area. The per genome nucleotide diversity measures were different using the Jukes and Cantor correction model measuring Pi(2)JC 0.07 in the East, and Pi(2)JC 0.05 in the West. Genetic diversity was greatest among genomes from Switzerland and its neighboring countries (Fig. 2b). The SNP patterns were further explored in F. tularensis strains from Western Europe, aiming to infer underlying biological processes. There were 207 SNPs in predicted coding regions (145 non-synonymous and 62 synonymous) amongst the total of 251 SNPs, and we identified a prominent AT mutational bias in these genomes already containing 77.8 percent AT nucleotides. (Table 1). The total number of G or C to A or T changes was 147 and the number of A or T to G or C changes was 54. Notably, the most common changes were G→A, C→T transitions, accounting for 58 percent of the mutations in the total data, a result indicating that weak forces are acting to counteract an increase in AT-content.
Table 1. Number of substitutions of the six nucleotide pairs in the coding regions of the 67 genomes.
| Substitution | Number non-synonymous (percentage)* | Number synonymous (percentage) |
|---|---|---|
| C→G, G→C | 0 (0) | 1 (0.5) |
| A→C, T→G | 7 (3) | 1 (0.5) |
| A→T, T→A | 12 (6) | 2 (1) |
| C→A, G→T | 21 (10) | 5 (2) |
| A→G, T→C | 24 (12) | 13 (6) |
| G→A, C→T | 81 (39) | 40 (19) |
| Total | 145 | 62 |
*Percentages were calculated as the number of the type of substitution event divided by the total of 207 substitutions, e.g. (7÷207)×100=3.
Historical and current tularemia distribution
A comparison of the geographical distribution of the strains analyzed in this study from 1947 to 2012 with historical data on tularemia epidemics from 1926–1955 (Jusatz, 1952a, b) revealed that historic disease areas largely have persisted (Fig. 3). The distribution of strains in our analysis reflected that tularemia was first reported in 1964 in Italy and 1997 in Spain indicating that these countries are new endemic areas.
Fig. 3.

Circles in blue with dotted margins represent F. tularensis samples analyzed in this study (1947–2012) and the circles in red represents the historical focal points between 1926 and 1955 in the corresponding regions as reported by Jusatz et al. in the 1950s. The dotted red line was marked as the boundary beyond which no tularemia cases were reported between 1926 and 1950 as per Jusatz et al. The gray arrows show the direction of migration of F. tularensis in recent years.
Estimate of mutation rate
Comparing the whole-genome phylogeny and the years of isolation for the corresponding F. tularensis isolates indicates that there was little temporal mutation signal in the dataset as a whole. Within clade B.45, with the largest number of strains whole-genome sequenced, the branch lengths are not correlated with chronological time, as strains isolated 60 years apart in Switzerland and France differed at only six SNPs even with whole-genome comparisons (Fig. 1). Using Bayesian temporal mutational analysis, our 67-genome dataset from Western Europe 1952–2012 did not have sufficient temporal structure for rate estimation. Thus, to maximize the power of the temporal mutation rate analysis, we selected whole-genome sequences representing active outbreak areas in Spain from 1998–99 (n=12) and 2007–08 (n=12) and identified a mean rate of 0.4 mutations per genome per year (1.87×10-7 mutations per site per year; see Fig. S1 and Table S4). The majority of mutations (n=45) among these 24 genomes were found at terminal tips of the phylogenetic tree and only a minority (n=24) were shared among multiple strains.
Dispersal patterns
Overall, genetic distance correlated poorly with geographic distance among genomes within the star-like clade B.45 (Fig. 4). For example, there were small genetic distances (4–9 SNPs) between pairs of strains assigned to subclades B.49 and B.51 that were separated in geographic space by 0–1750 km. In addition, pairs of strains within subclade B.50 that were separated by very small genetic distances (≤6 SNPs) were separated in geographic space by distances ranging from 150 to over 1500 km. This pattern indicates that there are few barriers to dispersal within the study area, as very similar genomes were sometimes separated by large geographic distances. Only in the two subclades that are geographically restricted to the recently emerging areas for tularemia in Spain (subclades B.48 and B.52) was there a strong correlation between genetic distance and geographic distance.
Fig. 4.
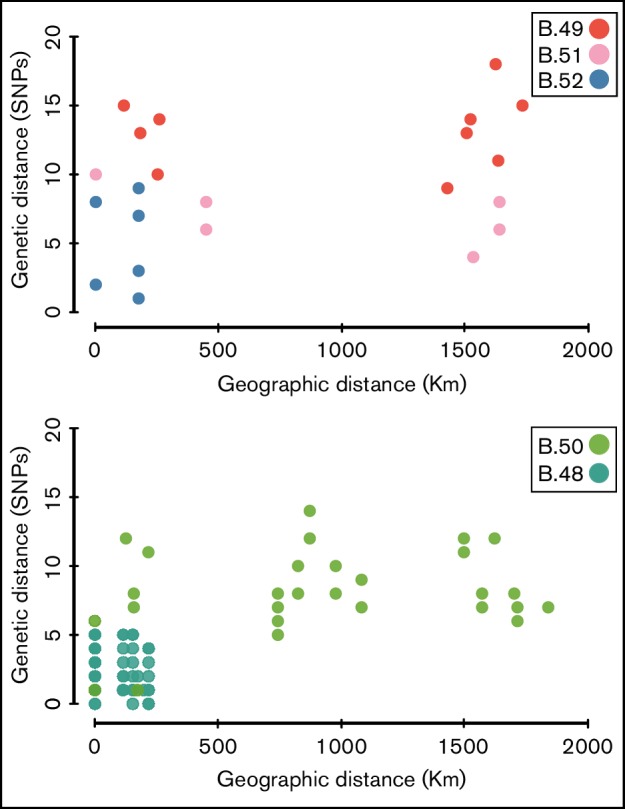
A Clade-wise comparison of genetic distance to geographic distance is plotted. The x-axis represents geographic distance between the strains in Kms and the y-axis represents genetic distance based upon SNP differences identified from whole-genome analyses. The colors of the circles are consistent with those of the clades in Fig. 1.
The genetic network analysis, which was conducted east to west across the study area using canSNP data for 205 strains, uncovered complex patterns of both local and long-distance dispersal events (Fig. 5). There were numerous examples of local dispersal events, with identical genotypes located at nearby geographic locations; this was the most abundant network pattern and was particularly common at the Eastern and Western boundaries of the study area. But there were also similar genotypes located across long geographic distances, which is consistent with past long-distance movement of recent bacterial ancestors. The network revealed many different long-distance connections between east and west locations and some intermediate-distance connections. Importantly, recent ancestors of several different genotypes appear to have been transported between the same locations (shown as thicker arcs in Fig. 5).
Fig. 5.

A geographical dispersal network of F. tularensis in continental Western Europe ordered from West to East. The pie charts at the bottom correspond to Fig. 2(a). An arc represents a possible movement of a genotype between two locations. The thickness of an arc is proportional to the number of shared genotypes at these two locations.
Discussion
Our study confirms that Western Europe was colonized by a monophyletic population of F. tularensis and indicates that this primarily occurred by clonal expansion of a specific population. The founder population originated in the Eastern boundary of Western Europe, and Western and Southern regions have been colonized by clonal descendants of this founder population. Our study also demonstrates that it is possible to translate large-scale genomic microbial population data into biological properties; we found that long-range dispersal is an important feature of tularemia ecology and that F. tularensis mutation rates are mostly slow.
There was higher genetic diversity among F. tularensis samples from around the Alps, indicating a longer evolutionary history in this region as compared with other areas of Western Europe. The presence of all the major clades – B.11, B.45, B.46 and B.47 – in Switzerland but in no other area is further evidence to indicate an evolutionarily older founder population from the Eastern boundary of the study region. In contrast, there was less genetic diversity in the Western regions of the study area. We note that these findings may be consistent with a recent colonization of Western Europe starting from the East and that this would fit with epidemiology records of the first tularemia outbreaks in Southwestern areas of Spain as recently as the 1990s (Gutiérrez et al., 2003; Instituto de Salud Carlos III, 1997). The star phylogeny of clade B.45 (i.e. a multi-furcation tree with many short branches connected at an internal node) contains the vast majority of all F. tularensis strains analyzed across Western Europe and is indicative of an evolutionary history with rapid expansion of a clone. We interpret this as a founder effect, meaning that the vast majority of F. tularensis in Western Europe was derived from a very small sample of an ancestral genetic pool.
Our findings support a model of F. tularensis biology involving outbreaks of disease being restricted to specific stationary ecosystems and landscapes, indicating that the pathogen is dependent on some specific local ecological conditions (Goethert & Telford, 2009; Svensson et al, 2009a). We found that locations of known historical tularemia outbreaks up to 1955 coincided with the distribution of strains investigated in this study, which is indicative of long-term persistence in these regions. Thus, it seems that F. tularensis has an ability to persist at certain locations and that this ability results in repeated outbreaks in those locations, as historically proposed by the Russian author Pavlovsky (Pavlovsky, 1966).
There was a puzzling mix of, on one hand, local genetic structure indicating micro-evolution with limited dispersal signified by identical or genetically very closely related strains from small geographic areas, and, on the other hand, clear deviations from these patterns. We found in several instances identical canSNP genotypes at distant locations, indicating that very long-distance and rapid movements of F. tularensis must have occurred that influenced the current genetic diversity of this bacterial population. There was also surprisingly weak correlation between genetic distance and geographic distance. We conclude that long-distance dispersal events have significantly influenced the current genetic diversity of F. tularensis, which would explain the observed patterns with canSNP identities at large distances (e.g. between Germany and Spain) and also the establishment of new regions of endemicity in Spain and Italy (Instituto de Salud Carlos III, 1997; Rinaldi et al., 1964). Our findings support the idea that the degree of dispersal limitation may be as important for microbes as it is for plants and mammals in determining the genetic diversity of populations (Pigot & Tobias, 2015).
The mechanisms of long-range dispersal of F. tularensis are unknown; possibly bacteria may move rapidly by infected domestic or wild animals, or via wind (Burrows et al., 2009). Infected hares may, for example, be imported from tularemia-endemic areas to previously tularemia-free areas, this has been suggested as a potential explanation for the emergence of the disease in Spain in the 1990s (Petersen & Schriefer, 2005); association with migratory birds is another possibility (Lopes de Carvalho et al., 2012). The European brown hare, Lepus europaeus, is recognized as an important game species throughout its distribution (Smith & Johnston, 2008). The local geographical migration of this hare is described to be restricted, but conservation actions and translocations of animals may have extended the geographical range of some hare populations including in Switzerland, France, Italy and Spain (Ferretti et al., 2010; Fischer & Tagand, 2012; Smith & Johnston, 2008). Wind-borne dispersal is another possible mechanism as F. tularensis is a prototype agent for infections acquired by inhalation of infectious aerosols (Dennis et al., 2001). Large outbreaks of natural infection have repeatedly occurred via inhalation of contaminated hay or straw dust generated in farming activities (Allue et al., 2008; Dahlstrand et al., 1971; Johansson et al., 2014; Syrjälä et al., 1985). Given the well-known propensity of F. tularensis to be part of aerosols, and its environmental survival properties, long-distance microbial dispersal may take place over vast distances like in other microbial populations (Nguyen et al., 2006; Smith et al., 2013). Notably, the occurrence of long-distance transport is not equal to the successful establishment of a new F. tularensis outbreak area; there may be high-frequency seeding of bacteria into new geographical areas but a low chance of bacterial survival and establishment due to unsuitable ecological conditions in these new areas.
The very small genetic diversity observed among the genomes collected over a 65-year time scale and, especially, the lack of correlation between mutation accumulation and time, is remarkable. It appears that the evolutionary rate for the F. tularensis genetic lineages investigated here compares with, or is lower, than the lowest rates found in recent analyses of a large set of genome collections representing a range of bacterial species (Duchêne et al., 2016). These results indicate that F. tularensis exhibits low but variable mutation rates over chronological time. We identified a mutation rate lower than one nucleotide substitution every second year per genome among a subset of strains recovered from an area in Spain; this region has emerging and recent outbreaks that should represent an area characterized by a high replication activity within the bacterial population. The overall very low or null rate of mutation in the total data set indicates a biology wherein the pathogen replicates during outbreaks and has a mechanism to survive long periods of inactivity with little replication between epidemics (Johansson et al., 2014), i.e. a resting phase for long-term survival (Romanova et al., 2000). Variable mutation rates related to higher replication rates during outbreaks have previously been suggested for Yersinia pestis (Cui et al., 2013). We acknowledge that it is problematic to assess recombination within this population due to an extensive genetic homogeneity but have found no evidence to question previous conclusions of a clonal population structure (Johansson et al., 2004, 2014; Larsson et al., 2009). In all populations with extremely little genetic diversity it is hard to know if a SNP resulted from a de novo mutation or was an import by allelic exchange of a continuous DNA stretch containing this SNP. Given the lack of homoplastic SNPs in our genomic data, however, possible events of homologous recombination are unlikely to have distorted our phylogenetic tree reconstruction (Hedge & Wilson, 2014). In future studies of possible recombination in F. tularensis, other types of mutations like indels, tandem repeats and inversions may provide additional information.
Our observations of nucleotide substitution patterns with an extreme AT-mutation bias amongst the F. tularensis genomes are in agreement with the idea that ecology and lifestyle influence genetic variation (Moran et al., 2008). It is likely that a recent host-adaptation of this pathogen confers strong genetic drift effects, because of repeated population bottlenecks in infected hosts, and a relaxed selection for many bacterial functions in an intracellular environment (Larsson et al., 2009). The large numbers of G→A or C→T transitions and C→A or G→T transversions in the F. tularensis population of Western Europe signify that selective forces acting to oppose the increase in AT content indeed are weak. An alternative explanation, AT-bias because of inefficient DNA-repair systems in F. tularensis, seems unlikely because DNA-repair genes were found to be intact in a strain from France (Sample ID FTNF002-00 in Table S1) (Larsson et al., 2009). Additional indirect evidence indicates we have captured strong genetic drift effects; the star phylogeny of the B.45 clade is probably a transient snapshot of evolution with its many subclades existing side by side in a polytomy. We have not seen such patterns in previous comparative whole-genome studies of F. tularensis (Afset et al., 2015; Johansson et al., 2014; Larsson et al., 2009) and it is expected that several of these subclades will become extinct after longer evolutionary time periods, by stochastic events or because of selection forces (Kryazhimskiy & Plotkin, 2008; Rocha et al., 2006; Wolf et al., 2009).
In conclusion, this study demonstrates how mutation-driven microbial evolution, and particularly, a biology with a resting survival phase, genetic drift effects and long-distance geographical dispersal, have interacted to form population variation in this species. The local diversity of the tularemia pathogen is influenced by two distinct components: first, a local component containing dispersal limitation wherein bacteria are accumulating genetic diversity and expanding locally; and, second, a component of long-distance movement with a very low degree of dispersal limitation resulting in genetic diversity imports and highly similar genotypes at large distances.
Acknowledgements
This work was supported by the Swedish Civil Contingencies Agency [grant number TA 014-2010-01] and the US Department of Homeland Security’s Science and Technology Directorate [award number HSHQDC-10-C-00139] pursuant to the agreement between the Kingdom of Sweden and the US government on Cooperation in Science and Technology for Homeland Security Matters. The authors R.E. and P.A. were supported by the Ministerio de Economía y Competitividad, Spain [grant number CGL2015-66962-C2-2-R]. We thank Johanna Thelaus, Per Stenberg, Andreas Sjödin, and Petter Lindgren for comments that improved the manuscript. We are grateful to the researchers, physicians, and veterinarians who contributed F. tularensis samples for this study.
Supplementary Data
Supplementary Data
Abbreviations:
- canSNP
canonical SNP
- JC
the Jukes and Cantor 1969 distance model of DNA evolution
- Indels
insertion or deletion mutation events
- SNP
Single-nucleotide polymorphism
References
- Afset J. E., Larssen K. W., Bergh K., Lärkeryd A., Sjödin A., Johansson A., Forsman M.(2015). Phylogeographical pattern of Francisella tularensis in a nationwide outbreak of tularaemia in Norway, 2011. Euro Surveill 209–14. 10.2807/1560-7917.ES2015.20.19.21125 [DOI] [PubMed] [Google Scholar]
- Alland D., Whittam T. S., Murray M. B., Cave M. D., Hazbon M. H., Dix K., Kokoris M., Duesterhoeft A., Eisen J. A., et al. (2003). Modeling bacterial evolution with comparative-genome-based marker systems: application to Mycobacterium tuberculosis evolution and pathogenesis. J Bacteriol 1853392–3399. 10.1128/JB.185.11.3392-3399.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allue M., Sopeña C. R., Gallardo M. T., Mateos L., Vian E., García M. J., Ramos J., Berjón A. C., Viña M. C., et al. (2008). Tularaemia outbreak in Castilla y León, Spain, 2007 an update. Euro Surveill 13http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=18948. [PubMed] [Google Scholar]
- Ariza-Miguel J., Johansson A., Fernández-Natal M., I, Martínez-Nistal C., Orduña A., Rodríguez-Ferri E. F., Hernández M., Rodríguez-Lázaro D.(2014). Molecular investigation of tularemia outbreaks, Spain, 1997–2008. Emerg Infect Dis 20754–761. 10.3201/eid2005.130654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birdsell D. N., Pearson T., Price E. P., Hornstra H. M., Nera R. D., Stone N., Gruendike J., Kaufman E. L., Pettus A. H., et al. (2012). Melt analysis of mismatch amplification mutation assays (Melt-MAMA): a functional study of a cost-effective SNP genotyping assay in bacterial models. PLoS One 7e32866. 10.1371/journal.pone.0032866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrows S. M., Elbert W., Lawrence M. G., Pöschl U.(2009). Bacteria in the global atmosphere – Part 1: Review and synthesis of literature data for different ecosystems. Atmos Chem Phys Discuss 910777–10827. 10.5194/acpd-9-10777-2009 [DOI] [Google Scholar]
- Correspondent (1947). Foreign letters – First Cases of Tularemia in France. J Am Med Assoc 135176. [Google Scholar]
- Cui Y., Yu C., Yan Y., Li D., Li Y., Jombart T., Weinert L. A., Wang Z., Guo Z., et al. (2013). Historical variations in mutation rate in an epidemic pathogen, Yersinia pestis. Proc Natl Acad Sci U S A 110577–582. 10.1073/pnas.1205750110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlstrand S., Ringertz O., Zetterberg B.(1971). Airborne tularemia in Sweden. Scand J Infect Dis 37–16. 10.3109/inf.1971.3.issue-1.02 [DOI] [PubMed] [Google Scholar]
- Dempsey M. P., Dobson M., Zhang C., Zhang M., Lion C., Gutiérrez-Martín C. B., Iwen P. C., Fey P. D., Olson M. E., et al. (2007). Genomic deletion marking an emerging subclone of Francisella tularensis subsp. holarctica in France and the Iberian Peninsula. Appl Environ Microbiol 737465–7470. 10.1128/AEM.00646-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis D. T., Inglesby T., V, Henderson D. A., Bartlett J. G., Ascher M. S., Eitzen E., Fine A. D., Friedlander A. M., Hauer J., et al. (2001). Tularemia as a biological weapon: medical and public health management. JAMA 2852763–2773. [DOI] [PubMed] [Google Scholar]
- Drummond A. J., Suchard M. A., Xie D., Rambaut A.(2012). Bayesian phylogenetics with BEAUti and the beast 1.7. Mol Biol Evol 291969–1973. 10.1093/molbev/mss075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchêne S., Holt K. E., Weill F.-X., Hello S. L., Hawkey J., Edwards D. J., Fourment M., Holmes E. C.(2016). Genome-scale rates of evolutionary change in bacteria. Microbial Genomics 2 10.1099/mgen.0.000094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferretti M., Paci G., Porrini S., Galardi L., Bagliacca M.(2010). Habitat use and home range traits of resident and relocated hares (Lepus europaeus, Pallas). Ital J Anim Sci 9e54 10.4081/ijas.2010.e54 [DOI] [Google Scholar]
- Finlay B. J.(2002). Global dispersal of free-living microbial eukaryote species. Science 2961061–1063. 10.1126/science.1070710 [DOI] [PubMed] [Google Scholar]
- Fischer C., Tagand R.(2012). Spatial behaviour and survival of translocated wild brown hares. Anim Biodivers Conserv 35189–196. [Google Scholar]
- Gelman A. C.(1961). The ecology of tularemia in May. Studies in Disease Ecology, 89–108. Edited by May J. M.New York: Hafner Publishing Company Inc. [Google Scholar]
- Goethert H. K., Telford S. R.3rd (2009). Nonrandom distribution of vector ticks (Dermacentor variabilis) infected by Francisella tularensis. PLoS Pathog 5e1000319. 10.1371/journal.ppat.1000319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez M. P., Bratos M. A., Garrote J., I, Dueñas A., Almaraz A., Alamo R., Rodríguez Marcos H., Rodríguez Recio M. J., Muñoz M. F., et al. (2003). Serologic evidence of human infection by Francisella tularensis in the population of Castilla y León (Spain) prior to 1997. FEMS Immunol Med Microbiol 35165–169. 10.1016/S0928-8244(03)00002-6 [DOI] [PubMed] [Google Scholar]
- Gyuranecz M., Birdsell D. N., Splettstoesser W., Seibold E., Beckstrom-Sternberg S. M., Makrai L., Fodor L., Fabbi M., Vicari N., et al. (2012). Phylogeography of Francisella tularensis subsp. holarctica, Europe. Emerg Infect Dis 18290–293. 10.3201/eid1802.111305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedge J., Wilson D. J.(2014). Bacterial phylogenetic reconstruction from whole genomes is robust to recombination but demographic inference is not. MBio 5e02158. 10.1128/mBio.02158-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Instituto de Salud Carlos III (1997). Brote de tularemia en Castilla y León. Boletín Epidemiológico Semanal 5249–256. [Google Scholar]
- Johansson A., Farlow J., Larsson P., Dukerich M., Chambers E., Byström M., Fox J., Chu M., Forsman M., et al. (2004). Worldwide genetic relationships among Francisella tularensis isolates determined by multiple-locus variable-number tandem repeat analysis. J Bacteriol 1865808–5818. 10.1128/JB.186.17.5808-5818.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson A., Petersen J. M.(2010). Genotyping of Francisella tularensis, the causative agent of tularemia. J AOAC Int 931930–1943. [PubMed] [Google Scholar]
- Johansson A., Lärkeryd A., Widerström M., Mörtberg S., Myrtännäs K., Öhrman C., Birdsell D., Keim P., Wagner D. M., et al. (2014). An outbreak of respiratory tularemia caused by diverse clones of Francisella tularensis. Clin Infect Dis 591546–1553. 10.1093/cid/ciu621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jusatz H. J.(1952a). [Second report on the propagation of tularemia into middle and western Europe in present time; geomedical investigations on the development in the last decade and epidemiological prognosis]. Z Hyg Infektionskr 134350–374. [PubMed] [Google Scholar]
- Jusatz H. J.(1952b). Tularemia in Europe, 1926–1951. Welt-Suchen Atlas, 7–16. Edited by Rodenwaldt E.Hamburg: Falk-Verlag. [Google Scholar]
- Jusatz H. J.(1955). [Incidence of tularemia in Mainfranken 1949–53; a geomedical analysis]. Arch Hyg Bakteriol 139189–199. [PubMed] [Google Scholar]
- Jusatz H. J.(1961). The geographical distribution of tularemia throughout the world, 1911–1959. Welt-Suchen Atlas, 7–12. Edited by Rodenwaldt E.Hamburg: Falk-Verlag. [Google Scholar]
- Kozarewa I., Turner D. J.(2011). 96-plex molecular barcoding for the Illumina Genome Analyzer. Methods Mol Biol 733279–298. 10.1007/978-1-61779-089-8_20 [DOI] [PubMed] [Google Scholar]
- Kryazhimskiy S., Plotkin J. B.(2008). The population genetics of dN/dS. PLoS Genet 4e1000304. 10.1371/journal.pgen.1000304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson P., Elfsmark D., Svensson K., Wikström P., Forsman M., Brettin T., Keim P., Johansson A.(2009). Molecular evolutionary consequences of niche restriction in Francisella tularensis, a facultative intracellular pathogen. PLoS Pathog 5e1000472. 10.1371/journal.ppat.1000472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado P., Rozas J.(2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 251451–1452. 10.1093/bioinformatics/btp187 [DOI] [PubMed] [Google Scholar]
- Lopes de Carvalho I., Zé-Zé L., Alves A. S., Pardal S., Lopes R. J., Mendes L., Núncio M. S.(2012). Borrelia garinii and Francisella tularensis subsp. holarctica detected in migratory shorebirds in Portugal. Eur J Wildl Res 58857–861. 10.1007/s10344-012-0617-3 [DOI] [Google Scholar]
- Low-Décarie E., Kolber M., Homme P., Lofano A., Dumbrell A., Gonzalez A., Bell G.(2015). Community rescue in experimental metacommunities. Proc Natl Acad Sci U S A 11214307–14312. 10.1073/pnas.1513125112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran N. A., McCutcheon J. P., Nakabachi A.(2008). Genomics and evolution of heritable bacterial symbionts. Annu Rev Genet 42165–190. 10.1146/annurev.genet.41.110306.130119 [DOI] [PubMed] [Google Scholar]
- Nemergut D. R., Schmidt S. K., Fukami T., O'Neill S. P., Bilinski T. M., Stanish L. F., Knelman J. E., Darcy J. L., Lynch R. C., et al. (2013). Patterns and processes of microbial community assembly. Microbiol Mol Biol Rev 77342–356. 10.1128/MMBR.00051-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T. M., Ilef D., Jarraud S., Rouil L., Campese C., Che D., Haeghebaert S., Ganiayre F., Marcel F., et al. (2006). A community-wide outbreak of legionnaires disease linked to industrial cooling towers-how far can contaminated aerosols spread? J Infect Dis 193102–111. 10.1086/498575 [DOI] [PubMed] [Google Scholar]
- Pavlovsky E. N.(1966). Natural nidality of transmissible diseases. Urbana Illinois: University of Illinois Press. [Google Scholar]
- Pearson T., Busch J. D., Ravel J., Read T. D., Rhoton S. D., U'Ren J. M., Simonson T. S., Kachur S. M., Leadem R. R., et al. (2004). Phylogenetic discovery bias in Bacillus anthracis using single-nucleotide polymorphisms from whole-genome sequencing. Proc Natl Acad Sci U S A 10113536–13541. 10.1073/pnas.0403844101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen J. M., Schriefer M. E.(2005). Tularemia: emergence/re-emergence. Vet Res 36455–467. 10.1051/vetres:2005006 [DOI] [PubMed] [Google Scholar]
- Pigot A. L., Tobias J. A.(2015). Dispersal and the transition to sympatry in vertebrates. Proc Biol Sci 28220141929. 10.1098/rspb.2014.1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilo P., Johansson A., Frey J.(2009). Identification of Francisella tularensis cluster in central and western Europe. Emerg Infect Dis 152049–2051. 10.3201/eid1512.080805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramette A., Tiedje J. M.(2007). Multiscale responses of microbial life to spatial distance and environmental heterogeneity in a patchy ecosystem. Proc Natl Acad Sci U S A 1042761–2766. 10.1073/pnas.0610671104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi A., Cervio C., Frittoli M., Mandelli G.(1964). Descrizione di un focolaio di tularemia in Italia (nota preliminare). [Description of an outbreak of tularaemia (preliminary note)]. Sel. Vet 5353–363. [Google Scholar]
- Rocha E. P., Smith J. M., Hurst L. D., Holden M. T., Cooper J. E., Smith N. H., Feil E. J.(2006). Comparisons of dN/dS are time dependent for closely related bacterial genomes. J Theor Biol 239226–235. 10.1016/j.jtbi.2005.08.037 [DOI] [PubMed] [Google Scholar]
- Romanova L., V, Mishan'kin B. N., Pichurina N. L., Vodop'ianov S. O., Saiamov S. R.(2000). [Noncultivatable forms of Francisella tularensis]. Zh Mikrobiol Epidemiol Immunobiol, 11–15. [PubMed] [Google Scholar]
- Simpson J. T., Wong K., Jackman S. D., Schein J. E., Jones S. J., Birol I.(2009). ABySS: a parallel assembler for short read sequence data. Genome Res 191117–1123. 10.1101/gr.089532.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. T., Johnston C. H.(2008). Lepus europaeus 2008. Available online: www.iucnredlist.org [Accessed date on 20 November 2016].
- Smith D. J., Timonen H. J., Jaffe D. A., Griffin D. W., Birmele M. N., Perry K. D., Ward P. D., Roberts M. S.(2013). Intercontinental dispersal of bacteria and archaea by transpacific winds. Appl Environ Microbiol 791134–1139. 10.1128/AEM.03029-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson K., Bäck E., Eliasson H., Berglund L., Granberg M., Karlsson L., Larsson P., Forsman M., Johansson A.(2009a). Landscape epidemiology of tularemia outbreaks in Sweden. Emerg Infect Dis 151937–1947. 10.3201/eid1512.090487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson K., Granberg M., Karlsson L., Neubauerova V., Forsman M., Johansson A.(2009b). A real-time PCR array for hierarchical identification of Francisella isolates. PLoS One 4e8360. 10.1371/journal.pone.0008360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syrjälä H., Kujala P., Myllylä V., Salminen A.(1985). Airborne transmission of tularemia in farmers. Scand J Infect Dis 17371–375. [DOI] [PubMed] [Google Scholar]
- Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S.(2011). mega5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 282731–2739. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vavrek M. J.(2011). Fossil: Palaeoecological and palaeogeographical analysis tools. Palaeontologia Electronica 141-16 http://palaeo-electronica.org/2011_1/238/index.html. [Google Scholar]
- Vellend M., Lajoie G., Bourret A., Múrria C., Kembel S. W., Garant D.(2014). Drawing ecological inferences from coincident patterns of population- and community-level biodiversity. Mol Ecol 232890–2901. 10.1111/mec.12756 [DOI] [PubMed] [Google Scholar]
- Vogler A. J., Birdsell D., Price L. B., Bowers J. R., Beckstrom-Sternberg S. M., Auerbach R. K., Beckstrom-Sternberg J. S., Johansson A., Clare A., et al. (2009). Phylogeography of Francisella tularensis: global expansion of a highly fit clone. J Bacteriol 1912474–2484. 10.1128/JB.01786-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Van Nostrand J. D., Deng Y., Lu X., Wang C., Zhou J., Han X.(2015). Scale-dependent effects of climate and geographic distance on bacterial diversity patterns across northern China's grasslands. FEMS Microbiol Ecol 91fiv133. 10.1093/femsec/fiv133 [DOI] [PubMed] [Google Scholar]
- Wolf J. B., Künstner A., Nam K., Jakobsson M., Ellegren H.(2009). Nonlinear dynamics of nonsynonymous (dN) and synonymous (dS) substitution rates affects inference of selection. Genome Biol Evol 1308–319. 10.1093/gbe/evp030 [DOI] [PMC free article] [PubMed] [Google Scholar]
Data Bibliography
- 1.Antwerpen, M. H., Schacht, E., Kaysser, P. & Splettstoesser, W. D. Complete Genome Sequence of a Francisella tularensis subsp. holarctica Strain from Germany Causing Lethal Infection in Common Marmosets. Genome Announc, 1. GenBank Accession #: PRJNA175244 (2013). [DOI] [PMC free article] [PubMed]
- 2.Atkins, L. M., Holder, M. E., Ajami, N. J., Metcalf, G. A., Weissenberger, G. M., Wang, M., Vee, V., Han, Y., Muzny, D. M., Gibbs, R. A. & other authors. High-Quality Draft Genome Sequence of Francisella tularensis subsp. holarctica Strain OR96-0246. Genome Announc. 3. GenBank Accession #: PRJNA30669 (2015). [DOI] [PMC free article] [PubMed]
- 3.Barabote, R. D., Xie, G., Brettin, T. S., Hinrichs, S. H., Fey, P. D., Jay, J. J., Engle, J. L., Godbole, S. D., Noronha, J. M., Scheuermann, R. H. & other authors. Complete genome sequence of Francisella tularensis subspecies holarctica FTNF002-00. PLoS One, 4, e7041. GenBank Accession #: PRJNA20197 (2009). [DOI] [PMC free article] [PubMed]
- 4.Baylor College of Medicine (BCM), USA. GenBank Accession #s: PRJNA30633 & PRJNA30635 (2008).
- 5.Coolen, J. P., Sjödin, A., Maraha, B., Hajer, G. F., Forsman, M., Verspui, E., Frenay, H. M., Notermans, D. W., de Vries, M. C., Reubsaet, F. A. & other authors. Draft genome sequence of Francisella tularensis subsp. holarctica BD11-00177. Stand Genomic Sci, 8, 539-47. GenBank Accession #: PRJNA177784 (2013). [DOI] [PMC free article] [PubMed]
- 6.Johansson, A., Lärkeryd, A., Widerström, M., Mörtberg, S., Myrtennäs, K., Öhrman, C., Birdsell, D., Keim, P., Wagner, D. M., Forsman, M. & other authors. An outbreak of respiratory tularemia caused by diverse clones of Francisella tularensis. Clin Infect Dis, 59, 1546-53. EMBL Nucleotide Sequence Database (ENA) Accession #s: ERS353713 & ERS353729 (2014). [DOI] [PMC free article] [PubMed]
- 7.Karlsson, E., Svensson, K., Lindgren, P., Byström, M., Sjödin, A., Forsman, M. & Johansson, A. The phylogeographic pattern of Francisella tularensis in Sweden indicates a Scandinavian origin of Eurosiberian tularaemia. Environmental Microbiology, 15, 634-645. GenBank Accession #: PRJNA89145 (2013). [DOI] [PubMed]
- 8.La Scola, B., Elkarkouri, K., Li, W., Wahab, T., Fournous, G., Rolain, J. M., Biswas, S., Drancourt, M., Robert, C., Audic, S. & other authors. Rapid comparative genomic analysis for clinical microbiology: the Francisella tularensis paradigm. Genome Res, 18, 742-50. GenBank Accession #: PRJNA19645 (2008). [DOI] [PMC free article] [PubMed]
- 9.Petrosino, J. F., Xiang, Q., Karpathy, S. E., Jiang, H., Yerrapragada, S., Liu, Y., Gioia, J., Hemphill, L., Gonzalez, A., Raghavan, T. M. & other authors. Chromosome rearrangement and diversification of Francisella tularensis revealed by the type B (OSU18) genome sequence. J Bacteriol, 188, 6977-85. GenBank Accession #: PRJNA17265 (2006). [DOI] [PMC free article] [PubMed]
- 10.Sjödin, A., Svensson, K., Öhrman, C., Ahlinder, J., Lindgren, P., Duodu, S., Johansson, A., Colquhoun, D. J., Larsson, P. & Forsman, M. Genome characterisation of the genus Francisella reveals insight into similar evolutionary paths in pathogens of mammals and fish. BMC Genomics, 13, 268. GenBank Accession #: PRJNA73369 (2012). [DOI] [PMC free article] [PubMed]
- 11.Svensson, K., Sjödin, A., Byström, M., Granberg, M., Brittnacher, M. J., Rohmer, L., Jacobs, M. A., Sims-Day, E. H., Levy, R., Zhou, Y. & other authors. Genome sequence of Francisella tularensis subspecies holarctica strain FSC200, isolated from a child with tularemia. J Bacteriol, 194, 6965-6. GenBank Accession #: PRJNA16087 (2012). [DOI] [PMC free article] [PubMed]
- 12.Swedish Defence Research Agency (FOI), Sweden. GenBank Accession #s: PRJNA285145 & PRJNA355394 (2016).
- 13.Wittwer, M. Spiez laboratory, Switzerland. GenBank Accession #: PRJNA286987 (2016).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.