

ABSTRACT
RNA interference (RNAi) is a gene-silencing mechanism that plays an important role in gene regulation in a number of eukaryotic organisms. Two core components, Dicer and Argonaute, are central in the RNAi machinery. However, the physiological roles of Dicer and Argonaute in the entomopathogenic fungus Metarhizium robertsii have remained unclear. Here, the roles of genes encoding Dicer (M. robertsii dcl1 [Mrdcl1] and Mrdcl2) and Argonaute (Mrago1 and Mrago2) proteins in M. robertsii were investigated. The results showed that the Dicer-like protein MrDCL2 and Argonaute protein MrAGO1 are the major components of the RNAi process occurring in M. robertsii. The Dicer and Argonaute genes were not involved in the regulation of growth and diverse abiotic stress response in M. robertsii under the tested conditions. Moreover, our results showed that the Dicer and Argonaute gene mutants demonstrated reduced abilities to produce conidia, compared to the wild type (WT) and the gene-rescued mutant. In particular, the conidial yields in the Δdcl2 and Δago1 mutants were reduced by 55.8% and 59.3%, respectively, compared with those from the control strains. Subsequently, for the WT and Δdcl2 mutant strains, digital gene expression (DGE) profiling analysis of the stage of mycelium growth and conidiogenesis revealed that modest changes occur in development or metabolism processes, which may explain the reduction in conidiation in the Δdcl2 mutant. In addition, we further applied high-throughput sequencing technology to identify small RNAs (sRNAs) that are differentially expressed in the WT and the Δdcl2 mutant and found that 4 known microRNA-like small RNAs (milRNAs) and 8 novel milRNAs were Mrdcl2 dependent in M. robertsii.
IMPORTANCE The identification and characterization of components in RNAi have contributed significantly to our understanding of the mechanism and functions of RNAi in eukaryotes. Here, we found that Dicer and Argonaute genes play an important role in regulating conidiation in M. robertsii. Our study also demonstrates that diverse small RNA pathways exist in M. robertsii. The study provides a theoretical platform for exploration of the functions of Dicer and Argonaute genes involved in RNAi in fungi.
KEYWORDS: Argonaute, Dicer, Metarhizium robertsii, RNA interference
INTRODUCTION
RNA interference (RNAi) is a gene silencing mechanism that plays an important role in regulating gene expression from fungi, plants, and animals, and it has become a valuable molecular tool for analyzing the functions of many genes (1, 2). The 20- to 30-nucleotide (nt) small RNA (sRNA) molecules have emerged as powerful regulators of gene expression and genome stability via conserved eukaryotic RNAi-related pathways (3). Dicer and Argonaute (AGO), as the two core proteins within the sRNA regulatory pathways, are involved in the initiation and maintenance of the mechanism central to this mode of gene regulation (4, 5). In the RNA-silencing pathway, Dicer cleaves long double-stranded RNA (dsRNA) into small interfering RNAs (siRNAs) (6–8) and also excises mature microRNAs (miRNAs) from pre-miRNAs in the cytoplasm (9). Generally, one strand of these sRNAs is then loaded into the RNA-induced-silencing complex (RISC), which contains AGOs. Under participation of AGOs with sequences complementary to the sRNAs, silencing then occurs by either cleavage or blocking translation of the target mRNA. Mature miRNAs and siRNAs are bound by AGOs, which participate in viral defense (10), posttranscriptional gene regulation (11), and transposon silencing (12, 13), processes which in turn affect somatic and germ line development. Both Dicer and AGO proteins are highly conserved from archaea to eukarya and expand largely in some plants and animals, and many organisms encode multiple members of the family (14). In distinct organisms, multiple AGOs and Dicers are involved in the RNAi pathway. Although these subtypes are similar to each other in amino acid sequence and structure, they participate in different sRNA regulatory pathways in regulating growth and development, as well as in response to abiotic and biotic stress (4, 15, 16). RNAi is mediated by sRNAs of about 20 to 30 nucleotides, and as the sRNAs regulators, Dicer and AGO have played an important role in this biological process (17).
In filamentous fungi, studies of Neurospora, Magnaporthe, Trichoderma, Botrytis cinerea, Aspergillus, and other species have provided a wealth of data supporting the notions that RNAi is widely conserved and that sRNA machinery components are involved in gene regulation, heterochromatin formation, and genome defense (18). In these fungi, gene regulation by RNAi has an important role in the growth, development, reproduction, and defense of cells against parasitic nucleotide sequences of viruses and transposons, as well as in the interactions with their hosts (19). M. robertsii, as a filamentous entomopathogenic fungus, is gaining prominence as an important pathogen used on a moderate scale for the biological control of several insect pests that cause important economic losses in agriculture. The application of M. robertsii as a biocontrol agent is a promising option in reducing the use of chemical acaricides, resulting in benefits for the environment. The technique of RNAi has been used successfully to suppress target gene expression in Metarhizium (20–23), and high-throughput sequencing of sRNAs in M. robertsii has led to the discovery of microRNA-like small RNAs (milRNAs) (24), both of which suggest the existence of the molecular machinery for the ribonucleoprotein complex known as the RISC in M. robertsii. However, as two core components in the RNAi machinery, Dicer and AGO in M. robertsii have physiological roles that remain unclear.
Currently, the genome sequence data on the assembled draft genome sequence of M. robertsii, which provides a basis of identifying RNA silencing-associated genes in the fungus, are available (25). In the present study, the characteristics and functions of Dicer and AGO genes in M. robertsii were systematically investigated.
RESULTS
Characteristics and expression profiles of Dicer and Argonaute genes.
We chose the sequences of Dicer-like 1/2 and QDE-1/2/3 of Neurospora crassa as reference sequences to retrieve their orthologs in M. robertsii, because Dicer and AGO were intensively studied in the model fungus N. crassa (26). Two Dicer-like (DCL) genes (GenBank accession numbers EFY96289.1 and EFY99337.1, named Mrdcl1 and Mrdcl2 [i.e., the M. robertsii dcl1 and dcl2 genes], respectively) and two AGO genes (accession numbers EFZ02610.1 and EFZ03314.1, named Mrago1 and Mrago2 [i.e., the M. robertsii ago1 and ago2 genes], respectively) were identified in the genome of M. robertsii. Bioinformatic analysis indicated that both the MrDCL1 and MrDCL2 proteins contain 5 conserved domains, including a DExD domain, a helicase C domain, a Duf283 domain, and two RNase III domains; MrDCL2 also includes a dsRNA-binding domain (DS-RBD) (Fig. 1). The MrDCL2 protein in M. robertsii is allied closely in the phylogenetic tree with the Cryphonectria parasitica DCL2 protein (see Fig. S1 in the supplemental material). Furthermore, both the MrAGO1 and MrAGO2 proteins contain a PAZ domain and a PIWI domain (Fig. 1). The PAZ domain of MrAGO1 is composed of 50 amino acids (from residues 438 to 487) and shares 49% homology with the PAZ domain of the QDE-2 protein in N. crassa. The C-terminal PIWI domain of MrAGO1 includes 316 amino acids (residues 665 to 980) and has 70.4% similarity with the domain in the N. crassa QDE-2 protein. The PIWI domain of MrAGO2 (residues 649 to 973) shares 63.3% homology with the PIWI domain of the QDE-2 protein in N. crassa. We aligned the PIWI domains of the two MrAGOs to detect whether MrAGO1 and MrAGO2 possessed the conserved catalytic residues and could potentially act as the slicer component of the RISC (Fig. S2). In the conserved DDH residues, H was replaced by aspartate in MrAGO1 and MrAGO2. Furthermore, all H798 sites in the MrAGO1/MrAGO2 group of fungi were replaced by proline, consistent with the previous studies that showed a variety of H798 residues in plants (5, 27–29) (Fig. S2). Moreover, the MrAGO1 and MrAGO2 proteins in M. robertsii are closely allied in the phylogenetic tree with the N. crassa QDE-2 protein and C. parasitica AGL2 protein, respectively (Fig. S3).
FIG 1.
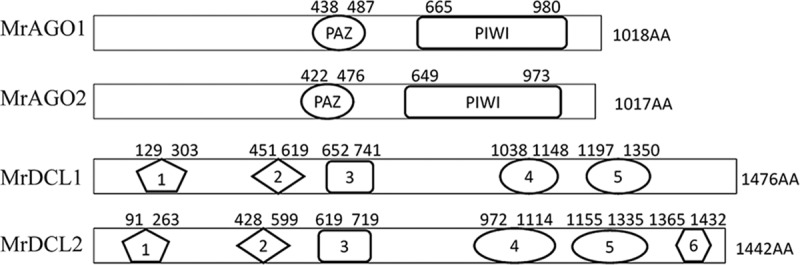
Peptide domain analysis of MrDCL and MrAGO proteins. 1, DExD domain; 2, helicase C domain; 3, Duf283 domain; 4, RNase III domain; 5, RNase III domain; 6, dsRNA-binding (DS-RBD) domain.
To further explore the possible roles of the genes Mrdcl1, Mrdcl2, Mrago1, and Mrago2 in different developmental stages, expression profiles of Mrdcl1, Mrdcl2, Mrago1, and Mrago2 in mycelial growth, early conidiogenesis, late conidiogenesis, and pure-spore stage were analyzed by quantitative reverse transcription-PCR (qRT-PCR), and the results show that the expression levels of Mrdcl1, Mrdcl2, Mrago1, and Mrago2 were detected in all investigated stages (Fig. 2). In particular, the expression trends of Mrdcl1, Mrdcl2, Mrago1, and Mrago2 were highly consistent in each stage, while their expression levels were stage specific. The expression of Mrdcl1 and Mrago1 increased from the mycelial growth stage to the pure-spore stage, and the highest expression levels of the genes were seen in the pure-spore stage, while the expression levels of Mrdcl2 and Mrago2 showed the relatively highest expression in the late-conidiogenesis stage.
FIG 2.

Expression profiles of Mrdcl1, Mrdcl2, Mrago1, and Mrago2 in different developmental stages of M. robertsii. Data are expressed as means ± SE of values from three independent experiments. Student’s t test was used to determine the statistical significance of differences between groups. Differences are considered significant where P < 0.05.
Disruption and complementation of Dicer and AGO.
To characterize the physiological functions of the Dicer and AGO genes in M. robertsii, we generated gene replacement mutants for each gene using different dominant selectable marker genes, BAR or BEN, as described in Materials and Methods.
The Δdcl1, Δdcl2, Δago1, and Δago2 gene deletion mutants, in which the named genes were inserted into the binary vectors pBar-dcl1, pBen-dcl2, pBar-ago1, and pBen-ago2, respectively, were constructed by replacing the coding regions with the BAR or BEN gene cassette.
We also constructed a Δago1 Δago2 double mutant strain and a Δdcl1 Δdcl2 double mutant strain for further functional analysis of Dicer and Argonaute genes. The Δdcl1 Δdcl2 mutant was constructed by replacing the coding region of Mrdcl1 in the ΔMrdcl2 mutant with the binary vector pBar-dcl1, and the Δago1 Δago2 mutant was constructed by replacing the coding region of Mrago1 in the ΔMrago2 mutant with the binary vector pBar-ago1.
Complementation strains (the cpΔdcl1, cpΔdcl2, cpΔago1, and cpΔago2 strains) were obtained by transforming cpBen-dcl1, cpBar-dcl2, cpBen-ago1, and cpBar-ago2 into the Δdcl1, Δdcl2, Δago1, and Δago2 mutants, respectively. All the recombinant strains were confirmed by PCR and reverse transcription-PCR (RT-PCR) (Fig. S4A to C).
Roles of Dicers and AGOs in asexual reproduction.
The growth rates of Dicer and AGO mutants were examined by incubating wild-type (WT) and mutant cultures at 25°C on Sabouraud-dextrose-yeast extract agar (SDAY) medium. All deletion mutants exhibited growth rates and phenotypic characteristics similar to those of the WT (data not shown). Osmotic, oxidative, and cell wall effects were examined on plates containing NaCl, H2O2, and SDS, respectively, with the data presented as the percentage of growth inhibition (GI). Contrary to our expectations, the GI of the Dicer and AGO mutants in the presence of stressful chemicals was not significantly different from that shown by the control strains (data not shown).
However, when conidia from the WT, the mutants, and the complemented mutants (control strains) were quantified, the results showed that the conidial yields in the Δdcl1, Δdcl2, Δdcl1 Δdcl2, Δago1, Δago2, and Δago1 Δago2 mutants were reduced by 27.0%, 55.8%, 40.2%, 59.3%, 46.0%, and 56.8%, respectively, compared with those from the control strains, which indicated a significant reduction of conidial yield in each deletion mutant, especially in the Δdcl2 and Δago1 mutants, compared with the yield of the WT (Fig. 3). The data may suggest that Mrdcl2 and Mrago1 are the major genes responsible for regulating conidiation compared with Mrdcl1 and Mrago2, respectively. These results indicate a role for Dicer and AGO in the regulation of asexual development in M. robertsii.
FIG 3.

Production of conidia of the WT and the Mrdcl1, Mrdcl2, Mrago1, and Mrago2 deletion mutants. Data are expressed as means ± SE of values from three independent experiments. Student’s t test was used to determine the statistical significance of differences between groups. Differences are considered significant where P < 0.05.
Mrdcl2 regulates the transcriptional response during conidiation.
Because Dicers are upstream in the RNAi pathway in charge of generating sRNAs by processing double-stranded RNA precursors, the conidial yield of the Δdcl2 mutant is significantly decreased. Therefore, to further elucidate the possible mechanism involved in the sporulation of M. robertsii affected by the Mrdcl2 gene, we examined the set of differentially expressed genes (DEGs) for mycelial growth (MY; conidia grown on SDAY for 1 day) and conidiogenesis (CO; mycelia grown on SDAY for 2.5 days) in the WT and the Δdcl2 mutant. The results show that a total of 4,264 differentially expressed genes (3,065 genes upregulated and 1,199 genes downregulated) for the WT and 4,397 genes (3,082 genes upregulated and 1,315 genes downregulated) for the Δdcl2 mutant were detected (Table S1). The expression of a number of these downregulated genes, such as those for cyclin-like protein (GenBank accession number MAA_00500), ATP-dependent DNA helicase PIF1 (MAA_10537), pyruvate carboxylase subunit A (MAA_01657), and the propionyl-coenzyme A (propionyl-CoA) carboxylase beta chain (MAA_01658), is increased in the Δdcl2 mutant, suggesting that those drastically suppressed genes were likely responsible for the sporulation defections in the Δdcl2 mutant.
To validate the differentially expressed genes detected by RNA sequencing (RNA-seq) data, we tested them by qRT-PCR in a different biological replicate. Overall, the 18 selected genes were successfully validated (Table 2).
TABLE 2.
Verification of DGE results by qRT-PCR analysis
| Gene ID | Gene product descriptiona | Primer sequence (5′–3′) | Value fromb: |
|||
|---|---|---|---|---|---|---|
| DGEc | qRT-PCRd | DGEe | qRT-PCRf | |||
| MAA_06313 | Developmental regulator flbA | ACTCCAAAGGGCATCACG | 0.97 | 3.27 | −0.02 | −1.02 |
| CAACAAAGCGGCGGAATA | ||||||
| MAA_01254 | Subtilisin-like serine protease | CGAACGATAGCCGTGGTC | 14.35 | 18.76 | −1.50 | −5.01 |
| GAAAGCCGCATTGAGCAG | ||||||
| MAA_09336 | Glutamine synthetase | CCGATTATTGACAACCACG | 1.26 | 4.46 | 0.55 | 2.10 |
| AAGGCATTTCCGAGTCCA | ||||||
| MAA_04342 | Zinc finger domain-containing protein | TTTCCAATCTACGACGACA | 2.37 | 5.96 | 0.56 | 4.21 |
| AGTCCGATTGATGGTCTTC | ||||||
| MAA_06808 | Secretion pathway protein Sls2/Rcy1 | ACGGAGGCGGTTTGAAGA | 0.86 | 2.85 | −1.41 | −4.72 |
| AATGTCGTCGCTTGTGGC | ||||||
| MAA_07188 | FAD-binding protein | AACCTGGCTATTAAAGTCACGC | 5.87 | 14.30 | −2.25 | −3.62 |
| CACCCGACTTGGCTTGGAC | ||||||
| MAA_02988 | APSES transcription factor | GCAAGGCACCAACCCACT | 2.03 | 3.12 | 1.05 | 4.41 |
| TGCTGCTCCGTAGGCTGA | ||||||
| MAA_02740 | Chitin synthase A | TGCGAACAGGGAAACCAG | 0.92 | 4.01 | −0.44 | −1.56 |
| GACCCGAGTTCGAGAAGAG | ||||||
| MAA_03529 | MAPKK kinase | ACCGAAAGTCGTATGTATCCT | 0.78 | 3.14 | −0.17 | −1.06 |
| CCACTGATTGTCATCCCAC | ||||||
| MAA_03342 | Integral membrane protein | ACTGATGTTGTTGCGGTGAT | 12.99 | 10.32 | −3.43 | −5.72 |
| AAATCTGCCAACCAAAGG | ||||||
| MAA_03181 | Serine/threonine protein kinase domain protein | ATCTTCACATCTGGCACGAC | 0.85 | 2.76 | 0.02 | 1.01 |
| GTTTCCGTGGGCAACATAC | ||||||
| MAA_03488 | Guanine nucleotide-binding protein alpha-subunit | TCGTGGTGTTGAGGAGTG | 0.43 | 1.15 | −0.52 | −1.09 |
| ATGCGGTAGGTCAAGTCG | ||||||
| MAA_02691 | Conidiophore development protein HymA | GCGGCTGTCATCCTCTAT | 0.29 | 1.42 | −0.10 | −1.03 |
| ATCGGCTGCTACCTCAA | ||||||
| MAA_03203 | Catalase A | TTTGGCACCTTTCGTCTCC | −3.68 | −10.20 | −1.16 | −4.12 |
| CAGTTGCCCTCTTGCGTGT | ||||||
| MAA_05842 | rRNA-processing protein Efg-1 | TTCAACCCAAAGCGTCAG | −3.01 | −6.51 | −1.52 | −3.27 |
| TTTGTCCGAGACGGTAGAT | ||||||
| MAA_08075 | Carboxylesterase, type B | ACCTCCAGTCGGAAACCT | −6.76 | −10.34 | −3.33 | −7.87 |
| GGTGCTCGTTGGAGTCTTA | ||||||
| MAA_10232 | Ankyrin repeat-containing protein | GTTTCACAAGAATCCTCCTC | 14.15 | 19.02 | −1.12 | −2.89 |
| AACGGATTGACGAGATGACT | ||||||
| MAA_02493 | Ribosomal protein L13 | TCGCCTCGCCTATTCACG | −1.85 | −6.26 | −0.03 | −1.78 |
| CCAGATCGGCTTGTGCTT | ||||||
APSES, ASM-1, Phd1, StuA, EFG1, and Sok2 family; MAPKK, mitogen-activated protein kinase kinase.
Positive numbers indicate that the genes were upregulated; negative numbers indicate that the genes were downregulated.
Log2(FPKM of WT CO/FPKM of WT MY), where FPKM is fragments per kilobase of transcript per million mapped reads.
Gene expression ratio of WT CO to WT MY.
Log2(FPKM of the Δdcl2 mutant CO/FPKM of the Δdcl2 mutant MY).
Gene expression ratio of the Δdcl2 mutant CO to the Δdcl2 mutant MY.
Enrichment analysis of gene ontology (GO) terms for DEGs in the WT and the Δdcl2 mutant was conducted to clarify which processes are more relevant to conidiation in M. robertsii. The results indicated that the upregulated genes are significantly enriched (P < 0.05) in 31 GO categories in both the WT and the Δdcl2 mutant. Interestingly, the downregulated genes in the WT are significantly enriched in 54 GO categories (23 in biological processes, 18 in molecular functions, and 13 in cellular components), while those in the Δdcl2 mutant are significantly enriched in 85 GO terms (39 in biological processes, 21 in molecular functions, and 25 in cellular components) (Table S2). Furthermore, we found that 2,263 upregulated and 713 downregulated genes are shared by the two strains by comparing the DEGs from the WT and the Δdcl2 strains (Fig. 4A and Table S3). The upregulated genes are involved mostly in processes such as the oxidation-reduction process (GO:0055114), transmembrane transport (GO:0055085), and the carbohydrate metabolic process (GO:0005975), while the downregulated genes are involved mainly in processes such as translation (GO:0006412), ribosomal biogenesis (GO:0042254), rRNA processing (GO:0006364), and mycelium development (GO:0043581). The results indicated that the DEGs in the Δdcl2 mutant might affect the regulation of the conidiation in M. robertsii (Fig. 4A and Table S3).
FIG 4.
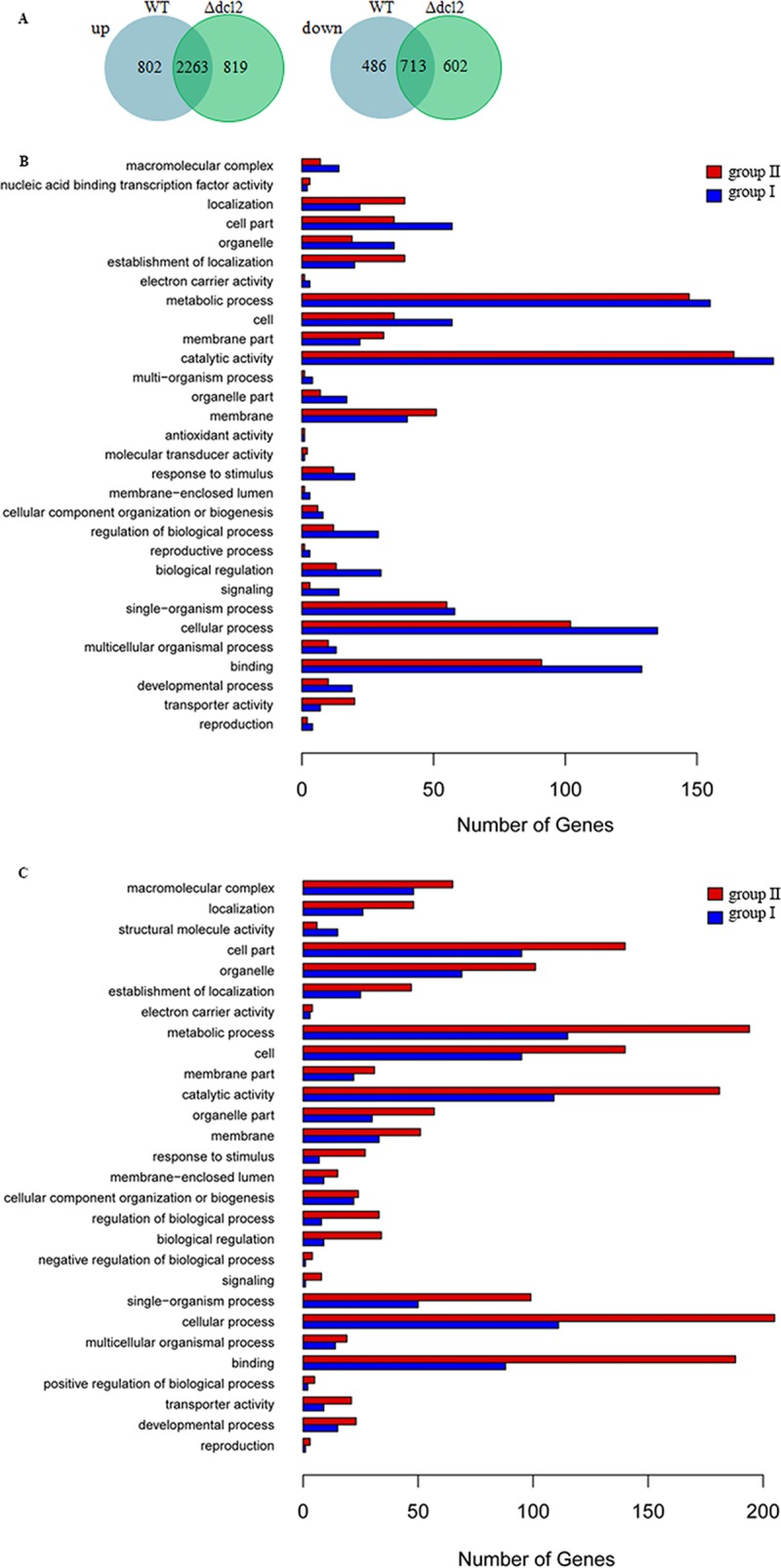
Analysis of the differentially expressed genes in the WT and Δdcl2 strains. (A) Overlap between upregulated (up) and downregulated (down) genes in the WT and Δdcl2 mutant strains; (B and C) GO terms of upregulated (B) and downregulated (C) genes of group I (specific for the WT) and group II (specific for the Δdcl2 mutant).
As a result of surveying genes, 1,288 genes (802 upregulated and 486 downregulated genes) were found to be differentially expressed only in the WT (group I), and at least some of them, such as those for cytochrome P450 CYP5148B3 (GenBank accession number MAA_01512; fold change, 11.0), flavin adenine dinucleotide (FAD) binding domain-containing protein (MAA_07499; fold change, 11.95), and fungus-specific transcription factor (MAA_09943; fold change, 11.1), may be required for conidiation in M. robertsii. To a certain extent, the DEGs (819 upregulated and 602 downregulated genes) found only in the Δdcl2 mutant (group II), including those for cytochrome b5 reductase protein (MAA_08854; fold change, 12.4), polyketide synthase (MAA_08549; fold change, −10.2), and TRI14-like protein (MAA_00154; fold change, 12.1), might inhibit the sporulation of M. robertsii. It is interesting to note that the number of group II downregulated genes that were involved in enrichment in GO terms was much larger than that in group I (Fig. 4B and C).
Mrdcl2 is involved in the biogenesis of small RNAs.
Combined with the results of phenotype and bioinformatic analyses, we speculated that Mrdcl2 may be the major gene involved in sRNA biosynthesis, compared with Mrdcl1 in M. robertsii.
Here, to further examine sRNAs that may be generated by the Mrdcl2 gene of M. robertsii, we constructed cDNA libraries of sRNAs from both the WT and Δdcl2 mutant strains. After removal of the barcode and exclusion of sequences that were shorter than 18 nucleotides, totals of 11,404,976 and 12,622,473 clean reads were obtained from the WT and the Δdcl2 mutant, respectively (Table S4). The unique sRNAs in the WT differed significantly from those in the Δdcl2 mutant, and only 7.39% of unique sRNAs were shared by both the WT and Δdcl2 mutant strains (Fig. 5A). These reads were mapped to the M. robertsii genome, and they coincided with a variety of genomic features, including tRNAs, rRNAs, small nuclear RNAs (snRNAs), and small nucleolar RNAs (snoRNAs) (Table S5). We found that the sRNAs identified from both the WT and Δdcl2 mutant strains were 18 to 30 nt in length, consistent with the range of lengths previously observed for sRNAs in fungi. The sRNAs of the selected population peaked at 21 nt in length, reaching 14.27% in the WT and only 8.88% in the Δdcl2 mutant (Fig. 5B).
FIG 5.

Characteristics of small RNAs from the WT and Δdcl2 mutant strains. (A) Proportion of the public and special sequences of sRNAs; (B) size distribution of small RNAs.
In addition, by analyzing the 5′-nucleotide bias in sRNAs, especially sRNAs of 21 nt, we observed that uracil (U) was more frequently found at their 5′ end in the WT and that this bias was guanine (G) in the Δdcl2 mutant (Fig. 6A and B). As shown in Fig. 6C and D, it is clear that the nucleotide biases at every position of the sRNAs differ significantly between the WT and the Δdcl2 mutant. Furthermore, analysis of the distributions of sRNAs on chromosomes showed that the sRNAs from the WT and the Δdcl2 mutant have distinct chromosomal distributions (Fig. S5). These results showed that the sRNAs differ in their modes of biogenesis in the Δdcl2 mutant and the WT, which indicated that Mrdcl2 plays an important role in sRNA biogenesis in M. robertsii.
FIG 6.

Analysis of nucleotide biases of sRNAs. (A and B) 5′-end nucleotide biases of 18- to ∼30-nt sRNAs in the Δdcl2 mutant (A) and WT strain (B); (C and D) nucleotide bias at each position of sRNAs in the Δdcl2 mutant (C) and WT strain (D).
It has been reported that several milRNAs were identified in M. robertsii by our laboratory (24); therefore, the expression of these milRNAs was further investigated in the WT and the Δdcl2 mutant. As shown in Fig. 7 and Table S6, 4 known milRNAs and 8 novel milRNAs were not detected in the Δdcl2 mutant, indicating that these milRNAs might be Mrdcl2 dependent. The expression levels of milRNA-1 and milRNA-8 did not change significantly between the Δdcl2 mutant and the WT. Interestingly, 1 known milRNA (man-milR-12) and 4 novel milRNAs were identified in the Δdcl2 mutant but not in the WT. These results suggested that Mrdcl2 was involved in milRNA biosynthesis in M. robertsii.
FIG 7.

Comparisons of expression levels of milRNAs in the WT and Δdcl2 mutant.
DISCUSSION
RNAi is a conserved eukaryotic gene silencing mechanism in the regulation of gene expression at both the posttranscriptional and transcriptional levels. Here, Dicers and AGOs, as core proteins of RNAi, were analyzed and characterized from M. robertsii.
Phylogenetic trees were constructed using the neighbor-joining method and plotted with MEGA, showing that MrDCL2 in M. robertsii was clustered in the phylogenetic tree with Dicer2 of N. crassa, an RNase III-like enzyme central to the majority of sRNA generation pathways (Fig. S1). Moreover, the MrAGO1 protein in M. robertsii is allied closely in the phylogenetic tree with N. crassa QDE-2, a canonical AGO protein (Fig. S3). Many studies have revealed that Dicer-like proteins are evolutionarily conserved in many eukaryotic organisms. Previous studies indicated that MDL-2 has a distinct role in siRNA accumulation, and no functional redundancy exists between MDL-1 and MDL-2 in the RNA-silencing pathway in Magnaporthe oryzae, which is similar to the results obtained with Drosophila melanogaster (30, 31). However, the two Dicer-like proteins appear redundant in transgene-induced posttranscriptional gene silencing in N. crassa (32). Many genes, such as Dicer, AGO, and RNA-dependent RNA polymerase (RdRp), involved in the sRNA regulatory RNAi pathway were expressed at different development stages or in specific organs, or both, which provides information regarding their functions (27). In the present study, we analyzed the expression of Dicers by relative qRT-PCR at different developmental stages of M. robertsii. The upregulation expression of Mrdcl2 at the late-conidiogenesis stage may suggest that Mrdcl2 is involved in the RNAi pathway to regulate gene expression in that stage. Based on the results of sequence analysis and qRT-PCR analysis, we speculated that Mrdcl2, not Mrdcl1, is the major Dicer gene that processes dsRNA into siRNA in the RNAi mechanism in vegetative growth and development, which is consistent with what occurs in most fungi (30, 32, 33).
Phenotypic analyses of yeasts and filamentous fungi with mutations in RNAi pathway components have revealed that although RNAi may play important roles in various aspects of development, homeostasis, and disease, its functions may vary significantly among different fungal species. In this study, to further investigate the potential roles of Dicers and AGOs (including Mrago1, Mrago2, Mrdcl1, and Mrdcl2), gene deletion and complementation were performed via Agrobacterium-mediated transformation, and the resultant strains were analyzed by PCR and RT-PCR. The results showed that all deletion mutants exhibited growth rates and colony morphology on SDAY or various environmental-stress media (including NaCl, H2O2, and SDS) similar to those of the WT strain (data not shown). This suggests that in M. robertsii, the RNAi machinery is not involved in the regulation of vegetative growth and diverse stress responses under the tested conditions. Moreover, we found that sporulation of all mutants was impaired. In particular, the conidial yields in the Δdcl2 and Δago1 mutants were reduced by 55.8% and 59.3%, respectively, compared with those of control strains. This is consistent with what has been observed in Trichoderma atroviride (34).
Transcriptomic analysis at the genome-wide level is a valuable tool to identify differentially expressed genes that are regulated by signaling pathways. For instance, digital gene expression (DGE) profiling revealed that genes involved in cell wall construction, conidiation, stress tolerance, cell cycle control, and calcium transport were downregulated in a mutant of Metarhizium acridum with a deletion of Cna (35). In the present study, to explore the underlying mechanism of effect on the sporulation by Mrdcl2, we used RNA-seq to systematically investigate differentially expressed genes that are regulated by Mrdcl2 during the MY and CO stages of M. robertsii. The results show that altered expression levels of genes, such as cyclin-like protein (GenBank accession number MAA_00500), cytochrome b5 reductase protein (MAA_08854), and TRI14-like protein (MAA_00154), may explain the phenotypes observed in reproductive growth in Mrdcl2 mutants. It has been reported that conidiation was associated with a coordinated downregulation of the expression of genes encoding ribosomal proteins in Aspergillus fumigatus (36). In agreement with this, many genes, including ribosomal protein L13 (MAA_02493), 50S ribosomal protein L14 (MAA_07112), 30S ribosomal protein S13 (MAA_06819), ribosomal protein S19 (MAA_08323), and ribosomal protein L10e (MAA_03051), encoding ribosomal proteins were repressed significantly during conidiation only in the WT and not in the Δdcl2 mutant. This suggests that during conidiation, there is a correlation with the synthesis of those proteins necessary for conidiation. Especially, since many genes involved in conidiation, including flbC, fluG, flbA, phiA, stuA, medA, wetA, brlA, and abaA, are repressed in the Δdcl2 mutant compared with the WT, this suggests that the mutant is affected during conidiation. Furthermore, genes such as wetA, brlA, and abaA have been identified as part of a central regulatory pathway that functions with other genes to control conidiation-specific gene expression and to determine the order of gene activation during conidiophore development and spore maturation in Aspergillus nidulans (37). In addition, previous research revealed that two developmental regulatory genes, stuA and medA, were necessary for spore formation in A. nidulans (38, 39). Therefore, differential expression of several genes between the Δdcl2 mutant and the WT were analyzed by qRT-PCR. The results show that the same genes were repressed in the Δdcl2 mutant, which is consistent with the previous findings for A. nidulans. It suggests that these genes may also function in promoting conidiation in M. robertsii.
Combined with the results of bioinformatic and qRT-PCR analyses, we speculated that Mrdcl2, not Mrdcl1, is the major Dicer gene whose enzyme processes dsRNA into sRNA (miRNA or siRNA), which is consistent with what occurs in most fungi (30, 32, 33). Therefore, characteristics and expression profiles of sRNAs (18 to 30 nt in length) from the Δdcl2 mutant were further analyzed by high-throughput sequencing and compared to those of the WT. Our results indicated that the Δdcl2 mutant lost the main characteristic features of the sRNAs found in the WT. Furthermore, the 5′-nucleotide bias was different from that of the WT, which contrasts with those observed for AGO-bound guide sRNAs in other fungi (40–42). Moreover, several sRNAs show differential expression in the Δdcl2 mutant and WT, which suggests that the sRNAs generated by Mrdcl2 may be associated with the regulation of conidiation in M. robertsii (see Table S6 in the supplemental material). In particular, milRNAs, including those in N. crassa (40), Penicillium marneffei (43), Sclerotinia sclerotiorum (44), and Fusarium graminearum (45), have been identified, and they have an important function in regulating different biological processes in these fungi. Previously, it was discovered by our group that milRNAs may play an important role in the regulation of conidiation in M. robertsii (24). In the present study, differential expression of milRNAs in WT and the Δdcl2 mutant was further investigated, and the results show that a total of 4 known milRNAs and 8 novel milRNAs dependent on Mrdcl2 were identified from sRNAs. Intriguingly, 4 novel milRNAs were found in the Δdcl2 mutant but not in the WT; that is, biosynthesis of the 4 milRNAs is independent of Mrdcl2, which indicates that a diverse small RNA pathway exists in M. robertsii. Indeed, a previous report indicated that there are at least four different pathways for milRNA production in the model fungus N. crassa, which required a distinct combination of Dicers, QDE-2, the exonuclease QIP, and an RNase III domain-containing protein, MRPL3 (40). Thus, further investigations should be considered to explore the biological functions of these proteins in milRNAs through experimental approaches in M. robertsii.
MATERIALS AND METHODS
Fungal strains and culture conditions.
M. robertsii strain ARSEF 23 was kindly provided by Chengshu Wang. Conidia were obtained from potato dextrose agar at 25°C for 12 days, cultured (107 conidia/ml; the same below unless mentioned otherwise) onto cellophane (0.5-μm pore) on SDAY, and incubated at 25°C. Fungal biomass was harvested at 1.5 days (mycelial growth), 2 days (early conidiogenesis), 3 days (late conidiogenesis), and 12 days (pure-spore stage) after incubation.
Sequence retrieval and phylogenetic analyses of Dicer and AGO families.
M. robertsii genome and transcriptome sequence data were downloaded from the NCBI (http://www.ncbi.nlm.nih.gov/). The BLASTp algorithm, underpinned by the Pfam and CDD databases, was used for searches of conserved protein domains or motifs.
The amino acid sequences of Dicer and AGO proteins from M. robertsii and other organisms obtained from GenBank were aligned using CLUSTALW (46). Phylogenetic trees were constructed using the neighbor-joining method and plotted with MEGA (47).
qRT-PCR analysis of Mrdcl1, Mrdcl2, Mrago1, and Mrago2 transcription in different developmental stages of M. robertsii.
Total RNAs from different developmental stages of M. robertsii were isolated using TRIzol reagent (Invitrogen, USA). Nucleic acid concentrations were measured with an ND-1000 spectrophotometer (NanoDrop, USA), and 1 μg of RNA was reverse transcribed using the PrimeScript first-strand cDNA synthesis kit (TaKaRa, China). Reverse-transcribed cDNA samples were used as the template for qRT-PCR amplification using SYBR green (TaKaRa, China) and a 7500 real-time PCR system (Applied Biosystems). Specific primers for the M. robertsii glyceraldehyde 3-phosphate dehydrogenase (gpd) gene (GenBank accession number MAA_07675) were used as an internal control for normalization (48). The cDNAs from three biological samples were used for analysis, and all the reactions were run in triplicate. qRT-PCR was performed using 1 μl of cDNA in a final volume of 10 μl containing 5 μl of 2× SYBR Premix Ex Taq II, 0.2 μl of ROX reference dye II, and 0.4 μl of specific primers for each AGO and Dicer (10 μM) (Table 1), with the following thermal cycling profile: 95°C for 30 s, followed by 40 cycles of amplification (95°C for 5 s, 60°C for 34 s). The threshold cycle (CT) was determined using the default threshold settings. Melting curves were also measured for the specificity of primers. Relative expression was calculated according to the 2−ΔΔCT method (49).
TABLE 1.
Sequences of primers used
| Category and gene | Primer name | Sequence (5′ to 3′) | Notes |
|---|---|---|---|
| Gene replacement | |||
| Mrdcl1 | Mrdcl1-5F | CGGAATTCGCGAAACAACAGCCAGAGTA | |
| Mrdcl1-5R | CGGGATCCGCAAACAACCCAGACCAGA | ||
| Mrdcl1-3F | GGACTAGTTCGATTTAGTGGGAATGAGC | ||
| Mrdcl1-3R | GCTCTAGACATCGCTTGAATCTCCTGTG | ||
| Mrdcl1-NF | TTGATTGCGATTCGGGTAG | PCR identification of Mrdcl1 deletion transformants | |
| Mrdcl1-NR | CGTCAACATCCTCCTCACC | ||
| Mrdcl2 | Mrdcl2-5F | AACTGCAGCAAAGGTTCCTCGCCAGTCC | |
| Mrdcl2-5R | AACTGCAGATCCCGCCATTCAACTAACG | ||
| Mrdcl2-3F | GGACTAGTCAGAAACAGCCGTTACATCC | ||
| Mrdcl2-3R | GCTCTAGAGCTCTGCCATTATTCTTGTCG | ||
| Mrdcl2-NF | CCGACACCTGGGATACGAT | PCR identification of Mrdcl2 deletion transformants | |
| Mrdcl2-NR | AGTTGCTCGCTCCTTTACG | ||
| Mrago1 | Mrago1-5F | CGGAATTCATCGCTTCCCGACTGTGA | |
| Mrago1-5R | CGGGATCCTGGCCGCTGCTGCTAAAT | ||
| Mrago1-3F | GGACTAGTTGGTCTCGTCGCTTCTGT | ||
| Mrago1-3R | GCTCTAGATGCTTGGGCATCGACTTG | ||
| Mrago1-NF | TCCCGAGACACTGATAGGC | PCR identification of Mrago1 deletion transformants | |
| Mrago1-NR | CAACGACTTATGGACCTG | ||
| Mrago2 | Mrago2-5F | AACTGCAGTTGACCTCGGTGCTTTCG | |
| Mrago2-5R | AACTGCAGGGCGCTGAGGGTTGCTAT | ||
| Mrago2-3F | GGACTAGTAGTCGGATATTGTGAAAGCG | ||
| Mrago2-3R | GCTCTAGACCTTTGGCTTGTATGTCTGC | ||
| Mrago2-NF | ATGTCTAGTGGGCATAGAGGTG | PCR identification of Mrago2 deletion transformants | |
| Mrago2-NR | AGCTGCTGAGCTGTCTGG | ||
| BAR | bar-F | GGAGGTCAACAATGAATGCC | |
| bar-R | CCACGTCATGCCAGTTCC | ||
| BEN | ben-F | ATGGCTACCTACTCCGTCGTG | |
| ben-R | CTCGTCCATACCCTCACCA | ||
| Gene complementation | |||
| Mrdcl1 | Mrdcl1cp5F | GCTCTAGAGCGAAACAACAGCCAGAG | |
| Mrdcl1cp3R | GCTCTAGATCCCGTCGTGTTCTTCTT | ||
| Mrdcl2 | Mrdcl2cp5F | GCTCTAGAGCCAAGGCAGTGAAGTAT | |
| Mrdcl2cp3R | GCTCTAGACTGCCATTATTCTTGTCG | ||
| Mrago1 | Mrago1cp5F | GCTCTAGAGACGAAAGAGGCATACACT | |
| Mrago1cp3R | GCTCTAGACTGCTTGGGCATCGACTT | ||
| Mrago2 | Mrago2cp5F | GCTCTAGAGGTCACCAAATTGCACCTA | |
| Mrago2cp3R | GCTCTAGAGAGCCACTGCCAGTATAAGT | ||
| Gene expression analysis and RT-PCR identification of deletion transformants | |||
| Mrdcl1 | Mrdcl1F | GAAGGCGGTCACTGTAATGG | |
| Mrdcl1R | ATGGGTATGACGGATGCTTG | ||
| Mrdcl2 | Mrdcl2F | CCGACTTCCTTCATCCTGTT | |
| Mrdcl2R | TTCGCAATCAATGTGACCTC | ||
| Mrago1 | Mrago1F | GAGAAGACCGAAGTAAAGGATG | |
| Mrago1R | CGGACGGAATGTTGATAGTG | ||
| Mrago2 | Mrago2F | GCATCAATCAGACGGTGGAG | |
| Mrago2R | ATTAGAGGAGGAGCCAGGTGAG | ||
| gpd | gpdF | GACTGCCCGCATTGAGAAG | |
| gpdR | AGATGGAGGAGTTGGTGTTG |
Gene deletion and complementation.
Targeted gene deletion of Mrdcl1, Mrdcl2, Mrago1, and Mrago2 genes was performed by homologous recombination via Agrobacterium-mediated fungal transformation, according to the procedure of Wang et al. (50), and pDHt-SK-bar/pDHt-SK-ben vectors were also kindly provided by Chengshu Wang (50, 51). Briefly, the 5′- and 3′-end-flanking regions of Mrdcl1 were amplified using the Dream Taq DNA polymerase (Fermentas, USA). The genomic DNA was used as the template in PCR along with the primer pairs Mrdcl1-5F/Mrdcl1-5R and Mrdcl1-3F/Mrdcl1-3R (Table 1), respectively. The purified 5′- and 3′-end-flanking fragments were subsequently cloned into the EcoRI/BamHI and SpeI/XbaI restriction enzyme sites in the binary vector pDHt-SK-bar (conferring resistance against ammonium glufosinate) to produce the binary vector pBar-dcl1 for Agrobacterium-mediated fungal transformation. M. robertsii transformation mediated by Agrobacterium tumefaciens was according to the protocol of Fang et al., with modifications, as described previously (52). A. tumefaciens strain AGL-1, containing pBar-dcl1, was cultured at 28°C for 16 to 20 h in yeast extract broth (YEB) medium (0.5% sucrose, 0.1% yeast extract, 0.5% peptone, 0.05% MgSO4·7H2O) supplemented with kanamycin (50 μg/ml) and carbenicillin (50 μg/ml), with shaking at 220 rpm. The A. tumefaciens cells were diluted to an optical density at 660 nm (OD660) of 0.15 in induction medium (IM) with or without 200 μM acetosyringone (AS). The cells were then grown in IM at 28°C with shaking at 220 rpm until an OD660 value of 0.5 to 0.8 was reached. M. robertsii conidia were harvested in 0.05% Tween 80 aqueous solution, and the conidial suspension was filtered through sterile nonwoven fabrics to remove mycelia. The working conidial concentrations were adjusted to 106 conidia/ml with sterile double-distilled water (ddH2O). One hundred microliters of a conidial suspension was mixed with an equal volume of A. tumefaciens cells in IM (OD660 = 0.5 to 0.8) with or without AS. The mixture (200 μl per plate) was spread on IM agar plates in the presence or absence of AS. After cocultivation at 28°C for 48 h, the cellophane sheet was transferred to M-100 plates containing ammonium glufosinate (200 μg/ml) as a selection agent for transformants and cefotaxime (300 μg/ml) to kill the A. tumefaciens cells and was incubated at 28°C. Putative transformants were visible at approximately 7 days. Individual transformants were transferred into SDAY plates for further analysis.
For mutant complementation, the Mrdcl1 gene was amplified together with the promoter region and terminator region using the primer pair Mrdcl1cp5F/Mrdcl1cp3R, and the product was digested with XbaI and then inserted into the binary vector pDHt-SK-ben (conferring resistance against benomyl) to transform the Δdcl1 mutant for obtaining the complement strain.
Transformants were verified by PCR, and RT-PCR analysis was performed using the primer pair Mrdcl1F/Mrdcl1R (Table 1). The gpd gene was used as an internal control.
Deletions of the Mrdcl2, Mrago1, and Mrago2 genes were performed by using the same method, and all primers used are listed in Table 1. To generate Dicer and AGO double mutants, the gene replacement constructs for Mrdcl1 and Mrago1 were transformed to the Δdcl2 and Δago2 mutants, respectively. The double-knockout mutants were also verified by PCR and RT-PCR analyses with the primers (Table 1).
Assays for cellular responses to chemical stresses.
To investigate the cellular responses on chemical stresses, 2-μl aliquots of conidial suspension were centrally spotted onto the plates of SDAY alone (control) or supplemented with a sensitive concentration of H2O2 (2 mM) for oxidative stress, SDS (0.025%) for cell wall perturbation, and NaCl (1 M) for hyperosmotic stress. After cultivation at 25°C for 7 days, the mean diameter of each fungal colony was cross-measured in each stress treatment plate and the control (chemical-free) plates.
Sporulation assay.
To assay the sporulation capacity of the WT and each mutant, 40-μl aliquots of conidial suspension were evenly spread on SDAY plates (35-mm diameter) and incubated for 7 days at 25°C and 12-h/12-h light/dark cycle. The conidia on each plate were harvested in 20 ml of 0.05% Tween 80 and vortexed adequately, and the conidial suspension was filtered through sterile nonwoven fabrics to remove mycelial debris. The conidial concentration in the suspension was counted with a hemocytometer and converted to the number of conidia per square millimeter of colony. This experiment was carried out in triplicate and repeated, with similar results obtained.
Digital gene expression profile analysis of the WT and Δdcl2 mutant.
Conidia of the WT and the Δdcl2 mutant were inoculated into SDAY medium at 1 × 107 conidia/ml. Fungal samples were collected after 1 day (mycelial growth [MY]) and 2.5 days (conidiogenesis [CO]). Total RNA was extracted using TRIzol reagent (Invitrogen, New York, NY, USA), according to the manufacturer's instructions. Total RNA (5 μg) was prepared for Illumina RNA-Seq, as we described previously (53). Briefly, poly(A) mRNA from the total RNA was enriched using oligo(dT) beads. Following purification, fragmentation buffer was added to break mRNA into small pieces. Using these cleaved RNA fragments as the template, the first cDNA strand was synthesized by reverse transcription with random hexamer primers. The second-strand cDNA was synthesized using buffer, dinucleoside triphosphates (dNTPs), RNase H, and DNA polymerase I, and the sequencing library was constructed according to the manufacturer's instructions (Illumina, San Diego, CA, USA). The libraries were sequenced on the Illumina HiSeq 2000 apparatus (Beijing Genomics Institute, Shenzhen, China) to produce 50-bp paired-end reads. Prior to mapping to the M. robertsii reference genome and annotated genes, adaptor tags, unknown, or low-quality tags were filtered to get clean tags. To compare levels of gene expression between the WT and a mutant, the number of raw clean tags in each library was normalized to the number of reads per kilobase per million reads (RPKM) to normalize gene expression levels. DEGs were identified using a false-discovery rate (FDR) of ≤0.001 and a fold change of ≥2 as the threshold. To further characterize the biological functions and metabolic pathways of DEGs, the DEGs were subjected to a GO functional analysis (Blast2GO; BioBam Bioinformatics) (54). A GO enrichment analysis of functional significance uses a hypergeometric test to map all DEGs to terms in the GO database, looking for significantly enriched GO terms among the genes differentially expressed from those in the background genome.
To validate the DGE results, 18 candidate genes were analyzed by qRT-PCR. The selected genes and primers are listed in Table 2. RNA samples used for qRT-PCR were the same as those used for DGE sequencing. First-strand cDNA synthesis and qRT-PCR were performed as described above. Three technical replicates of qRT-PCR were performed for each biological replicate.
Solexa sequencing and analysis of M. robertsii sRNAs.
Total RNAs were extracted from mycelia of the WT and the Δdcl2 mutant grown in SDAY for 2.5 days at 25°C. RNA concentrations were then evaluated using an ND-1000 spectrophotometer (NanoDrop, USA). A 2-μg aliquot was enriched for sRNA using the polyethylene glycol 8000 (PEG 8000) precipitation method (55). First, low-molecular-weight RNA was isolated from the total RNA. Next, the sRNA segments between 18 and 30 nt were separated using denatured 15% polyacrylamide gel electrophoresis (PAGE) gels and ligated sequentially to specific adaptors at the 5′ and 3′ ends. Then, the products were reverse transcribed to cDNA using RT-PCR. PCR products (75-nt size range) were obtained after amplification of cDNA by a low number of PCR cycles (15 to 20 cycles). The purified PCR products were checked for amount and quality with an Agilent 2100 Bioanalyzer and sequenced with an Illumina genome analyzer (BGI, Shenzhen, China).
Sequencing and bioinformatic analysis were performed as we described previously (24). Briefly, the final clean reads were obtained by getting rid of the adaptor contaminants from the raw reads. The sRNA sequences were mapped to the M. robertsii genome using the Short Oligonucleotide Alignment Program (SOAP) (25, 56). We used the Rfam database to remove the sRNAs that originated from rRNA, tRNA, snRNA, and snoRNA. The prediction of milRNAs was carried out using an algorithm named MIREAP (http://sourceforge.net/projects/mireap), which could identify all candidate precursors with canonical hairpin structures that were perfectly mapped by sequencing data.
Accession number(s).
The mRNA raw sequence data were deposited in NCBI's GEO database (http://www.ncbi.nlm.nih.gov/geo/) with the accession no. GSE80731.
Supplementary Material
ACKNOWLEDGMENTS
We thank Chengshu Wang for providing fungus material and gene deletion vectors.
This work was supported by the National Natural Science Foundation of China (grants 31272096, 31471821, and 31572060), the Anhui Provincial Natural Science Foundation (grants 1308085QC61 and 1408085MKL37), and the Natural Science Foundation of the Higher Education Institutions of Anhui Province (grants KJ2015A081).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.03230-16.
REFERENCES
- 1.Catalanotto C, Nolan T, Cogoni C. 2006. Homology effects in Neurospora crassa. FEMS Microbiol Lett 254:182–189. doi: 10.1111/j.1574-6968.2005.00037.x. [DOI] [PubMed] [Google Scholar]
- 2.Meister G, Tuschl T. 2004. Mechanisms of gene silencing by double-stranded RNA. Nature 431:343–349. doi: 10.1038/nature02873. [DOI] [PubMed] [Google Scholar]
- 3.Dang Y, Yang Q, Xue Z, Liu Y. 2011. RNA interference in fungi: pathways, functions, and applications. Eukaryot Cell 10:1148–1155. doi: 10.1128/EC.05109-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakayashiki H, Kadotani N, Mayama S. 2006. Evolution and diversification of RNA silencing proteins in fungi. J Mol Evol 63:127–135. doi: 10.1007/s00239-005-0257-2. [DOI] [PubMed] [Google Scholar]
- 5.Kapoor M, Arora R, Lama T, Nijhawan A, Khurana JP, Tyagi AK, Kapoor S. 2008. Genome-wide identification, organization and phylogenetic analysis of Dicer-like, Argonaute and RNA-dependent RNA polymerase gene families and their expression analysis during reproductive development and stress in rice. BMC Genomics 9:451. doi: 10.1186/1471-2164-9-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zamore PD, Tuschl T, Sharp PA, Bartel DP. 2000. RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell 101:25–33. doi: 10.1016/S0092-8674(00)80620-0. [DOI] [PubMed] [Google Scholar]
- 7.Bernstein E, Caudy AA, Hammond SM, Hannon GJ. 2001. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409:363–366. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- 8.Tang G, Reinhart BJ, Bartel DP, Zamore PD. 2003. A biochemical framework for RNA silencing in plants. Gene Dev 17:49–63. doi: 10.1101/gad.1048103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nelson P, Kiriakidou M, Sharma A, Maniataki E, Mourelatos Z. 2003. The microRNA world: small is mighty. Trends Biochem Sci 28:534–540. doi: 10.1016/j.tibs.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 10.Ding SW, Voinnet O. 2007. Antiviral immunity directed by small RNAs. Cell 130:413–426. doi: 10.1016/j.cell.2007.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fabian MR, Sonenberg N, Filipowicz W. 2010. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem 79:351–379. doi: 10.1146/annurev-biochem-060308-103103. [DOI] [PubMed] [Google Scholar]
- 12.Siomi MC, Saito K, Siomi H. 2008. How selfish retrotransposons are silenced in Drosophila germline and somatic cells. FEBS Lett 582:2473–2478. doi: 10.1016/j.febslet.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 13.Klattenhoff C, Theurkauf W. 2008. Biogenesis and germline functions of piRNAs. Development 135:3–9. doi: 10.1242/dev.006486. [DOI] [PubMed] [Google Scholar]
- 14.Carmell MA, Xuan Z, Zhang MQ, Hannon GJ. 2002. The Argonaute family: tentacles that reach into RNAi, developmental control, stem cell maintenance, and tumorigenesis. Gene Dev 16:2733–2742. doi: 10.1101/gad.1026102. [DOI] [PubMed] [Google Scholar]
- 15.He L, Hannon GJ. 2004. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 16.Tijsterman M, Plasterk RHA. 2004. Dicers at RISC: the mechanism of RNAi. Cell 117:1–3. doi: 10.1016/S0092-8674(04)00293-4. [DOI] [PubMed] [Google Scholar]
- 17.Li L, Chang SS, Liu Y. 2010. RNA interference pathways in filamentous fungi. Cell Mol Life Sci 67:3849–3863. doi: 10.1007/s00018-010-0471-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang SS, Zhang Z, Liu Y. 2012. RNA interference pathways in fungi: mechanisms and functions. Annu Rev Microbiol 66:305–323. doi: 10.1146/annurev-micro-092611-150138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Villalobos-Escobedo JM, Herrera-Estrella A, Carreras-Villasenor N. 2016. The interaction of fungi with the environment orchestrated by RNAi. Mycologia 108:556–571. doi: 10.3852/15-246. [DOI] [PubMed] [Google Scholar]
- 20.Liu J, Cao Y, Xia Y. 2010. Mmc, a gene involved in microcycle conidiation of the entomopathogenic fungus Metarhizium anisopliae. J Invertebr Pathol 105:132–138. doi: 10.1016/j.jip.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 21.Cao Y, Li M, Xia Y. 2011. Mapmi gene contributes to stress tolerance and virulence of the entomopathogenic fungus, Metarhizium acridum. J Invertebr Pathol 108:7–12. doi: 10.1016/j.jip.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 22.Leng Y, Peng G, Cao Y, Xia Y. 2011. Genetically altering the expression of neutral trehalase gene affects conidiospore thermotolerance of the entomopathogenic fungus Metarhizium acridum. BMC Microbiol 11:32. doi: 10.1186/1471-2180-11-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tiago PV, Fungaro MH, de Faria MR, Furlaneto MC. 2004. Effects of double-stranded RNA in Metarhizium anisopliae var. acridum and Paecilomyces fumosoroseus on protease activities, conidia production, and virulence. Can J Microbiol 50:335–339. doi: 10.1139/w04-023. [DOI] [PubMed] [Google Scholar]
- 24.Zhou Q, Wang Z, Zhang J, Meng H, Huang B. 2012. Genome-wide identification and profiling of microRNA-like RNAs from Metarhizium anisopliae during development. Fungal Biol 116:1156–1162. doi: 10.1016/j.funbio.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 25.Gao Q, Jin K, Ying SH, Zhang Y, Xiao G, Shang Y, Duan Z, Hu X, Xie XQ, Zhou G, Peng G, Luo Z, Huang W, Wang B, Fang W, Wang S, Zhong Y, Ma LJ, St Leger RJ, Zhao GP, Pei Y, Feng MG, Xia Y, Wang C. 2011. Genome sequencing and comparative transcriptomics of the model entomopathogenic fungi Metarhizium anisopliae and M. acridum. PLoS Genet 7:e1001264. doi: 10.1371/journal.pgen.1001264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu Y, Stenlid J, Elfstrand M, Olson A. 2013. Evolution of RNA interference proteins Dicer and Argonaute in Basidiomycota. Mycologia 105:1489–1498. doi: 10.3852/13-171. [DOI] [PubMed] [Google Scholar]
- 27.Meng F, Jia H, Ling N, Xue Y, Liu H, Wang K, Yin J, Li Y. 2013. Cloning and characterization of two Argonaute genes in wheat (Triticum aestivum L.). BMC Plant Biol 13:18. doi: 10.1186/1471-2229-13-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bai M, Yang GS, Chen WT, Mao ZC, Kang HX, Chen GH, Yang YH, Xie BY. 2012. Genome-wide identification of Dicer-like, Argonaute and RNA-dependent RNA polymerase gene families and their expression analyses in response to viral infection and abiotic stresses in Solanum lycopersicum. Gene 501:52–62. doi: 10.1016/j.gene.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 29.Qian Y, Cheng Y, Cheng X, Jiang H, Zhu S, Cheng B. 2011. Identification and characterization of Dicer-like, Argonaute and RNA-dependent RNA polymerase gene families in maize. Plant Cell Rep 30:1347–1363. doi: 10.1007/s00299-011-1046-6. [DOI] [PubMed] [Google Scholar]
- 30.Kadotani N, Nakayashiki H, Tosa Y, Mayama S. 2004. One of the two Dicer-like proteins in the filamentous fungi Magnaporthe oryzae genome is responsible for hairpin RNA-triggered RNA silencing and related small interfering RNA accumulation. J Biol Chem 279:44467–44474. doi: 10.1074/jbc.M408259200. [DOI] [PubMed] [Google Scholar]
- 31.Lee YS, Nakahara K, Pham JW, Kim K, He Z, Sontheimer EJ, Carthew RW. 2004. Distinct roles for Drosophila Dicer-1 and Dicer-2 in the siRNA/miRNA silencing pathways. Cell 117:69–81. doi: 10.1016/S0092-8674(04)00261-2. [DOI] [PubMed] [Google Scholar]
- 32.Catalanotto C, Pallotta M, ReFalo P, Sachs MS, Vayssie L, Macino G, Cogoni C. 2004. Redundancy of the two dicer genes in transgene-induced posttranscriptional gene silencing in Neurospora crassa. Mol Cell Biol 24:2536–2545. doi: 10.1128/MCB.24.6.2536-2545.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Haro JP, Calo S, Cervantes M, Nicolas FE, Torres-Martinez S, Ruiz-Vazquez RM. 2009. A single Dicer gene is required for efficient gene silencing associated with two classes of small antisense RNAs in Mucor circinelloides. Eukaryot Cell 8:1486–1497. doi: 10.1128/EC.00191-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carreras-Villaseñor N, Esquivel-Naranjo EU, Villalobos-Escobedo JM, Abreu-Goodger C, Herrera-Estrella A. 2013. The RNAi machinery regulates growth and development in the filamentous fungus Trichoderma atroviride. Mol Microbiol 89:96–112. doi: 10.1111/mmi.12261. [DOI] [PubMed] [Google Scholar]
- 35.Cao Y, Du M, Luo S, Xia Y. 2014. Calcineurin modulates growth, stress tolerance, and virulence in Metarhizium acridum and its regulatory network. Appl Microbiol Biotechnol 98:8253–8265. doi: 10.1007/s00253-014-5876-3. [DOI] [PubMed] [Google Scholar]
- 36.Twumasi-Boateng K, Yu Y, Chen D, Gravelat FN, Nierman WC, Sheppard DC. 2009. Transcriptional profiling identifies a role for BrlA in the response to nitrogen depletion and for StuA in the regulation of secondary metabolite clusters in Aspergillus fumigatus. Eukaryot Cell 8:104–115. doi: 10.1128/EC.00265-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park HS, Yu J-H. 2012. Genetic control of asexual sporulation in filamentous fungi. Curr Opin Microbiol 15:669–677. doi: 10.1016/j.mib.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 38.Sigl C, Haas H, Specht T, Pfaller K, Kurnsteiner H, Zadra I. 2011. Among developmental regulators, StuA but not BrlA is essential for penicillin V production in Penicillium chrysogenum. Appl Environ Microbiol 77:972–982. doi: 10.1128/AEM.01557-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adams TH, Wieser JK, Yu JH. 1998. Asexual sporulation in Aspergillus nidulans. Microbiol Mol Biol Rev 62:35–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee HC, Li L, Gu W, Xue Z, Crosthwaite SK, Pertsemlidis A, Lewis ZA, Freitag M, Selker EU, Mello CC, Liu Y. 2010. Diverse pathways generate microRNA-like RNAs and Dicer-independent small interfering RNAs in fungi. Mol Cell 38:803–814. doi: 10.1016/j.molcel.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bühler M, Spies N, Bartel DP, Moazed D. 2008. TRAMP-mediated RNA surveillance prevents spurious entry of RNAs into the Schizosaccharomyces pombe siRNA pathway. Nat Struct Mol Biol 15:1015–1023. doi: 10.1038/nsmb.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee HC, Chang SS, Choudhary S, Aalto AP, Maiti M, Bamford DH, Liu Y. 2009. qiRNA is a new type of small interfering RNA induced by DNA damage. Nature 459:274–277. doi: 10.1038/nature08041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu T, Hu J, Zuo Y, Jin Y, Hou J. 2015. Identification of microRNA-like RNAs from Curvularia lunata associated with maize leaf spot by bioinformation analysis and deep sequencing. Mol Genet Genomics 291:587–596. doi: 10.1007/s00438-015-1128-1. [DOI] [PubMed] [Google Scholar]
- 44.Bai Y, Lan F, Yang W, Zhang F, Yang K, Li Z, Gao P, Wang S. 2015. sRNA profiling in Aspergillus flavus reveals differentially expressed miRNA-like RNAs response to water activity and temperature. Fungal Genet Biol 81:113–119. doi: 10.1016/j.fgb.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 45.Chen Y, Gao Q, Huang M, Liu Y, Liu Z, Liu X, Ma Z. 2015. Characterization of RNA silencing components in the plant pathogenic fungus Fusarium graminearum. Sci Rep 5:12500. doi: 10.1038/srep12500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thompson JD, Higgins DG, Gibson JT. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumar S, Tamura K, Nei M. 2004. MEGA3: integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief Bioinform 5:150–163. doi: 10.1093/bib/5.2.150. [DOI] [PubMed] [Google Scholar]
- 48.Fang W, Bidochka MJ. 2006. Expression of genes involved in germination, conidiogenesis and pathogenesis in Metarhizium anisopliae using quantitative real-time RT-PCR. Mycol Res 110:1165–1171. doi: 10.1016/j.mycres.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 49.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 50.Wang C, St. Leger RJ. 2006. A collagenous protective coat enables Metarhizium anisopliae to evade insect immune responses. Proc Natl Acad Sci U S A 103:6647–6652. doi: 10.1073/pnas.0601951103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang W, Shang Y, Chen P, Gao Q, Wang C. 2015. MrpacC regulates sporulation, insect cuticle penetration and immune evasion in Metarhizium robertsii. Environ Microbiol 17:994–1008. doi: 10.1111/1462-2920.12451. [DOI] [PubMed] [Google Scholar]
- 52.Fang W, Pei Y, Bidochka MJ. 2006. Transformation of Metarhizium anisopliae mediated by Agrobacterium tumefaciens. Can J Microbiol 52:623–626. doi: 10.1139/w06-014. [DOI] [PubMed] [Google Scholar]
- 53.Wang ZX, Zhou XZ, Meng HM, Liu YJ, Zhou Q, Huang B. 2014. Comparative transcriptomic analysis of the heat stress response in the filamentous fungus Metarhizium anisopliae using RNA-Seq. Appl Microbiol Biotechnol 98:5589–5597. doi: 10.1007/s00253-014-5763-y. [DOI] [PubMed] [Google Scholar]
- 54.Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, Wang J, Li S, Li R, Bolund L, Wang J. 2006. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res 34:W293–W297. doi: 10.1093/nar/gkl031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu C, Meyers BC, Green PJ. 2007. Construction of small RNA cDNA libraries for deep sequencing. Methods 43:110–117. doi: 10.1016/j.ymeth.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 56.Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J. 2009. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25:1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.