

Abstract
Background
Flowers have an amazingly diverse display of colors and shapes, and these characteristics often vary significantly among closely related species. The evolution of diverse floral form can be thought of as an adaptive response to pollination and reproduction, but it can also be seen through the lens of morphological and developmental constraints. To explore these interactions, we use RNA-seq across species and development to investigate gene expression and sequence evolution as they relate to the evolution of the diverse flowers in a group of Neotropical plants native to Mexico—magic flowers (Achimenes, Gesneriaceae).
Results
The assembled transcriptomes contain between 29,000 and 42,000 genes expressed during development. We combine sequence orthology and coexpression clustering with analyses of protein evolution to identify candidate genes for roles in floral form evolution. Over 25% of transcripts captured were distinctive to Achimenes and overrepresented by genes involved in transcription factor activity. Using a model-based clustering approach we find dynamic, temporal patterns of gene expression among species. Selection tests provide evidence of positive selection in several genes with roles in pigment production, flowering time, and morphology. Combining these approaches to explore genes related to flower color and flower shape, we find distinct patterns that correspond to transitions of floral form among Achimenes species.
Conclusions
The floral transcriptomes developed from four species of Achimenes provide insight into the mechanisms involved in the evolution of diverse floral form among closely related species with different pollinators. We identified several candidate genes that will serve as an important and useful resource for future research. High conservation of sequence structure, patterns of gene coexpression, and detection of positive selection acting on few genes suggests that large phenotypic differences in floral form may be caused by genetic differences in a small set of genes. Our characterized floral transcriptomes provided here should facilitate further analyses into the genomics of flower development and the mechanisms underlying the evolution of diverse flowers in Achimenes and other Neotropical Gesneriaceae.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3623-8) contains supplementary material, which is available to authorized users.
Keywords: Comparative transcriptomics, Flower evolution, Gesneriaceae, Coexpression clustering, RNA-seq
Background
Flowers are a common way that humans connect to nature and the variety of colors and shapes remains one of the most visible and amazing products of evolution. Innovations in floral form have been proposed as one of the primary mechanisms of angiosperm diversification [1] and the phenotypic diversity of flowers is both visually striking and evolutionarily intriguing. Flower evolution is often thought about from an adaptive perspective with the evolution of floral form viewed as a function of reproductive biology or pollination biology [2]. However, developmental constraints and morphological potential can also be viewed as a function of floral organogenesis, morphology, and development rather than strictly an adaptive response [3]. In recent years, studies of flower morphology in an evolutionary and comparative context have been lifted by genetic analyses of developmental pathways underlying flower morphogenesis and biochemistry [4]. However, understanding the macroevolutionary consequences of flower modification through genetic and microevolutionary processes remains difficult. The difficulty arises from the multitude of possible genetic changes available to produce these phenotypic adaptations. Combining the power of transcriptome sequencing with comparative floral morphology allows for the exploration of the possible evolutionary genetic mechanisms involved in flower development and diversification.
We provide a first characterization of the floral transcriptomes in four species of magic flowers, Achimenes. This small genus of ~26 species is a member of the African violet family (Gesneriaceae), a large family distributed in the New World and Old World tropics. The family is renowned for its enormous diversity in habit, desiccation tolerance, leaf morphology, and, particularly, floral form [5–7]. Flower shape, color, and presentation are hypothesized to be important for diversification and speciation events in the family [7–11]. Convergence in floral form is found across the family as well as in individual genera and is likely tied to pollinator preferences and pollinator availability [7, 11]. In Achimenes, floral form appears to be quite variable among closely related species and similar corolla shapes and colors are found among species that occur in different clades [10] (Fig. 1). Multiple derivations of flower shape, color, and the presence of a petal spur appear across the genus [10] (Fig. 1). Populations of Achimenes are largely concentrated in central Mexico south to Costa Rica, with some populations existing in the Caribbean. General distributions of many closely related species often overlap with many populations found growing in the same habitat and elevation ranges [12]. Pollinator studies have been limited with observations recorded for only four species of Achimenes [13]. The major pollinator observed for each of the four Achimenes species corresponds tightly with the hypothesized pollination syndrome that was identified using combinations of floral traits thought to be important for pollinator attraction, such as color, shape, size, and orientation of the open flower [10]. The young age of the genus (~12 Ma) [7], coupled with a large number of shifts in flower shape, color, and pollination syndrome [10], makes Achimenes an ideal lineage to begin understanding the ecological, evolutionary, and molecular forces contributing to speciation and diversification of floral form.
Fig. 1.
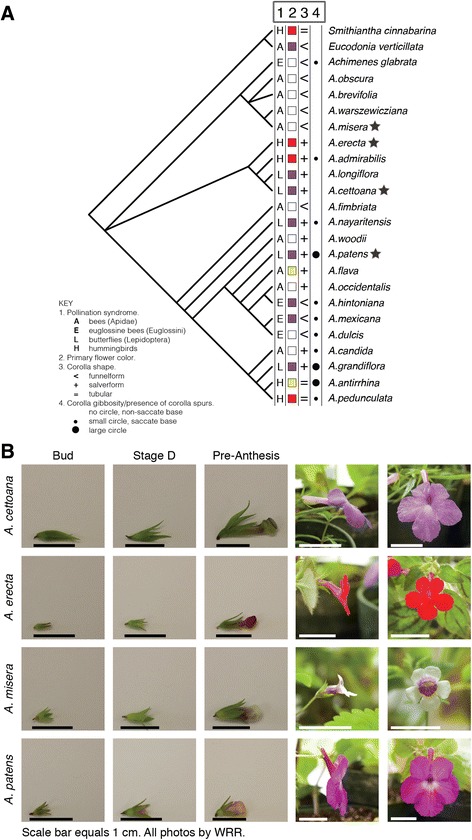
Phylogenetic relationships (modified from Roalson et al. [10]) and flower developmental stages sampled in Achimenes. a Floral morphological characters of interest are mapped onto the tips of the cladogram, including: pollination syndrome, primary flower color, corolla shape, and corolla gibbosity/presence of petal spurs. Four species were sampled for this study from across the genus and are indicated by a star. b The time-points sampled were: Bud, Stage D, and Pre-Anthesis flower buds. Bud stage was defined as pigmentation is largely absent and cells are beginning to elongate. Stage D was defined as pigmentation beginning to accumulate and the corolla begins to elongate. Pre-Anthesis stage was defined as flowers are nearly fully pigmented, the final size and shape of the flower has been determined, and the petal spur has developed from the corolla tube (as in A. patens). Scale bar equals 1 cm. All photos provided by W.R.R
Here, we present de novo floral transcriptome assemblies of four species of Neotropical Achimenes (Gesneriaceae) that vary in floral form, pigmentation patterns, and pollination syndrome. Diversity of flower shape and color among sister species in Achimenes present intriguing questions about the ecological and genetic forces contributing to these phenotypic divergences. We sampled flowers in three developmental stages from A. cettoana, A. erecta, A. misera, and A. patens. This sampling strategy allows inter- and intraspecies comparisons of gene expression during development and comparisons of sequence structure in order to begin investigating evolutionary and developmental mechanisms contributing to speciation and diversification. Utilizing high-throughput technologies has allowed researchers in both animal [14, 15] and plant [16–18] systems to sequence entire genomes, transcriptomes, and proteomes in order to understand fine-scale patterns of genetics and evolution. Our study takes advantage of these genomic approaches and provides resources that will serve as the basis for future studies into flower development, evolution, and plant-pollinator interactions.
Comparative transcriptomic studies in plants have seen an increasing publication rate in recent years as sequencing technologies keep increasing data output for lower cost. Many studies have taken a focused look at comparing developmental stages in a single species across different tissues [19–21], comparing gene expression in different organs [22, 23], or simply to generate preliminary genomic data that will guide more detailed studies [24–27]. Evolutionary questions have also been investigated using genome-wide expression data in plants, such as the evolution of gene expression patterns [16], parasitism [18], self-fertilization [28], or mass flowering [29]. Our study aims to bridge the gap between these different areas. We took a developmental approach by sampling several stages of flower development and an evolutionary approach by comparing transcriptome data across multiple species. This evolutionary-developmental approach to comparative transcriptomics presents a novel way to investigate the patterns and processes of flower diversification at the genomic level. This study provides annotated reference transcriptomes for four species of Achimenes and uses them for analyses of sequence orthology, coexpression clustering of genes during development, and selection tests to identify protein sites undergoing positive selection. We also use data from the transcriptomes to begin investigating the genetics of flower color, particularly the production of anthocyanin pigments. It is our goal that the resources and results provided herein will serve as the basis for future studies. This study is among the first explorations of Neotropical Gesneriaceae flower transcriptomes using large-scale sequencing, and the results described here may serve to guide further gene expression and functional genomic studies in Achimenes and other members of the Gesneriaceae.
Results
Assembly of high-quality achimenes floral transcriptomes
Sequencing the floral transcriptomes from three developmental stages in four species of Achimenes yielded over 270 million reads (Table 1). Each species had between 63 and 72 million paired-end reads sequenced (Table 1).
Table 1.
Sequencing and summary statistics for Achimenes reference floral transcriptome assemblies and annotation
| A. cettoana | A. erecta | A. misera | A. patens | |
|---|---|---|---|---|
| A. Sequencing | ||||
| Total reads | 67,428,998 | 63,582,836 | 69,588,964 | 71,960,488 |
| Bud | 21,112,016 | 18,680,312 | 24,016,214 | 22,585,994 |
| Stage D | 22,382,106 | 24,084,300 | 19,391,388 | 28,579,042 |
| Pre-Anthesis | 23,934,876 | 20,818,224 | 26,181,362 | 20,795,452 |
| Total length (bp) | 6,742,899,800 | 6,358,283,600 | 6,958,896,400 | 7,196,048,800 |
| B. Final merged assembly | ||||
| Primary transcripts | 29,065 | 41,381 | 41,285 | 37,898 |
| Alternate transcripts | 23,332 | 94,172 | 105,442 | 65,115 |
| N50 | 2,113 | 2,061 | 1,990 | 2,109 |
| Mean length (bp) | 1,417 | 1,268 | 1,260 | 1,304 |
| Total bases, Primary set | 41,202,771 | 52,511,722 | 52,038,201 | 49,447,956 |
Trinity assemblies using a k-mer size of 25 produced between 139,806 (A. cettoana) and 199,502 (A. erecta) contigs for each reference transcriptome (Additional file 1). These assembled contigs had N50 values between 1444 (A. misera) and 1794 (A. cettoana) bps, with mean lengths between 868 (A. misera) and 1027 (A. cettoana) bps (Additional file 1). Velvet and Oases assemblies were also performed using a range of k-mer sizes from 25 to 75 (Additional file 1). Generally, these assemblies produced higher numbers of contigs, with higher N50 values, and higher mean values than the Trinity assemblies (Additional file 1). The number of contigs ranged from 46,189 in A. cettoana using a k-mer size of 75 to 247,516 in A. erecta using a k-mer size of 35 (Additional file 1). N50 values were also showed some variation consistent with larger k-mers producing lower values (1,385 in A. misera) and smaller k-mers producing higher values (2,334 in A. erecta; Additional file 1). Assemblies for A. cettoana always produced far fewer contigs than the other species (e.g., using Velvet/Oases, 126,317 in A. cettoana versus 247,516 in A. erecta, see Additional file 1). The number of contigs assembled does not appear to negatively affect other assembly metrics; the mean length and N50 values were similar across all species assemblies (Additional file 1).
Merging the separate de novo assemblies reduced redundancy and provided useful sets of contigs for further analyses (Table 1; Additional file 1). Between 29,065 and 41,381 primary transcripts were obtained with N50 lengths between 1,990 and 2,113 bps (Table 1). The merging process also provided between 23,332 and 105,442 alternate transcripts, which are composed of possible isoforms (Table 1; Additional file 1).
Functional annotation and classification
The primary floral transcriptomes of A. cettoana, A. erecta, A. misera, and A. patens were annotated by BLASTx searches against the SwissProt [30] and the NCBI non-redundant (Nr) protein database [31]. For A. cettoana, 18,364 (63.18%) sequences had hits in the SwissProt database; A. erecta, 23,534 (56.87%) sequences had hits; A. misera, 23,120 (56.00%) sequences had hits; and A. patens, 20,838 (54.98%) sequences had hits (Table 2). The numbers of sequences with at least 75% coverage by their best protein hits were 10,281 (35.37%), 12,372 (29.90%), 11,420 (27.66%), and 11,097 (29.28%), for each transcriptome respectively. Against the Nr database, A. cettoana had 23,012 (79.17%) sequences with hits; A. erecta had 29,794 (72.00%) sequences with hits; A. misera had 29,783 (72.14%) sequences with hits; and A. patens had 26,776 (70.65%) sequences with hits (Table 2). Additionally, we performed BLASTn searches against a collection of Arabidopsis thaliana long non-coding RNA (lncRNA) sequences acquired from the Plant Non-coding RNA Database [32]. Against this set, A. cettoana had 76 (0.0026%) sequences with hits; A. erecta had 96 (0.0023%) sequences with hits; A. misera had 85 (0.0021%) sequences with hits; and A. patens had 117 (0.0031%) sequences with hits (Table 2). Non-coding ribosomal RNAs and tRNAs formed a small number of the total contigs (Additional file 2).
Table 2.
Overview of BLAST hits to primary transcript set and functional annotation output of the four reference transcriptomes
| A. cettoana | A. erecta | A. misera | A. patens | |
|---|---|---|---|---|
| SwissProt | 18,365 (63.18%) | 23,534 (56.78%) | 23,120 (56.00%) | 20,838 (54.98%) |
| Nr | 23,012 (79.17%) | 29,794 (72.00%) | 29,783 (72.14%) | 26,776 (70.65%) |
| PNRD | 76 (0.0026%) | 96 (0.0023%) | 85 (0.0021%) | 117 (0.0031%) |
| GO | 11,826 (40.69%) | 14,996 (36.24%) | 14,683 (35.56%) | 13,179 (34.78%) |
Abbreviations: GO gene ontology, Nr NCBI non-redundant protein database, PNRD plant non-coding RNA database
The sequences with matches in the SwissProt [30] or Nr [31] databases were further annotated with Gene Ontology (GO) terms [33] based on the SwissProt database, InterProScan [34], and ANNEX augmentation [35]. GO terms were assigned to 11,826 (40.69%) transcripts in A. cettoana, 14,996 (36.24%) transcripts in A. erecta, 14,683 (35.56%) transcripts in A. misera, and 13,179 (34.78%) in A. patens (Table 2). Numbers and proportions of sequences attributed to level 2 GO for Biological Process (BP), Cellular Component (CC), and Molecular Function (MF) type terms were qualitatively similar with slight variations likely due to numbers of transcripts assembled for each species (Additional file 3). Representation was qualitatively very similar between the four species, with all level 2 GO categories exhibiting no significant differences across species even after accounting for the effects of multiple testing (Χ 2 ≥ 1.65, FDR-corrected p-value , α = 0.001). We looked at further GO levels (level 3, level 4, etc.) and found similar composition of category assignment for each transcriptome.
Core enzymes of the anthocyanin biosynthetic pathway (ABP) were identified using HMMER [36] against homologs downloaded from GenBank (Additional file 4). The HMMER searches identified 224 proteins with similarity to anthocyanidin synthase (ANS, Additional file 5), 122 proteins with similarity to dihydroflavonol 4-reductase (DFR, Additional file 6), and 730 proteins with similarity to both F3′H (flavonoid 3′-hydroxylase, Additional file 7) and F3′5′H (flavonoid 3′,5′-hydroxylase, Additional file 7). These large groups of proteins represent putative gene families for each of these enzymes. Aligning the sequences of these proteins with the sequences of known proteins from other studies and constructing neighbor-joining trees allowed us to identify putative proteins from Achimenes involved in the ABP. We identified single copies of ANS (Additional file 5), DFR (Additional file 6; Additional file 8), F3′H (Additional file 7; Additional file 8), and F3′5′H (Additional file 7; Additional file 8) in each transcriptome, with the exception of A. misera where 6 copies of F3′5′H were identified (Additional file 7). Five of the six A. misera copies have very low normalized expression estimates and may represent genes that are expressed at too low of level to be detected at the current sequencing depth or may be artifacts of our assembly process. Expression estimates for each of the identified single copy enzymes generally increases from B to A stages (Fig. 2) as pigments accumulate in the floral tissue.
Fig. 2.
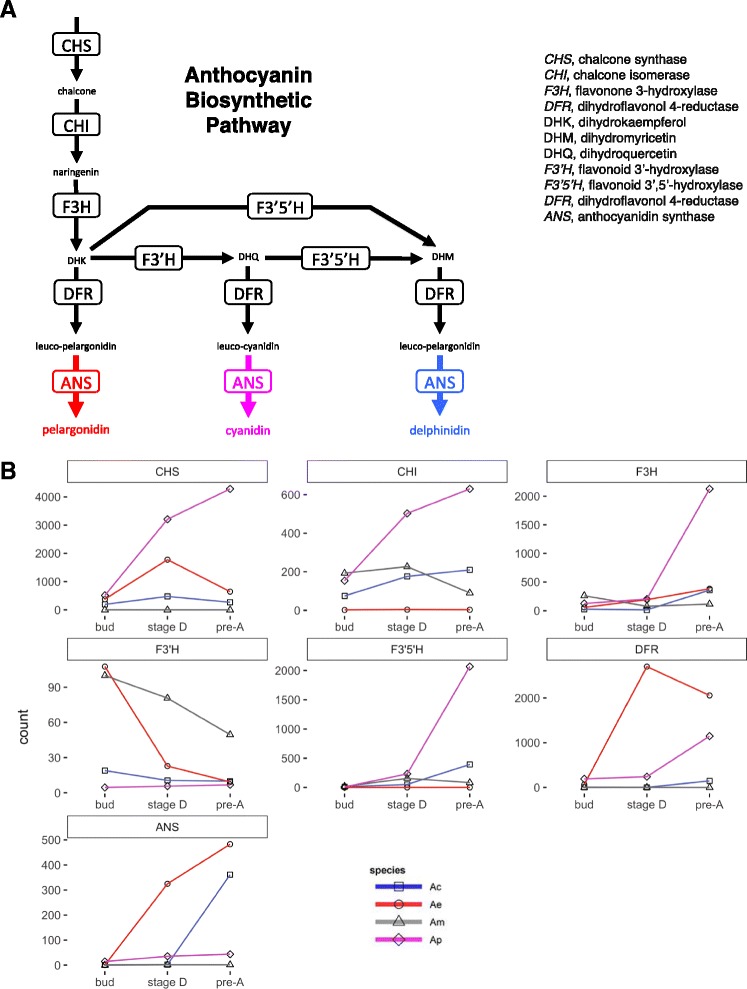
Expression estimates for core enzymes of the anthocyanin biosynthetic pathway in Achimenes. LEGEND: a. Schematic outline of the core anthocyanin biosynthetic pathway in plants. b. Expression (TPM) for the core enzymes of the anthocyanin biosynthetic pathway during flower development in Achimenes. Expression for A. cettoana, A. erecta, A. misera, and A. patens are in blue, red, grey, and pink lines, respectively
Putative enzymes of the carotenoid biosynthetic pathway (CBP) were identified from each Achimenes transcriptome using BLASTx. Both bit scores and E-values were used to identify best-hit transcripts. Using homologs from Arabidopsis as query, there were 12 proteins identified to be involved in carotenoid biosynthesis (Fig. 3). Proteins identified included ones belonging to both the α-carotene and β-carotene branches (Fig. 3).
Fig. 3.
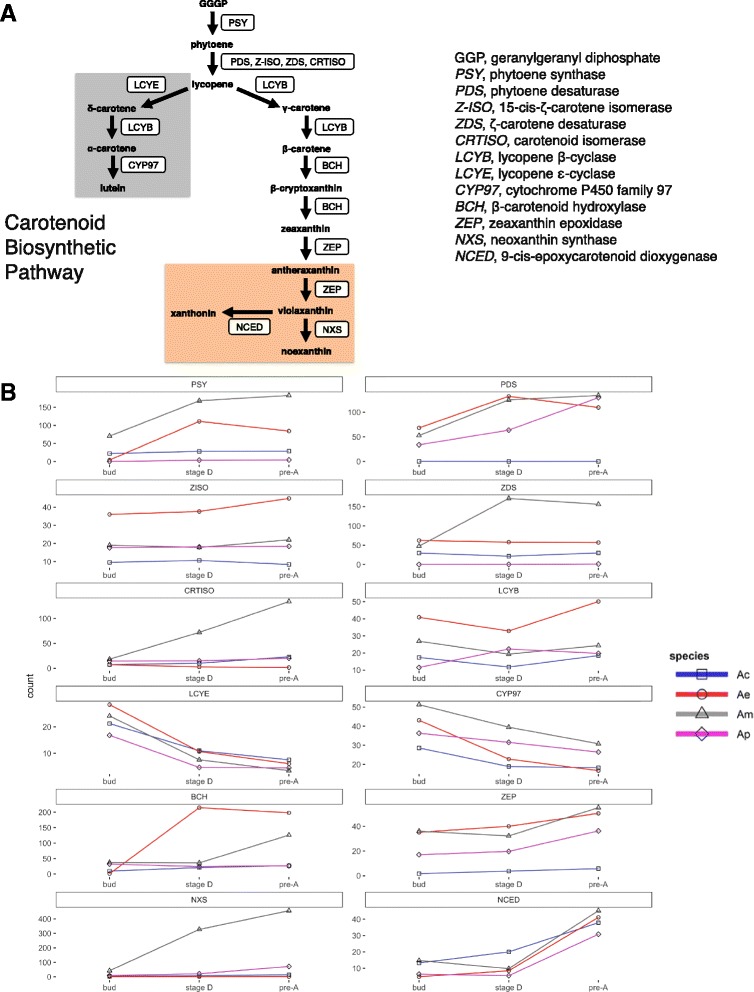
Expression estimates for core enzymes of the plant carotenoid biosynthetic pathway in Achimenes. a Schematic outline of the plant carotenoid biosynthetic pathway. The enzymes are shown in boxes to the side of the arrows. Grey and orange boxes indicate the α-carotene and β-carotene branches, respectively. b Expression (TPM) for the core enzymes of the carotenoid biosynthetic pathway during flower development in Achimenes. Expression for A. cettoana, A. erecta, A. misera, and A. patens are in blue, red, grey, and pink lines, respectively
Proteins related to flower development were additionally identified from each Achimenes transcriptome using BLASTx. We used both bit scores and E-values to identify putative proteins. Using homologs from Arabidopsis as query, there were 101 putative proteins identified that may be involved in flower development (Additional file 9). These included proteins involved in flowering transition, organ development, and floral repression (Additional file 9, Additional file 10). Among the proteins identified were A-, B-, C-, and E-class MADS-box genes, members of the AP2/ERF family, numerous homeobox genes, and many others (Additional file 9, Additional file 10). Each of these proteins has a distinct expression domain during development and may be expressed in floral organs (sepals, petals, etc.), in the floral meristem, or in the inflorescence (Additional file 9, Additional file 10).
Several genes involved in cell proliferation and hormone signaling were recently identified to be important for petal spur development in Aquilegia [37]. We identified homologs of these genes from each transcriptome using both bit scores and E-values to select likely candidate transcripts. The Achimenes transcripts identified include homologs of TCP4 and GIF1, both involved in cell division control (Fig. 4). TCP4 distinctly shows very high expression in A. patens and not the other Achimenes species, a similar pattern to that observed in Aquilegia [37] (Fig. 4). Other genes identified include STM involved in meristem indeterminacy [38], STY1 that regulates auxin biosynthesis [39], ARF3 and ARF8 that are auxin response factors, YUC6 and CYP71 both involved in auxin biosynthesis, and DWARF4 and BEH4 that function in the brassinosteroid pathway [40, 41] (Fig. 4).
Fig. 4.
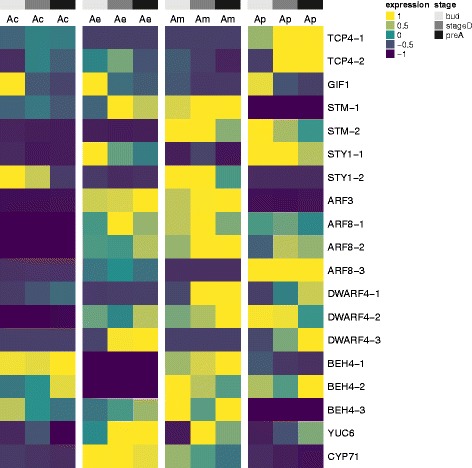
Expression of putative genes involved in petal spur development in Achimenes. Heatmap of scaled expression estimates for 10 Achimenes homologs of Aquilegia genes hypothesized to be important for petal spur development according to [37]. Rows and columns are not clustered
Lastly, we identified candidate R2R3-Myb transcription factors that may be involved in regulating anthocyanin and carotenoid biosynthesis in flowers. Using HMM profiles built from R2R3-Mybs shown to be involved in these pathways, we identified several candidate proteins. There are 8 Achimenes sequences identified that are closely related to R2R3-Mybs from Erythranthe and Antirrhinum that regulation floral anthocyanin production (Additional file 11). Nine Achimenes sequences were identified and related to an R2R3-Myb transcription factor in Erythranthe that regulated floral carotenoid production (Additional file 11).
Core, shared, and unique genes
We found a set of gene clusters that were common to all four Achimenes species and the outgroup Erythranthe lewisii (collectively termed the “Core transcriptome”). This core set of proteins consisted of 12,126 gene clusters (Fig. 5), which comprised 59%, 48%, 50%, and 49% of the total predicted proteins in A. cettoana, A. erecta, A. misera, and A. patens, respectively (Fig. 5). There were an additional 1,776 gene clusters (Fig. 5) that were unique and shared among all four gesneriad species (“Shared Achimenes”). These clusters comprised 7.4%, 6.2%, 6.5%, and 6.7% of the total predicted proteins, respectively (Fig. 5). In addition to the shared clusters within Achimenes, each species also contained unique protein sequences (unassigned to any cluster) that were not found in any of the other five transcriptomes; these unique sequences comprised 21–32% of the transcriptomes (Fig. 5). Approximately 14% of the transcriptomes were comprised of protein orthogroups shared between at least two of the five species (“Shared others”, Fig. 5).
Fig. 5.
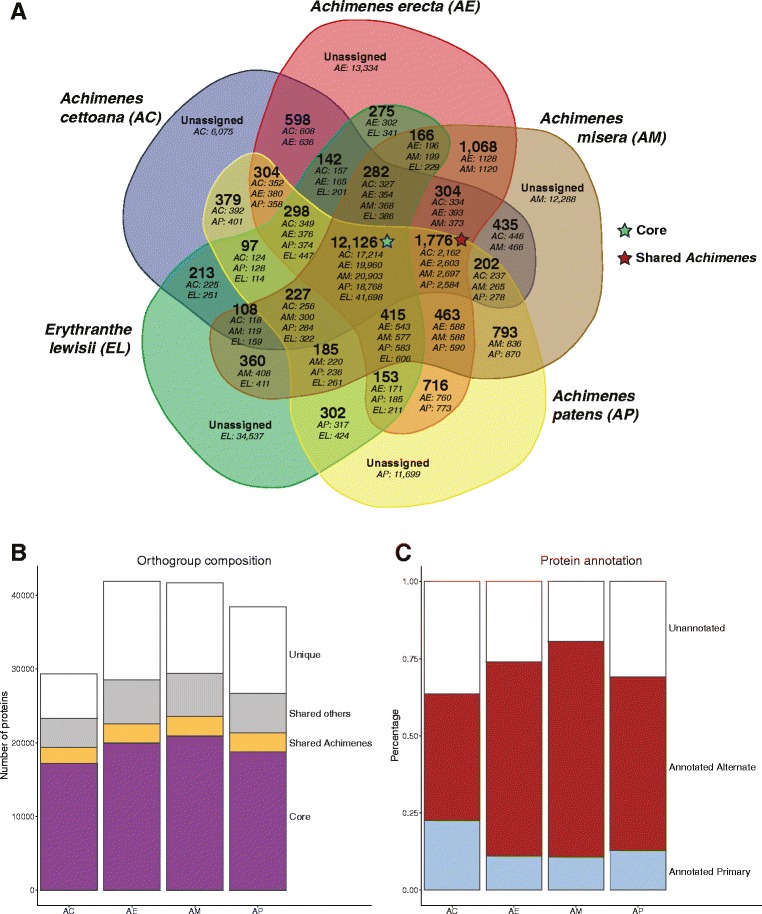
Sequence orthology and the proportion of annotated sequences in the Achimenes transcriptomes. a Venn diagram showing the number of shared or unique genes (in italics) and gene clusters (bold) among the five Achimenes species and Erythranthe as classified by the OrthoFinder program. “Core” and “Shared Achimenes” orthogroups are indicated with blue and red stars, respectively. Abbreviations: AC, Achimenes cettoana; AE, Achimenes erecta; AM, Achimenes misera; AP, Achimenes patens; EL, Erythranthe lewisii. b Proportion of the transcripts that comprised core, shared Achimenes, shared others, and unique genes. “Core” orthogroups were common to all four Achimenes and Erythranthe. “Shared Achimenes” are orthogroups that contain sequences from all four Achimenes species. “Shared other” are orthogroups present in two or three of the four Achimenes species. “Unique” genes are genes that are only present in one species and were unassigned to a specific orthogroup. c Proportion of annotated and non-annotated genes in the primary and alternate transcriptomes
Among the 12,126 orthogroups that were shared by all five species in the five-way comparison, there were 78 GO terms significantly enriched (FDR-corrected p-value < 0.05). As expected, most of these terms were related to primary metabolism, cellular components and structure, signaling, reproduction, and response to stimulus, among many others (Additional file 12). Within the protein clusters that were shared among all four Achimenes species (“Shared Achimenes”), 27 GO terms were significantly overrepresented (FDR-corrected p-value <0.05) in all species (Additional file 13). When comparing protein sequences that each species contributed to the “Shared Achimenes” orthogroup, there were 7 overrepresented GO terms identified in all four species individually. Interestingly, each of these terms were involved in DNA binding, including chromatin binding and transcription factor activity (Table 3). Among the sequences that were unassigned to any clusters, there were some differences in the number and type of GO terms that were significantly over- or underrepresented in each species, with 4 terms identified in A. patens and 26 terms identified in A. erecta. (Additional file 14).
Table 3.
Gene ontology terms overrepresented in the “Shared Achimenes” orthogroups
| Term | Description | Type | FDR | Single-test p-value | Number in test group | Number in reference group |
|---|---|---|---|---|---|---|
| GO:0003676 | Nucleic acid binding | MF | 4.20e-46 | 2.30e-48 | 977 | 7373 |
| GO:0003677 | DNA binding | MF | 2.10e-44 | 2.30e-46 | 612 | 3951 |
| GO:0003682 | Chromatin binding | MF | 3.10e-28 | 6.80e-30 | 157 | 619 |
| GO:0044877 | Macromolecular complex binding | MF | 3.10e-28 | 6.80e-30 | 157 | 619 |
| GO:0003700 | Transcription factor activity, sequence-specific DNA binding | MF | 6.30e-20 | 2.00e-21 | 200 | 1112 |
| GO:0001071 | Nucleic acid binding transcription factor activity | MF | 6.30e-20 | 2.00e-21 | 200 | 1112 |
| GO:1901363 | Heterocyclic compound binding | MF | 2.70e-08 | 1.20e-09 | 1517 | 16173 |
| GO:0097159 | Organic cyclic compound binding | MF | 2.70e-08 | 1.20e-09 | 1517 | 16173 |
| GO:0005618 | Cell wall | CC | 3.10e-06 | 1.50e-07 | 43 | 199 |
| GO:0030312 | External encapsulating structure | CC | 4.50e-06 | 2.40e-07 | 43 | 203 |
| GO:0005488 | Binding | MF | 1.30e-04 | 8.00e-06 | 2737 | 31646 |
| GO:0090304 | Nucleic acid metabolic process | BP | 2.70e-04 | 1.90e-05 | 145 | 1210 |
| GO:0006259 | DNA metabolic process | BP | 2.70e-04 | 1.90e-05 | 145 | 1210 |
| GO:0030246 | Carbohydrate binding | MF | 2.10e-03 | 1.60e-04 | 63 | 456 |
| GO:0019825 | Oxygen binding | MF | 5.60e-03 | 4.60e-04 | 5 | 5 |
| GO:0071944 | Cell periphery | CC | 5.60e-03 | 4.90e-04 | 58 | 430 |
| GO:0009653 | Anatomical structure morphogenesis | BP | 7.00e-03 | 6.50e-04 | 7 | 14 |
| GO:0015979 | Photosynthesis | BP | 9.40e-03 | 9.20e-04 | 35 | 229 |
| GO:0006725 | Cellular aromatic compound metabolic process | BP | 1.90e-02 | 2.30e-03 | 590 | 6399 |
| GO:1901360 | Organic cyclic compound metabolic process | BP | 1.90e-02 | 2.30e-03 | 590 | 6399 |
| GO:0046483 | Heterocycle metabolic process | BP | 1.90e-02 | 2.30e-03 | 590 | 6399 |
| GO:0006139 | Nucleobase-containing compound metabolic process | BP | 1.90e-02 | 2.30e-03 | 590 | 6399 |
| GO:0005576 | Extracellular region | CC | 2.10e-02 | 2.60e-03 | 31 | 209 |
| GO:0005634 | Nucleus | CC | 2.80e-02 | 3.70e-03 | 236 | 2391 |
| GO:0009607 | Response to biotic stimulus | BP | 3.80e-02 | 5.20e-03 | 19 | 113 |
| GO:0005615 | Extracellular space | CC | 4.20e-02 | 6.20e-03 | 6 | 17 |
| GO:0044421 | Extracellular region part | CC | 4.20e-02 | 6.20e-03 | 6 | 17 |
Abbreviations: BP biological process, CC cellular component, FDR false discovery rate corrected p-value, MF molecular function
Quantifying expression and coexpression clustering
We estimated gene expression by mapping RNA-seq reads from each developmental stage (B, Immature Bud; D, Stage D; A, Pre-Anthesis) back to the respective reference ‘primary’ transcriptome using bowtie [42] and RSEM [43]. In each of the four species (A. cettoana, A. erecta, A. misera, and A. patens), the mapping rate averaged 93.59%, 93.39%, 95.1%, and 92.07%, respectively. Additionally, mapping reads from one species onto another species reference produced successful mapping rates of >85%.
Over 5 independent runs, we used HTSCluster [44] and the EM algorithm [45] to fit a sequence of Poisson mixture models with K = 1, 2, …, 60 clusters for the expression estimates of each reference transcriptome. Using slope heuristics (Djump, dimension jump; DDSE, data driven slope estimation) [46], the number of clusters was determined to be K = 34, 30, 29, 25 for the A. cettoana, A. erecta, A. misera, and A. patens expression estimates, respectively. Visualization of the clustering displays numerous clusters with very high or very low expression levels during specific stages in development and also many clusters where expression is not qualitatively different between the three stages (Fig. 6; Additional file 15). Visualization of the maximum conditional probabilities of cluster membership for each species indicates confidence in cluster assignment (Additional file 16), particularly among clusters that have distinct high or low expression during a single developmental stage (Fig. 6; Additional file 15, Additional file 16). Examining what, if any, GO terms may be over- or underrepresented in specific coexpression clusters may be useful to determine any temporal patterns of gene expression during flower development. In A. cettoana, 22 of 34 (65%) clusters had significantly over-enriched GO terms associated. Likewise, A. erecta had 23 of 30 (77%), A. misera had 21 of 29 (72%), and A. patens had 21 of 25 (84%) clusters with significantly over-enriched GO terms (Additional file 17).
Fig. 6.
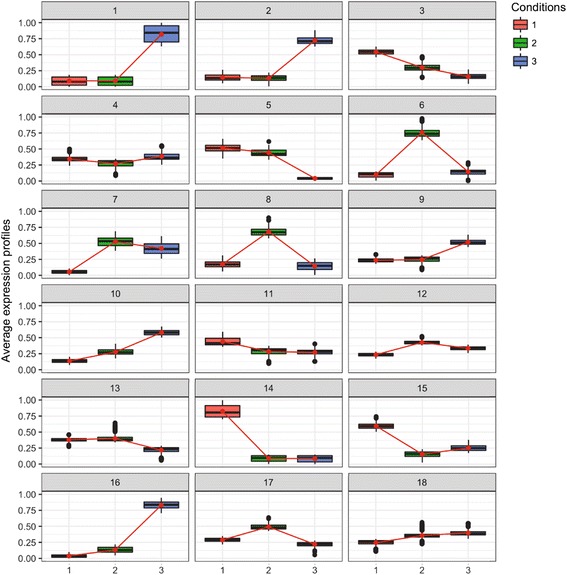
Coexpression cluster profiles of Achimenes cettoana transcripts using Poisson mixture models. Thirty-four coexpression clusters were determined for A. cettoana with Poisson mixture models using slope heuristics as implemented in [44]. Clusters 1 to 18 are presented here to provide an example of the dynamic patterns of gene coexpression seen during flower development in A. cettoana. The full figure showing all 34 clusters is included in Additional file 15. Boxplots indicate average gene expression profiles for each cluster. Conditions refer to the sampled stages of flower development: 1, Bud stage; 2, Stage D; and 3, Pre-Anthesis stage
GO term enrichment tests were performed for each cluster to identify general patterns of gene coexpression (Additional file 18). Trends in gene coexpression were apparent and what we expect for developing flowers. For instance, genes involved in photosynthesis tended to have higher expression in the B stage, while genes involved in primary metabolism and biosynthetic processes tended to be enriched in clusters without qualitative differences between stages (Additional file 18). While considering broad-scale patterns of gene categories that tend to be coexpressed together provided important results, we additionally wanted to investigate which clusters contained genes involved in flower shape and pigment production.
Many members of the ABP were coexpressed together (Additional file 19). In three species (A. cettoana, A. erecta, and A. patens), several of the downstream enzymes were found in the same coexpression cluster, including F3H, F3′H, F3′5′H, DFR, and ANS (Additional file 19). In A. misera, all enzymes were put into different coexpression clusters with the exception of CHI and F3′H (Additional file 19). Several of the candidate R2R3-Mybs identified were also coexpressed with enzymes of the ABP (Additional file 19). One R2R3-Myb was coexpressed in A. cettoana with F3′5′H; one was coexpressed in A. misera with ANS; and one was coexpressed in A. patens with F3H, F3′5′H, and ANS (Additional file 19).
There were very few enzymes of the CBP that were found in the same coexpression cluster (Additional file 19). The downstream enzymes of the β-carotene branch tended to be found in the same coexpression cluster in some species, particularly BCH, ZEP, NXS, and NCED (Additional file 19). Of the 9 candidate R2R3-Mybs identified, only one in A. erecta was coexpressed with any of the CBP enzymes, namely CYP97 (Additional file 19).
The genes identified to be involved in flower development did not show any clear coexpression patterns. For instance, genes that are involved in petal or carpel development are found across many different clusters likely due to very different temporal patterns of gene expression (Additional file 9, Additional file 10). Likewise, the candidate genes we looked at for involvement in petal spur development show very few coexpression patterns (Additional file 19, Additional file 10). Some transcripts of particular genes were coexpressed together, TCP4 in A. patens for example, while most others were found in different coexpression clusters (Additional file 19).
Detecting proteins under selection
As detection of positive selection requires a minimum of five species to obtain reliable estimates [47], orthogroups from the five-way analysis were stringently filtered. These filtering steps provided 2,930 orthogroups, containing 26,141 total sequences, for selection analyses. Sequence alignments were visually inspected to identify spurious alignments that could produce false positives in our selection analyses. After inspection, no clusters were removed from the subsequent analyses. Likelihood ratio tests comparing four models (M1a vs. M2a, M7 vs. M8) [48, 49] were employed to identify proteins and amino acids within those proteins potentially displaying signatures of selection. Comparison of M1a versus M2a (m12) identified 339 orthogroups containing proteins with signatures of selection, while M7 versus M8 comparisons (m78) identified 642 orthogroups (FDR-corrected p-values ≤ 0.05). Three hundred thirty-five orthogroups were identified by both m12 and m78 comparisons. The numbers of proteins identified in m12 were 64, 80, 68, and 76 for A. cettoana, A. erecta, A. misera, and A. patens, respectively (Additional file 20). In the m78 comparison, there were 125, 144, 133, and 143 proteins identified, respectively (Additional file 21).
Enrichment tests did not show any GO terms significantly over- or underrepresented in the list of proteins with sites undergoing positive selection. Comparisons were made both for the combined set of proteins, as well as protein sets for each individual species (including Erythranthe). However, several GO categories of interest to the current study were found in the protein set, including terms including flower development, anatomical structure morphogenesis, anthocyanin pigmentation, and transcription factor activity (Table 4).
Table 4.
Genes under positive selection in Achimenes and Erythranthe related to flower development and pigmentation
| Gene | Description | UniProd ID | Species | Test |
|---|---|---|---|---|
| 5MAT1 | Malonyl-coenzyme:anthocyanin 5-O-glucoside-6”’-O-malonyltransferase | Q8W1W9 | Ac | m12,m78 |
| SFH13 | Phosphatidylinositol/phosphatidylcholine transfer protein SFH13 | Q501H5 | Ac | m78 |
| UBC28 | Ubiquitin-conjugating enzyme E2 28 | Q94F47 | Ac | m78 |
| Y5241 | Probable receptor-like protein kinase at5g24010 | Q9FLW0 | Ac | m12 |
| LECRK91 | L-type lectin-domain containing receptor kinase | Q9LXA5 | Ac | m12 |
| GAUT14 | Galactouronosyltransferase 14 | Q8GWT1 | Ac | m12 |
| ABCB19 | ABC transporter B family member 19 | Q9LJX0 | Ae | m12 |
| ACR4 | ACT domain-containing protein ACR4 | Q8LJW3 | Ae | m12 |
| CYP90A1 | Cytochrome P450 90A1 | Q42569 | Ae | m12 |
| DCR | BAHD acyltransferase DCR | Q9FF86 | Ae | m12 |
| DFRA | Dihydroflavonol 4-reductase | P51102 | Ae | m78 |
| MAP70.2 | Microtubule-associated protein 70-2 | Q8L7S4 | Ae | m12 |
| TA14B | Transcription initiation factor TFIID subunit 14b | Q9FH40 | Ae | m12 |
| TKPR1 | Tetraketide alpha-pyrone reductase 1 | Q500U8 | Ae | m12 |
| GPPL2 | Haloacid dehalogenase-like hydrolase domain-containing protein at3g48420 | Q94K71 | Ae | m12 |
| HAT | Zinc finger bed domain-containing protein DAYSLEEPER | Q9M2N5 | Ae | m12 |
| KDSB | 3-deoxy-manno-octulosonate | Q9C920 | Ae | m12 |
| BIG1 | Brefeldin A-inhibited guanine nucleotide-exchange protein 1 | FAJSZ5 | Ae | m12 |
| PRMT13 | Probable histone-arginine methyltransferase 1.3 | Q84W92 | Am | m12 |
| ATX1 | Copper transport protein ATX1 | Q94BT9 | Am | m12 |
| CYP71A1 | Cytochrome P450 71A1 | P24465 | Am | m12 |
| FLXL1 | Protein FLX-like 1 | Q93V84 | Am | m12,m78 |
| FRI | Protein FRIGIDA | P0DH90 | Am | m12,m78 |
| DTX41 | Protein DETOXIFICATION 41 | Q9LYT3 | Am | m12 |
| Y1301 | BTB/POZ domain-containing protein at1g03010 | Q9SA69 | Am | m12 |
| AKR2A | Ankyrin repeat domain-containing protein 2A | Q9SAR5 | Ap | m78 |
| CAF2M | CRS2-associated factor 2 | Q9FFU1 | Ap | m12 |
| CKB2 | Casein kinase II subunit beta-2 | P40229 | Ap | m12 |
| GN | ARF guanine-nucleotide exchange factor GNOM | Q42510 | Ap | m12 |
| WNK1 | Serine/threonine-protein kinase WNK1 | Q9CAV6 | Ap | m12 |
| HDA19 | Histone deactylase 19 | O22446 | Ap | m12 |
| PRXQ | Peroxiredoxin chloroplastic | Q6UBI3 | Ap | m12 |
| CPK13 | Calcium-dependent protein kinase 13 | Q8W4I7 | Ap | m12 |
| LOL2 | Protein LOL2 | O65426 | Ap | m12 |
| EXO70A1 | Exocyst complex component EXO70A1 | Q9LZD3 | Ap | m12 |
| PRMT11 | Protein arginine N-methyltransferase 1.1 | Q9SU94 | El | m12 |
| BLH8 | BEL1-like homeodomain protein 8 | Q9SJJ3 | El | m12 |
| GDL15 | GDSL esterase/lipase at1g29670 | Q9C7N4 | El | m12 |
| GK-2 | Guanylate kinase 2 | Q9M682 | El | m12 |
| CUT1 | 3-ketoacyl-CoA synthase 6 | Q9XF43 | El | m12 |
| AATL1 | Lysine histidine transporter-like 8 | Q9SX98 | El | m12,m78 |
| MAA3 | Probable helicase MAGATAMA 3 | B6SFA4 | El | m12,m78 |
| RH27 | DEAD-box ATP-dependent RNA helicase 27 | Q9SB89 | El | m12 |
| SOBIR1 | Leucine-rich repeat receptor-like serine/threonine/tyrosine-protein kinase SOBIR1 | Q9SKB2 | El | m12 |
| BRM | ATP-dependent helicase BRM | Q6EVK6 | El | m12 |
| AGO2 | Protein argonaute 2 | Q9SHF3 | El | m12 |
| ARID5 | AT-rich interactive domain-containing protein 5 | Q0WNR6 | El | m12 |
Abbreviations: Ac Achimenes cettoana, Ae Achimenes erecta, Am Achimenes misera, Ap Achimenes patens, El Erythranthe lewisii, m12 PAML model comparison m1a vs. m2a, m78 PAML model comparison m7 vs. m8
Discussion
This study is among the first to employ RNA sequencing for comparative studies both between species and between developmental stages in flowering plants [16, 18]. This study is also among the first to characterize and annotate floral transcriptomes in Neotropical Gesneriaceae, a lineage well known for diverse and colorful flowers [50]. Achimenes offers a unique opportunity to study the genomics of flower diversification in a comparative context because very closely related species display an extraordinary range of morphological diversity likely tied to pollinator preferences and shifting patterns of gene expression. Rather than using a candidate-gene approach to understand patterns of speciation and diversification, we utilize high-throughput sequencing to begin searching for the potential pathways involved. We assembled between 29,000 and 42,000 putatively unique primary and alternate transcripts for four species of Achimenes that display many of the most common floral forms found in the genus. Orthogroup detection among Achimenes and against an Erythranthe corolla transcriptome revealed numerous conserved and distinct transcript clusters expressed among species (Fig. 5). Coexpression clustering revealed distinct patterns of gene expression in different stages of development (Fig. 6; Additional file 15). Assessing protein sequences for signatures of positive selection revealed numerous protein sites under selection in proteins involved in flower development, pollination, and transcription factor activity (Table 4, Additional file 20, Additional file 21). To further explore each of these analytical approaches, we annotated all transcriptomes with gene ontology terms for quantitative comparison. The overall GO representation of each transcriptome qualitatively matches that of other floral transcriptomes [28, 51] and there were no significant deviations in the GO representations between the four species of Achimenes. Comparisons of expression patterns for genes involved in anthocyanin and carotenoid biosynthesis, as well as flower development, also allow for further understanding of the temporal and evolutionary patterns of the expressed genes.
Assembly and consensus transcriptome
Experiments that use transcriptome sequencing have several considerations, including how many replicates to sequence and how much sequencing to perform. The aims of our study were to generate preliminary transcriptome data for four species with three developmental time points in each. Our experiment produced sequenced between 6.3 and 7.2 Gb pairs for each species we sampled (Table 1). From recent transcriptome analyses in other non-model plants, the read generation per sample is commonly 2 to 5 Gb [24, 52–54]. By combining the time point samples in each species, we hoped to provide a large set of reads for de novo reference assembly. After combining reads for each time point, the average number of base pairs used for assembly was 6.8 billion (Table 1), similar to these other studies [24, 52–54]. We believe this provides us with an adequate number of reads for initial characterization of our non-model plant subjects. As would be expected, increasing the sequencing depth for a given sample will greatly improve the ability to identify novel and unique transcripts. Future experiments in Achimenes will add additional sequencing depth and replicates. We additionally attempted to assemble the best set of transcripts with our data in order to perform comparative analyses relevant to floral developmental processes. Our approach to do numerous assemblies using different parameter settings was an attempt to generate as many complete transcripts as possible. Quality of our assemblies was confirmed by sequence comparison through orthology-based analyses and annotation of transcripts to known genes from model plant species. BLASTx hits to SwissProt proteins that had >80% coverage constituted between 34% and 40% of our assembled transcripts. These factors provide confidence that our experimental approach was able to meet the aims of our study and to provide initial characterization of the floral transcriptomes in non-model plants.
Our study is among the few that use a multiple assembler approach [55–57]. Rather than relying on a single de novo assembly program for all contig assembly, we used a combination of Trinity, Velvet, and Oases, to create seven assemblies for each transcriptome that we then merged into a single reference set of contigs. This approach has been used by other studies with success in increases contig length, recovering more unique transcripts, and minimizing sequence redundancy [55–57]. Our approach additionally took advantage of multiple k-mer lengths for assembly in Velvet and Oases. Multiple k-mer sizes have been demonstrated to assemble more lowly and highly expressed full-length transcripts than using a single k-mer size alone [58]. Our Trinity assemblies produced fewer contigs with lower N50 and mean lengths than the Velvet/Oases assemblies (Additional file 1). As the k-mer size increases, from 25 to 75, the Velvet and Oases assemblies produced fewer contigs with lower N50 and mean lengths (Additional file 1). Larger k-mer sizes also appeared to assemble the largest contigs even though the mean length overall was lower.
Although summaries of the distribution of contig lengths are informative, the goal of transcriptome assembly is not longer sequences, but rather accurate sequences. One metric that remains informative is the proportion of contigs that have significant similarities to known proteins. The difficulty in this measure stems from studies reporting slightly different results using different BLAST parameters and databases. However, nearly 80% of our combined assembly of primary and alternate transcripts had matches in SwissProt or Nr and this value is as high or higher than all other comparable statistics reported in other de novo assemblies [20, 24, 28]. Another useful metric is the proportion of the contig and its corresponding best BLAST hit that align to one another. Between 11,420 (27.66%) and 10,281 (35.37%) contigs are covered by at least 75% of their best BLAST hit. These results provide strong evidence that the contigs we assembled in absence of a reference genome largely represent real transcripts and not assembly error.
Core, shared, and unique genes
Our results indicate that the four Achimenes species in our study share a core set of genes expressed during flower development that may also be more broadly shared among other gesneriads. These transcripts code for proteins involved in essential cellular and metabolic functions, such as glycolysis, photosynthesis, and amino acid metabolism (Additional file 12). The transcriptomes also contained “shared” genes, which were observed in two or three of the four target species. There is limited data on how much physiological diversity might be present among such closely related gesneriad species because these taxa have been traditionally defined based on morphological features alone [10, 12]. Therefore, we were interested in what our data may reveal about the relatedness of these closely related taxa. Within the cluster that was unique to all four species (“Shared Achimenes”), there was significant overrepresentation of proteins involved in DNA binding and transcription factor activity (Table 3, Additional file 13). This may represent an artifact of our orthogroup clustering approach because our chosen comparison (Erythanthe) was a corolla-specific transcriptome rather than whole developing flower as in our samples. We expect that our sampling would capture additional transcripts representing transcription factors involved in calyx, stamen, and ovule development that may be missing from the Erythranthe transcriptome. The Erythranthe transcriptome is from corolla tissue; therefore, a more complete sampling of the flower would provide a more complete comparison. The overrepresentation of DNA binding activity may also represent an expansion and specialization of transcription factor gene families in Achimenes that may have a role in determining many of the unique phenotypes seen. Additional sampling of whole flowers in related species may provide insight into these two possibilities. The remainder of transcripts (approximately one quarter) in each of our four Achimenes transcriptomes was found in a single species (Fig. 5). The numbers of transcripts that were putatively species-specific is higher than what we would expect given the close phylogenetic relationships of the four species. Enrichment analyses also did not indicate large numbers of GO terms over- and underrepresented in each species (Additional file 14). Even with the large number of these unassigned transcripts, our assembly pipeline reduced nearly all redundancy by removing identical and closely related sequences.
Coexpression clustering
Coexpression clustering allows us to identify biological entities (e.g., genes) that share similar profiles across several developmental stages and may help identify groups of genes that are involved in the same biological processes [59, 60]. While we are unable to perform standard analyses of differential expression in the current study (no biological replicates), coexpression clustering provides interesting and useful information on the dynamic temporal changes in gene expression that occur during flower development. Future studies will include additional replicates to perform statistical analyses of differential expression both within and between species of Achimenes. Clustering analyses based on metric criteria, such as k-means [61] or hierarchical clustering [62], have been broadly used to cluster microarray-based measures of gene expression, as they are rapid, simple, and stable. These approaches require the user to decide on the metric and criterion to be optimized, as well as selecting the appropriate number of clusters, which may not be biologically relevant [63]. We chose an alternative approach, namely probabilistic clustering that uses Poisson mixture models that allowed us a straightforward approach for parameter estimation and model selection for cluster assignment, as well as a per-gene conditional probability of belonging to each cluster. Other model based clustering approaches may also utilize negative binomial (NB) algorithms (such as MBCluster.Seq) [64]. Poisson models have been shown to fit well to data without biological replicates [65] and NB models to data with biological replicates [66]. We therefore use Poisson models to explore patterns of coexpression in our transcriptomes.
Clustering selected between 25 and 34 groups for our transcriptomes that represented genes with shared expression profiles (Fig. 6; Additional file 15; Additional file 17). Enrichment tests validated our approach by identifying significant GO terms that were overrepresented in numerous clusters. A majority of clusters in each species had overrepresented GO terms (Additional file 17, Additional file 18). This clustering approach provides us with groups of genes that are expressed in similar stages that may be linked with particular metabolic or biosynthetic pathways of interest. Coexpression clustering has often been combined in other systems with experimental data or metabolic profiling [67, 68]. Combining clustering data with other approaches has the ability to provide additional support for specific patterns or processes detected from clustering. Obtaining lists of GO terms enriched in coexpression clusters is another useful approach to find patterns within large datasets that can then be used to guide experimental approaches to validate and provide additional support for the patterns seen. Our approach to coexpression clustering differs from commonly used coexpression network approaches that also seek to find biologically interesting clusters of genes sharing similar functional roles. Network analyses, which often use the Weighted Gene Correlation Network Analysis method (WGCNA) [69], usually require at least 15 samples to produce reliable results. Network approaches have been used in other floral transcriptomes to uncover gene networks involved in developing organs [70], floral bud development [71], and pistillate flowering [72]. In future analyses of Achimenes, additional replicates and sampling will allow us to perform network-based analyses that may uncover additional gene network modules involved in flower diversification.
Flower development: spurs
Numerous molecular genetic studies have demonstrated the crucial role of transcription factors in reproductive development of plants. The homologs of many of the genes identified in our study are well known to regulate aspects of flower development in model systems, such as Arabidopsis. As expected, we observed an abundance of genes involved in various processes related to flower development, such as the transition to flowering and floral organ identity (Additional file 10). Clear patterns are apparent for genes showing high or low levels of expression during the different developmental time points we sampled. Many studies that have used transcriptome sequencing to understand flower development have focused on sequencing individual floral organs (e.g., petals, stamens, etc.) and comparing them to identify genes differentially expressed between organs [22, 25, 54, 73]. Comparing expression between different tissues has the advantage of being able to identify where individual genes show high or low expression levels. Often these studies focus on a single species. Our aims for the current study were instead to investigate and compare the floral transcriptome in many closely related species that exhibit very diverse flowers. The advantage of our approach is the ability to begin understanding how gene expression differences may contribute to phenotypic differences among closely related species. We identified over 100 transcripts likely involved in flower developmental processes (Additional file 10). These transcripts in Achimenes largely show similar expression patterns seen in other flowering plants [22, 25, 74]. The orthologs of many well-known MADS-box genes (e.g., AP1, AP3, PI, and AG) are crucial for orchestrating floral organ identify [75, 76]. The expression patterns of these genes follow what we might expect given when the different floral organs are developing in Achimenes flowers (Additional file 10). For instance, the A-class genes AP1 shows high expression during the bud stage when sepals are developing and the B-class genes AP3 and PI have increased expression during D stage when petals are developing (Additional file 10). Elaboration of the petals to produce different shapes and widths likely involves genes outside these MADS-box genes [77].
Some species of Achimenes (including A. patens) exhibit a unique spur-like outgrowth of the petal tube that extends in opposition to the tube opening (Fig. 1). This petal spur has evolved independently at least three times in Achimenes, mostly in butterfly-pollinated species where the flower is presented at a downward angle (Fig. 1). The purpose of this petal spur in Achimenes has yet to be elucidated; it differs from the spurs in other lineages (such as columbines, Aquilegia) by not containing nectary tissue [10]. The genetic factors influencing the development of spurs have not yet been fully understood. Recent transcriptome sequencing of developing spur tissue in Aquilegia identified several candidate genes for this process, including homologs of TCP4, GRF1, and many other genes that contribute to cell proliferation and auxin signaling [37]. We see an increased level of gene expression for TCP4 in A. patens in the stages where spur growth is seen while this gene in the other three species remains much lower (Fig. 4). We also see an increase in gene expression of STY1 and ARF8 in A. patens, similar to what was reported in Aquilegia (Fig. 4). With the patterns seen in A. patens relative to the other species, we can hypothesize that TCP4 may be playing a significant role in the development of the petal spur. KNOX genes, particularly STM, have also been hypothesized to be important players in petal spur development in Antirrhinum and Linaria [78, 79]. Overexpression of KNOX genes in Antirrhinum produced spur-like outgrowths in the floral tube [78], while KNOX genes in Linaria displayed increased expression in petal spur tissue [79]. Our expression estimates for STM across Achimenes do not offer as clear a pattern as TCP4; STM gene expression patterns are similar across several species (Fig. 4). The pattern of STM expression is similar in both A. patens and A. misera (Fig. 4). Testing the functional roles of TCP4 and STM will be important in future work to determining which is more likely to be important for petal spur growth in Achimenes.
Flower color: anthocyanins
Differences in flower color are one of the most distinguishing characters that separate Achimenes species. Flowers across the genus display an amazing array of colors and color patterns, including species with white, yellow, red, blue, and purple pigmentation [10, 12] (Fig. 1). The primary pigment in flowers of Achimenes and most angiosperms are anthocyanins, a class of flavonoids that represent a large group of secondary metabolites [80]. The types of pigments present in floral tissue vary across Achimenes species, with all taxa containing anthocyanins and several containing a mix of anthocyanins and carotenoids. Anthocyanins contribute hues of blues, purples, and reds due primarily to production of pelargonidins, cyanidins, and delphinidins [80]. In plants, the biochemistry of the ABP is very well studied and understood in both model systems (e.g., Arabidopsis) [81] and non-model systems (e.g., Aquilegia, Mimulus, and Iochroma) [82–86]. While the biochemical reactions involved in the ABP are well understood, further research aims at understanding how the genetics of the pathway contributes to species differences in pigment production and the role it plays in adaptive evolution. The ABP is composed of 7 structural loci, with many of the earliest steps highly conserved in plants due to their role in producing precursor products involved in defense and UV protection [80, 81] (Fig. 2). The downstream pathway splits into 3 branches that lead to production of red pelargonidins, purple cyanidins, and blue delphinidins [80]. Flux down any of these branches is largely determined by the activity of two enzymes: F3′H and F3′5′H. Downregulation or inactivation of these enzymes can cause flux to be redirected down a different branch, resulting in a different flower color.
Several possible routes to produce variation in anthocyanin production exist, including gene loss or transcriptional regulation. One predominant example seen numerous times across flowering plants is the shift from blue-colored flowers to red-colored flowers that is closely associated with a shift from bee pollination to bird pollination [84, 85, 87–90]. These studies have implicated the downstream enzymes of the ABP (particularly ANS, DFR, F3′H, and F3′5′H) being involved in flower color transitions. Primarily, two often predictable routes have been suggested for the transition from blue to red anthocyanin pigment production: acquisition of mutations in DFR that alter its substrate specificity [84–86] or altered expression of F3′H and F3′5′H resulting from cis- of trans-regulatory mutations [84, 88–90]. Given the constrained structure of the ABP and the few demonstrated genetic changes involved in flower color transitions, our focus in Achimenes lays in genetic changes involving the enzymes DFR, F3′H, and F3′5′H, as well as the R2R3-Myb transcription factors that regulate the ABP [86, 91].
In Achimenes, multiple transitions from blue to red exist [10], and there also exists at least one likely red-to-blue flower color transition on the branch leading to A. cettoana (Fig. 1). This type of transition is exceedingly rare in plants and has few documented explanations. The transition of blue-to-red is more common and often involves predictable changes to key enzymes of the ABP, including DFR, F3′H, and F3′5′H (see Discussion above). One such case of red-to-blue flower color transition involves a gene duplication of F3′H and neofunctionalization to regain the role of F3′5′H in Asteraceae [92, 93]. A similar gene duplication event is not found when the gene trees are examined for F3′H and F3′5′H (Additional file 7), suggesting that changes in gene expression are more likely involved in a red-to-blue color transition in Achimenes.
We captured transcripts of core downstream enzymes of the ABP from all 4 transcriptomes, each with appreciable expression levels that show an increase from B to A stage (Fig. 2). Several patterns of expression emerge from the data. Both A. cettoana and A. patens have increased expression of F3′5′H, the enzyme responsible for directing the flux of the pathway toward delphinidin production (Fig. 2). These flowers are blue and purple, so this pattern is what we might expect to see. Expression levels of the enzymes in A. misera are much lower, which we might also expect given that this flower produces very little pigment except in areas of the corolla throat (Fig. 2). Expression of F3′5′H is much lower in A. erecta, the red-flowered species (Table 3) and this pattern follows the pattern seen in other systems [83, 85]. The possible explanation for how the red-to-blue color transition could have occurred in Achimenes will require more detailed studies than those presented here, but given that we see expression of all ABP enzymes, it is possible that differences in anthocyanin production are due to genetic changes in the transcription factors that regulate the pathway, not in loss of function mutations as found in other systems [83–85, 94]. Additionally, Achimenes species tend to produce anthocyanins in both floral and vegetative tissue [12]. This coupled with the captured expression of the ABP enzymes may suggest that flower color transitions may involve a change to substrate specificity in DFR or in the downregulation of F3′H and F3′5′H enzymes in red flowers through trans-activating mutations.
It is interesting to find that several of the ABP enzymes are coexpressed together and in three species (A. cettoana, A. misera, and A. patens) they are coexpressed with candidate R2R3-Mybs that we identified (Additional file 19). In A. cettoana, the candidate R2R3-Myb is coexpressed with F3′5′H, the enzyme that directs the metabolic flux of the pathway toward the production of delphinidins (Fig. 2). Another candidate R2R3-Myb in A. patens was coexpressed with F3H, F3′5′H, and ANS (Additional file 19). With this pattern in these two species, we might hypothesize that the candidate R2R3-Mybs are involved in transcriptional regulation of the ABP to produce delphinidin pigments. This is what we would expect given the blue and purple flower color in these species. In A. misera, one candidate R2R3-Myb was coexpressed with ANS and might be involved in regulating more downstream parts of the ABP (Additional file 19).
The role of R2R3-Myb transcription factors in regulating various steps of the ABP has been well studied in numerous plants [86, 91, 95] and the possible role of these transcription factors in Achimenes will need to be studied further. We identified putative proteins in Achimenes with high-similarity to R2R3-Mybs that have experimental evidence indicating their role in regulating anthocyanin accumulation (Additional file 11). These Achimenes R2R3-Mybs are closely related to homologs recently identified in Erythranthe [86] as well as homologs from Petunia [96] and Antirrhinum [97]. We can hypothesize that these R2R3-Mybs from Achimenes may function similarly to regulate expression of the ABP given their close similarity to other homologs as well as their coexpression patterns.
Flower color: carotenoids
Carotenoids are important pigments that carry out functions in protecting the photosynthetic apparatus from photooxidative damage and acting as accessory pigments in light harvesting [98]. In non-photosynthetic tissues, carotenoids are usually synthesized as secondary metabolites and accumulate in chromoplasts, providing the yellow, orange, and red colors in many flowers, thus serving an important function in the ecology and evolution of plants by attracting pollinators and seed dispersers [99]. In many Achimenes species, carotenoids are found throughout the corolla; while in other species carotenoid production is limited to the corolla throat (as in A. erecta and A. misera). Few species, including A. cettoana and A. patens, do not appear to produce carotenoids in the corolla tissue and only produce anthocyanins.
We identified putative enzymes in the plant carotenoid biosynthetic pathway (CBP) in each of our transcriptomes (Fig. 3). The CBP splits into two branches: the α-carotene branch (Fig. 3) and the β-carotene branch (Fig. 3). Biochemical studies of floral carotenoids are lacking in Gesneriaceae, therefore we cannot confidently assess which carotenoids are present in Achimenes corollas without doing biochemical experiments. Our expression estimates of the CBP enzymes indicate activity of all the core enzymes in each species (Fig. 3). Some species of Achimenes, including A. cettoana and A. patens, contain no carotenoids in the corolla and lower expression of the CBP enzymes in these species may reflect carotenoid accumulation in sepals and pollen. In other systems, particularly Erythranthe, all floral carotenoids are on the β-carotene branch [100]. We find lower levels of 2 enzymes exclusive to the α-carotene branch (LCYE and CYP97) compared to the other enzymes found on the β-carotene branch (LCYB, BCH, ZEP, NXS, and NCED; Fig. 3). These results may indicate that Achimenes and other gesneriad species are primarily producing floral carotenoids via the β-carotene branch, but further biochemical characterization and experimental studies will need to be undertaken to support this conclusion.
In general, expression estimates of CBP enzymes are lower in A. cettoana and A. patens (Fig. 3) and both of these butterfly-pollinated species contain little to no visible carotenoid pigment accumulation in their corolla. Flavonoids (like anthocyanins) absorb UV light and carotenoids reflect UV light. Presence of anthocyanins in the petal lobes and absence in the petal tube may reflect the common use of a ‘bulls-eye’ UV pattern to attract insect pollinators. In contrast, A. erecta and A. misera contain visible amounts of carotenoids in the corolla tube. Bee-pollinated A. misera flowers have a clear nectar guide on the ventral petal formed by the accumulation of carotenoids, an important trait for successful bee pollination [101, 102]. Bird-pollinated flowers, like A. erecta, often contain combinations of anthocyanins and carotenoids, with red anthocyanins preventing visitation by bees [103]. Taken together, the pigments contributing to flower color in Achimenes are important for determining what pollinators visit. Despite butterfly- and bee-pollinated flowers likely containing a nectar guide, in A. cettoana and A. patens it appears to be due to flavonoids, while in A. misera it appears from both flavonoids and carotenoids.
The regulation of carotenoid pigmentation in flowers is less well understood than the regulation of the ABP. An R2R3-Myb transcription factor, Reduced Carotenoid Pigmentation 1 (RCP1), has been the only transcription factor identified to be involved in flower-specific carotenoid biosynthesis [95]. Our analyses identified 9 transcripts with similarity to RCP1 (Additional file 11). However, when we look at patterns of coexpression we only find one candidate (in A. erecta) being coexpressed with any of the enzymes of the CBP (Additional file 19). Future genetic experiments will be important to elucidating the transcriptional regulation of this network in Achimenes flowers. So far, we have identified potential candidate transcription factors, but their specific function will need to be further explored.
Adaptive evolution
The evolution of floral form among the four Achimenes species is likely influenced by differences in pollinator availability and preferences. Within the group, there are distinct floral forms that correspond closely with different pollination syndromes [10]. Highly dimensional quantitative data of floral morphology and qualitative data of color and petal spur size can be reduced into groups that correspond closely to different pollinators. Flowers of Achimenes are visited by a number of insects (bees, Apidae; euglossine bees, Euglossini; butterflies, Lepidoptera) and hummingbirds (Trochilidae) [13]. Observations of pollinator visitation to four Achimenes species provide evidence for the use of pollination syndromes to separate floral form into unique groups [13]. Linking protein evolution to the convergent evolution of these different pollination syndromes may provide evidence for shared or different genetic routes to these forms. Previous studies have suggested the pathways involved in pigment production, particularly anthocyanins, are involved in pollination syndrome transitions [45, 74, 75].
Our selection analyses found numerous genes showing significant signs of molecular evolution (Table 4, Additional files 18, Additional file 21). However, our analyses did not provide statistical over- or underrepresentation of any GO terms within the set of proteins with sites under positive selection. We do find a number of proteins involved in various processes during flower development that might be involved with floral diversification (Table 4). Many genes have GO terms associated with them involving the regulation of flower development, anatomical structure development, and transcription factor activity, among others (Additional files 18, Additional file 21).
None of the core enzymes of the ABP or the CBP that we identified were under positive selection. However, a protein annotated as DFR was identified from A. erecta (Table 4). The sequence of this protein shares similar motifs with the DFR enzyme we identified above, but is not the same transcript (Additional file 6). Given its annotation and similarities it is likely involved in anthocyanin production, but possibly in a different step of the ABP than the core part of the pathway we considered here. Another protein was identified in A. erecta and annotated as ABCB19 (ABC transporter B family member 19; Table 4), an auxin efflux transporter with roles in mediating anthocyanin accumulation in floral tissue [104]. Additionally, in A. cettoana, a protein annotated as 5MAT1 (malonyl-coenzyme:anthocyanin 5-O-glucoside-6”’-O-malonyltransferase; Table 4) was also identified with a role in catalyzing the transfer of a malonyl group to the pelargonidin pigment classes [105]. Like DFR, both ABCB19 and 5MAT1 are likely involved in anthocyanin biosynthesis, albeit outside of the core pathway. Other studies have found signatures of positive selection in the core ABP enzymes [106], but in the current study we do not detect any significant evidence.
Some interesting genes involved in flower development were additionally identified to be under positive selection. In A. patens, HDA19 (histone deacetylase 19) is a protein involved in epigenetic repression and plays an important role in transcriptional regulation, particularly the repression of several A- and E-class MADS-box genes that control sepal and petal identity [107]. The role of this histone deacetylase in the epigenetic modification of floral developmental programs in A. patens is not immediately apparent; therefore, additional studies will be useful to understand the potential myriad roles this gene may play in development. Another protein under selection identified from A. misera, a homolog of FRI (frigida), is involved in flowering time transition [108]. Allelic variation in FRI was demonstrated in Arabidopsis to be important for natural variation in flowering time across different latitudes [108]. Flowers are produced on A. misera nearly constantly during the growing season and the potential role of FRI in development will need to be assessed in further experiments.
With expanded sampling of additional Achimenes species, our analyses of positive selection will be more robust than those presented here. We were able to include sequences from five species (4 ingroup and 1 outgroup) and compare gene families that contained members from each of those species. Our use of site-models allows our detection of specific amino acids within the protein that may be undergoing positive selection [48, 49]. Evolutionary change can also happen in the regulatory region of genes, which may affect the level, timing, and location of gene expression. Without a genome reference to look for upstream and downstream mutations that may affect particular genes, we are unable to currently look at these regions for their effect on genes involved in floral diversification.
Conclusions
The newly sequenced, assembled, and annotated floral transcriptomes for Achimenes. cettoana, A. erecta, A. misera, and A. patens provide valuable genomic resources to study the molecular mechanisms of development, adaptation, and speciation between closely related species. Comparative analyses of closely related taxa are important for understanding the molecular mechanisms involved in the evolution and diversification of lineages. The diversity of floral forms in Achimenes is hypothesized to correspond to pollinator-driven preferences toward different shapes, colors, and orientations to provide successful pollination and fertilization [7, 10]. Large similarities between the floral transcriptomes in closely related species with diverse floral phenotypes suggests that these visible differences are, in part, due to changes in a small set of genes. Combining analyses of sequence orthology, gene expression, and molecular evolution have provided initial candidates for future analyses into the diversification of floral form. Exploration of the expression patterns for genes relating to flower color and flower shape has provided interesting patterns corresponding to the floral form of each species. Patterns of expression for genes involved in anthocyanin and carotenoid biosynthesis indicate that flower color transitions may be due to changes in a small set of genes, some of which are coexpressed together. The datasets presented here also contribute to the growing number of available genomic resources for species in the family Gesneriaceae [50, 109–112] that are study organisms for desiccation tolerance, flower development, and leaf development. Together, these newly developed genomic tools provide a valuable resource for ecological and evolutionary genomics projects, serving as a starting point to begin understanding phenotypic variation and the evolutionary genetic forces driving variation across species and populations in the Gesneriaceae and other tropical plant lineages.
Methods
Plant material
Flower shape in Achimenes can take many forms, including funnelform, salverform, tubular, and a number of intermediate forms (Fig. 1). Primary flower color is also quite variable and is represented by flowers of white, purple, pink, red, blue, and yellow colors (Fig. 1). We chose to sample species broadly across Achimenes for the present study in order to develop initial resources for understanding the genomic basis for flower diversification. Our sampling includes A. cettoana, a butterfly pollinated species with purple-blue salverform flowers (Fig. 1), A. erecta, a hummingbird pollinated species with red salverform flowers (Fig. 1), A. misera, a bee pollinated species with small, white funnelform flowers with a purple throat (Fig. 1), and A. patens, a butterfly pollinated species with large, purple-pink salverform flowers and a noticeable petal spur (Fig. 1). These four species represent most of the common flower shapes and colors seen in the genus, and while they do not represent all of the possible floral forms, they present us with a starting point to guide future studies. Vouchers of each sampled species are deposited in the WR herbarium with the following identification numbers: A. cettoana, WR0155; A. erecta, WR0156; A. misera, WR0157; A. patens, WR0158.
Three stages of flower development were sampled so that temporal changes in gene expression could be studied. ‘Immature Bud’ (B) stage was the smallest flower buds that could be distinguished from vegetative buds (Fig. 1). ‘Stage D’ (D) were larger flower buds that were beginning to accumulate pigmentation, the cells in the corolla tube are elongating, and the petal spur (as in A. patens) is beginning to develop (Fig. 1). ‘Pre-Anthesis’ (A) flower buds were the largest and fully pigmented and were collected one-day before anthesis (Fig. 1). Given that the different species have different flowering times, these stages are determined from qualitative observations. Plants were grown in greenhouse conditions under natural daylight, controlled temperature ranging from 27 to 32 °C, and >80% humidity. For all experiments, plant material was harvested directly into liquid nitrogen and subsequently stored at -80 °C. To obtain enough fresh material for RNA extraction, between 2 and 5 flower buds were sampled from an individual plant.
Library preparation and sequencing
Total RNA was isolated from developing flower buds of Achimenes by grinding 50–100 mg of tissue frozen in liquid nitrogen. RNA was then extracted using the Qiagen RNeasy Plant Mini Kit (Qiagen, Valencia, CA) following the manufacturers instructions. To avoid genomic DNA contamination, RNA was treated with Rnase-free Dnase I (Thermo Fisher Scientific, Waltham, MA). The RNA integrity was assessed by visualization in 1.0% agarose gels and RNA Integrity Number (RIN) as measured by an Agilent 2100 BioAnalyzer (Agilent, Santa Clara, CA). Ribosomal-depleted RNA samples were prepared using the Ribo-Zero rRNA Removal Kit for plant leaf material (Illumina, San Diego, CA). Sequencing libraries were constructed using the TruSeq RNA-seq sample prep kit from Illumina (Illumina, San Diego, CA) according to manufacturers instructions. All stages of library preparation were performed at the Genome Sequencing and Analysis Facility (GSAF) at the University of Texas (Austin, TX). RNAseq libraries were quantified using a BioAnalyzer 2100 High Sensitivity DNA chip and pooled based on nM concentrations. Individual libraries were uniquely barcoded, multiplexed, and sequenced for 100 bp paired-end reads (2 x 100 bp) using one lane on the Illumina HiSeq2500 at the GSAF.
De novo assembly
Raw 100 bp paired-end Illumina reads were sorted by barcode and assessed for quality using the tools implemented in FastQC [113]. The 3′-ends of the reads were quality trimmed using FASTX-Toolkit [114], removing any reads that contained bases with Phred scores less than 20. We also discarded any low quality reads less than 50 bp long or with less than 80% of bases having a Phred score greater than 20. Contaminating Illumina adapter sequences and primers were also trimmed.
Three de novo assemblers were used to construct a robust set of contigs using different algorithms and k-mer sizes: Trinity (Tr), Velvet (Vt), and Oases (Oa) [115–117]. Data from the three developmental stages in each species were concatenated prior to de novo reference assembly. To provide sets of assembled transcripts, we employed multiple assemblers using a range of k-mer sizes. For Tr assembly, we used forward-reverse read orientation (--SS_lib_type FR) with the default k-mer size of 25. For Vt assembly, we utilized a multiple k-mer approach, with separate assemblies performed for k-mer sizes 25, 35, 45, 55, 65, and 75, and specifying a library insert size of 150 (-ins_length 150). Each Vt k-mer assembly was further assembled using Oa under the default settings.
In order to reduce the redundancy of assemblies and create sets of primary and secondary transcripts, all assemblies were subjected to the EvidentialGene tr2aacds pipeline [118]. Merged assemblies were produced using the seven de novo assemblies generated previously. Each de novo assembly for each species was generated using the three tissue samples from the same species. The EvidentialGene pipeline selects a ‘best’ set of de novo assembled transcripts, based on coding potential, from a pool of such sequences. The algorithm first infers the coding DNA sequences (CDS) and amino acid sequences for each sequence, and then removes redundant sequences using the amino acid information by choosing the best coding sequences from amongst identical sequences with fastanrdb (exonerate-2.2.0) [119] and CD-HIT-EST [120]. Self-on-self BLASTn is then implemented to identify highly similar sequences. The alignment data and CDS/protein identities are then used to select and output transcripts classified as ‘main’ (primary; the best transcripts with unique CDS) or ‘alternate’ (possible isoforms), and another set classified as ‘dropped’ which did not pass the internal filters of the pipeline. The chosen primary and alternate contigs were used for further analyses and annotation.
Functional annotation
To annotate transcripts, we conducted a BLAST search of all unique ‘primary’ transcripts against the SwissProt database (BLASTx, E-value = 1e-06) [30], NCBI non-redundant (Nr) protein database (BLASTx, E-value = 1e-06) [31], and Plant Non-coding RNA Database (BLASTn, E-value = 1e-06) [32]. Additionally, the ‘alternate’ transcripts sets were searched against the SwissProt database for annotation. For each sequence we retained the top five BLAST hits for subsequent analysis. We placed first priority to the SwissProt database hits for annotation, followed by the Nr and PNRD databases because the SwissProt database contains more GO identities associated with the protein hits than either the Nr or PNRD databases. Sequences with a match in either the SwissProt or Nr database were subsequently annotated with GO terms [33] as implemented in Blast2GO v.3.0 [121]. InterProScan was used to scan transcripts for domain and motif information that may provide additional GO identities not attributed using blastx hits alone [33]. GO terms were assigned based on BLAST hits and InterProScan results to cover three types of terms: BP, CC, and MF. We additionally integrated the Second Layer Concept of Myhre et al. [35] (ANNEX augmentation) to identify, given the molecular function, biological processes where the molecular functions are involved, and cellular components where they are active. Finally, GO terms were simplified to a smaller set of high-level GO terms (GO slims) [122]. We obtained GO slims through Blast2GO with the plant slims developed by the Arabidopsis Information Resource [122]. Additionally, non-coding ribosomal RNAs and transfer RNAs were detected using RNAmmer [123] and tRNAscan [124], respectively. We tested for significant differences in sequence representation for GO categories between all species with a Chi-square test followed by using False Discovery Rate (FDR, α = 0.05) adjusted p-values [125].
Specific enzymes related to anthocyanin pigment production were identified through hidden markov models (HMM) built and trained in HMMER [36]. We searched our assemblies for proteins identified as homologs to ANS, DFR, F3′H, and F3′5′H. Protein sequences from other studies were downloaded from GenBank (Additional file 4), aligned using MUSCLE [126], and used to create HMM profiles. These HMM profiles were then used to search our reference transcriptome to identify possible candidates proteins. These candidate proteins were then aligned with candidates from other studies (Additional file 4) using MUSCLE and visually inspected to identify and correct misaligned regions. Finally, these alignments were used to construct neighbor-joining trees in Geneious version R9 [127] with branch support assessed by performing 100 bootstrap replicates.
Putative proteins involved in flower development, carotenoid biosynthesis, and petal spur development (taken from [37]) were identified by BLASTp searches against Arabidopsis homologs downloaded from the UniProt database (Additional file 4; www.uniprot.org). The criteria used to determine the best-hit transcript were (in order): bit score, E-value, and percent identity.
Members of the R2R3-Myb transcription factor family that may be involved in floral pigmentation were identified using HMM models built and trained in HMMER [36]. Proteins with experimental evidence supporting their role in the transcriptional regulation of floral pigmentation were downloaded from GenBank (Additional file 4; https://www.ncbi.nlm.nih.gov/genbank/). The proteins were first aligned using MUSCLE [126], and then the conserved Myb domains were extracted, re-aligned using ClustalW [128], and used to construct a neighbor-joining tree in Geneious version R9 [127] with branch support assessed by performing 100 bootstrap replicates.
Orthogroup identification
We next identified conserved orthogroups from the sets of translated proteins identified in each Achimenes species using OrthoFinder v.0.3.0 [129]. This method solves the problem of gene length bias in BLAST searches by normalizing the bit scores by both gene length and phylogenetic distance and outperforms the more commonly used OrthoMCL in accuracy and speed [129]. Orthologs and paralogs were determined for each species individually as well as in five-way comparisons. In the comparative analyses, we used a corolla transcriptome from Erythranthe lewisii LF10 (15 mm corolla; available from http://www.monkeyflower.uconn.edu/resources) as comparison [86]. We chose E. lewisii for comparison because it is a flower-specific transcriptome that is phylogenetically close to Achimenes (both are members of the Order Lamiales). Protein coding sequences were produced for E. lewisii using TransDecoder v.2.0 [130], under default settings.
Quantifying and comparing gene expression patterns
Trimmed, high-quality reads from individual stage-specific samples (B, D, and A) were independently mapped onto each primary reference transcriptome using the ungapped alignment software bowtie [42]. We used the abundance of reads derived from each locus to estimate gene expression and calculate transcripts per kilobase million (TPM) values with the program RSEM (RNA-Seq by Expectation Maximization) [43]. The numbers of reads mapped per library were normalized by the trimmed mean of M-values normalization method (TMM) [131]. Genes were considered expressed in a developmental stage if they had a normalized TPM ≥0.01 in that stage. Expression estimates for floral developmental genes in individual species were transformed to Z-scores for heatmap representations.
Transcripts with estimated expression values ≤0.01 were removed prior to clustering. To cluster sets of co-expressed genes within each species, we performed clustering using HTSCluster [44]. Unlike other commonly used clustering algorithms (e.g., k-means, hierarchical), HTSCluster is a model based clustering approach that uses Poisson mixture models to cluster sequences using expression estimates and selects the appropriate number of clusters using slope heuristics (Djump and DDSE) [46]. We ran HTSCluster using the EM [45] algorithm for parameter estimation and tested cluster numbers ranging from K = 1, 2, …, 60. From 5 independent runs, we selected the model and associated cluster number that had the highest log-likelihood. We used both the Djump and DDSE criteria to select the number of clusters for each run. The degree of certitude in cluster assignment was additionally evaluated using the maximum conditional probabilities of cluster membership for the genes assigned to each cluster.
Detecting genes under selection
Each orthogroup identified with the OrthoFinder five-way analysis was run through a pipeline to identify protein sites potentially undergoing selection. The pipeline first takes the CDS sequences and inferred homology relationships and filtered these based on numeric, phylogenetic, and quality criteria to remove spurious data. We chose to keep proteins having a complete coding region (strings in multiples of 3), a minimum of 5 species and 5 sequences, and mean sequence divergence of ≤60%. Each satisfactory orthogroup then undergoes multiple sequence alignment using MUSCLE [126], protein-guided codon alignment using TrimAl [132], and phylogenetic tree reconstruction using dnaml from Phylip [133]. Finally these orthogroups are analyzed for signatures of selection using the site-models implemented in PAML v.4.6 [134]. For our analyses, we used the M1a (neutral), M2a (selection), M7 (beta), and M8 (beta + ω) models implemented in codeml [48, 49]. Model M1a was compared to M2a and M7 was compared to M8. Significance differences in model fit for each comparison were assessed using a likelihood ratio test followed by FDR correction for multiple hypothesis testing (α = 0.05).
Gene ontology enrichment analyses
We used the FatiGO [135] package as integrated with Blast2GO to assess enrichment of GO terms in the proteins identified during 1) orthogroup clustering, 2) coexpression clustering, or 3) detection of sites under positive selection. Previously for each Achimenes transcriptome, we obtained a list of annotated transcripts with associated GO identities. This information was then divided into three GO maps based on the three GO domains: 1) BP, 2) CC, and 3) MF. Each analysis was performed using a two-tailed Fisher’s Exact Test using FDR-corrected p-values (α ≤ 0.05). Both over- and underrepresented GO terms were identified for each cluster or group relative to the whole transcriptome background.
Acknowledgements
Joanna L. Kelley, Andrew McCubbin, and Amit Dhingra provided helpful discussion for this project. Brian W. Davis provided helpful comments on figure construction. The staff at the Genomic Sequencing and Analysis Facility (GSAF) at the University of Texas at Austin provided library preparation and sequencing services. We are grateful to Mohammed Bakkali and one anonymous reviewer for their critical review and constructive comments.
Funding
A Global Plant Sciences Initiative Fellowship awarded to WRR and funding from the School of Biological Sciences (WSU) to EHR supported this work. These funding sources had no role in the design, data collection, analyses, interpretation of data, and writing of the manuscript.
Availability of data and materials
The datasets generated during and analyzed during the current study are available in the NCBI Short Read Archive repository, available from accession SRP083265. The contig assemblies generated during the current study are available from the corresponding author upon request. The datasets supporting the conclusions of this article are included within the article and its additional files.
Authors’ contributions
WRR collected grew plant material, extracted RNA, performed all analyses, and wrote the manuscript. WRR and EHR conceived the project, designed experiments, and interpreted the findings. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Non-commercial plant material used in this study was acquired from established horticultural collections of Gesneriaceae plants cultivated in the School of Biological Sciences at Washington State University. Vouchers of each species have been placed in the WS herbarium.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- ABP
Anthocyanin biosynthetic pathway
- ANS
Anthocyanidin synthase
- BP
Biological process
- CBP
Carotenoid biosynthetic pathway
- CC
Cellular component
- CDS
Coding DNA sequences
- DDSE
Data driven slope estimation
- DFR
Dihydroflavonol 4-reductase
- Djump
Dimension jump
- EM
Expectation maximization algorithm
- F3′5′H
Flavonoid 3′,5′-hydroxylase
- F3′H
Flavonoid 3′-hydroxylase
- FDR
False discovery rate
- GO
Gene ontology
- HMM
Hidden markov models
- MF
Molecular function
- NB
Negative binomial
- Nr
NCBI non-redundant protein database
- Oa
Oases assembly
- PNRD
Plant Non-coding RNA Database
- TMM
Trimmed mean of M-values normalization method
- TPM
Transcripts per kilobase million
- Tr
Trinity assembly
- Ve
Velvet assembly
Additional files
Detailed sequencing and assembly statistics. A. Trinity assembly. B. Velvet and Oases assemblies. C. EvidentialGene merged assembly. (XLSX 41 kb)
Number of contigs detected as rRNAs and tRNAs in each transcriptome. (XLSX 33 kb)
Counts and proportion of level 2 Gene Ontology annotations for Achimenes transcriptomes. (PDF 361 kb)
Protein homologs downloaded from GenBank used for HMM profile searches. A. ANS. B. DFR. C. F3′H and F3′5′H. D. R2R3-Mybs. (XLSX 48 kb)
Neighbor-joining tree of anthocyanidin synthase (ANS) gene family. Putative Achimenes ANS orthologs are highlighted in red. Bootstrap support >50 are indicated above branches. (PDF 455 kb)
Neighbor-joining tree of dihydroflavonol 4-reductase (DFR) gene family. Putative Achimenes DFR orthologs are highlighted in red. Boostrap support >50 are indicated above branches. (PDF 330 kb)
Neighbor-joining tree of flavonoid 3′-hydroxylase (F3′H) and flavonoid 3′,5′-hydroxylase (F3′5′H) gene family. Putative Achimenes F3′H and F3′5′H orthologs are highlighted in red and blue, respectively. Bootstrap support >50 are indicated above branches. (PDF 1175 kb)
xAligned protein sequences for Achimenes F3′H, F3′5′H, and DFR. Amino acid substitutions between the sequences of A. cettoana, A. erecta, A. misera, and A. patens are highlighted in blue, red, black, and pink, respectively. A, F3′H; B, F3′5′H; C, DFR. (PDF 3121 kb)
Homologs of flower development genes identified in Achimenes. Included are the expression domain, primary role, and gene family. (XLSX 60 kb)
Expression of genes involved in flower development in Achimenes. (PDF 346 kb)
Neighbor-joining tree of R2R3-Mybs in Achimenes. Putative orthologs involved in anthocyanin and carotenoid biosynthesis are highlighted in blue and orange, respectively. Bootstrap support >50 are indicated above branches. (PDF 921 kb)
Significantly enriched Gene Ontology terms for sequences in the “Core” transcriptome after orthogroup classification. Terms are enriched if they have FDR-corrected p-values < 0.05 (Fisher’s Exact Test). Those terms that are overrepresented when all species are analyzed together (“Combined” column) are in bold. (XLSX 16 kb)
Significantly enriched Gene Ontology terms for sequences in the “Shared Achimenes” clusters after orthogroup classification. Terms are enriched if they have FDR-corrected p-values < 0.05 (Fisher’s Exact Test). Those terms that are overrepresented when all species are analyzed together (“Combined” column) are in bold. (XLSX 48 kb)
Significantly enriched Gene Ontology terms for sequences that were unassigned during orthogroup classification. Terms are enriched if they have FDR-corrected p-values < 0.05 (Fisher’s Exact Test). (XLSX 38 kb)
Coexpression clusters for Achimenes determined using Poisson mixture models. Gene profiles are depicted as boxplots. Conditions are as follows: 1, Bud stage; 2, Stage D; and 3, Pre-Anthesis stage. A, A. cettoana; B, A. erecta; C, A. misera; D, A. patens. (PDF 4192 kb)
Maximum conditional probability of cluster membership assigned by coexpression clustering using Poisson mixture models. A, Achimenes cettoana; B, A. erecta; C, A. misera; D, A. patens. (PDF 1176 kb)
Detailed model selection statistics and Gene Ontology enrichment for coexpression clustering. A. A. cettoana. B. A. erecta. C. A. misera. D. A. patens. (XLSX 54 kb)
Gene Ontology enrichment for coexpression clustering. A. A. cettoana. B. A. erecta. C. A. misera. D. A. patens. (XLSX 228 kb)
Coexpression clusters for candidates involved in anthocyanin biosynthesis, carotenoid biosynthesis, and spur development. (XLSX 40 kb)
Annotated proteins with sites under selection using PAML for M1a vs. M2a comparison. A. Achimenes cettoana. B. Achimenes erecta. C. Achimenes misera. D. Achimenes patens. E. Erythranthe lewisii. (XLSX 60 kb)
Annotated proteins with sites under selection using PAML for M7 vs. M8 comparison. A. Achimenes cettoana. B. Achimenes erecta. C. Achimenes misera. D. Achimenes patens. E. Erythranthe lewisii. (XLSX 75 kb)
Contributor Information
Wade R. Roberts, Email: wade.roberts@wsu.edu
Eric H. Roalson, Email: eric_roalson@wsu.edu
References
- 1.Barrett CH, Harder LD, Worley AC. The comparative biology of pollination and mating in flowering plants. Philos T Roy Soc B. 1996;351:1271–1280. doi: 10.1098/rstb.1996.0110. [DOI] [Google Scholar]
- 2.Regel PJ. Ecology and evolution of flowering plant dominance. Science. 1977;196:622–629. doi: 10.1126/science.196.4290.622. [DOI] [PubMed] [Google Scholar]
- 3.Endress PK, Matthews ML. First steps toward a floral structural characterization of the major Rosid subclades. Plant Syst Evol. 2006;260:223–251. [Google Scholar]
- 4.Becker A, Thiessen G. The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol Biol Evol. 2003;29:464–489. doi: 10.1016/s1055-7903(03)00207-0. [DOI] [PubMed] [Google Scholar]
- 5.Harrison J, Möller M, Cronk QCB. Evolution and development of floral diversity in Streptocarpus and Saintpaulia. Ann Bot-London. 1999;84:49–60. doi: 10.1006/anbo.1999.0887. [DOI] [Google Scholar]
- 6.Clark JL, Roalson EH, Pritchard RA, Coleman CL, Teoh V-H, Matos J. Independent origin of radial floral symmetry in the Gloxinieae (Gesnerioideae: Gesneriaceae) is supported by the rediscovery of Phinaea pulchella in Cuba. Syst Bot. 2011;36:757–767. doi: 10.1600/036364411X583709. [DOI] [Google Scholar]
- 7.Roalson EH, Roberts WR. Distinct processes drive diversification in different clades of Gesneriaceae. Syst Biol. 2016;65:662–684. doi: 10.1093/sysbio/syw012. [DOI] [PubMed] [Google Scholar]
- 8.Wiehler H. A report on the classification of Achimenes, Eucodonia, Gloxinia, Goyazia, and Anetanthus (Gesneriaceae) Selbyana. 1976;1:374–404. [Google Scholar]
- 9.Wiehler H. A synopsis of the neotropical Gesneriaceae. Selbyana. 1983;6:1–219. [Google Scholar]
- 10.Roalson EH, Skog LE, Zimmer EA. Phylogenetic relationships and the diversification of floral form in Achimenes (Gesneriaceae) Syst Bot. 2003;28:593–608. [Google Scholar]
- 11.Perret M, Chautems A, Spichiger R, Barraclough TG, Savolainen V. The geographical pattern of speciation and floral diversification in the Neotropics: the Tribe Sinningieae (Gesneriaceae) as a case study. Evolution. 2007;61:1641–1660. doi: 10.1111/j.1558-5646.2007.00136.x. [DOI] [PubMed] [Google Scholar]
- 12.Ramírez Roa MA. Revision de Achimenes (Gesneriaceae) Thesis: Universidad Nacional Autonoma de Mexico D.F; 1987. [Google Scholar]
- 13.Martén-Rodríguez S, Quesada M, Castro AA, Lopezaraiza-Mikel M, Fenster CB. A comparison of reproductive strategies between island and mainland Caribbean Gesneriaceae. J Ecol. 2015;103:1190–1204. doi: 10.1111/1365-2745.12457. [DOI] [Google Scholar]
- 14.Marra NJ, Romero A, DeWoody JA. Natural selection and the genetic basis of osmoregulation in heteromyid rodents as revealed by RNA-seq. Mol Ecol. 2014;23:2699–2711. doi: 10.1111/mec.12764. [DOI] [PubMed] [Google Scholar]
- 15.Davies KTJ, Bennett NC, Tsagkogeorga G, Rossiter SJ, Faulkes CG. Family-wide molecular adaptations to underground life in African mole-rats revealed by phylogenomic analysis. Mol Biol Evol. 2015;32:3089–3107. doi: 10.1093/molbev/msv175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davidson RM, Gowda M, Moghe G, Lin H, Vaillancourt B, Shiu SH, Jiang N, Robin BC. Comparative transcriptomics of three Poaceae species reveals patterns of gene expression evolution. Plant J. 2012;71:492–502. doi: 10.1111/j.1365-313X.2012.05005.x. [DOI] [PubMed] [Google Scholar]
- 17.Li QG, Zhang L, Li C, Dunwell JM, Zhang YM. Comparative genomics suggests that an ancestral polyploidy event leads to enhanced root nodule symbiosis in the Papilionoideae. Mol Biol Evol. 2013;30:2602–2611. doi: 10.1093/molbev/mst152. [DOI] [PubMed] [Google Scholar]
- 18.Yang Z, Wafula EK, Honaas LA, Zhang H, Das M, Fernandez-Aparicio M, Huang K, Bandaranayake PCG, Wu B, Der JP, Clarke CR, Ralph PE, Landherr L, Altman NS, Timko MP, Yoder JI, Westwood JH, de Pamphilis CW. Comparative transcriptome analyses reveal core parasitism genes and suggest gene duplication and repurposing as sources of structural novelty. Mol Biol Evol. 2015;32:767–790. doi: 10.1093/molbev/msu343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rowland LJ, Alkharouf N, Darwish O, Ogden EL, Polashock JJ, Bassil NV, Main D. Generation and analysis of blueberry transcriptome sequences from leaves, developing fruit, and flower buds from cold acclimation through deacclimation. BMC Plant Biol. 2012;12:46. doi: 10.1186/1471-2229-12-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang XM, Zhao L, Larson-Rabin Z, Li DZ, Guo ZH. De novo sequencing and characterization of the floral transcriptome of Dendrocalamus latiflorus (Poaceae: Bambusoideae) PLoS One. 2012;7 doi: 10.1371/journal.pone.0042082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Díaz-Riquelme J, Martínez-Zapater JM, Carmona MJ. Transcriptional analysis of tendril and inflorescence development in grapevine (Vitis vinifera L.) PLoS One. 2014;9 doi: 10.1371/journal.pone.0092339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zahn LM, Ma X, Altman NS, Zhang Q, Wall PK, Tian D, Gibas CJ, Gharaibeh R, Leebens-Mack JH, de Pamphilis CW, Ma H. Comparative transcriptomics among floral organs of the basal eudicot Eschscholzia californica as reference for floral evolutionary developmental studies. Genome Biol. 2010;11:R101. doi: 10.1186/gb-2010-11-10-r101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qiu WM, Zhu AD, Wang Y, Chai LJ, Ge XX, Deng XX, Guo WW. Comparative transcript profiling of gene expression between seedless Ponkan mandarin and its seedy wild type during floral organ development by suppression subtractive hybridization and cDNA microarray. BMC Genomics. 2012;13:397. doi: 10.1186/1471-2164-13-397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Logacheva MD, Kasianov AS, Vinogradov DV, Samigullin TH, Gelfand MS, Makeev VJ, Penin AA. De novo sequencing and characterization of floral transcriptome in two species of buckwheat (Fagopyrum) BMC Genomics. 2011;12:30. doi: 10.1186/1471-2164-12-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vining KJ, Romanel E, Jones RC, Klocko A, Alves-Ferreira M, Hefer CA, Aamarasinghe V, Dharmawardhana P, Naithani S, Ranik M, Wesley-Smith J, Solomon J, Jaiswai P, Myburg AA, Straus SH. The floral transcriptome of Eucalyptus grandis. New Phytol. 2014;206:1406–1422. doi: 10.1111/nph.13077. [DOI] [PubMed] [Google Scholar]
- 26.Wei W, Qi X, Wang L, Zhang Y, Hua W, Li D, Lv H, Zhang X. Characterization of the sesame (Sesamum indicum) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers. BMC Genomics. 2011;12:451. doi: 10.1186/1471-2164-12-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhide A, Schliesky S, Reich M, Weber APM, Becker A. Analysis of the floral transcriptome of Tarenaya hassleriana (Cleomaceae), a member of the sister group to the Brassicaceae: towards understanding the base of morphological diversity in Brassicales. BMC Genomics. 2014;15:140. doi: 10.1186/1471-2164-15-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ness RW, Siol M, Barrett SCH. De novo sequence assembly and characterization of the floral transcriptome in cross- and self-fertilizing plants. BMC Genomics. 2011;12:298. doi: 10.1186/1471-2164-12-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kobayashi MJ, Takeuchi Y, Kenta T, Kume T, Diway B, Shimizu KK. Mass flowering of the tropical tree Shorea beccariana was preceded by expression changes in flowering and drought-responsive genes. Mol Ecol. 2013;22:4767–4782. doi: 10.1111/mec.12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.The UniProt Consortium UniProt: a hub for protein information. Nucleic Acids Res. 2015;43:D204–D212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pruitt KD, Tatusova T, Maglott DR. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005;33:D501–D504. doi: 10.1093/nar/gki025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yi X, Zhang Z, Ling Y, Xu W, Su Z. PNRD: a plant non-coding RNA database. Nucleic Acids Res. 2015;43:D982–D989. doi: 10.1093/nar/gku1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweller R, Lopez R. InterProScan: protein domains identifier. Nucleic Acids Res. 2005;33(Suppl 2):W116–W120. doi: 10.1093/nar/gki442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Myhre S, Tveit H, Mollestad T, Lægreid A. Additional gene ontology structure for improved biological reasoning. Bioinformatics. 2006;22:2020–2027. doi: 10.1093/bioinformatics/btl334. [DOI] [PubMed] [Google Scholar]
- 36.Eddy SR. Profile Hidden Markov Models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 37.Yant L, Collani S, Puzey J, Levy C, Kramer EM. Molecular basis for three-dimensional elaboration of the Aquilegia petal spur. P Roy Soc B-Biol Sci. 2015;282:20142778. doi: 10.1098/rspb.2014.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hay A, Tsiantis M. KNOX genes: versatile regulators of plant development and diversity. Development. 2010;137:3153–3165. doi: 10.1242/dev.030049. [DOI] [PubMed] [Google Scholar]
- 39.Eklund DM, Ståldal V, Valsecchi I, Cierlik I, Eriksson C, Hiratsu K, Ohme-Takagi M, Sundström JF, Thelander M, Ezcurra I, Sundberg E. The Arabidopsis thaliana STYLISH1 protein acts as a transcriptional activator regulating auxin biosynthesis. Plant Cell. 2010;22:349–363. doi: 10.1105/tpc.108.064816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choe S. Signal-transduction pathways toward the regulation of brassinosteroid biosynthesis. J Plant Biol. 2007;50:225–229. doi: 10.1007/BF03030649. [DOI] [Google Scholar]
- 41.Yin Y, Vafeados D, Tao Y, Yoshida S, Asami T, Chory J. A new class of transcription factors mediates brassinosteroid-regulated gene expression in Arabidopsis. Cell. 2005;120:249–259. doi: 10.1016/j.cell.2004.11.044. [DOI] [PubMed] [Google Scholar]
- 42.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rau A, Maugis-Rabusseau C, Martin-Magniette ML, Celeux G. Co-expression analysis of high-throughput transcriptome sequencing data with Poisson mixture models. Bioinformatics. 2015;31:1420–1427. doi: 10.1093/bioinformatics/btu845. [DOI] [PubMed] [Google Scholar]
- 45.Dempster AP, Laird NM, Rubin DB. Maximum likelihood from incomplete data via the EM algorithm. J Roy Stat Soc B Met. 1977;39:1–38. [Google Scholar]
- 46.Baudry JP, Maugis C, Bertrand M. Slope heuristics: overview and implementation. Stat Comput. 2012;22:455–470. doi: 10.1007/s11222-011-9236-1. [DOI] [Google Scholar]
- 47.Anisimova M, Bielawski JP, Yang Z. Accuracy and power of the likelihood ratio test in detecting adaptive molecular evolution. Mol Biol Evol. 2001;18:1585–1592. doi: 10.1093/oxfordjournals.molbev.a003945. [DOI] [PubMed] [Google Scholar]
- 48.Yang Z, Nielsen R, Goldman N, Pedersen AMK. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155:431–449. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang Z, Wong WSW, Nielsen R. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol. 2005;22:1107–1118. doi: 10.1093/molbev/msi097. [DOI] [PubMed] [Google Scholar]
- 50.Alexandre H, Vrignaud J, Mangin B, Joly S. Genetic architecture of pollination syndrome transition between hummingbird-specialist and generalist species in the genus Rhytidophyllum (Gesneriaceae) Peer J. 2015;3 doi: 10.7717/peerj.1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tanase K, Nishitani C, Hirakawa H, Isobe S, Tabata S, Ohmiya A, Onozaki T. Transcriptome analysis of carnation (Dianthus caryophyllus L.) based on next-generation sequencing technology. BMC Genomics. 2012;13:292. doi: 10.1186/1471-2164-13-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.von Bakel H, Stout JM, Cote AG, Tallon CM, Sharpe AG, Hughes TR, Page JE. The draft genome and transcriptome of Cannabis sativa. Genome Biol. 2011;12:R102. doi: 10.1186/gb-2011-12-10-r102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parchman TL, Geise KS, Grahnen JE, Benkman CW, Buerkle CA. Transcriptome sequencing in an ecologically important tree species: assembly, annotation, and marker discovery. BMC Genomics. 2010;11:180. doi: 10.1186/1471-2164-11-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang J, Wu K, Zeng S, da Silva JA T, Zhao X, Tian CE, Xia H, Duan J. Transcriptome analysis of Cymbidium sinense and its application to the identification of genes associated with floral development. BMC Genomics. 2013;14:279. doi: 10.1186/1471-2164-14-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Melicher D, Torson AS, Dworkin I, Bowsher JH. A pipeline for the de novo assembly of the Themira biloba (Sepsidae: Diptera) transcriptome using a multiple k-mer length approach. BMC Genomics. 2014;15:188. doi: 10.1186/1471-2164-15-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nakasugi K, Crowhurst R, Bally J, Waterhouse P. Combining transcriptome assemblies from multiple de novo assemblers in the allo-tetraploid plant Nicotiana benthamiana. PLoS One. 2014;9 doi: 10.1371/journal.pone.0091776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen S, McElroy JS, Dane F, Peatman E. Optimizing transcriptome assemblies for Eleusine indica leaf and seedling by combining multiple assemblies from three de novo assemblers. Plant Genome. 2015;8. [DOI] [PubMed]
- 58.Zhao QY, Wang Y, Kong YM, Luo D, Li X, Hao P. Optimizing de novo transcriptome assembly from short-read RNA-Seq data: a comparative study. BMC Genomics. 2011;12(Suppl 14):S2. doi: 10.1186/1471-2105-12-S14-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. P Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jiang D, Tang C, Zhang A. Cluster analysis for gene expression data: a survey. IEEE T Knowl Data En. 2004;16:1370–1386. doi: 10.1109/TKDE.2004.68. [DOI] [Google Scholar]
- 61.MacQueen JB. Some methods for classification and analysis of multivariate observations. In: Le Cam LM, Neyman J, editors. Proceedings of the 5th Berkeley Symposium on Mathematical Statistics and Probability, number 1. Berkeley: Univ Calif Press; 1967. pp. 281–297. [Google Scholar]
- 62.Ward JH. Hierarchical grouping to optimize and objective function. J Am Stat Assoc. 1963;58:236–244. doi: 10.1080/01621459.1963.10500845. [DOI] [Google Scholar]
- 63.Guzzi PH, Masciari E, Mazzeo GM, Zaniolo C. A discussion on the biological relevance of clustering results. In: Bursa M, Khuri S, Renda ME, editors. International Conference on Information Technology in bio- and medical informatics. Gewerbestrasse: Springer Intl Pub; 2014. pp. 30–44. [Google Scholar]
- 64.Si Y, Liu P, Li P, Brutnell TP. Model-based clustering for RNA-seq data. Bioinformatics. 2014;30:197–205. doi: 10.1093/bioinformatics/btt632. [DOI] [PubMed] [Google Scholar]
- 65.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fukushima A, Nishizawa T, Hayakumo M, Hikosaka S, Saito K, Goto E, Kusano M. Exploring tomato gene functions based on coexpression modules using graph clustering and differential coexpression approaches. Plant Physiol. 2012;158:1487–1502. doi: 10.1104/pp.111.188367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Coneva V, Simopoulos C, Casaretto JA, El-Kereamy A, Guevara DR, Cohn J, Zhu T, Guo L, Alexander DC, Bi YM, McNicholas PD, Rothstein SJ. Metabolic and co-expression network-based analyses associated with nitrate response in rice. BMC Genomics. 2014;15:1056. doi: 10.1186/1471-2164-15-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Langfelder P, Horvath S. WGCNA: an R package for weighted correlated network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hollender CA, Kang C, Darwish O, Geretz A, Matthews BF, Slovin J, Alkharouf N, Liu Z. Floral transcriptomes in woodland strawberry uncover developing receptacle and anther gene networks. Plant Physiol. 2014;165:1062–1075. doi: 10.1104/pp.114.237529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fan Z, Li J, Li X, Wu B, Wang J, Liu Z, Yin H. Genome-wide transcriptome profiling provides insights into floral bud development of summer-flowering Camellia azalea. Sci Reports. 2015;2015:5. doi: 10.1038/srep09729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huang YJ, Liu LL, Huang JQ, Wang ZJ, Chen FF, Zhang QX, Zheng BS, Chen M. Use of transcriptome sequencing to understand the pistillate flowering in hickory (Carya cathayensis Sarg.) BMC Genomics. 2013;14:691. doi: 10.1186/1471-2164-14-691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yoo MJ, Chanderbali AS, Altman NS, Soltis PS, Soltis DE. Evolutionary trends in the floral transcriptome: insights from one of the basalmost angiosperms, the water lily Nuphar advena (Nymphaeaceae) Plant J. 2010;64:687–698. doi: 10.1111/j.1365-313X.2010.04357.x. [DOI] [PubMed] [Google Scholar]
- 74.Annick D, Remay A, Raymond O, Balzergue S, Chauvet A, Maene M, Pécrix Y, Yang SH, Jeauffre J, Thouroude T, Boltz V, Martin-Magniette ML, Janczarski S, Legeai F, Renou JP, Vergne P, Le Bris M, Foucher F, Bendahmane M. Genomic approach to study floral development genes in Rosa sp. PLoS One. 2011;6 doi: 10.1371/journal.pone.0028455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kater MM, Dreni L, Colombo L. Functional conservation of MADS-box factors controlling floral organ identity in rice and Arabidopsis. J Exp Bot. 2006;57:3433–3444. doi: 10.1093/jxb/erl097. [DOI] [PubMed] [Google Scholar]
- 76.Urbanus SL, Dinh QD, Angenent GC, Immink RG. Investigation of MADS domain transcription factor dynamics in the floral meristem. Plant Signal Behav. 2010;5:1260–1262. doi: 10.4161/psb.5.10.12949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ding B, Mou F, Sun W, Chen S, Peng F, Bradshaw HD, Yuan YW. A dominant-negative actin mutation alters corolla tube width and pollinator visitation in Mimulus lewisii. New Phytol. In press. [DOI] [PMC free article] [PubMed]
- 78.Golz JF, Keck EJ, Hudson A. Spontaneous mutations in KNOX genes give rise to a novel floral structure in Antirrhinum. Curr Biol. 2002;12:515–522. doi: 10.1016/S0960-9822(02)00721-2. [DOI] [PubMed] [Google Scholar]
- 79.Box MS, Dodsworth S, Rudall PJ, Bateman RM, Glover B. Characterization of Linaria KNOX genes suggest a role in petal-spur development. Plant J. 2011;68:703–714. doi: 10.1111/j.1365-313X.2011.04721.x. [DOI] [PubMed] [Google Scholar]
- 80.Grotewald E. The genetics and biochemistry of floral pigments. Annu Rev Plant Biol. 2006;57:761–780. doi: 10.1146/annurev.arplant.57.032905.105248. [DOI] [PubMed] [Google Scholar]
- 81.Saito K, Yonekura-Sakakibara K, Nakabayashi R, Higashi Y, Yamazaki M, Tohge T, Fernie AR. The flavonoid biosynthetic pathway in Arabidopsis: structural and genetic diversity. Plant Physiol Bioch. 2013;72:21–34. doi: 10.1016/j.plaphy.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 82.Whittall JB, Voelckel C, Kliebenstein DJ, Hodges SA. Convergence, constraint and the role of gene expression during adaptive radiation: floral anthocyanins in Aquilegia. Mol Ecol. 2006;15:4645–4657. doi: 10.1111/j.1365-294X.2006.03114.x. [DOI] [PubMed] [Google Scholar]
- 83.Streisfeld MA, Rausher MD. Altered trans-regulatory control of gene expression in multiple anthocyanin genes contributes to adaptive flower color evolution in Mimulus aurantiacus. Mol Biol Evol. 2009;26:433–444. doi: 10.1093/molbev/msn268. [DOI] [PubMed] [Google Scholar]
- 84.Smith SD, Rausher MD. Gene loss and parallel evolution contribute to species differences in flower color. Mol Biol Evol. 2011;28:2799–2810. doi: 10.1093/molbev/msr109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Smith SD, Rausher MD. Functional evolution of an anthocyanin pathway enzyme during a flower color transition. Mol Biol Evol. 2013;30:602–612. doi: 10.1093/molbev/mss255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yuan YW, Sagawa JM, Frost L, Vela JP, Bradshaw HD. Transcriptional control of floral anthocyanin pigmentation in monkeyflowers (Mimulus) New Phytol. 2014;204:1013–1027. doi: 10.1111/nph.12968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Streisfeld MA, Rausher MD. Genetic changes contributing to the parallel evolution of red floral pigmentation among Ipomoea species. New Phytol. 2009;183:751–763. doi: 10.1111/j.1469-8137.2009.02929.x. [DOI] [PubMed] [Google Scholar]
- 88.Des Marais DL, Rausher MD. Parallel evolution at multiple levels in the origin of hummingbird pollinated flowers in Ipomoea. Evolution. 2010;64:2044–2054. doi: 10.1111/j.1558-5646.2010.00972.x. [DOI] [PubMed] [Google Scholar]
- 89.Wessinger CA, Hileman LC, Rausher MD. Identification of major quantitative trait loci underlying floral pollination syndrome divergence in Penstemon. Philos T Roy Soc B. 2014;369:20130349. doi: 10.1098/rstb.2013.0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wessinger CA, Rausher MD. Ecological transition predictably associated with gene degeneration. Mol Biol Evol. 2015;32:347–354. doi: 10.1093/molbev/msu298. [DOI] [PubMed] [Google Scholar]
- 91.Lowry DB, Sheng CC, Lasky JR, Willis JH. Five anthocyanin polymorphisms are associated with an R2R3-MYB cluster in Mimulus guttatus (Phrymaceae) Am J Bot. 2012;99:82–91. doi: 10.3732/ajb.1100285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Seitz C, Eder C, Deimi B, Kellner S, Martens S, Forkmann G. Cloning, functional identification and sequence analysis of flavonoid 3′-hydroxylase and flavonoid 3′,5′-hydroxylase cDNAs reveals independent evolution of flavonoid 3′,5′-hydroxylase in the Asteraceae family. Plant Mol Biol. 2006;61:365–381. doi: 10.1007/s11103-006-0012-0. [DOI] [PubMed] [Google Scholar]
- 93.Seitz C, Ameres S, Schlangen K, Forkmann G, Halbwirth H. Multiple evolution of flavonoid 3′,5′-hydoxylase. Planta. 2015;242:561–573. doi: 10.1007/s00425-015-2293-5. [DOI] [PubMed] [Google Scholar]
- 94.Stankowski S, Streisfeld MA. Introgressive hybridization facilitates adaptive divergence in a recent radiation of monkeyflowers. P Roy Soc B-Biol Sci. 2015;282:20151666. doi: 10.1098/rspb.2015.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sagawa JM, Stanley LE, LaFountain AM, Frank HA, Liu C, Yuan YW. An R2R3-MYB transcription factor regulates carotenoid pigmentation in Mimulus lewisii flowers. New Phytol. 2015;209:1049–1057. doi: 10.1111/nph.13647. [DOI] [PubMed] [Google Scholar]
- 96.Quattrocchio F, Wing J, van der Woude K, Souer E, de Vetten N, Mol J, Koes R. Molecular analysis of the anthocyanin2 gene of Petunia and its role in the evolution of flower color. Plant Cell. 1999;11:1433–1444. doi: 10.1105/tpc.11.8.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schwinn K, Venail J, Shang Y, Mackay S, Alm V, Butelli E, Oyama R, Bailey P, Davies K, Martin C. A small family of MYB-regulatory genes controls floral pigmentation intensity and patterning in the genus Antirrhinum. Plant Cell. 2006;18:831–851. doi: 10.1105/tpc.105.039255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dall’Osto L, Fiore A, Cazzinga S, Giuliano G, Bassi R. Different roles of alpha- and beta-branch xanthophylls in photosystem assembly and photoreception. J Biol Chem. 2007;282:35056–35068. doi: 10.1074/jbc.M704729200. [DOI] [PubMed] [Google Scholar]
- 99.Glover BJ. Understanding flowers and flowering: an integrated approach. Oxford, UK: Oxford University Press; 2014. [Google Scholar]
- 100.LaFountain AM, Frank HA, Yuan Y-W. Carotenoid composition of the flowers of Mimulus lewisii and related species: implications regarding the prevalence and origin of two unique, allenic pigments. Arch Biochem Biophys. 2015;573:32–39. doi: 10.1016/j.abb.2015.03.006. [DOI] [PubMed] [Google Scholar]
- 101.Bradshaw HD, Wilbert M, Otto KG. Genetic mapping of floral traits associated with reproductive isolation in monkeyflowers (Mimulus) Nature. 1995;375:31. [Google Scholar]
- 102.Owen CR, Bradshaw HD. Induced mutations affecting pollinator choice in Mimulus lewisii (Phrymaceae) Arthropod-Plant Inte. 2011;5:235–244. doi: 10.1007/s11829-011-9133-8. [DOI] [Google Scholar]
- 103.Cronk Q, Ojeda I. Bird-pollinated flowers in an evolutionary and molecular context. J Exp Bot. 2008;59:715–727. doi: 10.1093/jxb/ern009. [DOI] [PubMed] [Google Scholar]
- 104.Lin R, Wang H. Two homologous ATP-binding cassette transporter proteins, AtMDR1 and AtPGP1, regulate Arabidopsis photomorphogenesis and root development by mediating polar auxin transport. Plant Physiol. 2005;138:949–964. doi: 10.1104/pp.105.061572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Suzuki H, Nakayama T, Yonekura-Sakakibara K, Fukui Y, Nakamura N, Nakao M, Tanaka Y, Yamaguchi MA, Kusumi T, Nishino T. Malonyl-CoA:anthocyanin 5-O-glucoside-6”’-O-malonyltransferase from scarlet sage (Salvia splendens) flowers. Enzyme purification, gene cloning, expression, and characterization. J Biol Chem. 2001;276:49013–49019. doi: 10.1074/jbc.M108444200. [DOI] [PubMed] [Google Scholar]
- 106.Ho WW, Smith SD. Molecular evolution of anthocyanin pigmentation genes following losses of flower color. BMC Evol Biol. 2016;16:98. doi: 10.1186/s12862-016-0675-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Krogan NT, Hogan K, Long JA. APETALA2 negatively regulates multiple floral organ identity genes in Arabidopsis by recruiting the co-repressor TOPLESS and the histone deacetylase HDA19. Development. 2012;139:4180–4190. doi: 10.1242/dev.085407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Johanson U, West J, Lister C, Michaels S, Amasino R, Dean C. Molecular analysis of FRIGIDA, a major determinant of natural variation in Arabidopsis flowering time. Science. 2000;290:344–347. doi: 10.1126/science.290.5490.344. [DOI] [PubMed] [Google Scholar]
- 109.Chiara M, Horner DS, Spada A. De novo assembly of the transcriptome of the non-model Streptocarpus rexii employing a novel heuristic to recover locus-specific transcript clusters. PLoS One. 2013;8 doi: 10.1371/journal.pone.0080961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ai B, Gao Y, Zhang X, Tao J, Kang M, Huang H. Comparative transcriptome resources of eleven Primulina species, a group of ‘stone plants’ from a biodiversity hot spot. Mol Ecol Resour. 2014;15:619–632. doi: 10.1111/1755-0998.12333. [DOI] [PubMed] [Google Scholar]
- 111.Xiao L, Yang G, Zhang L, Yang X, Zhao S, Ji Z, Zhou Q, Hu M, Wang Y, Chen M, Xu Y, Jin H, Xiao X, Hu G, Bao F, Hu Y, Wan P, Li L, Deng X, Kuang T, Xiang C, Zhu JK, Oliver MJ, He Y. The resurrection genome of Boea hygrometrica: a blueprint for survival of dehydration. P Natl Acad Sci USA. 2015;112:5833–5837. doi: 10.1073/pnas.1505811112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhu Y, Wang B, Phillips J, Zhang ZN, Du H, Xu T, Huang LC, Zhang XF, Xu GH, Li WL, Wang Z, Wang L, Liu YX, Deng X. Global transcriptome analysis reveals acclimation-primed processes involved in the acquisition of desiccation tolerance in Boea hygrometrica. Plant Cell Physiol. 2015;56:1429–144. doi: 10.1093/pcp/pcv059. [DOI] [PubMed] [Google Scholar]
- 113.FastQC. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 24 June 2014.
- 114.Hannon Lab. FASTX Toolkit. 2011. http://hannonlab.cshl.edu/fastx_toolkit/. Accessed 24 June 2014.
- 115.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotech. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schulz MH, Zerbino DR, Vingron M, Birney E. Oases: Robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics. 2012;28:1086–1092. doi: 10.1093/bioinformatics/bts094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gilbert D. EvidentialGene: tr2aacds, mRNA transcript assembly software. 2013. Accessed 30 June 2014http://arthropods.eugenes.org/EvidentialGene/. Accessed 30 June 2014.
- 119.Slater GS, Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 121.Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 122.The Gene Ontology Consortium Gene Ontology Consortium: going forward. Nucl Acids Res. 2015;43:D1049–D1056. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lagesen K, Hailin P, Rødland EA, Staerfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc B Met. 1995;57:289–300.
- 126.Edgar RC. MUSCLE: a multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 129.Emms DM, Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015;16:157. doi: 10.1186/s13059-015-0721-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, MacManes MD, Ott M, Orvis J, Pochet N, Strozzi F, Weeks N, Westerman R, William T, Dewey CN, Henschel R, LeDuc RD, Friedman N, Regev A. De novo transcript sequence reconstruction from RNA-Seq Reference Generation and Analysis with Trinity. Nature Protoc. 2013;8:1494–1512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Felsenstein J. PHYLIP – Phylogeny Inference Package (Version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- 134.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 135.Al-Shahrour F, Díaz-Uriarte R, Dopazo J. Fatigo: a web tool for finding significant associations of gene ontology terms with groups of genes. Bioinformatics. 2004;20:578–580. doi: 10.1093/bioinformatics/btg455. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated during and analyzed during the current study are available in the NCBI Short Read Archive repository, available from accession SRP083265. The contig assemblies generated during the current study are available from the corresponding author upon request. The datasets supporting the conclusions of this article are included within the article and its additional files.