

Abstract
Resistance (R) genes of plants are responsible for pathogen recognition and encode proteins that trigger a cascade of responses when a pathogen invades a plant. R genes are assumed to be under strong selection, but there is limited knowledge of the processes affecting R gene diversity in the wild. In this study, DNA sequence variation of Cf-2 homologs was surveyed in populations of Solanum pimpinellifolium, a wild relative of the cultivated tomato. The Cf-2 locus is involved in resistance to strains of the fungus Cladosporium fulvum. At least 26 different Cf-2 homologs were detected in natural populations of S. pimpinellifolium. These homologs differ by single base pair substitutions as well as indels in regions coding for leucine-rich repeats. Molecular population genetic analyses suggest that natural selection has acted heterogeneously on Cf-2 homologs, with selection against amino acid substitutions occurring in the 5′ portion of the genes, and possible restricted positive selection in the 3′ end. Balancing selection may have maintained haplotypes at the 5′ end of the genes. Limited sequence exchange between genes has also contributed to sequence variation. S. pimpinellifolium individuals differ in the number of Cf-2 homologs they contain, obscuring the relationships of orthology and paralogy. This survey of Cf-2 variation in S. pimpinellifolium illustrates the wealth of R gene diversity that exists in wild plant populations, as well as the complexity of interacting genetic and evolutionary processes that generate such diversity.
Keywords: Cf-2 locus, evolution, plant disease-resistant genes
Understanding the mechanisms governing the evolution of disease resistance in plants has been a goal of breeders and evolutionary ecologists. Genes involved in resistance to disease are attractive for the study of functional gene evolution, because pathogens are ubiquitous and may exert high selective pressure on their hosts. Attention has centered on R genes, the genes responsible for pathogen recognition. R genes encode proteins that trigger resistance responses when a pathogen invades the plant, possibly by interacting with Avr (avirulence) gene products expressed by pathogens (1). The interaction occurs in a “gene-for-gene” framework, in which the absence or lack of function of either member of a gene pair will lead to disease instead of resistance (2).
Most R gene evolutionary patterns have been deduced by the comparison of orthologous (allelic) haplotypes in a few accessions of a species (3, 4) or by comparisons of paralogs (genes related through duplication) from a single plant (5, 6). A few studies compare R genes throughout the genome of Arabidopsis thaliana (7, 8) or putative R genes across taxonomic groups (9–11). With the exception of A. thaliana, most studies of R gene variation have been on crop species, which may not contain the R gene variation present in the wild, and there has been little exploration of R genes in a population-level framework. The few studies addressing R gene evolution in populations either have not been carried out at the DNA sequence level, have sampled for genes with a predetermined resistance function, or have sampled germplasm collections, an approach that may undersample natural levels of diversity (12, 13; see ref. 14 for a recent exception). The lack of knowledge on the evolution of R genes in natural populations is exacerbated by fact that R genes often occur within multigene families, and little is known about their evolutionary dynamics.
To gain an understanding of the evolutionary processes affecting R gene diversity in natural populations, we studied the Cf-2 R gene family in native populations of Solanum pimpinellifolium L. [Lycopersicon pimpinellifolium (L.) Mill.]. S. pimpinellifolium is a wild relative of the cultivated tomato, Solanum lycopersicum L. (Lycopersicon esculentum Mill.); it grows as a perennial bush in the coastal lowlands of Peru and Ecuador (15) and shows intraspecific variation in the extent of outcrossing (16). The Cf-2 locus (17) mediates the resistance response to Avr2-carrying strains of the fungus Cladosporium fulvum Cooke, which causes leaf mold in tomatoes. Several R genes conferring resistance to strains of C. fulvum (Cf genes) have been isolated in at least two other species of the tomato clade (18–21), suggesting the fungus may have been important in the evolution of the group.
In the accession in which it was originally identified, Cf-2 comprises two tandem genes, Cf-2.1 and Cf-2.2 (18). The genes differ by three nonsynonymous nucleotides; both are functional and sufficient for resistance and are thought to be the result of a recent duplication event (18). A third distant paralogous gene, Hcr2-2A, which does not confer resistance to C. fulvum with Avr2, has been found ≈10 kb downstream from the Cf-2 locus (21).
All Cf genes found to date encode extracellular membrane-anchored proteins (22–24). The Cf-2 proteins consist of extracellular leucine rich repeats (LRRs) and a small transmembrane domain (18, 24). LRRs, a common motif in R proteins, are involved in protein–protein interactions (25) and are important for the determination of R gene specificity (24, 26). A subset of the Cf-2 LRRs can be grouped into two classes based on sequence, the A and B types, which form alternating repeats in the N-terminal region of the LRR domain (18).
We show that the evolutionary processes affecting the Cf-2 gene family are heterogeneous and complex. We have found evidence of intraspecific variation in gene number and sequence exchange among Cf-2 homologs. Moreover, we find that different modes of selection act on different portions of the genes. Our results indicate that Cf-2 is an actively evolving gene family within S. pimpinellifolium, and that the evolution of specific R genes cannot be separated from that of the entire gene family.
Materials and Methods
Sampling. Sixteen S. pimpinellifolium populations were sampled along ≈800 km of the northern coast of Peru (Fig. 3, which is published as supporting information on the PNAS web site). Populations occur in river valleys across the desert coast; when possible, one population per valley was sampled. Three to 10 individuals were collected per population, for a total of 138 individuals. Voucher herbarium specimens are deposited at the Museo de Historia Natural herbarium (USM) in Lima, Peru.
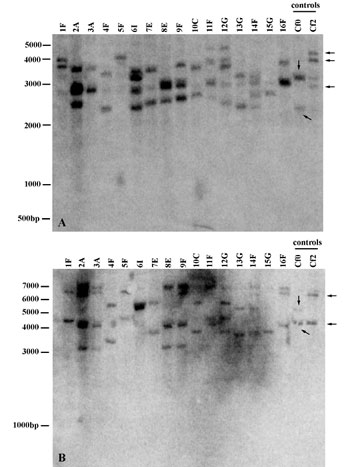
Molecular Procedures. Plants were grown in the greenhouse from a single seed for each collected individual. DNA was extracted from frozen leaf tissue by using a modified cetyltrimethylammonium bromide (CTAB) protocol (27) or Plant DNeasy Kits (Qiagen, Valencia, CA). For Southern blots, genomic DNA of one individual from each population was digested with DraI and BclI. Hybridization was carried out with an ≈1.6-kb radiolabeled DNA fragment of the 3′ end of the Cf-2 genes from the S. lycopersicum near-isogenic line Cf2. S. lycopersicum lines Cf0 and Cf2 were used as controls (see ref. 21). Conditions for Southern blot analysis are described in Fig. 4, which is published as supporting information on the PNAS web site.
Primers that would amplify the complete ORF of both Cf-2.1 and Cf-2.2, but not Hcr2-2A, were designed from GenBank accession nos. U42444, U42445, and AF053996 (Table 4, which is published as supporting information on the PNAS web site). Cf0 and Cf2 lines were used as amplification and sequencing controls. PCR reactions were carried out with Elongase (Invitrogen) under standard conditions. PCR products were separated on 0.8% agarose gels, and multiple bands were isolated and purified with a QIAquick Gel Extraction Kit (Qiagen). Purified PCR products were sequenced directly by primer walking along overlapping fragments (Table 4). Results were repeatable for individuals amplified more than once. Sequencing conditions are described in ref. 28.
Data Analysis. Gene networks, which show the mutational relationships among haplotypes, were constructed manually following a maximum parsimony criterion. Tree topology was verified with statistical parsimony by using the tcs program (29) and with heuristic searches using parsimony analyses in paup* version 4.0b3 (30). The limits of parsimony (31) were evaluated with tcs.
Levels of nucleotide diversity (π) (32), the number of polymorphic sites (S), and the number of synonymous substitutions per synonymous site (Ks) and nonsynonymous substitutions per non-synonymous site (Ka) (33) were calculated with dnasp (34). Pairwise comparisons of the number of changed and unchanged sites were tested for significance by using Fisher's exact test (35). Average Ka and Ks values were also calculated and tested for significant differences with t tests. Test statistics were corrected for multiple comparisons with the sequential Bonferroni test (36).
Evidence for positive selection was also evaluated by using a powerful maximum likelihood (ML) method (37, 38), which allows searches for individual amino acid sites under selection. The program codeml from PAML (39) was used to evaluate alternate codon substitution models that exclude (M7) and allow (M8) positive selection (37). A likelihood ratio (LR) test determined which model best fit the observed data. The significance of the LR statistic was evaluated by comparing it to a χ2 statistic. When positive selection was inferred, the putative positively selected sites were identified under M8, using a Bayesian approach to determine sites with a high probability of having a Ka/Ks ratio (ω) greater than one (37). Phylogenetic trees for the ML tests were obtained from heuristic searches in paup*, Ver. 4.0b3 (30). When a single most parsimonious tree was not available, a strict consensus tree was used.
To determine whether sequence exchange, such as recombination or gene conversion, has played a role in the generation of Cf-2 genes, the geneconv program (40) was used. Global P values were assigned based on 10,000 permutations and corrected for multiple comparisons. Analyses were carried out with no mismatches allowed (gscale = 0).
Results and Discussion
S. pimpinellifolium Individuals Vary in Cf-2 Copy Number. Although the Cf-2 gene family was initially characterized as consisting of two genes in S. pimpinellifolium, we find variation in gene number throughout the species. DNA gel blot analysis of 16 individuals revealed a diversity of large molecular-weight bands and variation in banding patterns among individuals (Fig. 4), indicating extensive intraspecific variation in Cf-2 copy number (one to four hybridization bands) and Cf-2 size. The results suggest that expansion and reduction of the gene family occurs rapidly enough to be seen at the level of the species. The intraspecific variation parallels that observed (21) for interspecific Cf-2 homologs.
PCR amplification patterns of Cf-2 homologs support the DNA gel blot analysis. Patterns vary widely between individuals, both within and among populations, with some individuals having several amplification products of different sizes (data not shown). From 120 successfully amplified plants, 191 2- to 4-kb PCR products were isolated and sequenced. Multiple sequences were isolated from some individuals. Levels of nucleotide divergence between all isolated genes are lower than between Cf-2.1/Cf-2.2 and the downstream paralog, Hcr2-2A; flanking regions are conserved, as is the position of the single intron occurring 42 nucleotides after the Cf-2.1/Cf-2.2 stop codons (18). Levels of variation in isolated genes are discussed fully below. These observations confirm that the isolated genes are Cf-2 homologs. Due to PCR bias against efficient amplification of very large bands, the sequenced products are a large, but not exhaustive, sample of the Cf-2 homolog diversity.
We discarded six of the sequences, which were unique in size and observed only once, to reduce the possibility of PCR artifacts. The remaining 185 products were classified into one of nine distinct categories based on size (Fig. 1). For this sample, the naming scheme suggested for Cf-2 intra- and interspecific homologs (41) was modified; each size class was designated Hcr2 (for homologous to Cladosporium resistance gene Cf-2), -p (for pimpinellifolium), and a final digit that identifies each size class (Table 1). The largest size class was left as Cf-2 and includes the originally identified Cf-2.1 and Cf-2.2 genes.
Fig. 1.

Schematic representation of the Cf-2 homolog size classes within S. pimpinellifolium. All genes are represented aligned to the Cf-2 size class. Regions left of the left vertical dashed line and right of the right vertical dashed line were used in sequence comparisons.
Table 1. Observed Cf-2 homolog size classes.
| Size class | ORF length, bp | ORF length, aa | No. of LRR | No. of A/B LRR | Frequency | Sequence* length, bp |
|---|---|---|---|---|---|---|
| Cf-2 | 3,339 | 1,112 | 38 | 20 | 47 | 3,566 |
| Hcr2-p1 | 2,976 | 991 | 33 | 15 | 7 | 3,203 |
| Hcr2-p2 | 2,445 | 814 | 27 | 9 | 19 | 2,672 |
| Hcr2-p3 | 2,547 | 848 | 27 | 9 | 13 | 2,774 |
| Hcr2-p4 | 2,403 | 800 | 25 | 7 | 4 | 2,630 |
| Hcr2-p5 | 2,259 | 752 | 23 | 5 | 5 | 2,486 |
| Hcr2-p6 | 1,113 | 370 | 12 | 3 | 7 | 1,340 |
| Hcr2-p7 | 1,464 | 487 | 17 | 13 | 35 | 1,691 |
| Hcr2-p8 | 849 | 282 | 8 | 4 | 48 | 1,076 |
Regions include the complete ORF, 132 bp upstream of the start codon and 95 bp downstream of the Cf-2 stop codon.
Although the dearth of intraspecific surveys of R loci makes it impossible to say whether the extensive copy number variation seen here for Cf-2 is typical, R genes often occur in gene families, and this type of variation may be a common phenomenon. Variation in copy number has also been observed for the RPP5 and RPP8 R loci in two ecotypes of A. thaliana (42, 43). Differences in gene copy number within species pose a challenge for determining orthology (the relationship between true alleles) and paralogy (genes related through a duplication event). If genes occur in tandem (as is probable for Cf-2, based on the tandem occurrence of Cf-2.1 and Cf-2.2 and of homologs in other Solanum species), extensive crosses and genetic analyses are needed to draw conclusions about homologs in different individuals. Sequence similarity is not a reliable indicator of orthology, as illustrated by the lack of significant divergence between Cf-2.1 and Cf-2.2. Further aspects of homology relationships among the Cf-2 genes are discussed below. The complexity inherent to a multigene family with varying number of members presents a challenge to traditional ortholog-based population analyses.
Size Differences Among Cf-2 Homologs Are Due to Variation in LRR Number. Size variation among Cf-2 homolog classes is due exclusively to variation in the number of LRR-coding units, principally of the A and B types (Table 1, Fig. 1). The size variation has consequences for the three smallest size classes, where deletion of LRRs has caused a frameshift mutation leading to an early stop codon (Fig. 1). In these homologs, the putative protein products have no membrane-spanning domain and may be pseudogenes. The region after the early stop remains alignable at the nucleotide level among truncated genes and all other Cf-2 homologs (nucleotide divergence: 0.96–2.49%). The remaining homolog size classes all have an intact membrane-spanning motif, despite variation in LRR number and other polymorphisms.
We examined the relationships of A type LRR units and B type units within each size class by comparing levels of similarity among units, within type, at the nucleotide and amino acid level (Table 5, which is published as supporting information on the PNAS web site). A and B type LRRs within each homolog have high levels of similarity and probably share common origins. Mantel tests were carried out to determine whether there is any correlation between repeat divergence and repeat position in the Cf-2 size class. No correlation with distance (measured in number of repeats separating two LRRs) was found for either type of repeat, indicating that similar repeats are not found closer together within the gene. Thus, the mechanisms generating LRR number diversity in Cf-2 homologs probably involve complex recombination events, rather than tandem duplications like those generated by slipped-strand mispairing.
The intraspecific variation in LRR number parallels that observed for interspecific homologs of Cf-2 in two other Solanum species (21). Both in the Hcr2-p genes, as well as in interspecific homologs, it is the number of A and B type repeats that varies the most between size classes. Differences in LRR number may have important consequences for pathogen recognition (24). Some studies have detected compromised R gene functionality due to experimental alteration (44) or naturally occurring variation (21) of LRR number. In S. pimpinellifolium, the insertion and deletion of LRRs in Cf-2 homologs have kept the length of each repeat intact for most size classes. The paucity of truncated size classes suggests selective maintenance of the reading frame and alternative functionality associated with LRR number for each size class. Regardless of functional consequences, the variation of LRR number in S. pimpinellifolium Cf-2 homologs implies that the generation of repeat combinations is a dynamic process that occurs within the lifespan of the species.
High Levels of Polymorphism Are Maintained in Cf-2 Homologs. SNPs were found within some Cf-2 size classes. Taking size class and SNP variation into account, a total of 26 different Cf-2 homologs were observed in natural populations of S. pimpinellifolium. Different homologs within a size class were designated by the size class name followed by a period and an identifying number (Tables 6 and 7, which are published as supporting information on the PNAS web site).
The variation Cf-2 homologs illustrates the wealth of R gene polymorphism that can exist in natural plant populations, including mainly inbreeding species such as S. pimpinellifolium. We aligned sequences to estimate levels of genetic diversity among Cf-2 homologs. The high levels of similarity within A and B type LRRs and the lack of a necessarily colinear relationship between LRRs of different genes precluded an unequivocal alignment of LRRs among homologs. Comparisons of genetic diversity were restricted to conserved areas in the 5′ and 3′ regions (Fig. 1). Comparisons among haplotypes in the 5′ region encompass the first 654 bp of the ORF (0–6.42% divergence), which includes two full-length A and B type LRRs. In the 3′ region, comparisons include 1,045 strongly conserved base pairs that do not contain A or B type LRRs (0–2.49% divergence). By contrast, divergence between the Cf-2 genes and the downstream paralog, Hcr2-2A, is 11.46% (5′ region) and 10.78% (3′ region). Sequences flanking the homologs' ORF 132 base pairs upstream of the start codon and 95 base pairs downstream of the Cf-2 stop codon were also included in diversity estimates.
We used nucleotide diversity among homologs as a measure of variation that includes frequency and divergence of sequences. Levels of nucleotide diversity in Cf-2 homologs vary greatly among gene regions (Table 2). Diversity is higher for the 5′ region than the 3′, and nucleotide diversity in flanking areas is between these two values. Both regions contain sequence encoding LRR and non-LRR motifs, and the 3′ comparison also includes noncoding sequence of Hcr2-p6, Hcr2-p7, and Hcr2-p8. High 5′ variation is observed even when the three truncated size classes are excluded (Table 2).
Table 2. Molecular evolutionary parameters for the gene segments examined.
| Gene segment | L* | A† | S‡ | η§ | π | Ka/Ks | Ka/KsP value¶ |
|---|---|---|---|---|---|---|---|
| 5′ | 654 | 15 | 72 | 76 | 0.0381 | 0.5638 | 8.869·10-11 |
| 6 | 48 | 50 | 0.0287 | 0.4797 | 0.0442 | ||
| 5′ non-LRR | 189 | 10 | 18 | 19 | 0.0335 | 1.0510 | 0.7301 |
| 4 | 6 | 6 | 0.0122 | 0.7230 | 0.7301 | ||
| 5′ LRR | 465 | 11 | 54 | 57 | 0.0400 | 0.4771 | 2.224·10-09 |
| 6 | 42 | 44 | 0.0354 | 0.4663 | 0.0338 | ||
| 3′ | 1045 | 17 | 55 | 58 | 0.0119 | ||
| 10 | 41 | 42 | 0.0115 | 0.8722 | 0.7301 | ||
| 3′ non-LRR | 360 | 11 | 19 | 21 | 0.0117 | ||
| 7 | 13 | 13 | 0.0130 | 1.2059 | 0.7301 | ||
| 3′ LRR | 685 | 13 | 36 | 37 | 0.0120 | ||
| 7 | 28 | 29 | 0.0107 | 0.6748 | 0.3920 | ||
| Flanking 5′ | 132 | 6 | 10 | 10 | 0.0185 | ||
| Flanking 3′ | 95 | 8 | 9 | 9 | 0.0239 |
Values in italics exclude size classes with early stop codons (Hcr2-p6, -p7, -p8).
Length of gene segment in base pairs.
No. of haplotypes.
No. of polymorphisms.
No. of mutations.
Bonferroni corrected; values in bold are significant.
Within the 5′ alignable region, nucleotide diversity is unequally partitioned between domains. The non-LRR region, which codes for a putative signal peptide and β domain with N-linked glycosylation site, has lower nucleotide diversity than the LRR-coding section, especially when truncated genes are removed from the analysis (Table 2). Such dichotomy is not observed in the 3′ portion of the genes.
The patterns of variation observed for the S. pimpinellifolium Cf-2 homologs are concordant with predicted protein motif functions. The resistance specificity of Cf-2, compared with Cf-5, a gene found in S. lycopersicum var cerasiforme, is confined to N-terminal LRRs 3–27 of the Cf-2 proteins (26). Fittingly, the 5′ LRR-coding region, which codes for the first six Cf-2 LRRs, is the most variable region of all within the Cf-2 homologs (Table 2). On the other hand, the 8.5–10 C-terminal LRRs of Cf-2.1, Cf-2.2, and three other Cf proteins, Cf-5, Cf-4, and Cf-9, are conserved and may interact with conserved signaling components of resistance pathways (24, 26). Levels of diversity are likewise low in the 3′ portion of the Cf-2 homologs. The pattern of low 3′ variation holds true for the candidate pseudogenes, even though in these sequences the 3′ region is noncoding and should be free of selective constraints.
Heterogeneous Modes of Selection Are Acting on Cf-2 Homologs. Patterns of Cf-2 homolog variation suggest that selection is acting on some protein-coding motifs. Pairwise values of Ks and Ka for all coding sequences indicate an overall scarcity of amino acid substitutions. Most Ka/Ks ratios are below one, and nonsynonymous mutations are never significantly more common than synonymous mutations (data not shown). Average Ks also exceeded average Ka for most comparisons (Table 2). Significant departures from one for average Ka/Ks ratios were seen only for the 5′ and 5′ LRR regions of the homologs (Table 2), suggesting purifying selection (selection against amino acid substitutions) in the 5′ LRR-coding section of the genes. Despite the action of purifying selection, the highest levels of nucleotide diversity and Ks occur in the 5′ LRR region (Table 2). This suggests a high mutation rate in this region filtered by selection against amino acid changes. The less variable 5′ non-LRR coding region and the 3′ region appear to have a neutral pattern of nucleotide substitution (Table 2).
Selection can often be highly localized within genes. For many R proteins, positive selection (selection for amino acid substitutions) has been detected primarily for solvent-exposed amino acids in the LRR regions (45). LRR units have a secondary structure of a β-strand connected to an α-helix (46). Within the β-strand, non-leucine residues are exposed to the solvent phase and are a protein-binding motif (25). β-Strands in R protein may interact with pathogen ligands (24), and the amino acids in the predicted β-strands are often hypervariable (6, 7, 42, 47).
We searched for individual sites under possible positive selection within the 5′ end of all Cf-2 genes and in the 3′ end of full-length coding homologs using an ML method (37). For the 3′ end, two analyses were carried out: one starting at base pair 1849 of the ORF (where only full-length coding homologs align) and at base pair 2296 (where all homologs are conserved) (see Fig. 5, which is published as supporting information on the PNAS web site, for trees used). No positive selection was detected at the 5′ end in any comparison, and the ω values found by the model allowing positive selection (M8) were low (Table 3), suggesting neutrality or purifying selection and supporting the Ka/Ks results.
Table 3. Likelihood ratio tests for positive selection and list of positively selected sites.
| Gene section | Length* | Codons† | P value M7:M8 | ω | p‡ |
|---|---|---|---|---|---|
| 5′ | 654 | 1-218 | 0.9323 | 0.00001 | 0.4540 |
| 654 | 1-218 | 0.6976 | 1.52 | 0.3252 | |
| 3′ | 1,044 | 766-1,113 | 0.0614 | 4.62 | 0.2306 |
| 1,491 | 617-1,113 | 0.0094 | 4.4 | 0.2037 |
Positively selected sites§: 627, 629, 632, 653, 666, 675, 679, 680, 696, 698, 710, 722, 746, 768, 780, 785, 806, 809, 833, 835, 836, 854, 877, 881, 882, 932, 953, 954, 960, 962, 999, 1007, 1029, 1044, 1067, 1070, 1079, 1086, 1093, 1111.
Length of gene section in base pairs; values in italics were obtained by excluding truncated size classes.
Codon positions covered.
Proportion of amino acid sites under positive selection.
Codon positions in the 3′ region with posterior probabilities >0.95; positions in bold are in the LRR region; underlined positions are solvent-exposed.
A significantly better fit with M8 was obtained for one comparison at the 3′ end, suggesting 40 sites potentially under positive selection (Table 3). Of these sites, 30 occur in the LRR-coding region and 10 in the non-LRR-coding region; however, this distribution does not differ from random (Fisher's exact test P = 0.8491). To determine whether the positively selected sites within the LRR region lie in β-sheets, the Cf-2.1 sequence was analyzed with the program sspro (48). The sspro2 and sspro8 models were used to determine the secondary structure of the protein. Residues classified as E (extended strand) by the program were interpreted as β-strands and possible exposed residues. Of the 30 putatively selected sites within the LRR region, eight fall within a β-sheet structure and 22 outside of it. There is no evidence that positively selected sites are more likely to be found in the putative solvent-exposed regions of the LRR (Fisher's exact test, P = 0.4792). Recently, it has been suggested that sites identified by ML methods may occasionally contain false positives (49); although we cannot rule out the possibility of false positives within the 3′ region of the Cf-2 homologs, it is unlikely that these would preferentially occur in given protein motifs and should not affect our conclusions.
The lack of strong evidence for positive selection primarily in solvent-exposed residues of the Cf-2 homologs is in contrast to most studies. Recently, Mauricio et al. (50) also reported lack of evidence for positive selection in A. thaliana Rps2 solvent-exposed residues and suggested that positive selection in these LRR regions is a hallmark of R gene paralogs. The S. pimpinellifolium Cf-2 homologs are most likely a mix of paralogous and orthologous genes, suggesting that positive selection on solvent-exposed amino acids may not be as ubiquitous as believed.
Evolutionary Relationships Among Cf-2 Homologs. The relationship among homologs was examined by constructing separate gene networks for the 5′ and 3′ regions of the genes, excluding the flanking regions (Fig. 2). In both gene networks, Cf-2 homologs are very divergent, with few intermediate sequences. However, the divergence is greater among homologs at the 5′ end. All nonsynonymous mutations in the 3′ Cf-2 haplotype tree correspond to amino acid sites under putative positive selection (Table 3 and Fig. 2), indicating that selected sites are distributed among all lineages of full-length homologs.
Fig. 2.

Gene networks of the alignable areas among homologs. Each step represents a mutational change between haplotypes. Connections exceeding the limits of parsimony were made based on parsimony analysis. Sites are numbered with respect to the Cf-2 size class ORF. The numbering scheme is maintained across deletions present in other haplotypes. Homoplasious mutations are marked with lowercase letters. Mutations leading to an amino acid change are marked with asterisks. Neither network is unique due to alternative placements of homoplasious mutations; alternative networks do not alter topological relationships among haplotypes. (A) Network of the first 654 bp of the 5′ portion of the genes. The limits of parsimony are 11 steps. There are 72 polymorphic sites and 76 mutations; 8 mutations are homoplasious. (B) Network of the 1,045 bp in the 3′ alignable portion of the genes. The spanned region includes the noncoding sequence for genes with early stop codons. The limits of parsimony are 14 steps. There are 55 polymorphic sites and 58 mutations; 8 mutations are homoplasious. A single 3-bp in-frame deletion occurs in size class Hcr2-p1 (position 2656–2658) and was counted as a single mutation for all analyses.
We evaluated differences in structural topology among the networks with a contingency test of homogeneity examining the distribution of interior and tip mutations in the 5′ and 3′ ends of the Cf-2 homologs. Because the gene regions belong to the same individuals, they share a demographic history. Differences between the structural conformations of the two networks may therefore be attributed to the action of selection, which, unlike demographic history, affects specific sites in the genome. Note that we are not evaluating differences in relationships among haplotypes but rather differences in distribution of mutations at each gene region.
Excluding homoplasious mutations, the 5′ region of the Cf-2 homologs has 53 interior and 15 tip mutations (Fig. 2A). The 3′ region has 28 interior and 22 tip mutations (Fig. 2B). The distribution of tip and interior mutations is not independent of gene region (Fisher's exact test, P = 0.0157), suggesting that different processes have shaped these regions. Both gene regions have more internal than tip mutations. The excess of internal mutations is greater for the 5′ end of the genes, translating into a tree with long internal branches (Fig. 2A). Long branch lengths are usually associated with balancing selection or structured populations with limited gene flow (51, 52). Because the 3′ end is not equally affected, this suggests that selection may be maintaining diverse haplotypes at the 5′ end of the Cf-2 homologs. Balancing selection and frequency-dependent selection are postulated as mechanisms for maintaining R gene diversity. Currently, there is limited knowledge on the resistance phenotypes associated with different members of R gene families. The persistence of related functions (e.g., resistance to alternative strains of C. fulvum) of Cf-2 homologs, could lead to the maintenance of orthologous and paralogous haplotypes favored by balancing selection.
Sequence Exchange Occurs Among Cf-2 Homologs. The preponderance of R genes in multigene families suggests that recombination and gene conversion may play an important role in R gene evolution. Interlocus gene conversion has been observed in herbivore resistance genes in A. thaliana (53). Sequence exchange within gene families is a putative mechanism for the generation of new pathogen recognition specificities (41, 54). The importance and frequency of sequence exchange, however, is still debated. The current paradigm is that R genes evolve through a birth-and-death model similar to that proposed for the vertebrate immune system (55, 56), where sequence exchange among alleles (orthologs), as well as point mutations, lead to the generation of most variation. New genes (paralogs) are formed through duplication, but the amount of recombination among them is limited and will not lead to sequence homogenization; thus orthologs will be more similar to each other than paralogs (56).
We evaluated the possibility of sequence exchange among ORFs of the Cf-2 homolog size classes by searching for fragments of matching sequences bounded by nonmatching sequences (40). Among all size classes, three possible sequence exchanges were detected among homologs (Table 8, which is published as supporting information on the PNAS web site). An additional exchange event is suggested for Hcr2-p8 with a sequence not detected in our sample. When only full ORF homologs (which share longer sequence due to fewer indels) were considered, three additional events were identified (Table 8). One exchange in the 5′ A/B-LRR-coding region (Hcr2-p3 and Hcr2-p5) is questionable due to alignment difficulties. The exchange between Hcr2-p7 and Hcr2-p8 is especially apparent in the gene networks (Fig. 2); sequences of these two size classes cluster in the 3′ portion of the genes and are divergent in the 5′ portion.
Evidence for sequence exchange among Cf-2 homologs suggests that some exchange must be occurring among paralogs. However, sequence homogenization, expected under rampant recombination and concerted evolution, has not occurred, because haplotypes are still distinguishable. The high variation in LRR number among the Cf-2 homologs is probably one of the most important factors in the evolutionary dynamics of this gene family; the presence of repeats may facilitate sequence exchange events between paralogs, the creation of chimeric genes, and the origin of new resistance specificities. Furthermore, the rate of generation and loss of R genes through duplication and deletion may be high, given the variability in Cf-2 gene number within S. pimpinellifolium.
One prediction of the birth-and-death model that cannot be evaluated for Cf-2 homologs is the greater similarity of orthologs. When gene copy number varies within a species, and occasional sequence exchange occurs among genes, distinguishing true alleles (orthologs) from genes related through duplication (paralogs) may not be possible. The situation is further confounded by the possibility of related genes retaining the same or similar functions, as seen for Cf-2.1 and Cf-2.2. In the case of Cf-2, orthologous relationships cannot be assumed for homologous genes within a size class, as illustrated by Cf-2.1 and Cf-2.2. Others have also remarked on the difficulties of attempting to distinguish orthologs from paralogs (42, 53, 57). The complex evolution of R genes within gene families challenges current available methods of population genetics analyses, originally created to evaluate alleles of single copy genes.
Conclusion
The Cf-2 homologs constitute an actively evolving gene family in natural populations of S. pimpinellifolium. A diversity of mechanisms maintain variation at various levels within this R gene family. Point mutations, filtered by the action purifying and positive selection in different gene regions, represent one level of variation in Cf-2 homologs. Balancing selection may also have played a role in maintaining the diverse haplotypes of the 5′ end of the genes. Cf-2 homolog variation is not exclusively attributed to natural selection, however. Deletion and duplication of LRR-coding units and limited sequence exchange events between genes have contributed to sequence variation among homologs. These mechanisms have created additional levels of variation: size polymorphism among different Cf-2 homologs, as well as copy-number variation among individuals. This survey of Cf-2 variation in S. pimpinellifolium illustrates the wealth of R gene diversity that exists in wild plant populations, as well as the complexity of interacting processes that can generate such diversity.
Supplementary Material
Acknowledgments
We thank Y.-C. Chiang, H. Sevener, and members of the Purugganan laboratory for comments on the manuscript. We are grateful to M. Dixon (Southampton University, Southampton, U.K.) and M. Parniske (The Sainsbury Laboratory, John Innes Centre, Norwich, U.K.) for supplying Cf0, Cf2, and Cf9 seed. This work was supported by National Science Foundation Dissertation Improvement Grant DEB-0073082.
Author contributions: A.L.C. and B.A.S. designed research; A.L.C. performed research; A.L.C. analyzed data; and A.L.C. and B.A.S. wrote the paper.
Abbreviations: LRR, leucine-rich repeats; ML, maximum likelihood.
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. AY793347–AY793372).
References
- 1.Hammond-Kosack, K. E. & Jones, J. D. G. (1997) Annu. Rev. Plant Physiol. Plant Mol. Biol. 48, 575–607. [DOI] [PubMed] [Google Scholar]
- 2.Flor, H. H. (1946) J. Agric. Res. 73, 335–357. [Google Scholar]
- 3.Bittner-Eddy, P. D., Crute, I. R., Holub, E. B. & Beynon, J. L. (2000) Plant J. 21, 177–188. [DOI] [PubMed] [Google Scholar]
- 4.Ellis, J. G., Lawrence, G. J., Luck, J. E. & Dodds, P. N. (1999) Plant Cell 11, 495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang, G.-L., Ruan, D.-L., Song, W.-Y., Sideris, S., Chen, L. L., Pi, L.-Y., Zhang, S., Zhang, Z., Fauquet, C., Gaut, B. S., et al. (1998) Plant Cell 10, 765–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meyers, B. C., Shen, K. A., Rohani, P., Gaut, B. S. & Michelmore, R. W. (1998) Plant Cell 11, 1833–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mondragon-Palomino, M., Meyers, B. C., Michelmore, R. W. & Gaut, B. S. (2002) Genome Res. 12, 1305–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Richly, E., Kurth, J. & Leister, D. (2002) Mol. Biol. Evol. 19, 76–84. [DOI] [PubMed] [Google Scholar]
- 9.Pan, Q. L., Wendel, J. & Fluhr, R. (2000) J. Mol. Evol. 50, 203–213. [DOI] [PubMed] [Google Scholar]
- 10.Vleeshouwers, V., Martens, A., van Dooijeweert, W., Colon, L. T., Govers, F. & Kamoun, S. (2001) Mol. Plant–Microbe Interact. 14, 996–1005. [DOI] [PubMed] [Google Scholar]
- 11.Cannon, S. B., Zhu, H., Baumgarten, A. M., Spangler, R., May, G., Cook, D. R. & Young, N. D. (2002) J. Mol. Evol. 54, 548–562. [DOI] [PubMed] [Google Scholar]
- 12.Stahl, E. A., Dwyer, G., Mauricio, R., Kreitman, M. & Bergelson, J. (1999) Nature 400, 667–671. [DOI] [PubMed] [Google Scholar]
- 13.Caicedo, A. L., Schaal, B. A. & Kunkel, B. N. (1999) Proc. Natl. Acad. Sci. USA 96, 302–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rose, L. E., Bittner-Eddy, P. D., Langley, C. H., Holub, E. B., Michelmore, R. W. & Beynon, J. L. (2004) Genetics 166, 1517–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luckwill, L. C. (1943) The Genus Lycopersicon: An Historical, Biological, and Taxonomic Survey of the Wild and Cultivated Tomatoes (Aberdeen Univ. Press, Aberdeen, U.K.).
- 16.Rick, C. M., Fobes, J. F. & Holle, M. (1977) Plant Syst. Evol. 127, 139–170. [Google Scholar]
- 17.Jones, D. A., Dickinson, M. J., Balint-Kurti, P. J., Dixon, M. S. & Jones, J. D. G. (1993) Mol. Plant–Microbe Interact. 6, 348–357. [DOI] [PubMed] [Google Scholar]
- 18.Dixon, M. S., Jones, D. A., Keddie, J. S., Thomas, C. M., Harrison, K. & Jones, J. D. G. (1996) Cell 84, 451–459. [DOI] [PubMed] [Google Scholar]
- 19.Jones, D. A., Thomas, C. M., Hammond-Kosack, K. E., Balint-Kurti, P. J. & Jones, J. D. G. (1994) Science 266, 789–793. [DOI] [PubMed] [Google Scholar]
- 20.Thomas, C. M., Jones, D. A., Parniske, M., Harrison, K., Balint-Kurti, P. J., Hatzixanthis, K. & Jones, J. D. G. (1997) Plant Cell 9, 2209–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dixon, M. S., Hatzixanthis, K., Jones, D. A., Harrison, K. & Jones, J. D. G. (1998) Plant Cell 10, 1915–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Piedras, P., Rivas, S., Dröge, S. & Jones, J. D. G. (2000) Plant J. 21. [DOI] [PubMed]
- 23.Van der Hoorn, R. A. L., Van der Ploeg, A., de Wit, P. J. G. M. & Joosten, M. H. A. J. (2001) Mol. Plant–Microbe Interact. 14, 412–415. [DOI] [PubMed] [Google Scholar]
- 24.Jones, D. A. & Jones, J. D. G. (1997) Adv. Bot. Res. 24, 89–167. [Google Scholar]
- 25.Kobe, B. & Kajava, A. V. (2001) Curr. Opin. Struct. Biol. 11, 725–732. [DOI] [PubMed] [Google Scholar]
- 26.Seear, P. J. & Dixon, M. S. (2003) Mol. Plant Pathol. 4, 199–202. [DOI] [PubMed] [Google Scholar]
- 27.Hillis, D. M., Maple, B. K., Larson, A., Davis, S. K. & Zimmer, E. A. (1996) in Molecular Systematics, eds. Hillis, D. M., Moritz, C. & Maple, B. K. (Sinauer, Sunderland, MA), pp. 321–381.
- 28.Caicedo, A. L. & Schaal, B. A. (2004) Mol. Ecol. 13, 1871–1882. [DOI] [PubMed] [Google Scholar]
- 29.Clement, M., Posada, D. & Crandall, K. A. (2000) Mol. Ecol. 9, 1657–1659. [DOI] [PubMed] [Google Scholar]
- 30.Swofford, D. L. (2000) paup*, Phylogenetic Analysis Using Parsimony (Sinauer, Sunderland, MA).
- 31.Templeton, A. R., Crandall, K. A. & Sing, C. F. (1992) Genetics 132, 619–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nei, M. (1987) Molecular Evolutionary Genetics (Columbia Univ. Press, New York).
- 33.Nei, M. & Gojobori, T. (1986) Mol. Biol. Evol. 3, 418–426. [DOI] [PubMed] [Google Scholar]
- 34.Rozas, J. & Rozas, R. (1999) Bioinformatics 15, 174–175. [DOI] [PubMed] [Google Scholar]
- 35.Zhang, J., Kumar, S. & Nei, M. (1997) Mol. Biol. Evol. 14, 1335–1338. [DOI] [PubMed] [Google Scholar]
- 36.Hochberg, Y. (1988) Biometrika 75, 800–802. [Google Scholar]
- 37.Yang, Z., Nielsen, R., Goldman, N. & Pederson, A.-M. K. (2000) Genetics 155, 431–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anisimova, M., Bielawski, J. P. & Yang, Z. (2001) Mol. Biol. Evol. 18, 1585–1592. [DOI] [PubMed] [Google Scholar]
- 39.Yang, Z. (1997) Comput. Appl. Biosci. 13, 555–556. [DOI] [PubMed] [Google Scholar]
- 40.Sawyer, S. A. (1999) geneconv (Dept. of Mathematics, Washington University, St. Louis, MO).
- 41.Parniske, M., Hammond-Kosack, K. E., Golstein, C., Thomas, C. M., Jones, D. A., Harrison, K., Wulff, B. B. H. & Jones, J. D. G. (1997) Cell 91, 821–832. [DOI] [PubMed] [Google Scholar]
- 42.Noel, L., Moores, T. L., van der Biezen, E. A., Parniske, M., Daniels, M. J., Parker, J. E. & Jones, J. D. G. (1999) Plant Cell 11, 2099–2111. [PMC free article] [PubMed] [Google Scholar]
- 43.McDowell, J. M., Dhandaydham, M., Long, T. A., Aarts, M. G. M., Goff, S., Holub, E. B. & Dangl, J. L. (1998) Plant Cell 10, 1861–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wulff, B. B. H., Thomas, C. M., Smoker, M., Grant, M. & Jones, J. D. G. (2001) Plant Cell 13, 255–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ellis, J., Dodds, P. & Pryor, T. (2000) Trends Plant Sci. 5, 373–379. [DOI] [PubMed] [Google Scholar]
- 46.Kobe, B. & Deisenhofer, J. (1993) Nature 366, 751–756. [DOI] [PubMed] [Google Scholar]
- 47.Bergelson, J., Kreitman, M., Stahl, E. A. & Tian, D. (2001) Science 292, 2281–2285. [DOI] [PubMed] [Google Scholar]
- 48.Baldi, P., Brunak, S., Frasconi, P., Pollastri, G. & Soda, G. (1999) Bioinformatics 15, 937–946. [DOI] [PubMed] [Google Scholar]
- 49.Suzuki, Y. & Nei, M. (2004) Mol. Biol. Evol. 21, 914–921. [DOI] [PubMed] [Google Scholar]
- 50.Mauricio, R., Stahl, E. A., Korves, T., Tian, D., Kreitman, M. & Bergelson, J. (2003) Genetics 163, 735–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hudson, R. R. & Kaplan, N. L. (1988) Genetics 120, 831–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nordborg, M. (2002) Curr. Opin. Plant Biol. 5, 69–73. [DOI] [PubMed] [Google Scholar]
- 53.Kroymann, J., Donnerhacke, S., Schnabelrauch, D. & Mitchell-Olds, T. (2003) Proc. Natl. Acad. Sci. USA 100, 14587–14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pryor, T. & Ellis, J. (1993) Adv. Plant Pathol. 10, 281–305. [Google Scholar]
- 55.Nei, M., Gu, X. & Sitnikova, T. (1997) Proc. Natl. Acad. Sci. USA 94, 7799–7806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Michelmore, R. W. & Meyers, B. C. (1998) Genome Res. 8, 1113–1130. [DOI] [PubMed] [Google Scholar]
- 57.Michelmore, R. (2000) Curr. Opin. Plant Biol. 3, 125–131. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}