

Abstract
Biliary atresia (BA) is a neonatal obstructive cholangiopathy that progresses to end‐stage liver disease, often requiring transplantation. The murine model of BA, employing rhesus rotavirus (RRV), parallels human disease and has been used to elucidate mechanistic aspects of a virus induced biliary cholangiopathy. We previously reported that the RRV VP4 gene plays an integral role in activating the immune system and induction of BA. Using rotavirus binding and blocking assays, this study elucidated how RRV VP4 protein governs cholangiocyte susceptibility to infection both in vitro and in vivo in the murine model of BA. We identified the amino acid sequence on VP4 and its cholangiocyte binding protein, finding that the sequence is specific to those rotavirus strains that cause obstructive cholangiopathy. Pretreatment of murine and human cholangiocytes with this VP4‐derived peptide (TRTRVSRLY) significantly reduced the ability of RRV to bind and infect cells. However, the peptide did not block cholangiocyte binding of TUCH and Ro1845, strains that do not induce murine BA. The SRL sequence within TRTRVSRLY is required for cholangiocyte binding and viral replication. The cholangiocyte membrane protein bound by SRL was found to be Hsc70. Inhibition of Hsc70 by small interfering RNAs reduced RRV's ability to infect cholangiocytes. This virus‐cholangiocyte interaction is also seen in vivo in the murine model of BA, where inoculation of mice with TRTRVSRLY peptide significantly reduced symptoms and mortality in RRV‐injected mice. Conclusion: The tripeptide SRL on RRV VP4 binds to the cholangiocyte membrane protein Hsc70, defining a novel binding site governing VP4 attachment. Investigations are underway to determine the cellular response to this interaction to understand how it contributes to the pathogenesis of BA. (Hepatology 2017;65:1278‐1292)
Abbreviations
- aa
amino acid
- BA
biliary atresia
- RRV
rhesus rotavirus
- siRNA
small interfering RNA
Biliary atresia (BA) is an inflammatory cholangiopathy unique to infancy and is the most common cause of pathologic jaundice in the pediatric population, leading to extrahepatic biliary obstruction. Surgical reconstruction of the extrahepatic biliary tree may restore drainage in some; however, many infants progress to cirrhosis and end stage liver disease. Due to the progressive nature of the disease, BA is the most common indication for pediatric liver transplantation in the United States.1
The etiology of BA remains unclear. In 1974, Landing proposed that a viral insult leads to a progressive inflammatory process, resulting in cholangiopathic disease.2 Subsequent studies found multiple viruses including rotavirus group C,3 reovirus type 3,4 Epstein‐Barr virus,5 cytomegalovirus,6 and human papillomavirus7 in explanted livers of infants with BA. In a murine model of BA, rhesus rotavirus (RRV) infection of newborn BALB/c mice resulted in obstructive cholangiopathy,8, 9 the symptoms of which included bilirubinuria, jaundice, acholic stools, and growth retardation that mirrored the human form of the disease.9 In this model, the morphologic and histologic changes in the biliary tree10 parallel those found in human BA.8
Rotaviruses, including RRV, are 65‐ to 75‐nm icosahedral, nonenveloped viruses containing 11 double‐stranded RNA gene segments encoding six structural proteins (VP1‐4, VP6, and VP7) and six nonstructural (NSP1‐6) proteins. The outer layer is comprised of a protein capsid consisting of VP4 and VP7 proteins, which are involved in cellular binding.11 In previous studies, we found that one of the cellular targets of RRV is the biliary epithelial cell (cholangiocyte).12 The mechanism by which RRV attaches to a host cell is a multistep process involving sialic acid labeled glycoproteins13, 14 and the integrins α2β1, α4β1, αVβ3, and αXβ2.14, 15, 16, 17, 18, 19, 20 We have shown previously that expression of the α2β1 integrin plays a role in cholangiocyte susceptibility to RRV infection.21 Although blocking of the α2β1 integrin reduced cholangiocyte vulnerability by 50%, RRV was still able to infect the cell, suggesting the existence of other, undefined cholangiocyte cell surface receptors that may govern susceptibility.
We have previously used reassortant viruses to study the specific RRV genes involved in cholangiocyte susceptibility and viral pathogenicity. Using single‐gene reassortants created from RRV and TUCH (a simian rotavirus strain capable of infecting cholangiocytes without causing murine BA), we found that the gene segment 4 of RRV encoding for the VP4 protein confers the ability of the virus to attach to and infect cholangiocytes, causing murine BA.18, 22 In a subsequent study, we found that virus strains with high homology to the RRV VP4 protein are capable of inducing murine BA.23
The primary goal of this study was to determine the mechanism by which RRV VP4 governs the murine model of BA. Computer‐aided analyses were performed to identify potential protein–protein interaction sites on the RRV VP4 protein. A specific sequence (SRL) was identified on this protein that binds to cholangiocytes. It was then determined that the cholangiocyte binding site for this peptide is the Hsc70 protein.
Methods
ETHICS STATEMENT
All animal research was performed in accordance with regulations and protocols approved by the Institutional Animal Care and Use Committee at Cincinnati Children's Hospital Medical Center (protocol number IACUC2013‐0039), which adheres to the National Institutes of Health OLAW regulation (Animal Assurance number A3108) and the Animal Welfare Act (certification number 31‐8‐001).
VIRUSES AND CELLS
The mouse cholangiocyte cell line generously provided by James Boyer (Yale Liver Care Center, Hartford, CT), MA104 cells (BioWhittaker, Walkersville, MD), and human cholangiocytes (H69 cells) kindly provided by Douglas Jefferson (New England Medical Center, Tufts University, Boston, MA) were cultured as described previously.21, 24 We used several rotavirus strains: RRV, a simian strain (kindly provided by H. Greenberg, Stanford University, Palo Alto, CA), TUCH (named after the locations where the strain was isolated: Tulane National Primate Research Center and Cincinnati Children's Hospital), a simian strain,23 Ro1845 a human strain (generously provided by Yasutaka Hoshino, NIAID) and GRV a goat strain (graciously provided by Osamu Nakagomi, Nagasaki University, Japan). Virus stocks were grown in MA104 cells as described previously and kept frozen at −80°C until thawed for use.12 Gene sequences of gene segment 4 from each strain were obtained from GenBank: RRV [AY033150], TUCH [AY596189], Ro1845 [EU708893], and GRV [AB055967]
STRUCTURAL MODELING AND INTERACTION SITES MAPPING
To map putative functional sites that may play a role in RRV VP4 interactions with cholangiocytes, a number of structural bioinformatics tools were employed to analyze and extrapolate from available structural data on entry apparatus. This included several solved conformations of the spike protein VP4 (PDB entries 1SLQ, 2B4H, 2B4I, 4V7Q). Additional protein–protein interaction sites were predicted using SPPIDER (http://sppider.cchmc.org) that combines sequence‐based solvent accessibility prediction and data on unbound as well as known bound structures to identify sites likely to get buried upon complex formation.25 These predictions were further analyzed in the context of existing structural and sequence data using POLYVIEW‐3D (http://polyview.cchmc.org), ConSurf (http://consurf.tau.ac.il/) and Evolutionary Trace Annotation Server (http://mammoth.bcm.tmc.edu/eta/index.html).
PEPTIDES
Synthetic peptides spanning different regions on the VP4 protein of RRV, TUCH, and Ro1845 strains were synthesized and purified by way of high‐performance liquid chromatography and kept lyophilized (GenScript, Piscataway, NJ). Unrelated peptides (GHRP) of the same size and random sequence peptides with identical amino acid (aa) compositions were synthesized for use as controls. Peptides were resuspended in Dulbecco's modified Eagle's medium before use in replication assays. Biotinylated TRTRVSRLY peptides were synthesized using Fmoc‐Lys(Fmoc)‐OH strategy where two biotin moieties are attached to each motif separated by hexanoic acid (GenScript).
VIRAL BINDING ASSAY
The ability of the virus to attach to the cholangiocytes was assessed by binding assays as described previously.21 Briefly, cells were grown to confluence in 24‐well plates. The cells, medium, and inoculating virus were cooled to 4°C. Cells were then inoculated with varying amounts of virus and incubated at 4°C for 1 hour. The inoculum was removed and cells were washed twice to remove unbound virus. The cells then underwent two freeze/thaw cycles, and the virus within the final cell fraction represented the attached virus. The washes and residual inoculum were combined. This combined solution represented all unbound virus. The amount of bound and unbound virus was determined by way of focus forming assay. The amount of attached virus was expressed as percent of total amount of virus used to inoculate cells.
QUANTIFICATION OF INFECTIOUS ROTAVIRUS
Focus forming assay was used to determine the presence of infectious rotavirus as described previously.26 Briefly, 96‐well plates were treated with serially diluted virus and incubated at 37°C for 14‐16 hours. After acetone fixation, Guinea pig anti‐rotavirus immunoglobulin G primary antibody (1:1000) was added and incubated for 30 minutes. Wells were aspirated and washed with phosphate‐buffered saline and fluorescein isothiocyanate–tagged goat anti‐guinea pig immunoglobulin G secondary antibody (1:500) was added for 30 minutes at 37°C. Wells were then washed twice, and plates were scored using ultraviolet microscopy (10 × objective). Quantities of infectious virus were reported as FFU per milligram of tissue.
IN VIVO USE OF TRTRVSRLY PEPTIDE IN THE MURINE MODEL OF BA
All animal studies were performed in accordance with institutional animal welfare guidelines using an approved animal protocol. The experimental model of biliary atresia was induced in BALB/c mice (Envigo Labs, Indianapolis, IN). Newborn pups were injected intraperitoneally with 500 μg of TRTRVSRLY or SSASAW peptide diluted in saline along with RRV at 7.5 × 105 ffu/pup. A subsequent injection of peptide was given on days 1 and 2 of life. Pups were monitored for 21 days for clinical signs of hepatobiliary injury (i.e., acholic stools, bilirubinuria, and jaundice in non–fur‐covered skin) and survival. Subsets of these mice were sacrificed 7 days after infection, and their extrahepatic biliary tract was harvested. The tissues were analyzed for the presence of replication‐competent virus by focus forming assay.
PROTEIN PRECIPITATION WITH BIOTINYLATED TRTRVSRLY PEPTIDES
Biotinylated TRTRVSRLY peptide was synthesized by GenScript. Peptides were designed with an N‐terminal biotin linker. For affinity pull‐downs, 2 mg of immobilized peptide was added to confluent monolayer of cholangiocytes and incubated at 4°C for 1 hour. After incubation, cells were lysed and the membrane fraction was isolated using 0.38 M NaCl buffer. This membrane fraction was later treated with streptavidin agarose (Peirce, Rockford, IL) to pull down any bound protein.27 Control samples for each lysis buffer used included a sample in which no peptide was added. After extensive washes, bound proteins were eluted from the immobilized peptides by boiling in SDS sample buffer and run in 4%‐20% polyacrylamide gel.
MASS SPECTROMETRY ANALYSIS OF CHOLANGIOCYTE MEMBRANE PROTEINS AS THEY INTERACT WITH THE TRTRVSRLY PEPTIDE
Silver‐stained bands were excised from the gels and tryptic‐digested as described by Eismann et al.28 The mass lists were generated by MALDI‐TOF mass spectrometry on a 4800 MALDI‐TOF (Applied Biosystems, Forest City, CA). The identity search was performed using the MASCOT search algorithm (Matrix Science, Boston, MA) by scanning the current version of the National Center for Biotechnology Information sequence database. The significance of the identity was judged from the search engine's scoring system.
TRANSFECTION OF SMALL INTERFERING RNA AGAINST Hsc70
Transfection of small interfering RNA (siRNA) to reduce Hsc70 expression was performed as described previously.21 The siRNA sequences were as follows: sequence 1, GCUUGAGUAAGGAAGAUAU; sequence 2, GAUUAAGUCAGUCCAAGAA; sequence 3, UCACGGACACAGAGAGAUU; sequence 4, CCACAGGGACCCAAAACAA (Dharmacon, Lafayette, CO). Nontargeting siRNA was purchased from Dharmacon. Immunohistochemistry and Western blot analysis was used to detect reduced membrane expression of Hsc70. Immunocytochemistry was performed by harvesting cholangiocytes before and after siRNA transfection and cytospin centrifugation of 10,000 cells onto slides. Slides were fixed in 4% paraformaldehyde and were blocked with 5% normal donkey serum. Slides were incubated at 4°C overnight with rat anti‐mouse Hsc70 antibody diluted 1:200 in DAKO antibody diluent. After incubation, the slides were washed with phosphate‐buffered saline and further incubated with anti‐rat Alexafluor 594–conjugated secondary antibody, followed by incubation for 1.5 hours at room temperature. The slides were washed and embedded with DAPI mounting media for visualization under a fluorescent microscope (Nikon Eclipse 90).
STATISTICAL ANALYSIS
Analysis of noncontinuous variables was performed using chi‐squared and Fisher exact tests. Results of continuous variables are expressed as the mean ± standard error and were analyzed using analysis of variance with post hoc testing where appropriate; P < 0.05 was considered statistically significant. All statistical analysis was performed using Prism 7 (GraphPad Software, La Jolla, CA).
Results
ANALYSIS OF RRV VP4 PROTEIN–PROTEIN INTERACTIONS
We used the SPPIDER program to identify possible protein–protein interaction sites on VP4, which predicted a number of putative protein interaction sites (http://onlinelibrary.wiley.com/doi/10.1002/hep.28947/suppinfo). Of the nine possible sites of interest, six were determined to have aa sequences analogous to those strains, which do not cause murine BA (TUCH and Ro1845) and were excluded from further analysis. Synthetic peptides were generated for the three remaining peptide sequences (PTAAGV, SSASAW, and TRTRVSRLY) for use in subsequent studies (Fig. 1A).
Figure 1.
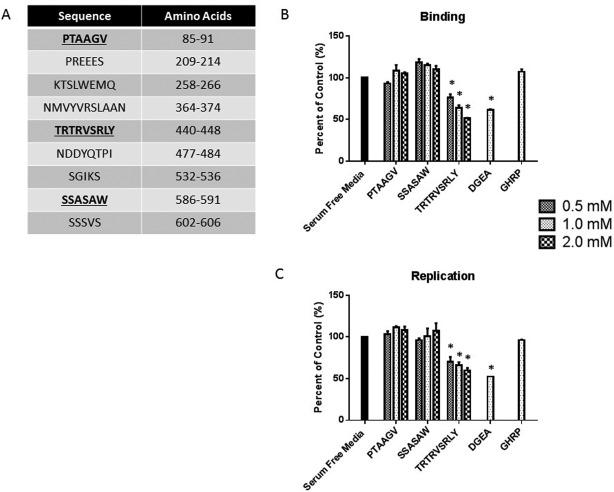
Viral binding and replication in cholangiocytes treated with synthetic peptides. (A) Table illustrating the nine potential protein–protein interaction sites on RRV VP4 as determined by the SPPIDER program. The bolded sequences (PTAAGV, SSASAW, and TRTRVSRLY) represent the region where RRV differs from TUCH and Ro1845. (B, C) Mouse cholangiocytes were treated with peptides PTAAGV, SSASAW, or TRTRVSRLY at varying concentrations followed by RRV infection at 4°C. DGEA and GHRP were used as a positive and negative control, respectively. (B) Cholangiocytes treated with TRTRSVRLY or DEGA showed a significant dose‐dependent decrease in RRV binding. (C) Cholangiocytes also revealed a significant dose‐dependent decrease in viral yield when treated with TRTRVSRLY, whereas PTAAGV and SSASAW had no effect on viral replication. *P < 0.05 versus serum‐free media.
BLOCKING OF RRV BINDING AND REPLICATION BY SYNTHETIC VP4 DERIVED PEPTIDE
Through in vitro peptide binding assays, the synthetic peptides of interest derived from the VP4 protein (PTAAGV, SSASAW, and TRTRVSRLY) were used to determine which specific sequence was involved in cholangiocyte binding. Investigation of cholangiocyte binding determined that the TRTRVSRLY peptide was able to block RRV binding in a dose‐dependent manner. Increased concentrations of the peptide (0.5 mM, 1 mM, 2 mM) resulted in blocking of the virus, as a percent of control (media without peptides), to be significantly decreased compared with treatment with media alone (76.33%, 64.17%, and 51.60%, respectively; P < 0.05). Conversely, the PTAAGV and SSASAW peptides did not interfere with RRV–cholangiocyte binding. DGEA is the collagen‐binding domain for the α2β1 cholangiocyte receptor and has been shown to be involved in RRV infection.29 It was therefore used as a positive control, resulting in decreased viral binding (61.40%). GHRP is a nonspecific peptide that was used as a negative control and has been shown to have no ability to inhibit RRV binding21 (Fig. 1B).
To determine whether cholangiocyte surface blocking with synthetic peptides had any effect on viral replication, blocking assays were performed followed by incubation at 37°C for 18 hours. Viral yield in cholangiocytes treated with increasing dosages of synthetic TRTRVSRLY peptide was significantly lower than media or GHRP peptide controls (69.97%, 66.23%, 59.90%; P < 0.05), whereas the PTAAGV and SSASAW peptides had no effect on viral yield (Fig. 1C). TRTRVSRLY peptide demonstrated similar blocking in MA104 cells (http://onlinelibrary.wiley.com/doi/10.1002/hep.28947/suppinfo).
INHIBITION OF VIRAL BINDING AND REPLICATION BY THE TRTRVSRLY PEPTIDE IS STRAIN SPECIFIC
We have shown that the induction of BA in the murine model was rotavirus strain‐specific.12 In these investigations, RRV and GRV (goat strain) were able to induce the murine model of BA,23 while TUCH (a simian strain) and Ro1845 (a human strain) could not. Blocking and replication assays were performed on cholangiocytes using the synthetic TRTRVSRLY peptide and these other rotavirus strains. The TRTRVSRLY peptide inhibited binding of RRV and GRV (BA‐inducing strains) by 45.13% and 41.27%, respectively, when compared with control (P < 0.05). Conversely, the peptide had no effect on viral binding of the TUCH and Ro1845 strains (Fig. 2A). Similarly, viral yield was reduced in RRV and GRV by 51.29% and 35.51% of control (P < 0.05), respectively, whereas cholangiocytes infected with Ro1845 and TUCH showed no reduction in viral yield after treatment with TRTRVSRLY peptide (Fig. 2B).
Figure 2.
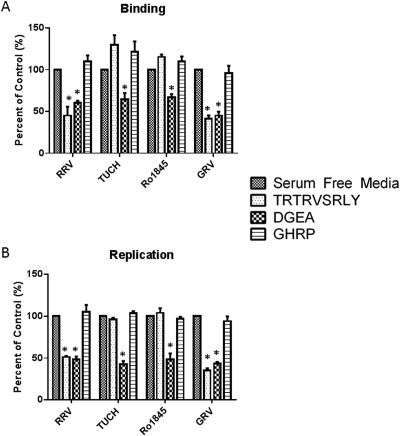
Viral binding and replication in cholangiocytes treated with TRTRVSRLY as a function of rotavirus strain. Mouse cholangiocytes were pretreated with 1 mM synthetic peptide TRTRVSRLY followed by infection with RRV, TUCH, Ro1845, or GRV. (A) Viral binding revealed significant inhibition of binding only in strains that induce murine biliary atresia (RRV and GRV). DGEA and GHRP are positive and negative controls, respectively. (B) A significant reduction in viral yield in RRV and GRV was measured, with no decrease in replication for TUCH or Ro1845. *P < 0.05 versus serum‐free media.
TUCH AND Ro1845 VP4 AMINO ACID SEQUENCES (440‐448) HAVE NO EFFECT ON VIRUS BINDING AND REPLICATION
TRTRVSRLY constitutes the 440‐448 aa sequence on RRV VP4. The 440‐448 aa sequences in other strains, including TUCH and Ro1845 differ from that of RRV (Fig. 3A). This information was used to synthesize peptides of those regions found in the respective viruses to determine whether the inhibition of binding and replication was sequence‐specific. Cholangiocytes were treated with MRTRVSGLY (Ro1845) and VRTRISGLY (TUCH) peptides. Binding and replication assays were performed as described previously. TRTRVSRLY peptide was able to inhibit binding and replication of RRV, with no effect on Ro1845 and TUCH. MRTRVSGLY and VRTRISGLY had no effect on any of the strains assessed (Fig. 3B,C), suggesting that BA‐inducing strains (RRV and GRV) use the TRTRVSRLY sequence as a strain‐specific cholangiocyte binding site.
Figure 3.
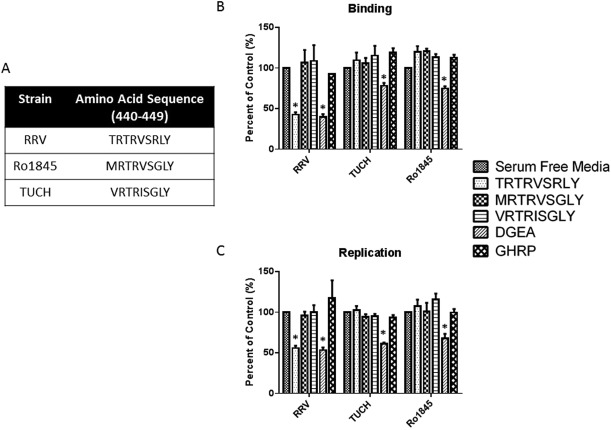
Corresponding peptides (aa 440‐448) on TUCH and Ro1845 have no effect on rotavirus binding and replication. (A) Amino acids 440‐448 of RRV, TUCH, and Ro1845. (B, C) Treatment of mouse cholangiocytes with 1 mM MRTRVSGLY and VRTRISGLY had no effect on binding or replication in RRV, TUCH, or Ro1845. In all strains, DGEA (positive control) significantly inhibited both viral binding and replication compared with media‐only control. Treatment with TRTRVSRLY significantly reduced RRV binding and viral yield with no effect on other rotavirus strains. *P < 0.05 versus serum‐free media pretreatment.
THE SRL PORTION OF THE TRTRVSRLY PEPTIDE IS INTEGRAL TO VIRAL BINDING AND REPLICATION
Once it was determined that the TRTRVSRLY peptide plays a role in RRV binding and cholangiocyte infection, the next objective was to identify the specific aa within this 9 aa peptide responsible for these functions. RRV and Ro1845 440‐448 peptides differ by two aa. We synthesized peptides with each of those single aa changes (MRTRVSRLY, TRTRVSGLY) to evaluate the region of the peptide critical in binding and replication. Altering aa 440 (T→M) had no impact on inhibition of binding and replication, with both TRTRVSRLY and MRTRVSRLY significantly inhibiting RRV function. However, alteration at peptide 446 (R→G) inhibited the peptide's ability to block binding and replication (Fig. 4A,B). This result suggests that the critical portion of the 9 aa sequence lay within the last five residues. We then investigated the latter half of the TRTRVSRLY sequence to determine which aa govern the peptide's function. Peptides measuring 5 aa were synthesized with single aa changes at each position of the VP4 VSRLY sequence. Assaying rotavirus binding and replication revealed that peptides in which the SRL aa sequence was preserved (VSRLY, ISRLY, and VSRLA) maintained the ability to inhibit virus binding and replication. Those peptides in which the S, R, or L peptide was modified (VVRLY, VSGLY, VSRAY) were unable to exhibit such inhibition (Fig. 4C,D). These results suggest that the presence of the SRL aa sequence is integral to RRV–cholangiocyte binding and viral replication.
Figure 4.
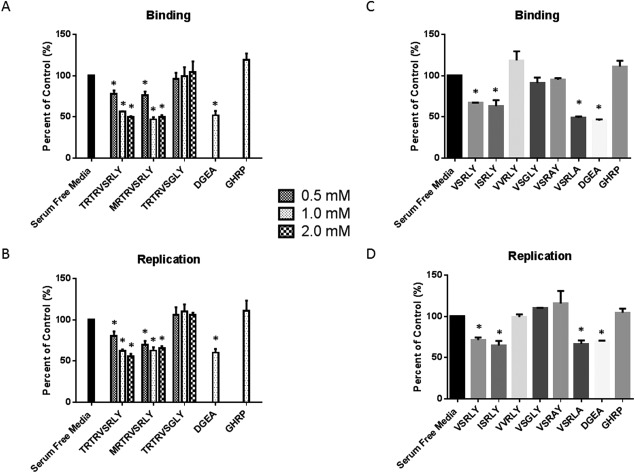
The SRL portion of TRTRVSRLY has an effect on viral binding and replication. (A, B) Mouse cholangiocytes pretreated with TRTRSVRLY or synthetic peptides with altered aa at positions 440 (T→M) or 446 (R→G). Altering aa 440 had a similar effect as TRTRVSRLY while a peptide containing an altered aa 446 could not inhibit RRV binding or replication. (C, D) Cholangiocytes pretreated with 1 mM of synthetic peptides with single aa changes from VSRLY. Peptides conserving the SRL portion maintained the peptide's ability to block RRV binding and replication. Peptides with an altered single aa between 445 and 447 lost the ability to block RRV binding and replication. *P < 0.05 versus serum‐free media.
TRTRVSRLY PEPTIDE INHIBITS RRV REPLICATION IN VIVO AND ATTENUATES BA PHENOTYPE
To prove that the TRTRVSRLY peptide played a similar role in vivo, pups were inoculated with the peptide and RRV at birth followed by peptide again on day of life 1 and 2. Symptoms (acholic stool, bilirubinuria, jaundice, and weight loss) were recorded for 21 days. Mice injected with TRTRVSRLY peptide along with RRV developed significantly less symptoms compared with those injected with RRV and SSASAW peptide or saline (21.1% versus 100.0% versus 100.0%, respectively; P < 0.05) (Fig. 5A). Survival was similarly improved when pups were given the TRTRVSRLY peptide, with only 26.3% mortality when compared with 92.9% with SSASAW peptide and 88.5% with saline (Fig. 5B).
Figure 5.
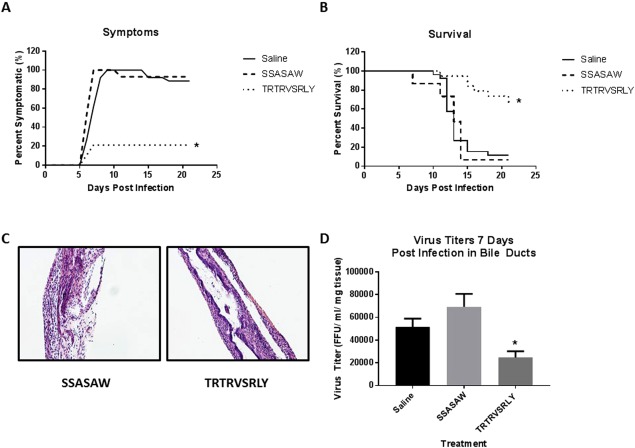
Effects of in vivo use of TRTRSVRLY peptide. Mice were treated with the TRTRVSRLY peptide along with RRV. Saline (no peptide) or SSASAW peptides with RRV injection were used as a control. (A) Symptoms were tracked in both sets of mice for 21 days. Symptoms in the TRTRVSRLY‐treated group were 21.1% versus 100.0% in the saline control group. (B) Survival was noted to be significantly improved (26.3%) in mice treated with the synthetic peptide versus saline control (88.5%). (C) Histologic evaluation of the extrahepatic biliary tract 12 days after infection with RRV and peptide treatment. Hematoxylin and eosin staining of extrahepatic bile ducts after infection with RRV and SSASAW peptide treatment resulted in complete obstruction of extrahepatic biliary duct. In contrast, those mice treated with TRTRVSRLY peptide had some mild inflammation with a patent bile duct. Magnification: × 10. (D) Viral titers from extrahepatic bile ducts harvested 7 days after infection were significantly lower in mice treated with the TRTRVSRLY peptide versus saline or those treated with SSASAW peptide. *P < 0.05.
Bile ducts were harvested 12 days after infection from SSASAW and TRTRVSRLY peptide–treated mice and stained with hematoxylin and eosin. Mice treated with SSASAW peptide exhibited a complete obstruction of their bile ducts, whereas those treated with TRTRVSRLY had a mild inflammation with a patent lumen (Fig. 5C). Viral titers were obtained from extrahepatic bile ducts 7 days after injection with RRV. As expected, bile ducts of mice treated with the TRTRVSRLY peptide had significantly lower amounts of virus than those treated with SSASAW and the no peptide controls (Fig. 5D), thus proving that the synthetic peptide (TRTRVSRLY) competitively inhibited viral replication and subsequently attenuated the disease phenotype.
INHIBITION OF VIRAL BINDING AND REPLICATION BY THE TRTRVSRLY PEPTIDE ON HUMAN CHOLANGIOCYTES
To test the relevance of the TRTRVSRLY peptide on human rotavirus infection, we next performed binding and replication assays on human cholangiocytes (H69 cells). Similar to the experiments performed in mouse cholangiocytes, treatment with 1 mM TRTRVSRLY peptide significantly reduced the ability of RRV to bind to and replicate within human cholangiocytes to 65.9% and 57.9% of control, respectively (Fig. 6A,B).
Figure 6.
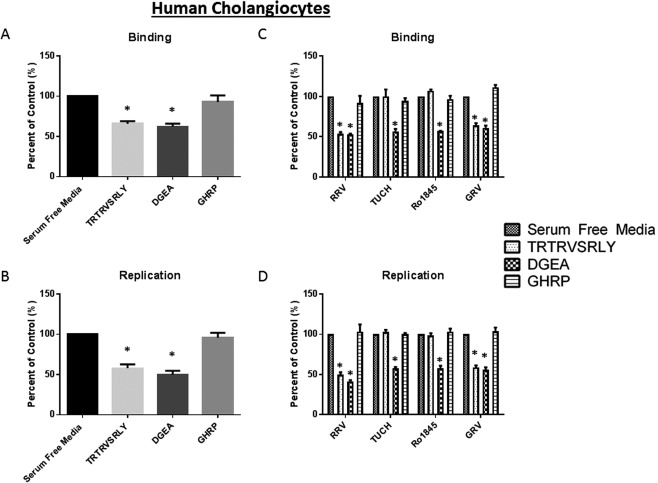
Viral binding and replication in human cholangiocytes treated with TRTRVSRLY as a function of rotavirus strain. (A, B) Human cholangiocytes (H69) pretreated with 1 mM TRTRVSRLY peptide demonstrated significant decrease in RRV binding and replication. DGEA and GHRP are positive and negative controls, respectively. (C) The TRTRVSRLY peptide significantly inhibited the binding of only those strains that induce murine biliary atresia (RRV and GRV) with no effect on TUCH or Ro1845. (D) Similarly, RRV and GRV viral yields were significantly reduced, with no decrease in yield for TUCH or Ro1845. *P < 0.05 versus serum‐free media control.
Previously, we reported strain‐specific infectability of human cholangiocytes mirroring that of mouse cholangiocytes.24 To prove that the TRTRVSRLY peptide strain‐specific blocking was consistent in human cholangiocytes, we performed binding and replication assays on H69 cells infected with RRV, TUCH, Ro1845, and GRV. In agreement with our mouse data, the TRTRVSRLY peptide was able to block the binding of RRV and GRV to H69 cells while having no effects on TUCH and Ro1845 binding (Fig. 6C). An identical effect was observed with replication, because RRV and GRV exhibited reduced viral yields after treatment with the TRTRVSRLY peptide (Fig. 6D).
THE Hsc70 PROTEIN ON THE CELL MEMBRANE IS INVOLVED IN RRV VP4 BINDING THROUGH SRL
To identify the SRL peptide's corresponding cholangiocyte receptor, cell membrane fractions were treated with a synthetic biotinylated TRTRVSRLY peptide as described previously. After isolation of the bound protein from a silver‐stained polyacrylamide gel, MALDI‐TOF analysis identified the protein of interest to be 70 KDa protein, Hsc70 (Fig. 7A).
Figure 7.
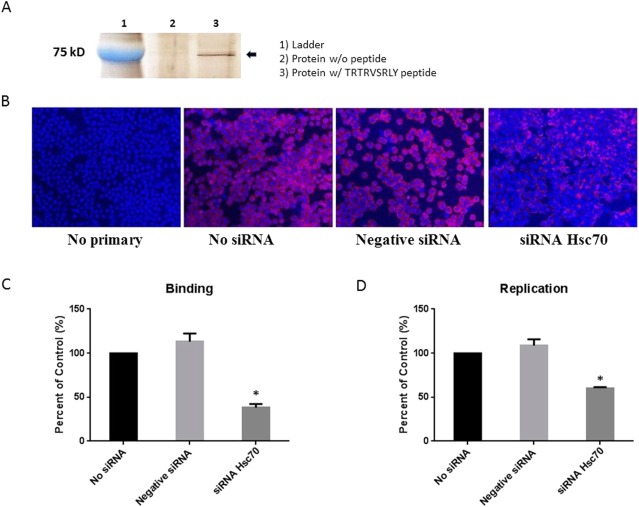
The role of Hsc70 in binding to TRTRVSRLY. (A) Silver‐stained band of protein immunoprecipitated from mouse cholangiocytes treated with no peptide (lane 2) and biotinylated TRTRVSRLY peptide (lane 3). (B) Cholangiocytes were treated with transfection agent in the absence of siRNA, negative (scrambled) siRNA, or Hsc70 siRNA. No siRNA (second image from left) and negative siRNA (third image from left) were negative controls. siRNA against Hsc70 was able to block Hsc70 expression on the cholangiocyte cell surface. (C, D) When infected with RRV, cholangiocytes treated with Hsc70 siRNA showed a significant reduction in virus binding and replication versus negative controls. *P < 0.05 versus no siRNA.
Interestingly, Hsc70 has been implicated in rotavirus cell entry through a secondary interaction involving the VP4 protein.30
Hsc70 KNOCKDOWN IN CHOLANGIOCYTE BLOCKS THE ACTION OF TRTRVSRLY PEPTIDE
To determine whether Hsc70 was a cholangiocyte receptor for the SRL sequence in RRV VP4, siRNA was used to knock down Hsc70 expression. An siRNA combination of four sequences targeting the Hsc70 gene was transfected into cholangiocytes, followed by immunostaining for Hsc70. “No siRNA” referred to cells treated with a transfection agent without siRNA and was used as a negative control. A combination of scrambled sequences–or negative siRNA–which are incapable of blocking any cellular function, were used as a second negative control. Forty‐eight hours after transfection of siRNA against Hsc70, a decrease of cholangiocyte surface expression of Hsc70 was detected (Fig. 7B). Binding assays were performed on cholangiocytes infected with RRV to validate the role of Hsc70 in viral binding. Only those treated with Hsc70 siRNA displayed a significant decrease in RRV binding and replication compared with cholangiocytes treated with negative controls (Fig. 7C,D).
Hsc70 siRNA‐transfected cholangiocytes underwent TRTRVSRLY peptide binding assays, revealing that down‐regulation of Hsc70 expression in cholangiocytes led to inability of TRTRVSRLY to block RRV binding, while those treated with negative control siRNA remained susceptible to inhibition of RRV attachment. All cells remained sensitive to DGEA blocking regardless of treatment (Fig. 8A). Percent of control is normalized to cells treated with serum free media in the absence of peptide (presented as 100%).
Figure 8.
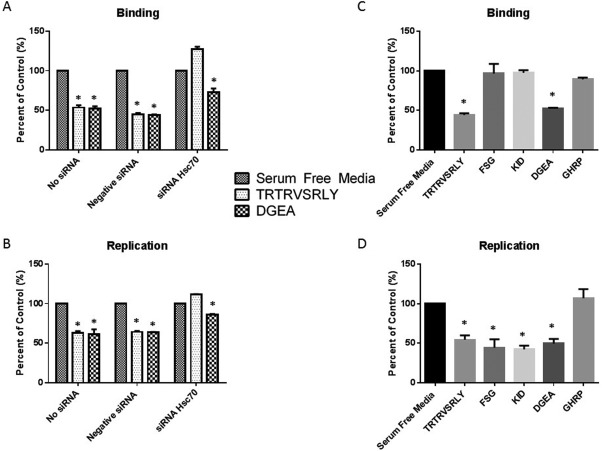
The role of Hsc70 in RRV binding and replication. (A, B) Hsc70 siRNA‐transfected mouse cholangiocytes treated with 1 mM TRTRVSRLY were unable to inhibit RRV binding or replication, whereas DGEA displayed significant inhibition versus serum‐free media in all treatments. (C) Cholangiocytes pretreated with synthetic 1 mM TRTRVSRLY, FSG, and KID were infected with RRV. A significant decrease in viral binding with TRTRVSRLY was seen, with no change in binding with FSG or KID. (D) Viral replication assays with synthetic peptides revealed an inhibition of viral replication in cholangiocytes treated with TRTRVSRLY, FSG, and KID. *P < 0.05 versus serum‐free media.
RRV replication was similarly assessed using negative, no siRNA, and Hsc70 siRNA. Once again, negative and no siRNA remained susceptible to both DGEA and synthetic TRTRVSRLY peptide viral blockade. Viral replication in Hsc70 siRNA–treated cells resulted in no change in viral yield for TRTRVSRLY peptide when normalized to serum‐free control. In the absence of Hsc70, viral binding and yield were lower in all treatment groups (Fig. 8B).
THE SRL PEPTIDE PLAYS A ROLE AS A PRIMARY BINDING SITE IN ROTAVIRUS ENTRY
Previous studies have shown that a secondary, post‐attachment interaction exists between some rotavirus strains and Hsc70. In these studies, the post‐attachment secondary interaction with Hsc70 was mediated by the aa sequence within VP4 spanning aa 531‐554, termed FSG (FSGIKSTIDAAKSMATNVMKKFKK)31 and 642‐658, termed KID (KTKIDRSTQISPNTLPD).32 In these experiments, MA104 cells treated with these peptides displayed reduced viral replication due to decreased viral entry.31, 32 We used the FSG and KID peptides to determine whether they were involved in primary Hsc70 binding and replication in cholangiocytes similarly to the SRL peptide. Blocking assays using the three different peptides demonstrated that only TRTRVSRLY was capable of significantly inhibiting primary viral binding (44.35%), whereas FSG and KID had no effect (Fig. 8C). In addition, all three peptides inhibited RRV replication (44.53%, 42.43%, and 54.50%). These results confirm that TRTRSVRLY blocks RRV primary attachment on cholangiocytes, resulting in reduced replication rates, whereas FSG and KID peptides had no effect on primary binding but reduced post‐attachment interaction, as reflected by reduced viral yield (Fig. 8D).
Discussion
BA is a devastating disease, affecting newborns in the first months of life. It is characterized by bile duct obstruction, liver injury, progressing to fibrosis and without treatment, death. Although the etiology of BA remains unclear, perinatal viral infection has been implicated in its pathogenesis.33 Several viruses, including rotavirus, may play a role in the activation of both inflammatory and autoimmune pathways.34 In the murine model of BA, RRV contributes to a release of pro‐inflammatory cytokines and immune cell activation.35, 36 Although the exact mechanism through which rotavirus plays a role has not yet been elucidated, current studies indicate that the virus targets biliary epithelium, leading to inflammation and extrahepatic biliary obstruction.12, 35
We have shown previously that the VP4 gene of RRV plays an integral role in the murine BA model induction through cholangiocyte infection22 and immune cell activation.23 In 1997, Coulson et al.15 discovered the collagen peptide‐binding sequence DGE on rotavirus. This was then determined to bind the α2β1 integrin on cholangiocytes. This receptor, used in primary rotavirus binding, was not found on hepatocytes or Cho cells, less susceptible lines, suggesting that other receptors may govern RRV infectivity.19
Using sequence and structure based modeling approaches, we determined three peptide sequences within the RRV VP4 protein that map into putative protein–protein binding sites. Through selective treatment with these peptides (PTAAGV, TRTRVSRLY, SSASAW), we determined that only TRTRVSRLY (aa 440‐448) was able to decrease RRV binding and subsequent replication in murine cholangiocytes along with MA104 cells, and that the cell type rotavirus replicates efficiently. This peptide was only capable of inhibiting RRV and GRV, two strains with homologous TRTRVSRLY sequences, both of which can induce murine BA. However, it was not capable of inhibiting binding of TUCH and Ro1845, viruses with dissimilar peptide sequences that cannot induce murine BA. This result suggests that TRTRVSRLY is an essential peptide that plays a role in RRV‐specific cellular binding. Within the TRTRVSRLY sequence, it was determined that the 3‐aa sequence SRL plays a critical role. This established an RRV‐specific peptide that governs binding and entry and is specific to rotavirus strains capable of inducing murine BA.15
To determine the role of the RRV TRTRVSRLY peptide in human BA, we performed similar experiments in human cholangiocyte cells (H69 cells). Through this analysis, we determined that the TRTRVSRLY peptide is also capable of diminishing both RRV and GRV binding and viral replication, both strains capable of inducing the murine model of BA. It is important to note that there has been human rotavirus strains isolated which contain the TRTRVSRLY sequence in their VP4 protein.37, 38 Unfortunately, these strains were not maintained and as a result, are not available for study.
Once we established the importance of the TRTRVSRLY peptide in the in vitro infection of cholangiocytes, we evaluated its function in the RRV‐induced in vivo model of BA. Through our investigations, we determined that the TRTRVSRLY peptide plays a role in the murine model of BA, lessening symptoms and mortality as well as resulting in lower viral titers in the bile ducts of mice.
Following determination of SRL specificity, we sought the corresponding cholangiocyte membrane receptor for this peptide. We found that in cholangiocytes, SRL attachment occurs through the Hsc70 protein. Hsc70 is a constitutively expressed chaperone within the Hsp70 family. This protein is involved in various cellular functions, including uncoating during clathrin‐mediated endocytosis,39 protein folding,40 protein degradation through ubiquitin‐proteasome degradation,41 and the import of proteins into organelles.42 This protein is also up‐regulated during inflammation, infection, and malignant processes.39 Interaction with antigen‐presenting cells through LRP1/CD91 (low density lipoprotein receptor‐related protein 1) may result in translocation of nuclear factor κB1 into the nucleus and dendritic cell maturation.43
Rotavirus cell entry has been shown to be a complex, multistep process with various cellular receptors, including integrins and Hsc70.44, 45 Hsc70 is present on the cell surface of multiple rotavirus‐susceptible and nonsusceptible cell types, including monkey MA104 and human Caco‐2 cells and is believed to be essential to infection in susceptible cells. When blocked with antibodies, the ability of rotavirus to infect the cell is diminished.30 The Hsc70 protein has been shown to be involved in secondary rotavirus binding through a post‐attachment step17, 30, 45 by way of the peptide sequences KID (642‐658) and FSG (531‐554).31, 32 The rotavirus capsid protein, composed of VP4 (later cleaved into VP5 and VP8) and VP7, is responsible for initial cell interaction. VP4 has been shown to play an essential role in cellular binding and attachment.45 Previous investigations of aa sequences within the VP4 protein by Zarate et al.32 provided knowledge that specific Hsc70 binding occurs through the KID (aa 642‐658) domain on VP5. Blocking this domain impedes viral replication, though not binding, suggesting that this is a secondary binding site. Similarly, Gualtero et al.31 showed the FSG peptide (aa 531‐554) within VP4 is implicated in Hsc70 binding and also plays a role in rotavirus replication, though not primary binding. Through our investigation of the TRTRVSRLY peptide, we determined that this unique peptide, unlike previously described peptides, interacts with Hsc70 during the primary binding phase of the VP4 domain of RRV, whereas the previously established peptides are involved in a post‐attachment step. Thus, blocking Hsc70 through siRNA had a significant effect in blocking RRV binding. Replication of RRV, however, was similar to that of previous reports and was inhibited not only by TRTRSVRLY but also the KID and FSG peptides.30, 32 Studies are underway to elucidate these differences.
Interestingly, through our analysis of the National Center for Biotechnology Information blastp database (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins&;PROGRAM=blastp&BLAST_PROGRAMS=blastp&;PAGE_TYPE=BlastSearch&DBSEARCH=true&;QUERY=&;SUBJECTS=), the presence of a VSRLY peptide was also found on other viruses including reovirus, cytomegalovirus, human papillomavirus, Epstein‐Barr virus, bluetongue virus, polyomavirus, coronavirus, respiratory syncytial virus, adenovirus, rodent paramyxovirus, cyprinid, canine picodicistrovirus, herpes simplex virus 1, torovirus, and chicken gallivirus 1. Several of these (cytomegalovirus, Epstein‐Barr virus, human papillomavirus, and reovirus) have been found in explanted livers of infants with BA.3, 4, 5, 6, 7 This aa sequence was found on viral attachment proteins in cytomegalovirus and reovirus. In Epstein‐Barr virus and human papillomavirus, it is also located on the viral capsid. Thus, this sequence may be involved in cholangiocyte binding in a similar fashion to the RRV VSRLY peptide. The role of Hsc70 binding and human BA induction as a function of these proteins has not yet been established.
In conclusion, our findings support the concept that the rotavirus binding and entry process is multifaceted and involves binding of several VP4 peptides in a primary and secondary fashion to Hsc70 and integrins. Further investigations are underway to identify the cellular response following interaction of the peptide/virus with Hsc70 to understand the pathogenesis of BA.
Supporting information
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep.28947/suppinfo.
Supporting Information
Acknowledgment
The sequence and structure analyses were performed with the help of the Cincinnati Children's Hospital Medical Center Protein Informatics Core and the University of Cincinnati Bioinformatics Core.
Potential conflict of interest: Mrs. McNeal received grants from Merck, GlaxoSmithKline, and Sanofi Pasteur.
This study was funded by National Institutes of Health grants R01 DK‐091566 and P30 DK‐078392.
REFERENCES
- 1. Balistreri WF, Grand R, Hoofnagle JH, Suchy FJ, Ryckman FC, Perlmutter DH, et al. Biliary atresia: current concepts and research directions. Summary of a symposium. Hepatology 1996;23:1682–1692. [DOI] [PubMed] [Google Scholar]
- 2. Landing BH. Considerations of the pathogenesis of neonatal hepatitis, biliary atresia and choledochal cyst—the concept of infantile obstructive cholangiopathy. Prog Pediatr Surg 1974;6:113–139. [PubMed] [Google Scholar]
- 3. Riepenhoff‐Talty M, Gouvea V, Evans MJ, Svensson L, Hoffenberg E, Sokol RJ, et al. Detection of group C rotavirus in infants with extrahepatic biliary atresia. J Infect Dis 1996;174:8–15. [DOI] [PubMed] [Google Scholar]
- 4. Glaser JH, Balistreri WF, Morecki R. Role of reovirus type 3 in persistent infantile cholestasis. J Pediatr 1984;105:912–915. [DOI] [PubMed] [Google Scholar]
- 5. Fjaer RB, Bruu AL, Nordbo SA. Extrahepatic bile duct atresia and viral involvement. Pediatr Transplant 2005;9:68–73. [DOI] [PubMed] [Google Scholar]
- 6. Domiati‐Saad R, Dawson DB, Margraf LR, Finegold MJ, Weinberg AG, Rogers BB. Cytomegalovirus and human herpesvirus 6, but not human papillomavirus, are present in neonatal giant cell hepatitis and extrahepatic biliary atresia. Pediatr Dev Pathol 2000;3:367–373. [DOI] [PubMed] [Google Scholar]
- 7. Drut R, Drut RM, Gomez MA, Cueto Rua E, Lojo MM. Presence of human papillomavirus in extrahepatic biliary atresia. J Pediatr Gastroenterol Nutr 1998;27:530–535. [DOI] [PubMed] [Google Scholar]
- 8. Petersen C, Grasshoff S, Luciano L. Diverse morphology of biliary atresia in an animal model. J Hepatol 1998;28:603–607. [DOI] [PubMed] [Google Scholar]
- 9. Riepenhoff‐Talty M, Schaekel K, Clark HF, Mueller W, Uhnoo I, Rossi T, et al. Group A rotaviruses produce extrahepatic biliary obstruction in orally inoculated newborn mice. Pediatr Res 1993;33:394–399. [DOI] [PubMed] [Google Scholar]
- 10. Czech‐Schmidt G, Verhagen W, Szavay P, Leonhardt J, Petersen C. Immunological gap in the infectious animal model for biliary atresia. J Surg Res 2001;101:62–67. [DOI] [PubMed] [Google Scholar]
- 11. Li Z, Baker ML, Jiang W, Estes MK, Prasad BV. Rotavirus architecture at subnanometer resolution. J Virol 2009;83:1754–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Allen SR, Jafri M, Donnelly B, McNeal M, Witte D, Bezerra J, et al. Effect of rotavirus strain on the murine model of biliary atresia. J Virol 2007;81:1671–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lopez S, Arias CF. Multistep entry of rotavirus into cells: a Versaillesque dance. Trends Microbiol 2004;12:271–278. [DOI] [PubMed] [Google Scholar]
- 14. Arias CF, Isa P, Guerrero CA, Mendez E, Zarate S, Lopez T, et al. Molecular biology of rotavirus cell entry. Arch Med Res 2002;33:356–361. [DOI] [PubMed] [Google Scholar]
- 15. Coulson BS, Londrigan SL, Lee DJ. Rotavirus contains integrin ligand sequences and a disintegrin‐like domain that are implicated in virus entry into cells. Proc Natl Acad Sci U S A 1997;94:5389–5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Graham KL, Halasz P, Tan Y, Hewish MJ, Takada Y, Mackow ER, et al. Integrin‐using rotaviruses bind alpha2beta1 integrin alpha2 I domain via VP4 DGE sequence and recognize alphaXbeta2 and alphaVbeta3 by using VP7 during cell entry. J Virol 2003;77:9969–9978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guerrero CA, Mendez E, Zarate S, Isa P, Lopez S, Arias CF. Integrin alpha(v)beta(3) mediates rotavirus cell entry. Proc Natl Acad Sci U S A 2000;97:14644–14649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hewish MJ, Takada Y, Coulson BS. Integrins alpha2beta1 and alpha4beta1 can mediate SA11 rotavirus attachment and entry into cells. J Virol 2000;74:228–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Londrigan SL, Graham KL, Takada Y, Halasz P, Coulson BS. Monkey rotavirus binding to alpha2beta1 integrin requires the alpha2 I domain and is facilitated by the homologous beta1 subunit. J Virol 2003;77:9486–9501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zarate S, Espinosa R, Romero P, Guerrero CA, Arias CF, Lopez S. Integrin alpha2beta1 mediates the cell attachment of the rotavirus neuraminidase‐resistant variant nar3. Virology 2000;278:50–54. [DOI] [PubMed] [Google Scholar]
- 21. Jafri M, Donnelly B, Allen S, Bondoc A, McNeal M, Rennert PD, et al. Cholangiocyte expression of alpha2beta1‐integrin confers susceptibility to rotavirus‐induced experimental biliary atresia. Am J Physiol Gastrointest Liver Physiol 2008;295:G16–G26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang W, Donnelly B, Bondoc A, Mohanty SK, McNeal M, Ward R, et al. The rhesus rotavirus gene encoding VP4 is a major determinant in the pathogenesis of biliary atresia in newborn mice. J Virol 2011;85:9069–9077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Walther A, Mohanty SK, Donnelly B, Coots A, Lages CS, Lobeck I, et al. Rhesus rotavirus Vp4 sequence‐specific activation of mononuclear cells is associated with cholangiopathy in murine biliary atresia. Am J Physiol Gastrointest Liver Physiol 2015;309:G466–G474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Coots A, Donnelly B, Mohanty SK, McNeal M, Sestak K, Tiao G. Rotavirus infection of human cholangiocytes parallels the murine model of biliary atresia. J Surg Res 2012;177:275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Porollo A, Meller J. Prediction‐based fingerprints of protein‐protein interactions. Proteins 2007;66:630–645. [DOI] [PubMed] [Google Scholar]
- 26. Mohanty SK, Donnelly B, Bondoc A, Jafri M, Walther A, Coots A, et al. Rotavirus replication in the cholangiocyte mediates the temporal dependence of murine biliary atresia. PLoS One 2013;8:e69069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Matsumoto Y, Shindo Y, Takakusagi Y, Takakusagi K, Tsukuda S, Kusayanagi T, et al. Screening of a library of T7 phage‐displayed peptides identifies alphaC helix in 14‐3‐3 protein as a CBP501‐binding site. Bioorg Med Chem 2011;19:7049–7056. [DOI] [PubMed] [Google Scholar]
- 28. Eismann T, Huber N, Shin T, Kuboki S, Galloway E, Wyder M, et al. Peroxiredoxin‐6 protects against mitochondrial dysfunction and liver injury during ischemia‐reperfusion in mice. Am J Physiol Gastrointest Liver Physiol 2009;296:G266–G274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Staatz WD, Fok KF, Zutter MM, Adams SP, Rodriguez BA, Santoro SA. Identification of a tetrapeptide recognition sequence for the alpha 2 beta 1 integrin in collagen. J Biol Chem 1991;266:7363–7367. [PubMed] [Google Scholar]
- 30. Guerrero CA, Bouyssounade D, Zarate S, Isa P, Lopez T, Espinosa R, et al. Heat shock cognate protein 70 is involved in rotavirus cell entry. J Virol 2002;76:4096–4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gualtero DF, Guzman F, Acosta O, Guerrero CA. Amino acid domains 280‐297 of VP6 and 531‐554 of VP4 are implicated in heat shock cognate protein hsc70‐mediated rotavirus infection. Arch Virol 2007;152:2183–2196. [DOI] [PubMed] [Google Scholar]
- 32. Zarate S, Cuadras MA, Espinosa R, Romero P, Juarez KO, Camacho‐Nuez M, et al. Interaction of rotaviruses with Hsc70 during cell entry is mediated by VP5. J Virol 2003;77:7254–7260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bezerra JA. Potential etiologies of biliary atresia. Pediatr Transplant 2005;9:646–651. [DOI] [PubMed] [Google Scholar]
- 34. Mack CL. The pathogenesis of biliary atresia: evidence for a virus‐induced autoimmune disease. Semin Liver Dis 2007;27:233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shivakumar P, Campbell KM, Sabla GE, Miethke A, Tiao G, McNeal MM, et al. Obstruction of extrahepatic bile ducts by lymphocytes is regulated by IFN‐gamma in experimental biliary atresia. J Clin Invest 2004;114:322–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Saxena V, Shivakumar P, Sabla G, Mourya R, Chougnet C, Bezerra JA. Dendritic cells regulate natural killer cell activation and epithelial injury in experimental biliary atresia. Sci Transl Med 2011;3:102ra194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Khamrin P, Maneekarn N, Peerakome S, Yagyu F, Okitsu S, Ushijima H. Molecular characterization of a rare G3P[3] human rotavirus reassortant strain reveals evidence for multiple human‐animal interspecies transmissions. J Med Virol 2006;78:986–994. [DOI] [PubMed] [Google Scholar]
- 38. Li K, Lin XD, Huang KY, Zhang B, Shi M, Guo WP, et al. Identification of novel and diverse rotaviruses in rodents and insectivores, and evidence of cross‐species transmission into humans. Virology 2016;494:168–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stricher F, Macri C, Ruff M, Muller S. HSPA8/HSC70 chaperone protein: structure, function, and chemical targeting. Autophagy 2013;9:1937–1954. [DOI] [PubMed] [Google Scholar]
- 40. Frydman J, Hartl FU. Principles of chaperone‐assisted protein folding: differences between in vitro and in vivo mechanisms. Science 1996;272:1497–1502. [DOI] [PubMed] [Google Scholar]
- 41. Ciechanover A. The ubiquitin‐proteasome pathway: on protein death and cell life. EMBO J 1998;17:7151–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Okuno Y, Imamoto N, Yoneda Y. 70‐kDa heat‐shock cognate protein colocalizes with karyophilic proteins into the nucleus during their transport in vitro . Exp Cell Res 1993;206:134–142. [DOI] [PubMed] [Google Scholar]
- 43. Srivastava P. Roles of heat‐shock proteins in innate and adaptive immunity. Nat Rev Immunol 2002;2:185–194. [DOI] [PubMed] [Google Scholar]
- 44. Lopez S, Arias CF. Early steps in rotavirus cell entry. Curr Top Microbiol Immunol 2006;309:39–66. [DOI] [PubMed] [Google Scholar]
- 45. Arias CF, Guerrero CA, Mendez E, Zarate S, Isa P, Espinosa R, et al. Early events of rotavirus infection: the search for the receptor(s). Novartis Found Symp 2001;238:47–60; discussion 60‐43. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep.28947/suppinfo.
Supporting Information