

Abstract
Cross-linking mass spectrometry (XL-MS) represents a recently popularized hybrid methodology for defining protein-protein interactions (PPIs) and analyzing structures of large protein assemblies. In particular, XL-MS strategies have been demonstrated to be effective in elucidating molecular details of PPIs at the peptide resolution, providing a complementary set of structural data that can be utilized to refine existing complex structures or direct de novo modeling of unknown protein structures. To study structural and interaction dynamics of protein complexes, quantitative cross-linking mass spectrometry (QXL-MS) strategies based on isotope-labeled cross-linkers have been developed. Although successful, these approaches are mostly limited to pair-wise comparisons. In order to establish a robust workflow enabling comparative analysis of multiple cross-linked samples simultaneously, we have developed a multiplexed QXL-MS strategy, namely QMIX (Quantitation of Multiplexed, Isobaric-labeled cross (X)-linked peptides) by integrating MS-cleavable cross-linkers with isobaric labeling reagents. This study has established a new analytical platform for quantitative analysis of cross-linked peptides, which can be directly applied for multiplexed comparisons of the conformational dynamics of protein complexes and protein-protein interactions at the proteome scale in future studies.
TOC image
INTRODUCTION
Protein-protein interactions (PPIs) are fundamental to the assembly, structure, and function of protein complexes. Disturbances in endogenous PPIs can negatively impact cellular activities, leading to various types of human disease. Characterization of the architectures of protein complexes and their protein interaction interfaces is critical towards unraveling the molecular mechanisms underlying human pathologies and providing insight on potential targets for drug therapies, exemplifying a new paradigm in disease treatment development1.
Due to its versatility, sensitivity, accuracy and speed, cross-linking mass spectrometry (XL-MS) has emerged as a powerful approach for mapping protein interaction networks2–7 and characterizing large protein complex structures8–14. The cross-linked peptides identified within and between proteins represent “peptide resolution” distance constraints that have been successfully used to derive and/or refine the structures of protein complexes8–14. One of the inherent challenges in XL-MS studies is the unambiguous identification of cross-linked peptides. To facilitate this process, we previously developed a new class of MS-cleavable cross-linkers, i.e. sulfoxide-containing MS-cleavable cross-linking reagents, that enable simplified and accurate identification of cross-linked peptides using multistage tandem mass spectrometry (MSn)4,15–17. These robust and reliable reagents have been successfully applied to define protein-protein interactions both in vitro9,16,18–20 and in vivo4. To probe structural dynamics of protein complexes, stable isotope-labeled cross-linking reagents (usually deuterium labeled) are often used to permit comparisons of two selected conformational states in a single experiment18,21–23. To allow unambiguous identification and quantification of cross-linked peptides simultaneously, we had developed a pair of sulfoxide-containing MS-cleavable, deuterium-labeled cross-linking reagents allowing quantitative comparison of cross-links identified by either light- or heavy-labeled reagent17,18. Although successful, the usage of isotope-labeled cross-linking reagents is currently limited to binary comparisons. In addition, deuterated and non-deuterated cross-linked peptides do not co-elute together perfectly, making their automated quantitation challenging17,24. Finally, the synthesis of stable isotope-labeled cross-linkers can be burdensome. Recently, SILAC-based quantitation has been incorporated with non-labeled cross-linkers for quantitative comparison of cross-linked peptides through labeling of targeting amino acids such as lysine25, thus eliminating the need for stable isotope-labeled cross-linkers. In contrast to deuterium-labeled cross-linking reagents, 13C/12C and 15N/14N labeled amino acids are typically used for SILAC labeling, which leads to labeled peptides co-eluting chromatographically. Although SILAC-based methods can be implemented for three-way comparisons, it is best suited for binary comparisons due to the limited variety of isotope-labeled amino acids that can produce sufficient mass differences among compared peptides. Regardless, when stable isotope labels are introduced through labeled amino acids in SILAC experiments or isotope-coded cross-linkers, quantitation is carried out based on differentially labeled peptides detected at the MS1 level, thus increasing sample complexity and decreasing the detection of low abundance cross-linked peptides. Therefore, it remains technically challenging to compare more than two cross-linked samples simultaneously, especially for very complex samples such as the proteome.
In recent years, isobaric labeling strategies including isobaric tags for relative and absolute quantification (iTRAQ)26 and tandem mass tags (TMT)27,28 have emerged as powerful quantitation methods for proteomics due to their unique multiplexing capability. Currently, commercially available TMT reagents have multiplexing capacity up to 10-plex, and have been widely used for various applications including proteome wide expression profiling29. Isobaric labeling-based multiplexed quantitation methods permit the parallel analysis of multiple proteome experiments, significantly increasing throughput without changing sample complexity. This is due to the fact that isobaric labeled peptides from compared samples carry the same m/z values and are measured as one mass spectral peak during MS1 analysis. Peptide/protein quantitation is achieved through the detection of unique reporter ions resulting from the fragmentation of isobaric labels at the MS2 level. However, it has been reported that quantitation accuracy and precision from such experiments are often compromised due to contaminating near-isobaric ions being isolated and fragmented together with the target ions, thus skewing reporter ion intensities30. Such peptide quantitation interference often results in underestimation of quantitative changes among compared samples, which can be effectively eliminated using triple-stage mass spectrometry (MS3)30. Recent advancements in instrumentation and software have ushered the development of more accurate and reproducible workflows, such as MS3-level synchronous precursor selection (SPS) to increase TMT reporter ion detection while minimizing reporter ion ratio distortion31,32. Given the fact that MSn analysis has been successfully implemented for unambiguous identification of peptides cross-linked by MS-cleavable cross-linking reagents4,16,17, we hypothesize that isobaric reagents can be perfectly integrated with such XL-MSn workflows, thus allowing us to establish a novel multiplexed quantitative XL-MS strategy for comparing multiple cross-linked samples simultaneously. To test this, we have coupled our previously developed MS-cleavable cross-linking reagent disuccinimidyl sulfoxide (DSSO) with the isobaric Tandem Mass Tag™ duplex (TMT2) labeling reagents for comparative cross-linking analysis using a model protein. This combinatory approach represents the first report on isobaric reagent-based quantitative cross-linking mass spectrometry. The results presented here demonstrate the feasibility of the proposed method and its potential for multiplexed quantitative XL-MS analysis to dissect protein structural and interaction dynamics at the protein complex and the proteome-wide scale in the future.
EXPERIMENTAL PROCEDURES
Materials and Reagents
General chemicals were purchased from Fisher Scientific or VWR International, bovine heart cytochrome c (98% purity) from Sigma-Aldrich. Tandem Mass Tag™ reagents purchased from Life Technologies (Thermo Fisher Scientific).
DSSO Cross-linking of Cytochrome c
200 μM cytochrome c in PBS buffer (pH 7.4) was reacted with DSSO in a molar ratio of 1:5 (protein: cross-linker) for 1 h at room temperature and quenched with excess hydroxylamine. Cross-linked proteins were then pelleted via TCA precipitation and re-suspended in 8 M urea. Re-suspended proteins were reduced with 15 mM TCEP for 30 min and alkylated with 30 mM chloroacetamide for 45 min in dark, and then diluted to 5 M urea. Cross-linked proteins were then digested with Lys-C for 4 h at 37° followed by dilution to 1.5 M urea and digestion by trypsin at 37° overnight. The resulting peptide mixtures were de-salted using Waters C18 Sep-Pak cartridges and fractionated by peptide size exclusion (SEC) as previously described by Leitner et al.10. The SEC fractions containing cross-linked peptides were used for subsequent TMT labeling and LC-MSn analysis.
TMT2 labeling of Cross-linked Cytochrome C Peptides
Approximately 80 μg of cross-linked cytochrome c peptides were used for TMT labeling. Peptides were diluted to 100 μL using 50 mM TEAB (triethyl ammonium bicarbonate) and split into equivalent 50 μL aliquots. To each aliquot was added 20 μL of 20 μg/μL of TMT2-126 or TMT2-127 isobaric labeling reagent in anhydrous ACN and incubated for 1 h at room temperature. 5% hydroxylamine was added to each sample to a final concentration of 0.25% and incubated for 15 min to quench the labeling reaction. Samples were cleaned and de-salted again using Waters C18 Sep-PAK cartridges and concentrated. Prior to LC-MSn analysis, TMT2-126 and TMT2-127 labeled peptides were mixed at five pre-determined molar ratios (10:1, 5:1, 1:1, 1:5, and 1:10).
Liquid Chromatography-Multistage Tandem Mass Spectrometric (LC-MSn) Analysis
Mixed peptide samples were analyzed utilizing a Thermo Scientific™ EASY-nLC™ 1000 UPLC system coupled on-line to a Thermo Scientific™ Orbitrap Fusion Lumos™ MS. A Thermo Scientific™ EASY-Spray™ source with a 25 cm × 75 μm PepMap EASY-Spray Column was used to separate peptides over a 55 min acetonitrile gradient of 6% to 35% at a flow rate of 300 nL/min. Each mixed peptide sample was analyzed using three individual acquisition methods: 1) a targeted ID-MS3 acquisition optimized for DSSO cross-linked peptide identification, 2) a MultiNotch MS3 acquisition featuring synchronous precursor selection (SPS)31, and 3) a combined ID-MS3 targeted acquisition with additional SPS-MS3 for all precursor ions selected for ID-MS3. For methods 1 and 3, mass-difference-dependent HCD-MS3 acquisitions were triggered if a unique mass difference (Δ=31.9721) was observed between fragment ions in the CID-MS2 spectrum. MS1 acquisition was performed in top speed mode with a cycle time of 5 s. MS1 and MS2 scans were acquired in the Orbitrap whereas MS3 scans were detected in the ion trap. For MS1 scans, the scan range was set from 375 to 1600 m/z, resolution set to 120,000, and the AGC target set to 4×105. For MS2 scans, the resolution was set to 30,000, the AGC target was set to 5e4, the precursor isolation width was 1.6 m/z, and the maximum injection time was 100 ms for CID. The CID-MS2 normalized collision energy was 25%. For MS3 scans, HCD was used with a collision energy of 35%, the AGC target was set to 2×104, and the maximum injection time was set to 120 ms. For methods 2 and 3 containing SPS-MS3, the AGC target was set up to 5e4, with MS1 isolation window to 1.6 m/z and MS2 isolation window to 2 m/z and 10 notches. The maximum injection time was set to 105 ms and resolution to 30,000.
Identification and Quantitation of TMT2 Labeled DSSO Cross-linked Peptides
Monoisotopic masses and charges of parent ions and corresponding fragment ions, and ion intensities from cross-linker and peptide fragmentation in ID-MS3 spectra were extracted as MGF files using ProteoWizard MSConvert. MS3 spectra were subjected to protein database searching using a developmental version of Protein Prospector (v. 5.17.0) using Batch-Tag against cytochrome c (SwissProt accession #: P62894) with mass tolerances for parent ions and fragment ions set as ± 20 ppm and 0.6 Da respectively. Trypsin was set as the enzyme with four maximum missed cleavages allowed. Cysteine carbamidomethylation was selected as a constant modification, while protein N-terminal acetylation, methionine oxidation, N-terminal conversion of glutamine to pyroglutamic acid, and asparagine deamidation were selected as variable modifications. In addition, four defined modifications on uncleaved lysines and free protein N-termini were selected: alkene (A: C3H2O, +54 Da), sulfenic acid (S: C3H4O2S, +104 Da), and unsaturated thiol (T: C3H2OS, +86 Da) modifications due to remnant moieties for DSSO, as well as a single modification for TMT2 labeling (+225 Da). Initial acceptance criteria for peptide identification required a reported expectation value ≤ 0.1.
The in-house software xl-Discoverer, designed to validate and summarize cross-linked peptides based on MSn data and database searching, was used to automatically generate and summarize identified cross-linked peptide pairs15. Peak intensities for TMT2-126 and TMT2-127 reporter ions were extracted directly from Lumos™ RAW files to obtain final TMT ratios after considering isotope purities of each isobaric reagent as instructed in the manufacturer’s protocol27.
RESULTS AND DISCUSSION
Development of A New Multiplexed QXL-MS Strategy
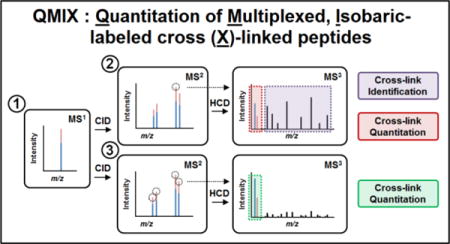
In order to increase throughput and facilitate the simultaneous quantitative analysis of differential protein complex topologies under multiple conditions, we have developed a novel multiplexed QXL-MS strategy called QMIX (Quantitation of Multiplexed, Isobaric-labeled cross (X)-linked peptides), which integrates our MS-cleavable cross-linking reagent-based XL-MSn workflow with isobaric label-based multiplexed quantitation (Figure 1). This strategy is established due to the fact that the identification of peptides cross-linked by MS-cleavable reagents is best achieved through MSn analysis4,16,17. Coincidentally, MSn analysis is most advantageous for multiplexed protein quantitation when using isobaric reagents due to peptide interference30,31. In theory, the QMIX strategy can be accomplished by combining any effective MS-cleavable cross-linker (e.g. sulfoxide-containing MS-cleavable reagents) with any types of isobaric reagents. To demonstrate the feasibility of our multiplexed quantitative strategy for cross-linked peptides, we have employed a sulfoxide-containing amine reactive cross-linker, disuccinimidyl sulfoxide (DSSO) and TMT2 labeling reagents. The DSSO-based XL-MSn workflow has been demonstrated to be effective and robust for fast and unambiguous identification of cross-linked peptides9,16, while TMT reagents have been widely and successfully used for multiplexing protein quantitation including proteomes and phosphoproteomes32,33. In contrast to previously reported QXL-MS strategies that rely on isotope-coded cross-linkers or SILAC-labeled lysines18,21,22,25, our proposed QMIX strategy has a unique multiplexing capability that enables simultaneous quantitation of multiple cross-linked peptides in a similar manner to multiplexing quantitation of non-cross-linked peptides. The maximum number of cross-linked samples that can be concurrently compared would be limited only by the number of available isobaric tags. Since TMT labeling reagents are isobaric and structurally identical, differentially labeled cross-linked peptides co-elute simultaneously in the LC chromatogram and are measured as a single peak in the survey MS1 scan, similar to TMT-labeled non-cross-linked peptides. Thus, in contrast to other types of isotope label-based QXL-MS strategies18,22,23,25, isobaric labeling does not increase sample complexity. In fact, the signal intensities of the same cross-linked peptides are augmented even if they are contributed through multiple labeled samples, thereby increasing the detectability of low abundance cross-linked peptides for their identification and quantitation. Moreover, TMT labeling would allow all types of peptides to be quantified at the same time, thus enabling thorough comparison of samples at different levels. Finally, the ability of multiplexing would permit the analysis of multiple compared samples in a single run to significantly improve throughput and provide the flexibility of performing biological replicates concomitantly. Collectively, QMIX represents a general strategy that is much more versatile and flexible than any existing QXL-MS strategies.
Figure 1. The general MSn analysis workflow for identifying and quantifying TMT-labeled, DSSO cross-linked peptides.
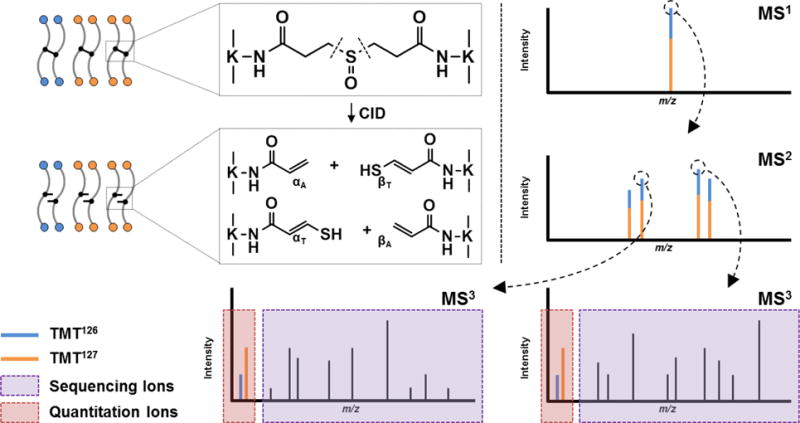
Fragment ions from MS2 are selected for subsequent HCD analysis in MS3, releasing both b and y ions for sequencing, as well as TMT reporter ions for quantitation.
Fragmentation of TMT-labeled, DSSO-cross-linked peptides
Incubation of TMT2 reagent with DSSO cross-linked peptides results in the covalent labeling of non-cross-linked lysine residues, as well as free N-terminal primary amines generated through enzymatic digestion. We found that the complete labeling of all available primary amines of cross-linked peptides was able to be achieved based on the instructions provided for labeling non-cross-linked peptides. Cross-linked lysine residues were unaffected by TMT labeling reagent, nor did they prevent the efficient labeling of nearby residues. Unambiguous identification of DSSO cross-linked peptides is accomplished through MSn analysis as previously described16. Because the MS-cleavable C-S bonds adjacent to the sulfoxide within the linker spacer region are significantly more labile than the amide bonds of the peptide backbone, MS2 fragmentation of a DSSO inter-linked peptide during collision induced dissociation leads to the physical separation of the two covalently linked peptides into single peptide chains. These MS2 fragments can then be subjected to subsequent MS3 for peptide sequencing. Along with mass fingerprinting of MS1 precursor ions and the characteristic fragmentation of cross-linked peptides in MS2, the sequences of individual peptides determined by MS3 are integrated to confidently determine the identities of cross-linked peptides. To evaluate whether TMT labeling interferes with the MSn analysis of DSSO cross-linked peptides, cytochrome c was cross-linked with DSSO, and digested prior to TMT labeling. The resulting TMT-labeled peptides were then analyzed by LC-MSn. As an example, a TMT2-labeled DSSO inter-linked peptide α-β (m/z 1050.07034+) detected in MS1 (Figure 2A) yielded two pairs of dominant fragment ions αA/βT (m/z 972.072+/m/z 1118.072+) and αT/βA (m/z 988.062+/m/z 1102.062+) during MS2 analysis (Figure 2B). This is expected, as the cleavage of one of the two MS-cleavable C-S bonds in a DSSO inter-linked heterodimeric peptide α-β would result in the observance of two predictive fragment pairs αA/βS or αS/βA that carry complementary alkene (A, +54.01 Da) or sulfenic acid (S, +103.99 Da) cross-linker remnant moieties16. The sulfenic acid moiety often undergoes dehydration to form a more stable unsaturated thiol moiety (T, +85.98 Da), generating dominant αA/βT or αT/βA peptide ion pairs in the MS2 spectra. MS3 sequencing of the αT and βA fragment ion pair yielded series of b and y ions that unambiguously identified them as 75Y*IPGTKTMIFAGIK*87, in which K80 was modified by a saturated thiol moiety, and 41T*GQAPGFSYTDANKANK*56, in which K54 was modified by the alkene moiety (Figures 2C and 2D). In addition, MS3 sequencing confirmed that both peptides were fully labeled by TMT2 reagent on free primary amines at their N-termini and C-terminal lysine residues. Along with mass fingerprinting of the MS1 precursor and its fragmentation pattern in MS2, the linkage was determined between K54 and K80 of cytochrome c (Figure 2). Collectively, our results indicate that TMT labeling does not interfere with the characteristic fragmentation of DSSO cross-linked peptides during MS2 analysis and their subsequent MS3 sequencing for unambiguous identification16.
Figure 2. MSn analysis of a selected TMT-labeled, DSSO cross-linked cytochrome c peptide.
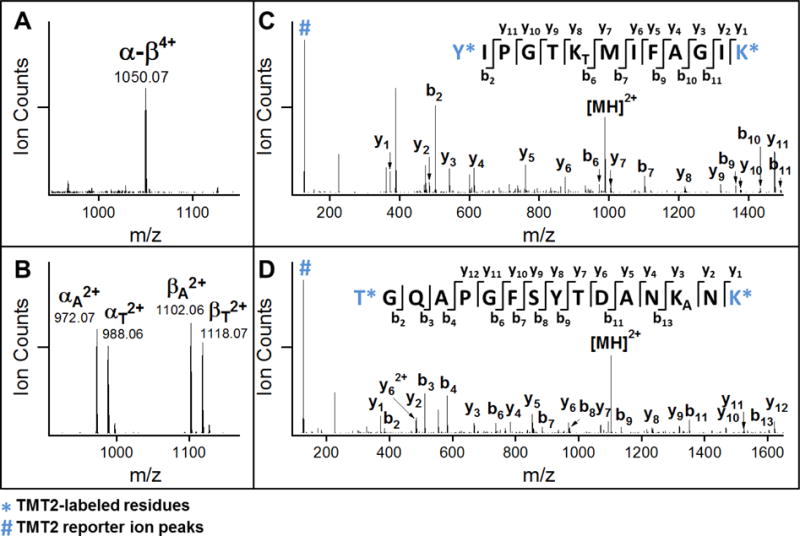
(A) MS1 spectrum of the TMT-labeled, DSSO cross-linked peptide α-β (m/z 1050.07034+). (B) MS2 spectrum of α-β, in which four dominant fragment ions were detected: αA2+/βT2+ and αT2+/βA2+. (C–D) MS3 spectra of MS2 fragment ions αT2+ (m/z 988.062+) and βA2+ (m/z 1102.062+), which were identified as 75Y*IPGTKTMIFAGIK*87 and 41T*GQAPGFSYTDANKANK*56 respectively and unambiguously confirming a fully TMT-labeled cross-link between K54 and K80 of cytochrome c. Note: * indicates the TMT2-labeled amino acids, KA: alkene modified lysine, and KT: unsaturated thiol modified lysine.
MSn Analysis of TMT-labeled Cytochrome c Cross-linked Peptides
Previously, data-dependent MSn acquisition methods were used to identify DSSO cross-linked peptides using an Orbitrap XL mass spectrometer, in which the top 3 MS2 fragment ions were often selected for MS3 sequencing4,16,18. This is based on the fact that DSSO inter-linked heterodimeric peptides (α-β) produce four dominant fragment pairs: αA/βT and αT/βA as illustrated in Figure 2B. Therefore, MS3 sequencing of the three most intense MS2 fragment ions is typically sufficient for the identification of DSSO cross-linked peptides. In this work, we have employed the Orbitrap Fusion™ Lumos™ Tribrid™ mass spectrometer as the Orbitrap XL does not have the capability of performing isobaric labeling-based quantitation due to poor sensitivity in HCD. In comparison, the Lumos™ not only has superior sensitivity, resolution, scanning rate and dynamic range, but also has multiple fragmentation techniques (CID, HCD, ETD and EThcD) and the flexibility of integrating them at any stage of MSn analysis. Therefore, we sought out the possibility of performing a targeted MSn method in which alkene- and thiol-modified fragment ion pairs of the same sequence (i.e. αA/αT or βA/βT) would be first identified on the fly based on their defined mass differences (i.e. Δ(αT − αA) or Δ(βT − βA)), which is equal to the mass of a sulfur atom, 31.9721 Da. The top 2 pairs, αA/αT and βA/βT fragment ions would then be selected for subsequent MS3 analysis. This approach potentially enables all of the four predicted MS2 fragment ions to be sequenced in a selective manner, thus increasing throughput and identification confidence. During our initial assessment, we found that this targeted MSn analysis appears to be more effective than conventional Top N data-dependent MSn method for these samples. Therefore, we have employed the targeted MSn acquisition method in this work, which resulted in the identification of a redundant total of 652 cytochrome c cross-linked peptides representing 79 unique K-K linkages across 5 samples of TMT2-labeled peptides mixed at known concentrations (Table S-1).
Quantitation of TMT-labeled, DSSO cross-linked peptides
Next, we investigated whether DSSO cross-linked peptides can be effectively quantified using TMT labeling and MSn analysis. To test this, we have labeled cross-linked cytochrome c peptides with TMT2-126 and TMT2-127 respectively, and then mixed them in 5 known ratios, i.e. 10:1, 5:1, 1:1, 1:5; 1:10. To quantify the relative abundances of TMT2-126 and TMT2-127 labeled cross-linked peptides, we have examined three different data acquisition methods as illustrated in Figure 3: 1) ID-MS3, 2) SPS-MS3, and 3) ID-SPS-MS3 (Figures 3A–C). It is noted that method 3 combines the features of methods 1 and 2. The first method ID-MS3 involves direct MS3 analysis in HCD that enables the detection of sequence ions for peptide identification and TMT reporter ions for quantitation simultaneously. Each MS3 spectrum of the selected MS2 fragment ions contributes TMT reporter ions for quantitation. For instance, Figure 4A illustrates the MS3 spectrum of the αA (m/z 599.702+) fragment of an inter-linked peptide of cytochrome c [7G*K*KIFVQK*14 (α) inter-linked to 40K*TGQAPGFSYTDANK*54 (β)] from the 1:1 sample, in which K9 cross-linked to K40. As shown, the two TMT2 reporter ions (m/z 126 and 127) were detected and used for ratio determination. Due to the abundance of natural 13C isotopes and impurities in TMT2 isobaric labeling reagents, a percentage of TMT2-126 contributes to TMT2-127 reporter ion detection, which must be corrected as previously described27. Based on reporter ions detected in MS3 spectra for each cross-linked peptide, their TMT ratios (126:127) were first calculated, which were then used to obtain the final average ratios for each sample. As a result, their average TMT ratios for the five premixed samples were determined as 11.64, 5.74, 1.11, 0.22, and 0.11 respectively (Figure 4C), correlating well with the expected ratios. The results suggest that it is feasible to identify and quantify TMT labeled and DSSO cross-linked peptides simultaneously using the ID-MS3 method, i.e. normal MSn analysis. With this method, the ratio deviations in the five selected samples vary from 14% ~ 35%. The larger variations appear to be particularly associated with samples containing more TMT2-126 relative to TMT2-127 (e.g. 10:1). This is more likely due to the fact that the observed reporter ions (m/z 126 and 127) are much smaller in abundance than sequence ions using the ID-MS3 method (Figure 4A) and different amounts of isotope impurities in TMT2-126 and TMT2-127 reagents contribute to ratio correction. Thus, the relative quantitation of cross-links in these situations could potentially be compromised.
Figure 3. Three MSn acquisition methods utilized for analyzing TMT-labeled, DSSO cross-linked peptides.
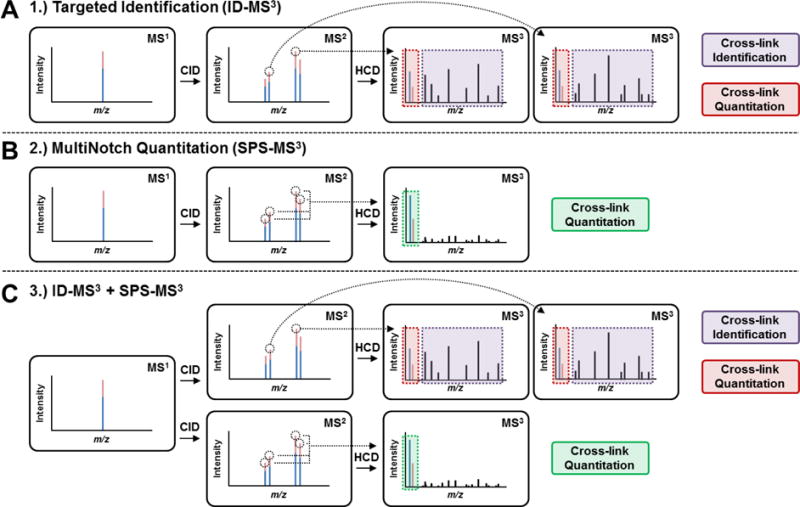
(A) Method 1: targeted identification method, ID-MS3. (B) Method 2: MultiNotch quantitation using synchronous precursor selection (SPS), SPS-MS3. (C) Method 3: combined acquisition method consisting of ID-MS3 with SPS-MS3, ID-SPS-MS3.
Figure 4. Quantitation of TMT-labeled, DSSO cross-linked peptides from the five premixed samples.
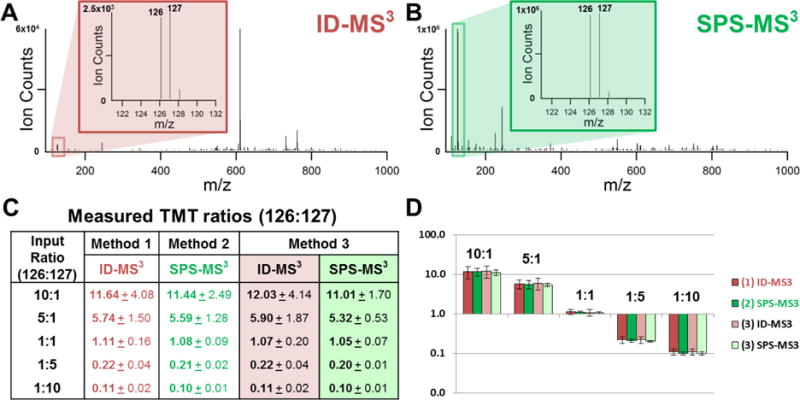
(A) MS3 spectrum of the αA (m/z 599.702+) of the cross-linked peptide [7G*K*KIFVQK*14 (α) and 40K*TGQAPGFSYTDANK*54 (β)] (m/z 763.83645+). αA sequence was determined as 7G*K*KAIFVQK*14. Inset illustrates the two report ions. (B) SPS-MS3 spectrum of the same cross-linked peptide (m/z 763.83645+) described in (A). Inset illustrates the two report ions. (C) Average TMT ratios (126:127) for the five selected samples using the three acquisition methods. (D) Corresponding plot of average TMT ratios obtained using the three acquisition methods from the five premixed samples.
To improve the accuracy in TMT-based quantitation, a MultiNotch-based SPS-MS3 acquisition method has been previously developed to allow the integration of multiple MS3 signals from up to 10 MS2 fragment ions, significantly boosting up the relative intensities of TMT reporter ions for quantitation31. Therefore, we employed a similar SPS-MS3 acquisition method to evaluate its feasibility for quantitation of cross-linked peptides (Figure 3B). However, this type of experiment can only acquire quantitative information, and the correlated peptide identification has to be done from a separate experiment using the ID-MS3 method (Figure 3A). In comparison to Figure 4A, Figure 4B illustrates the SPS-MS3 spectrum of the same cross-linked peptide from the 1:1 sample. As shown, the intensities of TMT report ions (m/z 126 and 127) are significantly enhanced, which are more than 100 times higher than those obtained using the ID-MS3 method (Figure 4A). Using the entirety of the SPS-MS3 spectra without considering peptide identity, the average TMT ratios for the five samples were determined as 11.44, 5.59, 1.08, 0.21 and 0.10 respectively (Figure 4C), corroborating very well with the expected values. Importantly, there was a significant decrease in experimental variation compared to ID-MS3 acquisition, demonstrating that quantitation via MultiNotch MS3 acquisition is indeed much more accurate.
To enable the identification and quantification of TMT-labeled, DSSO cross-linked peptides simultaneously with better accuracy, we employed an acquisition method, ID-SPS-MS3, utilizing both ID-MS3 and SPS-MS3 for each precursor ion selected for identification and quantification. In comparison to the 652 redundant cross-linked peptides identified from targeted ID-MS3 analyses, 600 redundant cytochrome c cross-linked peptides were identified from the same samples using ID-SPS-MS3 acquisition, representing 79 and 70 unique K-K linkages (Table S-1), respectively. This result suggests that the overall increase in duty cycle during the ID-SPS-MS3 experiment does not significantly impact the total number of identified cross-links. To compare, we have calculated TMT ratios (126:127) of each cross-link obtained from ID-MS3 and SPS-MS3 spectra resulted from ID-SPS-MS3 acquisition. The respective average ratios for the five pre-mixed samples are summarized in Figure 4C and plotted in Figures 4D and S-1. As shown, the average ratios determined using the two different acquisition methods in the same experiment are similar to those obtained in the two separate experiments as described above. This demonstrates that it is feasible to use standard MS3 identification methods to simultaneously identify and quantify DSSO cross-linked peptides. However, SPS-MS3 method indeed permits TMT-based quantitation of cross-linked peptides with much better accuracy and less variation (Figure 4C). This was evidenced by the tight clustering of individual reporter ion ratios from SPS-MS3 analysis around the average ion ratios, which resulted in lower standard deviations compared to those obtained from ID-MS3 (Figure S-1). This observation is consistent with significantly increased reporter ion signals from multiple MS2 ions in SPS-MS3 experiments, thereby increasing quantitation accuracy in general. Importantly, the results have shown the effectiveness of integrating ID-MS3 and SPS-MS3 method, thus permitting simultaneous identification and quantitation of DSSO cross-linked peptides, and enabling automated multiplexing quantitative analysis of cross-linked peptides. Collectively, these results have demonstrated the capability of quantifying cross-linked peptides using the QMIX approach, combining isobaric, MS-cleavable cross-linking reagents and multistage mass spectrometry.
CONCLUSION
Here we present a novel analytical platform, QMIX, integrating isobaric labeling with MS-cleavable cross-linking reagents for the identification and multiplexing quantitation of cross-linked peptides simultaneously using MSn analysis. The incorporation of isobaric tags enables multiplexed quantitation of cross-linked peptides in a scope that cannot be easily achieved by any existing stable isotope labeling based quantitative mass spectrometry. In addition, this general strategy is compatible with all cross-linking reagents regardless of their residue-targeting chemistries, or chemical functionalities. Although isobaric labeling in theory can be applied to conventional non-cleavable cross-linkers based XL-MS strategies, coupling this multiplexing strategy with MS-cleavable cross-linking reagents is the best combination due to simplified and accurate identification of cross-linked peptides using MSn analysis offered by MS-cleavable cross-linking reagents. With the QMIX strategy, the quantitation of cross-linked peptides is achieved at the MS3 level, thus eliminating peptide quantitation interference as commonly observed at the MS1 level using isotope-coded cross-linkers or targeting residues. Although MS3 sensitivity is much lower than MS1 and MS2, the ultrahigh sensitivity in MSn analysis provided by advanced instrumentation such as the Lumos™ mass spectrometer makes this strategy practical. Therefore, any new developments in isobaric labeling for quantitative proteomics, such as Dileu reagents34, can also be potentially employed to increase multiplexing ability of quantifying cross-linked peptides in future studies. In summary, this work represents a proof-of-principle of the QMIX strategy and establishes a solid foundation for future studies toward multiplexed comparison of protein complex conformational dynamics under various biological conditions. This will not only increase experimental throughput, but also advance our capability in QXL-MS studies beyond pair-wise comparisons.
Supplementary Material
Acknowledgments
We thank Drs. A.L. Burlingame and Robert Chalkley for the developmental version of Protein Prospector. This work was supported by National Institutes of Health grants RO1GM074830 to L.H, and R01GM106003 to L.H. and S. R.. E.J.N. was supported by an institutional Chemical and Structural Biology Training Grant predoctoral fellowship (T32-GM10856).
ABBREVIATIONS
- PPIs
protein-protein interactions
- XL-MS
cross-linking mass spectrometry
- QXL-MS
quantitative cross-linking mass spectrometry
- QMIX
quantitation of multiplexed, isobaric-labeled cross (X)-linked peptides
- DSSO
disuccinimidyl sulfoxide
- TMT
Tandem Mass Tag™ reagents
- MS
mass spectrometry
- MSn
multi-stage tandem mass spectrometry
- CID
collision induced dissociation
- HCD
higher energy collisional dissociation
- LC-MSn
liquid chromatography multistage tandem mass spectrometry
- XIC
extracted ion chromatogram
References
- 1.Wells JA, McClendon CL. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 2.Guerrero C, Tagwerker C, Kaiser P, Huang L. Mol Cell Proteomics. 2006;5:366–378. doi: 10.1074/mcp.M500303-MCP200. [DOI] [PubMed] [Google Scholar]
- 3.Chavez JD, Weisbrod CR, Zheng C, Eng JK, Bruce JE. Mol Cell Proteomics. 2013;12:1451–1467. doi: 10.1074/mcp.M112.024497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaake RM, Wang X, Burke A, Yu C, Kandur W, Yang Y, Novtisky EJ, Second T, Duan J, Kao A, Guan S, Vellucci D, Rychnovsky SD, Huang L. Mol Cell Proteomics. 2014;13:3533–3543. doi: 10.1074/mcp.M114.042630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu C, Yang Y, Wang X, Guan S, Fang L, Liu F, Walters KJ, Kaiser P, Huang L. Mol Cell Proteomics. 2016 doi: 10.1074/mcp.M116.058271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haupl B, Ihling CH, Sinz A. J Proteome Res. 2016 doi: 10.1021/acs.jproteome.6b00513. [DOI] [PubMed] [Google Scholar]
- 7.Tan D, Li Q, Zhang MJ, Liu C, Ma C, Zhang P, Ding YH, Fan SB, Tao L, Yang B, Li X, Ma S, Liu J, Feng B, Liu X, Wang HW, He SM, Gao N, Ye K, Dong MQ, Lei X. eLife. 2016;5 doi: 10.7554/eLife.12509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herzog F, Kahraman A, Boehringer D, Mak R, Bracher A, Walzthoeni T, Leitner A, Beck M, Hartl FU, Ban N, Malmstrom L, Aebersold R. Science. 2012;337:1348–1352. doi: 10.1126/science.1221483. [DOI] [PubMed] [Google Scholar]
- 9.Kao A, Randall A, Yang Y, Patel VR, Kandur W, Guan S, Rychnovsky SD, Baldi P, Huang L. Mol Cell Proteomics. 2012;11:1566–1577. doi: 10.1074/mcp.M112.018374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leitner A, Joachimiak LA, Bracher A, Monkemeyer L, Walzthoeni T, Chen B, Pechmann S, Holmes S, Cong Y, Ma B, Ludtke S, Chiu W, Hartl FU, Aebersold R, Frydman J. Structure. 2012;20:814–825. doi: 10.1016/j.str.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi Y, Fernandez-Martinez J, Tjioe E, Pellarin R, Kim SJ, Williams R, Schneidman D, Sali A, Rout MP, Chait BT. Mol Cell Proteomics. 2014;13:2927–2943. doi: 10.1074/mcp.M114.041673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeng-Elmore X, Gao XZ, Pellarin R, Schneidman-Duhovny D, Zhang XJ, Kozacka KA, Tang Y, Sali A, Chalkley RJ, Cote RH, Chu F. J Mol Biol. 2014 doi: 10.1016/j.jmb.2014.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robinson PJ, Trnka MJ, Pellarin R, Greenberg CH, Bushnell DA, Davis R, Burlingame AL, Sali A, Kornberg RD. eLife. 2015;4 doi: 10.7554/eLife.08719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen ZA, Jawhari A, Fischer L, Buchen C, Tahir S, Kamenski T, Rasmussen M, Lariviere L, Bukowski-Wills JC, Nilges M, Cramer P, Rappsilber J. EMBO J. 2010;29:717–726. doi: 10.1038/emboj.2009.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gutierrez CB, Yu C, Novitsky EJ, Huszagh AS, Rychnovsky SD, Huang L. Anal Chem. 2016 doi: 10.1021/acs.analchem.6b02240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kao A, Chiu CL, Vellucci D, Yang Y, Patel VR, Guan S, Randall A, Baldi P, Rychnovsky SD, Huang L. Mol Cell Proteomics. 2011;10:M110.002212. doi: 10.1074/mcp.M110.002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu C, Kandur W, Kao A, Rychnovsky S, Huang L. Anal Chem. 2014;86:2099–2106. doi: 10.1021/ac403636b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu C, Mao H, Novitsky EJ, Tang X, Rychnovsky SD, Zheng N, Huang L. Nature communications. 2015;6:10053. doi: 10.1038/ncomms10053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu F, Rijkers DT, Post H, Heck AJ. Nat Methods. 2015;12:1179–1184. doi: 10.1038/nmeth.3603. [DOI] [PubMed] [Google Scholar]
- 20.Liu J, Yu C, Hu X, Kim JK, Bierma JC, Jun HI, Rychnovsky SD, Huang L, Qiao F. Cell reports. 2015;12:2169–2180. doi: 10.1016/j.celrep.2015.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fischer L, Chen ZA, Rappsilber J. Journal of proteomics. 2013;88:120–128. doi: 10.1016/j.jprot.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmidt C, Zhou M, Marriott H, Morgner N, Politis A, Robinson CV. Nature communications. 2013;4:1985. doi: 10.1038/ncomms2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmidt C, Robinson CV. Nat Protoc. 2014;9:2224–2236. doi: 10.1038/nprot.2014.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boutilier JM, Warden H, Doucette AA, Wentzell PD. Journal of chromatography. B, Analytical technologies in the biomedical and life sciences. 2012;908:59–66. doi: 10.1016/j.jchromb.2012.09.035. [DOI] [PubMed] [Google Scholar]
- 25.Chavez JD, Schweppe DK, Eng JK, Zheng C, Taipale A, Zhang Y, Takara K, Bruce JE. Nature communications. 2015;6:7928. doi: 10.1038/ncomms8928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 27.Dayon L, Hainard A, Licker V, Turck N, Kuhn K, Hochstrasser DF, Burkhard PR, Sanchez JC. Anal Chem. 2008;80:2921–2931. doi: 10.1021/ac702422x. [DOI] [PubMed] [Google Scholar]
- 28.McAlister GC, Huttlin EL, Haas W, Ting L, Jedrychowski MP, Rogers JC, Kuhn K, Pike I, Grothe RA, Blethrow JD, Gygi SP. Anal Chem. 2012;84:7469–7478. doi: 10.1021/ac301572t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Isasa M, Rose CM, Elsasser S, Navarrete-Perea J, Paulo JA, Finley DJ, Gygi SP. J Proteome Res. 2015;14:5306–5317. doi: 10.1021/acs.jproteome.5b00802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ting L, Rad R, Gygi SP, Haas W. Nat Methods. 2011;8:937–940. doi: 10.1038/nmeth.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McAlister GC, Nusinow DP, Jedrychowski MP, Wuhr M, Huttlin EL, Erickson BK, Rad R, Haas W, Gygi SP. Anal Chem. 2014;86:7150–7158. doi: 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Erickson BK, Jedrychowski MP, McAlister GC, Everley RA, Kunz R, Gygi SP. Anal Chem. 2015;87:1241–1249. doi: 10.1021/ac503934f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Whiteaker JR, Zhao L, Yan P, Ivey RG, Voytovich UJ, Moore HD, Lin C, Paulovich AG. Mol Cell Proteomics. 2015;14:2261–2273. doi: 10.1074/mcp.O115.050351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frost DC, Greer T, Li L. Anal Chem. 2015;87:1646–1654. doi: 10.1021/ac503276z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.