

Abstract
The reactivity of mononuclear metal-hydroperoxo adducts has fascinated researchers in many areas due to their diverse biological and catalytic processes. In this study, a mononuclear cobalt(III)-peroxo complex bearing a tetradentate macrocyclic ligand, [CoIII(Me3-TPADP)(O2)]+ (Me3-TPADP = 3,6,9-trimethyl-3,6,9-triaza-1(2,6)-pyridinacyclodecaphane), was prepared by reacting [CoII(Me3-TPADP)(CH3CN)2]2+ with H2O2 in the presence of triethylamine. Upon protonation, the cobalt(III)-peroxo intermediate was converted into a cobalt(III)-hydroperoxo complex, [CoIII(Me3-TPADP)(O2H)(CH3CN)]2+. The mononuclear cobalt(III)-peroxo and -hydroperoxo intermediates were characterized by a variety of physicochemical methods. Results of electrospray ionization mass spectrometry clearly show the transformation of the intermediates: the peak at m/z 339.2 assignable to the cobalt(III)-peroxo species disappears with concomitant growth of the peak at m/z 190.7 corresponding to the cobalt(III)-hydroperoxo complex (with bound CH3CN). Isotope labeling experiments further support the existence of the cobalt(III)-peroxo and -hydroperoxo complexes. In particular, the O-O bond stretching frequency of the cobalt(III)-hydroperoxo complex was determined to be 851 cm−1 for 16O2H samples (803 cm−1 for 18O2H samples) and its Co-O vibrational energy was observed at 571 cm−1 for 16O2H samples (551 cm−1 for 18O2H samples; 568 cm−1 for 16O22H samples) by resonance Raman spectroscopy. Reactivity studies performed with the cobalt(III)-peroxo and -hydroperoxo complexes in organic functionalizations reveal that the latter is capable of conducting oxygen atom transfer with an electrophilic character, whereas the former exhibits no oxygen atom transfer reactivity under the same reaction conditions. Alternatively, the cobalt(III)-hydroperoxo complex does not perform hydrogen atom transfer reactions, while analogous low-spin Fe(III)-hydroperoxo complexes are capable of this reactivity. Density function theory calculations indicate that this lack of reactivity is due to the high free energy cost of O-O bond homolysis that would be required to produce the hypothetical Co(IV)-oxo product.
Graphical abstract
A cobalt(III)-hydroperoxo complex was prepared by the protonation of a cobalt(III)-peroxo complex. Reactivity studies reveal that the cobalt(III)-hydroperoxo complex is capable of conducting oxygen atom transfer with an electrophilic character. Alternatively, the cobalt(III)-hydroperoxo complex does not perform hydrogen atom transfer reactions, while analogous low-spin Fe(III)-hydroperoxo complexes are capable of this reactivity. Density function theory calculations indicate that this lack of reactivity is due to the high free energy cost of O-O bond homolysis.
Introduction
Mononuclear metal-oxygen species such as metal-superoxo, -peroxo, -hydroperoxo and -oxo complexes have important roles as key intermediates involved in catalytic oxygenation reactions and biological oxidation reactions.1–5 Heme and non-heme iron-oxygen intermediates have been invoked in the catalytic cycles of dioxygen activation by metalloenzymes.6 Among the iron-oxygen adducts, iron-hydroperoxo species have received considerable attention, since the intermediates have been implicated as reactive species in DNA cleavage activity of bleomycin, a glycopeptide that is effective as an antitumor drug in concert with iron and dioxygen.7
In the case of cobalt complexes, a large number of cobalt-O2 species have been synthesized as models of dioxygen carrier proteins and as oxidants in organic functionalizations.8,9 In models of biological oxygen carriers, Cavin et al., for example, have extensively examined the oxygen carrier properties of cobalt(II) complexes with SALEN ligands.10 In addition, the synthetic complexes have potential applications in dioxygen separation and storage.11
Recent advances in the characterization and reactivity of cobalt-O2 intermediates reveal that the cobalt-superoxo and -peroxo species are active oxidants in electrophilic and nucleophilic reactions.12 A notable example is the formation of side-on cobalt(III)-peroxo complexes bearing a series of tetraazamacrocyclic ligands.8a,8e The intermediates have nucleophilic character (e.g., aldehyde deformylation) toward organic substrates. The end-on cobalt(III)-superoxo species has been proposed as reactive species in electrophilic reactions (e.g., hydrogen atom transfer).13 Very recently, the reactivity of an end-on cobalt(II)-superoxo intermediate with a redox non-innocent ligand has been investigated in catalytic deformylation reactions.14
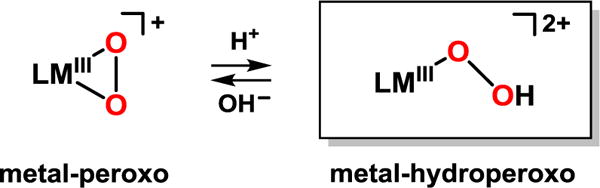
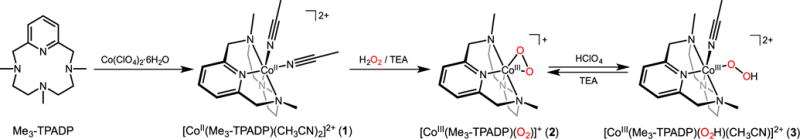
In contrast, there are few examples of cobalt(III)-hydroperoxo intermediates.15 Recently, the interconversion of cobalt(III)-peroxo and -hydroperoxo species via acid-base reactions has been reported where the cobalt(III)-hydroperoxo complex has shown reactivity in ligand oxidation (Scheme 1).16 Nevertheless, the reactivity of cobalt(III)-hydroperoxo species has been rarely explored in external substrate oxidation.17 Herein, we report the synthesis and characterization of a cobalt(III)-peroxo complex bearing a tetradentate macrocyclic ligand, 3,6,9-trimethyl-3,6,9-triaza-1(2,6)-pyridinacyclodecaphane (Me3-TPADP) (Scheme 2). Upon protonation, the cobalt(III)-peroxo complex, [CoIII(Me3- TPADP)(O2)]+ (2), was converted into a cobalt(III)-hydroperoxo complex, [CoIII(Me3-TPADP)(O2H)(CH3CN)]2+ (3). Complex 3 shows high reactivity in sulfoxidation reactions with electrophilic character, which was confirmed by a Hammett plot. Further, density function theory (DFT) calculations were applied to explain the reactivity difference between 3 and its hypothetical iron analogue in electrophilic reactions.
Scheme 1.
Acid-Base Reaction between the Metal-Peroxo and -Hydroperoxo Complexes.
Scheme 2.
Synthetic Procedures for Mononuclear Cobalt Complexes
Experimental Section
Materials
All chemicals obtained from Aldrich Chemical Co. were the best available purity and used without further purification unless otherwise indicated. Solvents were dried according to published procedures and distilled under Ar prior to use.18 H218O2 (95% 18O-enriched, 2.2% H218O2 in water) was purchased from ICON Services Inc. (Summit, NJ, USA).
Physical Methods
UV-vis spectra were recorded on a Hewlett Packard 8454 diode array spectrophotometer equipped with a UNISOKU Scientific Instruments for low-temperature experiments or with a circulating water bath. Electrospray ionization mass spectra (ESI-MS) were collected on a Waters (Milford, MA, USA) Acquity SQD quadrupole Mass instrument, by infusing samples directly into the source using a manual method. The spray voltage was set at 2.5 kV and the capillary temperature at 80 °C. Resonance Raman spectra were obtained using a liquid nitrogen cooled CCD detector (CCD-1024×256-OPEN-1LS, HORIBA Jobin Yvon) attached to a 1-m single polychromator (MC-100DG, Ritsu Oyo Kogaku) with a 1200 groovs/mm holographic grating. An excitation wavelength of 355-nm was provided by an Nd:YAG laser (Photonic Solutions, SNV-20F), with 10 mW power at the sample point. All measurements were carried out with a spinning cell (1000 rpm) at −30 °C. Raman shifts were calibrated with indene, and the accuracy of the peak positions of the Raman bands was ±1 cm−1. The effective magnetic moments was determined using the modified 1H NMR method of Evans at room temperature.19 A WILMAD® coaxial insert (sealed capillary) tubes containing the blank acetonitrile-d3 solvent (with 1.0 % TMS) only was inserted into the normal NMR tubes containing the complexes dissolved in acetonitrile-d3 (with 0.03 % TMS). The chemical shift of the TMS peak (and/or solvent peak) in the presence of the paramagnetic metal complexes was compared to that of the TMS peak (and/or solvent peak) in the inner coaxial insert tube. The effective magnetic moment was calculated using the equation, μ = 0.0618(ΔνT/2fM)1/2, where f is the oscillator frequency (MHz) of the superconducting spectrometer, T is the absolute temperature, M is the molar concentration of the metal ion, and Δv is the difference in frequency (Hz) between the two reference signals. CW-EPR spectra were taken at 5 K using an X-band Bruker EMX-plus spectrometer equipped with a dual mode cavity (ER 4116DM). Low temperatures were achieved and controlled using an Oxford Instruments ESR900 liquid He quartz cryostat with an Oxford Instruments ITC503 temperature and gas flow controller. Product analysis was performed on a Thermo Fisher Trace 1310 gas chromatograph (GC) system equipped with a flame ionization detector (FID) and mass spectrometer (GC-MS). 1H NMR, 13C NMR and 31P NMR spectra were measured with Bruker AVANCE III-400 spectrometer at CCRF in DGIST.
Generation and Characterization of [CoIII(Me3-TPADP)(O2)]+ (2)
Treatment of [CoII(Me3-TPADP)(CH3CN)2](ClO4)2 (0.5 mM) with 10 equiv of H2O2 in the presence of 2 equiv of triethylamine (TEA) in CH3CN (2 mL) afforded the formation of a blue solution at low temperature. Physicochemical data, including UV-vis and ESI-MS, were reported in Figure 2. [CoIII(Me3-TPADP)(18O2)]+ was prepared by adding 10 equiv of H218O2 (18 μL, 90% 18O-enriched, 2.2% H218O2 in water) to a solution containing [CoII(Me3-TPADP)(CH3CN)2](ClO4)2 (0.5 mM) and 2 equiv of TEA in CH3CN (2 mL) at 25 °C.
Figure 2.
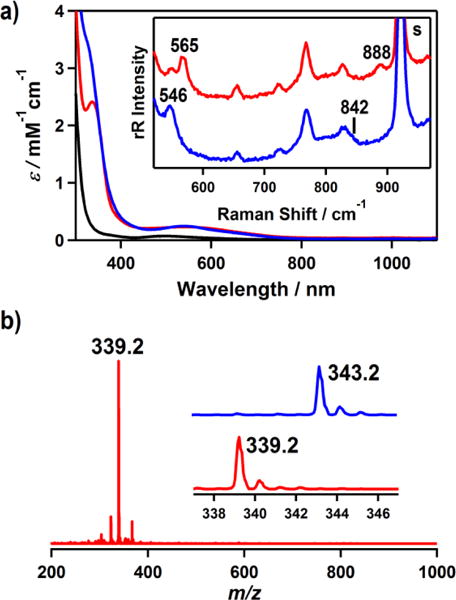
(a) UV-vis spectra of [CoII(Me3-TPADP)(CH3CN)2]2+ (1) (black line), [CoIII(Me3-TPADP)(O2)]+ (2) (red line), and [CoIII(Me3-TPADP)(O2H)(CH3CN)]2+ (3) (blue line) in CH3CN at −30 °C. Inset shows the resonance Raman spectra of 2 prepared with H216O2 (red line) and H218O2 (blue line) obtained upon excitation at 355 nm in CH3CN at −30 °C. (b) ESI-MS of 2 in CH3CN at −40 °C. Insets show the observed isotope distribution patterns for [Co(Me3-TPADP)(16O2)]+ (lower) and [Co(Me3-TPADP)(18O2)]+ (upper).
Generation and Characterization of [CoIII(Me3-TPADP)(O2H)(CH3CN)]2+ (3)
Treatment of [CoIII(Me3-TPADP)(O2)]+ (0.5 mM) with 3 equiv of perchloric acid (HClO4) in CH3CN (2 mL) at −20 °C afforded a magenta solution. Physicochemical data, including UV-vis, ESI-MS, and resonance Raman, were reported in Figures 2 and 3. [CoIII(Me3-TPADP)(18O2H)(CH3CN)]2+ was prepared by adding 3 equiv of HClO4 to a solution containing [CoIII(Me3-TPADP)(18O2)]+ (0.5 mM) in CH3CN (2 mL) at −20 °C. [CoIII(Me3-TPADP)(16O22H)(CH3CN)]2+ was prepared by adding 113.8 μL of D2O to a solution containing 3 (0.5 mM) in CH3CN (2 mL) at −20 °C.
Figure 3.
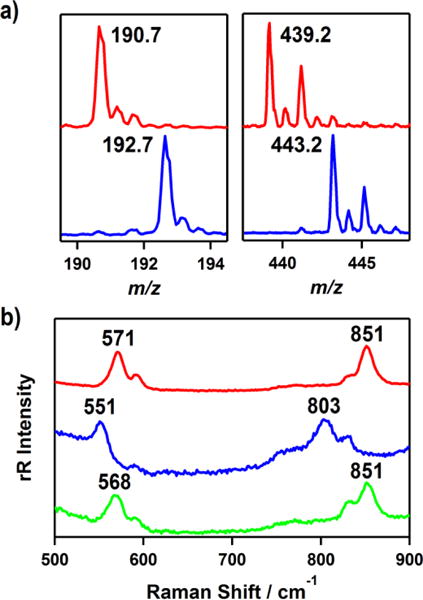
(a) ESI-MS spectra of [CoIII(Me3-TPADP)(O2H)(CH3CN)]2+ (3) in CH3CN at −40 °C, which show the observed isotope distribution patterns for [Co(Me3-TPADP)(16O2H)(CH3CN)]2+ (red line, left), [Co(Me3-TPADP)(18O2H)(CH3CN)]2+ (blue line, left), [Co(Me3-TPADP)(16O2H)(ClO4)]+ (red line, right), [Co(Me3-TPADP)(18O2H)(ClO4)]+ (blue line, right). (b) Resonance Raman spectra of 3 (16 mM) prepared with 16O (red line) and 18O (blue line) labeled samples of 2 obtained upon excitation at 355 nm in CH3CN at −30 °C. Green line shows the spectrum of 3 prepared with a 16O labeled sample of 2 and HClO4 diluted in D2O.
X-ray crystallography
Single crystal of [CoII(Me3-TPADP)(CH3CN)2](ClO4)2 was picked from solutions by a nylon loop (Hampton Research Co.) on a handmade copper plate mounted inside a liquid N2 Dewar vessel at ca. −40 °C and mounted on a goniometer head in a N2 cryostream. Data collections were carried out on a Bruker SMART APEX II CCD diffractometer equipped with a monochromator in the Mo Kα (λ = 0.71073 Å) incident beam. The CCD data were integrated and scaled using the Bruker-SAINT software package, and the structure was solved and refined using SHELXTL V 6.12.20 Hydrogen atoms were located in the calculated positions. All non-hydrogen atoms were refined with anisotropic thermal parameters. Crystal data for [CoII(Me3-TPADP)(CH3CN)2](ClO4)2: C18H30Cl2CoN6O8, Monoclinic, P21/n, Z = 8, a = 15.7400(3), b = 19.7237(4), c = 16.3257(3) Å, β = 97.6290(10)°, V = 5023.47(17) Å3, μ = 0.951 mm−1, ρcalcd = 1.556 g/cm3, R1 = 0.0345, wR2 = 0.0890 for 12456 unique reflections, 641 variables. The crystallographic data for [CoII(Me3-TPADP)(CH3CN)2](ClO4)2 is listed in Table S1, and Table S2 lists the selected bond distances and angles. CCDC-1448782 for [CoII(Me3-TPADP)(CH3CN)2](ClO4)2 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif (or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223-336-033; or deposit@ccdc.cam.ac.uk).
Computational Details
DFT calculations were performed using the Gaussian 09 computational package.21 All calculations used the B3LYP functional and the TZVP basis, and solvent effects were included using the polarized continuum model with acetonitrile as the solvent. Geometry optimizations were carried out using the default convergence criteria and were confirmed as minima by the absence of imaginary modes in a frequency calculation. Enthalpies and Gibbs free energies were calculated at 298.15 K. Mayer bond order analysis was performed using QMForge,22 and molecular orbital contours were obtained using LUMO.23
Reactivity studies
All reactions were run monitoring UV-vis spectral changes of reaction solutions, and rate constants were determined by fitting the changes in absorbance at 523 nm for [CoIII(Me3-TPADP)(O2H)(CH3CN)]2+ (3). Reactions were run at least in triplicate, and the data reported represent the average of these reactions. In situ-generated 3 were used in kinetic studies, such as the oxidation of triphenylphosphine (PPh3) and thioanisole in CH3CN at −20 and −40 °C. After the completion of reactions, pseudo-first-order fitting of the kinetic data allowed us to determine kobs values. Products formed in the oxidation of PPh3 by 3 in CH3CN at −20 °C were analyzed by 31P NMR. Products formed in the oxidation of thioanisole by 3 in CH3CN at −20 °C were analyzed by injecting the reaction mixture directly into GC and GC-MS. Products were identified by comparing with authentic samples, and product yields were determined by comparison against standard curves prepared with authentic samples.
Results and Discussion
Synthesis and Characterization
Me3-TPADP was synthesized by a modification of a previously reported procedure [see the Supporting Information (SI), Experimental Section].24 The Me3-TPADP was characterized by electrospray ionization mass spectrometry (ESI-MS) and 1H and 13C nuclear magnetic resonance (NMR) methods (see the SI, Experimental Section).
Synthetic procedures for Co(II) and Co(III) complexes used are outlined in Scheme 2. The purple starting Co(II) complex, [CoII(Me3-TPADP)(CH3CN)2](ClO4)2 (1-(ClO4)2) was synthesized by reacting Co(ClO4)2·6H2O with the Me3-TPADP ligand in CH3CN. Single crystals of 1-(ClO4)2 contained two crystallographically independent but virtually identical cations in the asymmetric unit (denoted “A” and “B”; see the SI, Table S1 and S2). Complex 1 has a six-coordinated Co(II) ion with four nitrogen atoms of the Me3-TPADP ligand and two nitrogen atoms of CH3CN solvent molecules (Figure 1a). The Co(II) geometry is best described as distorted octahedral.
Figure 1.
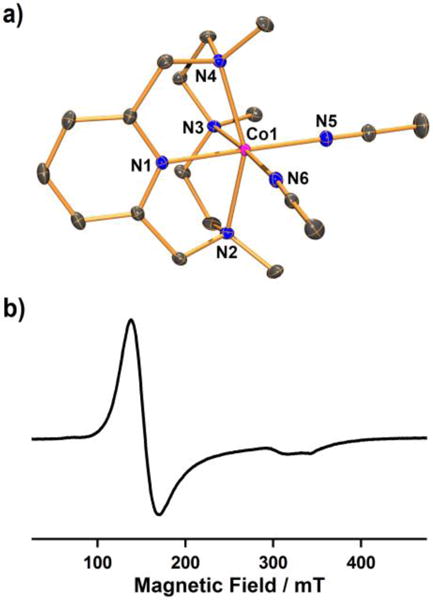
(a) ORTEP plot of [CoII(Me3-TPADP)(CH3CN)2]2+ (1A) with thermal ellipsoid drawn at the 30 % probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å): Co1-N1 2.0574(14), Co1-N2 2.1999(14), Co1-N3 2.2108(14), Co1-N4 2.2050(14), Co1-N5 2.0514(15), Co1-N6 2.1151(15) (b) X-band EPR spectrum of 1 in frozen CH3CN at 5 K. Spectroscopic settings: frequency = 9.646 GHz, microwave power = 0.998 mW, modulation frequency = 100 kHz, and modulation amplitude = 1 mT.
The UV-vis spectrum of 1 in CH3CN shows a broad absorption band at 498 nm (ε = 80 M−1 cm−1) (Figure 2a). The ESI-MS spectrum of 1 exhibits three signals at a mass-to-charge ratio (m/z) of 174.2, 194.7, and 406.2 (see the SI, Figure S1), corresponding to [Co(Me3-TPADP)(CH3CN)]2+ (calcd m/z 174.1), [Co(Me3-TPADP)(CH3CN)2]2+ (calcd m/z 194.6), and [Co(Me3-TPADP)(ClO4)]+ (calcd m/z 406.1), respectively. At room temperature, 1 exhibits a magnetic moment of 4.2 μB in CD3CN using the 1H NMR Evans method consistent with the presence of three unpaired electrons,19 and the X-band electron paramagnetic resonance (EPR) spectrum of 1 in CH3CN at 5 K shows an axial signal at g = 4.5 and 2.08 (Figure 1b). These results indicate an S = 3/2 ground state for 1.25 In addition, the redox potential of 1 was determined as E1/2 = 0.16 V (vs Fc+/Fc) by cyclic voltammetry (see the SI, Figure S2).26
The cobalt(III)-peroxo complex, [CoIII(Me3-TPADP)(O2)]+ (2), was prepared by adding 10 equiv of hydrogen peroxide (H2O2) to a reaction solution containing 1 in the presence of 2 equiv of triethylamine (TEA) in CH3CN at 25 °C (Scheme 2), where the color of the solution changed from purple to blue. Complex 2 could not be isolated due to its thermal instability; however it was generated and characterized at low temperature in solution. The UV-vis spectrum of 2 in CH3CN at −40 °C shows an intense absorption band at 338 nm (ε = 2400 M−1 cm−1) and a weak absorption band at 550 nm (ε = 230 M−1 cm−1) (Figure 2a). The ESI-MS spectrum of 2 exhibits a prominent signal at m/z 339.2 (Figure 2b), whose mass and isotope distribution pattern correspond to [Co(Me3-TPADP)(O2)]+ (calcd m/z 339.1) (see the SI, Figure S3). When the reaction was carried out with isotopically labeled H218O2, a mass peak corresponding to [Co(Me3-TPADP)(18O2)]+ appeared at m/z 343.2 (calcd m/z 343.1) (Figure 2b, inset). The four mass unit shit upon the substitution of 16O with 18O indicates that 2 has two oxygen atoms.
The resonance Raman spectrum of 2 was collected using 355 nm excitation in CH3CN at −30 °C. 2 prepared with H216O2 exhibits an isotope-sensitive band at 888 cm−1, which shifts to 842 cm−1 when H218O2 is used (Figure 2a, inset, see the SI, Figure S4). The observed isotopic shift of its 16Δ–18Δ value of 46 cm−1 is in agreement with the calculated value of 51 cm−1 for the O-O harmonic oscillator. This value is comparable to those reported for structurally and spectroscopically characterized side-on cobalt(III)-peroxo complexes, such as [CoIII(12-TMC)(O2)]+ (902 cm−1) and [CoIII(13-TMC)(O2)]+ (902 cm−1).8e In addition, the Co-O2 symmetric stretch of 2 was observed at 565 cm−1, which shifts to 546 cm−1 upon 18O-substitution (16Δ–18Δ = 19 cm−1; 16Δ–18Δ(calcd) = 21 cm−1) (Figure 2a, inset, see the SI, Figure S4).
The X-band EPR spectrum of 2 is silent at 4.3 K, suggesting either a low spin (S = 0) or an integer spin (S = 1 or 2) Co(III) d6 species. The 1H NMR spectrum of 2 recorded in CD3CN at −40 °C exhibits sharp features in the 0 – 10 ppm region (data not shown), indicating that 2 is a low-spin (S = 0) state.
Addition of 3 equiv of perchloric acid (HClO4) to a solution containing 2 in CH3CN at −20 °C immediately produced an EPR silent magenta intermediate 3 with electronic absorption bands at ~330 nm (ε = 3300 M−1 cm−1) and 528 nm (ε = 240 M−1 cm−1) (Figure 2a). 3 is thermally unstable. Even at −40 °C, the characteristic absorption bands of 3 decayed over the course of hours (18% decay for 1 h). The decay of 3 obeyed first-order reaction kinetics, and activation parameters, ΔH‡ = −115.9 kcal mol−1 and ΔS‡ = −15.7 cal mol−1 K−1, are obtained by the Eyring plot between 283 and 313 K (s). 3 could not be formed via simple oxidation of 1 with excess H2O2. The ESI-MS spectrum of the solution at −40 °C suggested the formation of Co(III)-hydroperoxo species, [Co(Me3-TPADP)(O2H)(CH3CN)]2+ at m/z 190.7 (calcd m/z 190.6) and [Co(Me3-TPADP)(O2H)(ClO4)]+ at m/z 439.2 (calcd m/z 439.1) (Figure 3a, see the SI, Figure S6), together with some unidentified species due to the thermal instability of 3 (see the s). When the reaction was carried out with 2 prepared with isotopically labeled H218O2, the mass peaks corresponding to 3 shifted to m/z 192.7 for [Co(Me3-TPADP)(18O2H)(CH3CN)]2+ (calcd m/z 192.6) and 443.2 for [Co(Me3-TPADP)(18O2H)(ClO4)]+ (calcd m/z 443.1) (Figure 3a). Intermediate 3 reverted back to 2 on addition of 3 equiv of TEA. The addition of HClO4 to the resulting solution regenerated 3, suggesting that 2 and 3 can be readily interconverted by the acid-base reaction depicted in Scheme 2. Such chemistry is well known for other metal-based peroxo and hydroperoxo systems.16,27a,28,29
The resonance Raman spectrum of 3, obtained upon 355 nm excitation in CH3CN at −30 °C, exhibits an isotope-sensitive band at 851 cm−1 which shifted to 803 cm−1 upon 18O-substitution (Figure 3b). The observed isotopic shift of its 16Δ–18Δ value of 48 cm−1 is in good agreement with the calculated value of 49 cm−1 for the O-O harmonic oscillator. The observed O-O frequency at 851 cm−1 is comparable to that of a low-spin bleomycin-Co(III)-hydroperoxo complex (828 cm−1).7h The Co-O vibrational frequency of 3 was observed at 571 cm−1, which shifts to 551 cm−1 on 18O-substitution (16Δ–18Δ = 20 cm−1; 16Δ–18Δ(calcd) = 26 cm−1) (Figure 3b). In order to support the formation of a hydroperoxide ligand in 3, we performed additional isotope labeling experiments. Upon 2H-substitution in 3, the 571 cm−1 feature exhibited a 3 cm−1 downshift (Figure 3b). The result is quite similar to those of previously reported Fe(III)-O2H species.7g,30 This deuterium isotope effect supports the presence of a hydroperoxide ligand. The 2H-substitution in 3 was further confirmed by ESI-MS (see the SI, Figure S7, inset). The 1H NMR spectrum of 3 recorded in CD3CN at −40 °C shows sharp features in the 0 – 10 ppm region (data not shown), indicating that 3 is a low-spin Co(III) d6 species. On the basis of the spectroscopic data presented above, intermediate 3 is assigned as a low-spin Co(III)-hydroperoxo complex.
Reactivity
The reactivity of 2 and 3 has been investigated in electrophilic reactions: oxygen atom transfer (i.e., the oxidation of triphenylphosphine (PPh3) and thioanisole) and hydrogen atom transfer (i.e., the oxidation of xanthene and cyclohexadiene (CHD)) reactions (Scheme 3). Upon addition of the substrates to the solution of 2 in CH3CN at −20 and −40 °C, the intermediate remained intact and product analysis of the reaction solution did not show oxygenated products (see the SI, Figure S8).
Scheme 3.
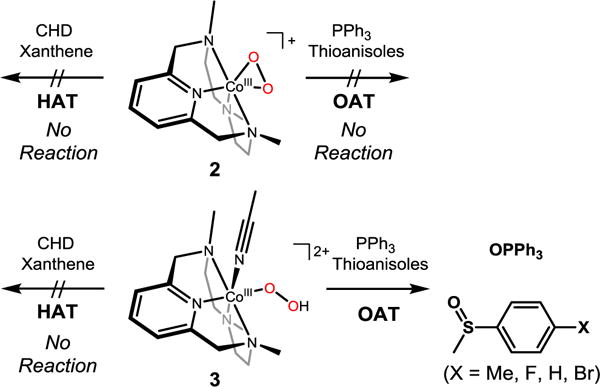
Overall reactivity of 2 and 3 in electrophilic reactions (e.g., HAT and OAT)
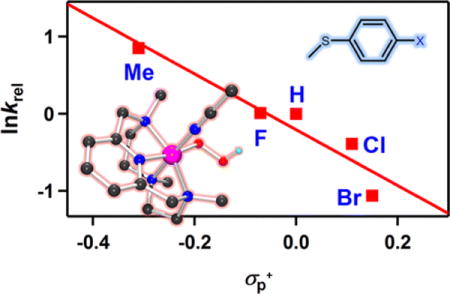
Unlike 2, addition of PPh3 to a solution of 3 in CH3CN at −20 °C shows the disappearance of the characteristic absorption band of 3 with a pseudo-first-order decay (see the SI, Figure S9). Product analysis of the reaction solution revealed the formation of triphenylphosphine oxide in a quantitative yield. Kinetic studies of 3 with thioanisole under the same reaction conditions also exhibit a pseudo-first-order reaction profile (Figure 4a, see the SI, Figure S10), and the first-order rate constants increased proportionally with the substrate concentration (k2 = 6.7(8) × 10−1 M−1 s−1) (Figure 4b). The rates were dependent on reaction temperature, where a linear Eyring plot was obtained between 233 and 263 K to give the activation parameters of ΔH‡ = 4.2 kcal mol−1 and ΔS‡ = −49.4 cal mol−1 K−1 (Figure 4c). The product analysis of the reaction solution of the oxidation of thioanisole by 3 revealed that methyl phenyl sulfoxide was produced with a yield of 75 ± 8%, and the oxygen source of the product was found to be the hydroperoxo ligand of 3 on the basis of an 18O-labeling experiment performed with 18O-labeled 3 (see the s). In addition, [CoIII(Me3-TPADP)(OH)(CH3CN)]2+ was found in the reaction solution as a decomposed product of 3 (see the SI, Figure S12). The FT-IR spectrum of the Co(III)-OH product obtained via precipitation had a peak at 3500 cm−1, which is consistent with an O-H vibration.31 The reactivity of 3 was further examined with para-substituted thioanisoles, para-X-Ph-SCH3 (X = Me, F, H, Cl, Br), to investigate the electronic effect of para-substituents on the oxidation of thioanisoles by 3 (Figure 4d). The Hammet plot of the pseudo-first-order rate constants versus σp+ gave a ρ value of −3.6(6). The negative ρ value indicates the electrophilic character of 3 in OAT reactions. Product analyses of the final reaction mixture revealed the formation of para-substituted methyl phenyl sulfoxides. It should be noted that there is a proton effect on the reactivity of 3 in the sulfoxidation reaction (see the SI, Figure S13). This result is in sharp contrast with the reactivity of [FeIII(TMC)(O2H)]2+, where no significant proton effect was observed.27c The origin of such a proton effect remains to be established in future experiments.
Figure 4.
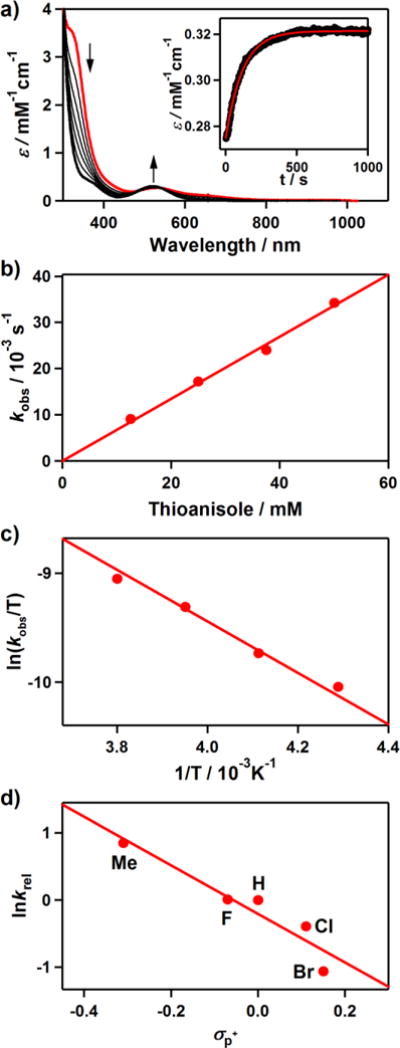
Reactions of [CoIII(Me3-TPADP)(O2H)(CH3CN)]2+ (3) with thioanisole in CH3CN. (a) UV-vis spectral changes of 3 (0.5 mM) upon addition of 25 equiv of thioanisole at −40 °C. Inset shows the time course of the absorbance at 523 nm. (b) Plot of kobs against thioanisole concentration to determine a second-order rate constant for 3 at −40 °C. (c) Plot of pseudo-first-order rate constants against 1/T to determine activation parameters for the reaction of 3 (0.5 mM) and 50 equiv of thioanisole. (d) Hammett plot of lnkrel against σp+ of para-substituted thioanisoles. The krel values were calculated by dividing kobs of para-X-Ph-SCH3 (X = Me, F, H, Cl, Br) by kobs of thioanisole at −40 °C.
It is worth noting that 3 is not capable of performing HAT reactions (data not shown). However, the reactivity of [FeIII(TMC)(O2H)]2+ in both OAT and HAT reactions has previously been reported.27b One possible explanation for the different reactivity of metal(III)-hydroperoxo species is the inability of cobalt to access its high-valent state. DFT calculations for thermodynamics of O-O homolysis for 3 relative to a low-spin Fe(III) analogue provide significant insight into their difference in reactivity (vide supra).
Density Functional Theory Studies
In a previous study evaluating the HAT reactivity of the low-spin Fe(III)-hydroperoxo complex [FeIII(N4Py)(O2H)]2+, it was found that the barrier for HAT was mostly due to homolysis of the O-O bond.27d In the Me3-TPADP ligand system in this study, the Fe(III)-hydroperoxo complex is too unstable to be isolated for experimental evaluation. To calibrate this ligand set to the [FeIII(N4Py)(O2H)]2+ results for correlation to 3, DFT calculations were performed on the hypothetical [FeIII(Me3-TPADP)(O2H)(CH3CN)]2+ complex to determine the thermodynamics of O-O bond homolysis relative to the results for the low-spin [FeIII(N4Py)(O2H)]2+ complex, which were supported by experiment.7g,27d The calculated ΔH of O-O homolysis for the Me3-TPADP on the S = 1/2 surface is 23.2 kcal mol−1, compared to 23.1 kcal mol−1 previously reported for N4Py, and the ΔG for Me3-TPADP was found to be 13.0 kcal mol−1, compared to 12.7 kcal mol−1 for N4Py. In the Me3-TPADP case, as in the N4Py case, the final products are an S = 1 FeIV=O and an hydroxyl radical. Given the good agreement between the present set of calculations and those previously reported,7g these calculations were extended to the Co(III)-hydroperoxo complex 3.
The geometry of 3 was optimized on the S = 0, S = 1, and S = 2 surfaces. The relative energies of these structures are given on the left of Figure 5. The lowest-energy structure for 3 was found to be S = 0, consistent with the 1H NMR data (vide supra); thus, we focus on the S = 0 calculation. Relevant geometric and vibrational parameters calculated for 3, as well as those for its iron analogue, are presented in Table 1. The structures are similar, with the main difference being the calculated M-O bond length, which is 1.88 Å for 3 and 1.80 Å for the hypothetical iron analogue. This difference arises due to the additional electron in the Co(III) relative to the low-spin Fe(III) complex, which occupies a metal d orbital π antibonding with the hydroperoxide. The calculated ν(Co-(O2H)) is 555 cm−1, in agreement with the experimental value of 571 cm−1 and 66 cm−1 lower in energy than the corresponding calculated stretch in the iron analogue, consistent with the longer Co-O bond length. The calculated ν(O-O) is 928 cm−1, which overestimates the experimental value of 851 cm−1 by 77 cm−1. A similar disagreement between calculation and experiment was observed in the previous study on the low-spin [FeIII(N4Py)(O2H)]2+ complex,7g where an equivalent DFT calculation overestimated the experimental ν(O-O) by 98 cm−1. This was attributed to the calculations underestimating the donation from hydroperoxy σ and π bonding orbitals to the metal. Given the similarity between the present DFT calculations for 3 and the previous experimentally-calibrated calculations for [FeIII(N4Py)(O2H)]2+, these calculations for 3 were used to evaluate the thermodynamics of its O-O bond homolysis. The enthalpies and Gibbs free energies of the O-O homolysis products of 3 on the S = 0, S = 1, and S = 2 surfaces are presented on the right-hand side of Figure 5. The S = 2 products are uphill by ΔG = 39.3 kcal mol−1 and are discounted. The S = 1 and S = 0 products are at 21.7 and 27.3 kcal mol−1 in Gibbs free energy and correspond to S = 3/2 CoIV=O and S = 1/2 CoIV=O complexes, respectively, antiferromagnetically aligned to the S = 1/2 hydroxyl radical. Figure 5 also presents the energetics of O-O homolysis for the iron analogue of 3, which is ~9 kcal mol−1 more favorable than the lowest energy O-O homolysis pathway for 3. Note that this pathway would further require a crossover from the S = 0 to the S = 1 surface.
Figure 5.
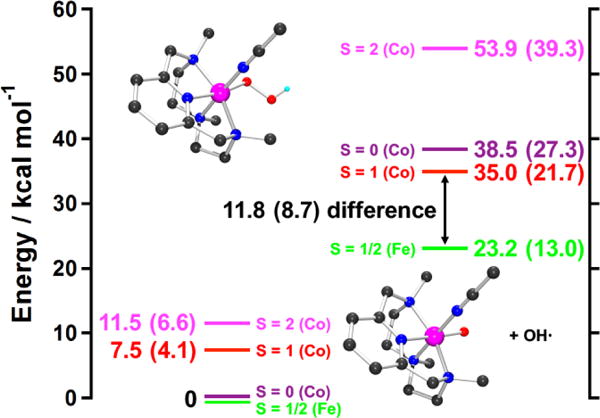
Thermodynamics of O-O homolysis for 3 on the S = 0 (violet line), S = 1 (red line), and S = 2 (magenta line) surfaces, as well as for the Fe analogue on the S = 1/2 (green line) surface. Energies are given as enthalpies, with Gibbs free energy at 298.15 K in parentheses. Homolysis of the O-O bond is 8.7 kcal mol−1 more unfavorable for 3 relative to the iron complex. Insets show the geometry optimized structures of 3 (upper) and [CoIV(Me3-TPADP)(O)(CH3CN)]2+ (lower) (black, C; blue, N; cyan, H; red, O; pink, Co).
Table 1.
Calculated Geometric and Vibrational Parameters for 3 and Its S=1/2 FeIII-O2H Analogue.
| Complex | 3 | S=1/2 FeIII-O2H |
|---|---|---|
| M-O (Å) | 1.88 | 1.80 |
| O-O (Å) | 1.44 | 1.44 |
| M-O-O (°) | 117 | 117 |
| M-Lequatorial, ave (Å) | 2.03 | 2.05 |
| ν(M-(O2H)) (cm−1) | 555 | 621 |
| ν(O-O) (cm−1) | 928 | 888 |
To understand the difference in O-O homolysis energy for 3 relative to its iron analogue, the electronic structures of the MIV=O reaction products were analyzed, and are summarized in Figure S14 in the SI. Geometric parameters, as well as Mayer bond orders, for the products are presented in s. An S = 1 FeIV=O has one σ-bond between Fe and O from dz2 interacting with the oxo pz orbital, and one net Fe-O π bond from dxz and dyz interacting with the oxo px and py, respectively. In going from an S = 1 FeIV=O to an S = 1/2 CoIV=O complex, a M-O π bonding interaction is lost through addition of an electron to the dxz orbital, which has a π* interaction with the O px orbital (see the SI, Figure S14, bottom to upper-left), leading to a weaker Co-O relative to Fe-O bond. This additional π* interaction is reflected both in its longer bond length (1.79 vs 1.64 Å) and lower Mayer bond order (1.14 vs 1.39). This weaker Co-O bond destabilizes the S = 1/2 CoIV=O complex relative to the S = 1 FeIV=O. The homolysis to form this CoIV=O product is 14.3 kcal mol−1 higher in energy than the comparable homolysis reaction for Fe (27.3 vs. 13.0 kcal mol−1). For the lower energy S = 3/2 CoIV=O case, the extra electron relative to FeIV=O S=1 is instead added to the dx2-y2 orbital (see the s, bottom to upper-right), which is σ antibonding to the equatorial ligands. In this case, the Co-O bond is not perturbed by the additional electron, and is in fact stronger than the Fe-O bond, as reflected in its shorter bond length (1.62 Å vs 1.64 Å for FeIV=O) and higher Mayer bond order (1.85 vs. 1.39). This is due to the greater Zeff on CoIV relative to FeIV, which stabilizes its d manifold and allows stronger mixing with the oxygen p orbitals. However, the equatorial Co-L bonds are greatly weakened relative to the iron complex (see the SI, Table S3; the average M-Lequatorial bond increases to 2.17 from 2.07 Å and the total Mayer bond order for these bonds decreases to 1.74 from 2.16). This overcomes the stronger Co-O bonding interaction and results in an S = 3/2 CoIV=O that is less stable than the S = 1 FeIV=O by 9 kcal mol−1. Note that the CoIII-O2H is also less stable than the FeIII-O2H due to weakened M-O2H bonding arising from the additional dπ* electron, but this involves loss of a relatively weak π bonding interaction (vide supra; also see differences in calculated M-O bond lengths and stretching frequencies in Table 1). Thus, 3 is much less reactive than equivalent low-spin Fe(III)-hydroperoxo complexes due to the higher barrier for O-O bond homolysis resulting from the additional electron in a equatorial sigma antibonding orbital of the CoIV=O S = 3/2 product.
Conclusions
The metal-hydroperoxo intermediates in organic functionalizations are of current interest in enzymatic processes, pharmaceutical research and industrial catalysis. We have synthesized mononuclear cobalt(III)-peroxo (2) and -hydroperoxo (3) complexes bearing a common macrocyclic Me3-TPADP ligand, where 3 was prepared by protonation of 2. The consecutive interconversion of 3 to 2 by addition of a base supports acid-base chemistry. The intermediates were characterized with a variety of physicochemical methods. Although the UV-vis spectra of 2 and 3 are not very different, ESI-MS spectra of 2 clearly exhibit the formation of the cobalt(III)-peroxo adduct at low temperature and resonance Raman spectra of 3 show an O-O stretching vibration at 851 cm−1 for 16O samples (803 cm−1 for 18O samples), which is assignable to that of hydroperoxo species. The reactivities of 2 and 3 were compared in electrophilic reactions; 3 is capable of conducting OAT reactions, whereas 2 exhibits no OAT reactivity. Alternatively, 3 does not perform HAT, whereas low-spin Fe(III)-hydroperoxo complexes do. DFT calculations show that this is due to the high O-O bond homolysis energy relative to the iron analogue. This energy difference is due to the additional electron in an antibonding orbital that would destabilize a high-valent Co-oxo product.
Supplementary Material
Acknowledgments
J. C. at DGIST acknowledges the financial support from the NRF (2014R1A1A2056051), the Ministry of Science, ICT and Future Planning (DGIST R&D Program 16-BD-0403, KCRC 2014M1A8A1049320, and KCGRC 2016M3D3A01913243), and the Ministry of oceans and Fisheries (Marine Biotechnology Program 20150220) of Korea. T. Og. acknowledges the support of “Strategic Young Researcher Overseas Visits Program for Accelerating Brain Circulation” and Grant-in-Aid for Scientific Research (No. 15H00960) both by JSPS. E. I. S. at Stanford University acknowledges finical support by the National Institutes of Health (Grant No. GM 40392).
Footnotes
Supporting Information. Experimental details, Figure S1–S14, Table S1–S3. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes: The authors declare no competing financial interest.
References
- 1.(a) Nam W. Dioxygen Activation by Metalloenzymes and Models. Acc Chem Res. 2007;40:465. and review articles in the special issue. [Google Scholar]; (b) Ray K, Pfaff FF, Wang B, Nam W. Status of Reactive Non-Heme Metal–Oxygen Intermediates in Chemical and Enzymatic Reactions. J Am Chem Soc. 2014;136:13942–13958. doi: 10.1021/ja507807v. [DOI] [PubMed] [Google Scholar]
- 2.(a) Sheldon RA, Kochi JK. Metal-Catalyzed Oxidations of Organic Compounds. Academic Press; New York: 1981. [Google Scholar]; (b) Patai S, editor. The chemistry of peroxides. Wiley; Chichester: 1983. [Google Scholar]; (c) Norman JA, Pez GP, Roberts DA. In: Oxygen Complexes and Oxygen Activation by Transition Metals. Martel AE, Sawyer DT, editors. Plenum Press; New York: 1988. [Google Scholar]; (d) Ando W, editor. Organic Peroxides. Wiley; Chich ester: 1992. [Google Scholar]
- 3.special issue on; (a) Metal–Dioxygen Complexes. Chem Rev. 1994;94:567–856. [Google Scholar]; (b) Strukul G. Transition Metal Catalysis in the Baeyer–Villiger Oxidation of Ketones. Angew Chem, Int Ed. 1998;37:1198–1209. doi: 10.1002/(SICI)1521-3773(19980518)37:9<1198::AID-ANIE1198>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]; (c) Murahashi S. Synthetic Aspects of Metal‐Catalyzed Oxidations of Amines and Related Reactions. Angew Chem, Int Ed. 1995;34:2443–2465. [Google Scholar]
- 4.Ortiz de Montellano PR. Cytochrome P450: Structure, Mechanism, and Biochemistry. 3rd. Kluwer Academic/Plenum Publishers; New York: 2005. [Google Scholar]
- 5.(a) Holm RH, Solomon EI. Introduction: Bioinorganic Enzymology II. Chem Rev. 2014;114:3367–3368. doi: 10.1021/cr500118g. and review articles in the special issue. [DOI] [PubMed] [Google Scholar]; (b) Kovaleva EG, Lipscomb JD. Versatility of Biological Non-heme Fe(II) Centers in Oxygen Activation Reactions. Nat Chem Biol. 2008;4:186–193. doi: 10.1038/nchembio.71. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ortiz de Montellano PR. Hydrocarbon Hydroxylation by Cytochrome P450 Enzymes. Chem Rev. 2010;110:932–948. doi: 10.1021/cr9002193. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) McQuarters AB, Wolf MW, Hunt AP, Lehnert N. 1958–2014: after 56 Years of Research, Cytochrome P450 Reactivity is Finally Explained. Angew Chem, Int Ed. 2014;53:4750–4752. doi: 10.1002/anie.201402404. [DOI] [PubMed] [Google Scholar]; (e) Tolman WB. Editorial for the Virtual Issue on Models of Metalloenzymes. Inorg Chem. 2013;52:7307–7310. doi: 10.1021/ic4013813. [DOI] [PubMed] [Google Scholar]
- 6.(a) Wertz DL, Valentine JS. Nucleophilicity of Iron-Peroxo Porphyrin Complexes. Struct Bonding. 2000;97:37–60. [Google Scholar]; (b) Gibson DT, Parales RE. Aromatic Hydrocarbon Dioxygenases in Environmental Biotechnology. Curr Opin Biotechnol. 2000;11:236–243. doi: 10.1016/s0958-1669(00)00090-2. [DOI] [PubMed] [Google Scholar]; (c) Wertz DL, Sisemore MF, Selke M, Driscoll J, Valentine JS. Mimicking Cytochrome P-450 2B4 and Aromatase: Aromatization of a Substrate Analogue by a Peroxo Fe(III) Porphyrin Complex. J Am Chem Soc. 1998;120:5331–5332. [Google Scholar]; (d) Goto Y, Wada S, Morishima I, Watanabe Y. Reactivity of Peroxoiron(III) Porphyrin Complexes: Models for Deformylation Reactions Catalyzed by Cytochrome P-450. J Inorg Biochem. 1998;69:241–247. [Google Scholar]; (e) Hashimoto K, Nagatomo S, Fujinami S, Furutachi H, Ogo S, Suzuki M, Uehara A, Maeda Y, Watanabe Y, Kitagawa T. A New Mononuclear Iron(III) Complex Containing a Peroxocarbonate Ligand. Angew Chem, Int Ed. 2002;41:1202–1205. doi: 10.1002/1521-3773(20020402)41:7<1202::aid-anie1202>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]; (f) Annaraj J, Suh Y, Seo MS, Kim SO, Nam W. Mononuclear Nonheme Ferric-Peroxo Complex in Aldehyde Deformylation. Chem Commun. 2005:4529–4531. doi: 10.1039/b505562h. [DOI] [PubMed] [Google Scholar]; (g) Kovaleva EG, Lipscomb JD. Crystal Structures of Fe2+ Dioxygenase Superoxo, Alkylperoxo, and Bound Product Intermediates. Science. 2007;316:453–457. doi: 10.1126/science.1134697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Chow MS, Liu LV, Solomon EI. Further Insights into the Mechanism of the Reaction of Activated Bleomycin with DNA. Proc Natl Acad Sci U S A. 2008;105:13241–13245. doi: 10.1073/pnas.0806378105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Neese F, Zaleski JM, Loeb-Zaleski K, Solomon EI. Electronic Structure of Activated Bleomycin: Oxygen Intermediates in Heme versus Non-Heme Iron. J Am Chem Soc. 2000;122:11703–11724. [Google Scholar]; (c) Sam JW, Tang X-J, Peisach J. Electrospray Mass Spectrometry of Iron Bleomycin: Demonstration that Activated Bleomycin is a Ferric Peroxide Complex. J Am Chem Soc. 1994;116:3250–5256. [Google Scholar]; (d) Westre TE, Loeb KE, Zaleski JM, Hedman B, Hodgson KO, Solomon EI. Determination of the Geometric and Electronic Structure of Activated Bleomycin Using X-ray Absorption Spectroscopy. J Am Chem Soc. 1995;117:1309–1313. [Google Scholar]; (e) Burger RM, Peisach J, Horwitz SB. Activated Bleomycin. A Transient Complex of Drug, Iron, and Oxygen that Degrades DNA. J Biol Chem. 1981;256:11636–11644. [PubMed] [Google Scholar]; (f) Hecht SM. The Chemistry of Activated Bleomycin. Acc Chem Res. 1986;19:383–391. [Google Scholar]; (g) Lehnert N, Neese F, Ho RYN, Que L, Jr, Solomon EI. Electronic Structure and Reactivity of Low-Spin Fe(III)-Hydroperoxo Complexes: Comparison to Activated Bleomycin. J Am Chem Soc. 2002;124:10810–10822. doi: 10.1021/ja012621d. [DOI] [PubMed] [Google Scholar]; (h) Rajani C, Kincaid JR, Petering DH. Resonance Raman Studies of HOO-Co(III) Bleomycin and Co(III) Bleomycin: Identification of Two Important Vibrational Modes, ν(Co-OOH) and ν(O-OH) J Am Chem Soc. 2004;126:3829–3836. doi: 10.1021/ja030622v. [DOI] [PubMed] [Google Scholar]
- 8.(a) Jo Y, Annaraj J, Seo MS, Lee Y-M, Kim SY, Cho J, Nam W. Reactivity of a Cobalt(III)-Peroxo Complex in Oxidative Nucleophilic Reactions. J Inorg Biochem. 2008;102:2155–2159. doi: 10.1016/j.jinorgbio.2008.08.008. [DOI] [PubMed] [Google Scholar]; (b) Hikichi S, Akita M, Moro-oka Y. New Aspects of the Cobalt-Dioxygen Complex Chemistry Opened by Hydrotris(pyrazoly)borate Ligands (TpR): Unique Properties of TpRCo-Dioxygen Complexes. Coord Chem Rev. 2000;198:61–87. [Google Scholar]; (c) Rahman AFMM, Jackson WG, Willis AC. The First Sideways-Bonded Peroxo Complex for a Tetraaminecobalt(III) Species. Inorg Chem. 2004;43:7558–7560. doi: 10.1021/ic040044z. [DOI] [PubMed] [Google Scholar]; (d) Hu X, Castro-Rodriguez I, Meyer K. Dioxygen Activation by a Low-Valent Cobalt Complex Employing a Flexible Tripodal N-Heterocyclic Carbene Ligand. J Am Chem Soc. 2004;126:13464–13473. doi: 10.1021/ja046048k. [DOI] [PubMed] [Google Scholar]; (e) Cho J, Sarangi R, Kang HYLee JY, Kubo M, Ogura T, Solomon EI, Nam W. Synthesis, Structural, and Spectroscopic Characterization and Reactivities of Mononuclear Cobalt(III)–Peroxo Complexes. J Am Chem Soc. 2010;132:16977–16986. doi: 10.1021/ja107177m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Smith TD, Pilbrow JR. Recent Developments in the Studies of Molecular Oxygen Adducts of Cobalt(II) Compounds and Related Systems. Coord Chem Rev. 1981;39:295–383. [Google Scholar]
- 9.(a) Terry NW, Amma EL, Vaska L. Molecular Oxygen Binding in a Monomeric Cobalt Complex. Crystal and Molecular Structure of Dioxygen-Bis[cis-1,2-bis(diphenylphosphino)ethylene]cobalt Tetrafluoroborate. J Am Chem Soc. 1972;94:653–655. [Google Scholar]; (b) Egan JW, Jr, Haggerty BS, Rheingold AL, Sendlinger SC, Theopold KH. Crystal Structure of a Side-On Superoxo Complex of Cobalt and Hydrogen Abstraction by a Reactive Terminal Oxo Ligand. J Am Chem Soc. 1990;112:2445–2446. [Google Scholar]; (c) Rodley GA, Robinson WT. Structure of a Monomeric Oxygen-Carrying Complex. Nature. 1972;235:438–439. doi: 10.1038/235438a0. [DOI] [PubMed] [Google Scholar]; (d) Gall RS, Rogers JF, Schaefer WP, Christoph GG. The Structure of a Monomeric Oxygen Carrying Cobalt Complex: Dioxygen-N,N′-(1,1,2,2-tetramethyl)ethylenebis(3-tert-butylsalicylideniminato)(1-benzylimidazole)cobalt(II) J Am Chem Soc. 1976;98:5135–5144. [Google Scholar]; (e) Tiné MR. Cobalt Complexes in Aqueous Solutions as Dioxygen Carriers. Coord Chem Rev. 2012;256:316–327. [Google Scholar]; (f) Schaefer WP, Huie BT, Kurilla MG, Ealick SE. Oxygen-Carrying Cobalt Complexes. 10. Structures of N,N′-Ethylenebis(3-tert-butylsalicylideniminato)cobalt(II) and Its Monomeric Dioxygen Adduct. Inorg Chem. 1980;19:340–344. [Google Scholar]; (g) Busch DH, Jackson PJ, Kojima M, Chmielewski P, Matsumoto N, Stevens JC, Wu W, Nosco D, Herron N, Ye N, Warburton PR, Masarwa M, Stephenson NA, Christoph G, Alcock NW. Dioxygen Adducts of Lacunar Cobalt(II) Cyclidene Complexes. Inorg Chem. 1994;33:910–923. [Google Scholar]; (h) Wang C-C, Chang H-C, Lai Y-C, Fang H, Li C-C, Hsu H-K, Li Z-Y, Lin T-S, Kuo T-S, Neese F, Ye S, Chiang Y-W, Tsai M-L, Liaw W-F, Lee W-Z. A structurally characterized Nonheme Cobalt–Hydroperoxo Complex Derived from Its Superoxo Intermediate via Hydrogen Atom Abstraction. J Am Chem Soc. 2016 doi: 10.1021/jacs.6b08642. [DOI] [PubMed] [Google Scholar]
- 10.(a) Calvin M, Bailes RH, Wilmarth WK. The Oxygen-Carrying Synthetic Chelate Compounds.1a I. J Am Chem Soc. 1946;68:2254–2256. [Google Scholar]; (b) Barkelew CH, Calvin M. Oxygen-Carrying Synthetic Chelate Compounds. II. The Rates of Oxygenation of the Solid Compounds1. J Am Chem Soc. 1946;68:2257–2262. [Google Scholar]; (c) Bailes RH, Calvin M. The Oxygen-Carrying Synthetic Chelate Compounds. VII. Preparation1. J Am Chem Soc. 1947;69:1886–1893. [Google Scholar]
- 11.Niederhoffer EC, Timmons JH, Martell AE. Thermodynamics of Oxygen Binding in Natural and Synthetic Dioxygen Complexes. Chem Rev. 1984;84:137–203. [Google Scholar]
- 12.(a) Bailey CL, Drago RS. Utilization of O2 for the Specific Oxidation of Organic Substrates with Cobalt(II) Catalysts. Coord Chem Rev. 1987;79:321–332. [Google Scholar]; (b) Cho J, Sarangi S, Nam W. Mononuclear Metal–O2 Complexes Bearing Macrocyclic N-Tetramethylated Cyclam Ligands. Acc Chem Res. 2012;45:1321–1330. doi: 10.1021/ar3000019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Zombeck A, Drago RS, Corden BB, Gaul JH. Activation of Molecular Oxygen. Kinetic Studies of the Oxidation of Hindered Phenols with Cobalt-Dioxygen Complexes. J Am Chem Soc. 1981;103:7580–7585. [Google Scholar]; (b) Hamilton DE, Drago RS, Zombeck A. Mechanistic Studies on the Cobalt(II) Schiff Base Catalyzed Oxidation of Olefins by O2. J Am Chem Soc. 1987;109:374–379. [Google Scholar]
- 14.Corcos AR, Villanueva O, Walroth RC, Sharma SK, Bacsa J, Lancaster KM, MacBeth CE, Berry JF. Oxygen Activation by Co(II) and a Redox Non-Innocent Ligand: Spectroscopic Characterization of a Radical–Co(II)–Superoxide Complex with Divergent Catalytic Reactivity. J Am Chem Soc. 2016;138:1796–1799. doi: 10.1021/jacs.5b12643. [DOI] [PubMed] [Google Scholar]
- 15.(a) Bayston JH, Winfield ME. A Mononuclear Hydroperoxo Complex and Its Significance in Catalytic Oxidation Mechanisms. J Catal. 1964;3:123–128. [Google Scholar]; (b) Guzei IA, Bakac A. Macrocyclic Hydroperoxocobalt(III) Complex: Photochemistry, Spectroscopy, and Crystal Structure. Inorg Chem. 2001;40:2390–2393. doi: 10.1021/ic001391l. [DOI] [PubMed] [Google Scholar]; (c) Wang W-D, Bakac A, Espenson JH. Oxidation and Reduction Reactions of Hydroperoxo Cobalt Macrocycles. Inorg Chem. 1995;34:4049–4056. [Google Scholar]
- 16.Kim D, Cho J, Lee Y-M, Sarangi R, Nam W. Synthesis, Characterization, and Reactivity of Cobalt(III)–Oxygen Complexes Bearing a Macrocyclic N-Tetramethylated Cyclam Ligand. Chem–Eur J. 2013;19:14112–14118. doi: 10.1002/chem.201300107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Mirza SA, Bocquet B, Robyr C, Thomi S, Williams AF. Reactivity of the Coordinated Hydroperoxo Ligand. Inorg Chem. 1996;35:1332–1337. doi: 10.1021/ic950950z. [DOI] [PubMed] [Google Scholar]; (b) Tcho W-Y, Wang B, Lee Y-M, Cho K-B, Shearer J, Nam W. A Mononuclear Nonheme Cobalt(III)–Hydroperoxide Complex with an Amphoteric Reactivity in Electrophilic and Nucleophilic Oxidative Reactions. Dalton Trans. 2016;45:14511–14515. doi: 10.1039/c6dt01194b. [DOI] [PubMed] [Google Scholar]
- 18.Armarego WLF, Perrin DD, editors. Purification of Laboratory Chemicals. Pergamon Press; Oxford: 1997. [Google Scholar]
- 19.(a) Evans DF. 400. The Determination of the Paramagnetic Susceptibility of Substances in Solution by Nuclear Magnetic Resonance. J Chem Soc. 1959:2003–2005. [Google Scholar]; (b) Löliger J, Scheffold R. Paramagnetic Moment Measurements by NMR. A Micro Technique. J Chem Educ. 1972;49:646–647. [Google Scholar]; (c) Evans DF, Jakubovic DA. Water-Soluble Hexadentate Schiff-Base Ligands as Sequestrating Agents for Iron(III) and Gallium(III) J Chem Soc, Dalton Trans. 1988:2927–2933. [Google Scholar]
- 20.Sheldrick GM. SHELXTL/PC. for Windows XP. Bruker AXS Inc.; Madison, WI: 2001. Version 6.12. [Google Scholar]
- 21.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision D.01. Gaussian, Inc; Wallingford CT: 2013. [Google Scholar]
- 22.Adam L. Tenderholt. QMForge; Version 2.2. http://qmforge.sourceforge.net. [Google Scholar]
- 23.Kieber-Emmons MT. LUMO. Version 1.0.1. [Google Scholar]
- 24.Wen J, Geng Z, Yin Y, Wang Z. A Mononuclear Mn2+ Complex Based on a Novel Tris-(ethyl acetate) Pendant-Armed Tetraazamacrocycle: Effect of Pyridine on Self-Assembly and Weak Interactions. Inorg Chem Commun. 2012;21:16–20. [Google Scholar]
- 25.Kang PC, Eaton GR, Eaton SS. Pulsed Electron Paramagnetic Resonance of High-Spin Cobalt(II) Complexes. Inorg Chem. 1994;33:3660–3665. [Google Scholar]
- 26.Complexes 2 and 3 did not give reversible or quasi-reversible behavior in cyclovoltammetry.
- 27.(a) Li F, Meier KK, Cranswick MA, Chakrabarti M, Heuvelen KMV, Münck E, Que L., Jr Characterization of a High-Spin Non-Heme FeIII–OOH Intermediate and Its Quantitative Conversion to an FeIV=O Complex. J Am Chem Soc. 2011;133:7256–7259. doi: 10.1021/ja111742z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cho J, Jeon S, Wilson SA, Liu LV, Kang EA, Braymer JJ, Lim MH, Hedman B, Hodgson KO, Valentine JS, Solomon EI, Nam W. Structure and Reactivity of a Mononuclear Non-Haem Iron(III)–Peroxo Complex. Nature. 2011;478:502–505. doi: 10.1038/nature10535. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kim YM, Cho K-B, Cho J, Wang B, Li C, Shaik S, Nam W. A Mononuclear Non-Heme High-Spin Iron(III)–Hydroperoxo Complex as an Active Oxidant in Sulfoxidation Reactions. J Am Chem Soc. 2013;135:8838–8841. doi: 10.1021/ja404152q. [DOI] [PubMed] [Google Scholar]; (d) Liu LV, Hong S, Cho J, Nam W, Solomon EI. Comparison of High-Spin and Low-Spin Nonheme FeIII–OOH Complexes in O–O Bond Homolysis and H-Atom Abstraction Reactivities. J Am Chem Soc. 2013;135:3286–3299. doi: 10.1021/ja400183g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.So H, Park YJ, Cho K-B, Lee Y-M, Seo MS, Cho J, Sarangi R, Nam W. Spectroscopic Characterization and Reactivity Studies of a Mononuclear Nonheme Mn(III)–Hydroperoxo Complex. J Am Chem Soc. 2014;136:12229–12232. doi: 10.1021/ja506275q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.(a) Jensen KB, McKenzie CJ, Nielsen LP, Pedersen JZ, Svendsen HM. Deprotonation of Low-Spin Mononuclear Iron(III)–Hydroperoxide Complexes Give Transient Blue Species Assigned to High-Spin Iron(III)–Peroxide Complexes. Chem Commun. 1999:1313–1314. [Google Scholar]; (b) Ho RYN, Roelfes G, Hermant R, Hage R, Feringa BL, Que L., Jr Resonance Raman Evidence for the Interconversion between an [FeIII–η1-OOH]2+ and [FeIII–η2-O2]+ Species and Mechanistic Implications Thereof. Chem Commun. 1999:2161–2162. [Google Scholar]; (c) Nebe T, Beitat A, Würtele C, Dücker-Benfer C, van Eldik R, McKenzie CJ, Schindler S. Reinvestigation of the Formation of a Mononuclear Fe(III) Hydroperoxido Complex Using High Pressure Kinetics. Dalton Trans. 2010;39:7768–7773. doi: 10.1039/c0dt00247j. [DOI] [PubMed] [Google Scholar]
- 30.(a) Roelfes G, Lubben M, Chen K, Ho RYN, Meetsma A, Genseberger S, Hermant RM, Hage R, Mandal SK, Young VG, Jr, Zang Y, Kooijman H, Spek AL, Que L, Jr, Feringa BL. Iron Chemistry of a Pentadentate Ligand That Generates a Metastable FeIII–OOH Intermediate. Inorg Chem. 1999;38:1929–1936. doi: 10.1021/ic980983p. [DOI] [PubMed] [Google Scholar]; (b) Ho RYN, Roelfes G, Feringa BL, Que L., Jr Raman Evidence for a Weakened O-O Bond in Mononuclear Low-Spin Iron(III)-Hydroperoxides. J Am Chem Soc. 1999;121:264–265. [Google Scholar]; (c) Simaan AJ, Döpner S, Banse F, Bourcier S, Bouchoux G, Boussac A, Hildebrandt P, Girerd J-J. FeIII-Hydroperoxo and Peroxo Complexes with Aminopyridyl Ligands and the Resonance Raman Spectroscopic Identification of the Fe–O and O–O Stretching Modes. Eur J Inorg Chem. 2000:1627–1633. [Google Scholar]; (d) Mairata i Payeras A, Ho RYN, Fujita M, Que L., Jr The Reaction of [FeII(tpa)] with H2O2 in Acetonitrile and Acetone-Distinct Intermediates and Yet Similar Catalysis. Chem–Eur J. 2004;10:4944–4953. doi: 10.1002/chem.200400480. [DOI] [PubMed] [Google Scholar]; (e) Ohta T, Liu J-G, Naruta Y. Resonance Raman Characterization of Mononuclear Heme-Peroxo Intermediate Models. Coord Chem Rev. 2013;257:407–413. [Google Scholar]
- 31.(a) Bergquist C, Fillebeen T, Morlok MM, Parkin G. Protonation and Reactivity towards Carbon Dioxide of the Mononuclear Tetrahedral Zinc and Cobalt Hydroxide Complexes, [TpBut,Me]ZnOH and [TpBut,Me]CoOH: Comparison of the Reactivity of the Metal Hydroxide Function in Synthetic Analogues of Carbonic Anhydrase. J Am Chem Soc. 2003;125:6189–6199. doi: 10.1021/ja034711j. [DOI] [PubMed] [Google Scholar]; (b) Lucas RL, Zart MK, Murkerjee J, Sorrell TN, Powell DR, Borovik AS. A Modular Approach toward Regulating the Secondary Coordination Sphere of Metal Ions: Differential Dioxygen Activation Assisted by Intramolecular Hydrogen Bonds. J Am Chem Soc. 2006;128:15476–15489. doi: 10.1021/ja063935+. [DOI] [PubMed] [Google Scholar]; (c) Singh UP, Babbar P, Tyagi P, Weyhermuller T. A Mononuclear Cobalt(II) Hydroxo Complex: Synthesis, Molecular Structure, and Reactivity Studies. Transition Met Chem. 2008;33:931–940. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.