

Abstract
There is a need for improved methods to image genetically engineered cells, including immune cells used for cell-based therapy. Given the genetic manipulation inherent to gene therapy, the use of a reporter protein is a logical solution and positron emission tomography (PET) can provide the desired sensitivity and spatial localization. We developed a broadly applicable PET imaging strategy based on the small bacterial protein E. coli dihydrofolate reductase (Ec dhfr) and its highly specific small molecule inhibitor, trimethoprim (TMP). The difference in TMP affinity for bacterial compared to mammalian DHFR suggests that a TMP radioligand would have a low background in unmodified mammalian tissues and high retention in Ec dhfr engineered cells, providing high contrast imaging. Here, we describe the in vitro properties of [11C]TMP and show over 10-fold increased signal in transgenic Ec dhfr cells compared to control. In a mouse xenograft model, [11C]TMP rapidly accumulated in Ec dhfr carrying cells within minutes of intravenous administration. Moreover, [11C]TMP can identify less than a million xenografted cells in a small volume in tissues other than the abdominal compartment. This limit of detection is a clinically relevant number and bodes well for clinical translation especially given that [11C]TMP is an isotopologue of clinically approved antibiotic.
Keywords: imaging, reporter gene, cell therapy, PET, molecular imaging, chemical biology
Graphical Abstract
Sellmyer and colleagues developed a new positron emission tomography (PET) reporter gene system based on the bacterial dihydrofolate reductase protein paired with [11C]trimethoprim. This technology shows high sensitivity and holds promise as a clinically translatable imaging tool for gene and cell therapies.
Introduction
As gene and cell therapy becomes routine in clinical practice, there is an imperative for improved methods to image genetically engineered cells, including immune cells used for cell-based therapy such as chimeric antigen receptor (CAR) T cells.1, 2, 3 Clinical questions such as whether the genetically modified cells have reached the target tissue have no simple answer and no single agent provides a facile and repeatable imaging tool for long term tracking of engineered cells for cancer and other diseases.4, 5 Given the genetic manipulation inherent to gene therapy, a genetic imaging handle/reporter protein is a logical solution and positron emission tomography (PET) can provide the desired sensitivity and spatial localization.6 Current nuclear imaging reporter proteins such as sodium iodide symporter (NIS) and herpes virus thymidine kinase (HSV1-tk) are limited by size, off-target effects, lack of trapping of the radioisotope (NIS), and immunogenicity (HSV1-tk).5, 7 Further, other agents targeting endogenous human receptor proteins also suffer from off-target binding, relatively poor sensitivity (limited to detecting 1 million cells in a small volume), or variable protein expression dependent on the type of cell transduced.8
Two of the most influential advances in modern biomedical imaging have borrowed proteins from another kingdom or phylum. The GFP from the jellyfish (A. victoria) has changed molecular and cellular biology enabling greater understanding cellular and protein function. Luciferase from the North American firefly (P. pyralis) has changed preclinical small animal imaging, leading to new insights on cancer, infection, and the immune system.9 We developed a PET strategy geared toward human imaging based on the bacterial protein E. coli dihydrofolate reductase (Ec dhfr) and its highly specific small molecule inhibitor, trimethoprim (TMP), which is often used clinically in combination with sulfamethoxazole (Bactrim/Septra, TMP-SMX) as an antimicrobial agent for treatment of bacterial infections.10 dhfr protein is an enzyme that reduces dihydrofolate to tetrahydrofolate and is critical to several steps in prokaryotic metabolism.11 The bacterial binding site of Ec dhfr is significantly different than the mammalian binding site, resulting in greater than three to four orders of magnitude difference in TMP binding affinity between the two proteins.11, 12 Thus, we suspected that TMP and radiochemical derivatives of TMP would be relatively orthogonal (biologically inert) in mammalian tissues, allowing accumulation in Ec dhfr engineered cells. As a transgene, Ec dhfr may also have potential for immunogenicity, however, several features of the protein may make that less likely, including that the protein size is far smaller (18 kDa) than HSV1-tk, leading to fewer possible immunogenic epitopes (see the Discussion). Additionally, the combination of radiolabeled TMP paired with Ec dhfr reporter protein has many attractive features such as a chemically modifiable small molecule, an approved and widely used parent compound that may be rapidly translated to the clinic without necessarily needing investigational new drug (IND) approval, and genetic portability of the transgene.13, 14
We describe the synthesis of 5-(3,5-dimethoxy-4-([11C]methoxy)benzyl)pyrimidine-2,4-diamine ([11C]TMP), characterize in vitro and in vivo properties of [11C]TMP, as well as show the biodistribution and sensitivity of [11C]TMP for Ec dhfr carrying cells (the lower limit of the number of Ec dhfr cells detectable) in a rodent model. These studies indicate that TMP radiotracers are a potential solution to allow non-invasively imaging of less than 1 million engineered cells in a small volume and advances current PET reporter gene technologies with broad basic science and clinical potential.8
Results
[11C]TMP was synthesized from phenol precursor using [11C]methyl iodide, and the precursor was prepared via a known method with yields of 50%–60% for both steps of the reaction (Figure 1A).13 There was high radiochemical purity (>98%) and specific activity ranged from 500–1,000 Ci/mmol. To assess [11C]TMP binding to recombinant Ec dhfr protein, a dot blot assay was performed that showed protein concentration-dependent uptake that was completely blocked with cold TMP compound (10 μM, Figure S1).
Figure 1.

Synthesis of [11C]TMP, Microscopy, Western Blot, and In Vitro Cellular Uptake in HCT116 and HEK293 Cell Lines
(A) TMP is converted to the phenol with heat and hydrobromic acid (HBr). The 11C methyl iodide under basic conditions with heating allows conversion to [11C]TMP that is then purified by HPLC. Typical yields for each step were 50%–60% and end product specific activity was 500–1,000 Ci/mmol. (B) Fluorescence microscopy of Ec-dhfr-YFP retrovirally transduced HCT116 and HEK293 cells. Scale bar, 20 μm. (C) Immunoblot probing for YFP of Ec-dhfr-YFP fusion proteins with GAPDH loading control. The expected fusion protein molecular weight is 45 kDa. (D) In vitro cell uptake studies. HCT116 cells and HEK293 cells were incubated with of [11C]TMP (2 million CPM) in PBS for 30 min, washed ×2, and assayed for uptake with and without cold TMP (10 μM). The experiment was performed in triplicate with a single experiment shown. Error bars, SD (n = 3).
Transgenic mammalian cell lines carrying Ec dhfr were made using the Phoenix amphotrophic retroviral transduction and selected for Ec-dhfr-YFP expression using yellow fluorescence-activated cell sorting (FACS). Bright field and fluorescent microscopy confirmed presence of the transgene in human embryonic kidney (HEK293) cells and human colon carcinoma (HCT116) cells and western blotting confirmed the correct molecular weight (Figures 1B and 1C, 45 kDa).
Radiotracer cell uptake studies were completed using both cell lines. Control (low uptake) and Ec dhfr (high uptake) HEK293 and HCT116 cells (8 million) were incubated with [11C]TMP as well as with competing cold TMP (10 μM) to block radiotracer uptake (Figure 1D). There was over 10-fold increased signal at the time of assay with HEK293 cells and 3-fold increased signal over background with HCT116 cells, which correlated with differences in Ec dhfr protein expression on the western blot.
Methotrexate (MTX) is a high affinity inhibitor of mammalian DHFR with sub-nanomolar dissociation constant that also binds to Ec dhfr (KI 2.4 nM).15, 16 Thus, MTX could be used to test the selectivity of uptake of [11C]TMP for Ec dhfr. Co-treatment with MTX (10 μM) uptake experiments showed no change dynamic range of uptake in the short incubation period (30 min) and only marginally affected the absolute uptake numbers in control HEK293 cells (Figure S2).
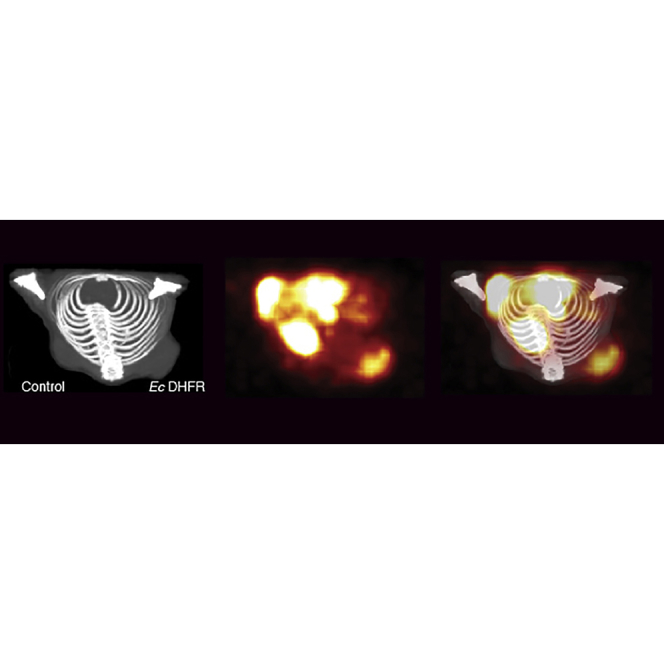
After successful in vitro uptake experiments, HCT116 tumor cells were xenografted into the posterior back/shoulder subcutaneous tissues of nude mice to assess in vivo tracer distribution and uptake. Tumors were grown over 10 days and animals were maintained on a low folate diet. Mice were anesthetized and imaged with small animal PET/computed tomography (CT). A representative image is shown at 90 min (5 min bin) after [11C]TMP injection that shows on-target signal coming from the Ec dhfr carrying tumor cells (Figure 2A). A time activity curve shows rapid and dynamic identification of Ec dhfr carrying cells and over 3-fold signal above background with HCT116 cells compared to control cells, which correlated with in vitro uptake data in HCT116 cells (Figure 2B). Other tissues with rapid uptake included the kidneys and bladder. There is decreased specific signal from dhfr tumors after pre-treatment with cold oral TMP competition (0.2 mg/mL via the drinking water, Figure 2C).
Figure 2.

Small Animal Micro PET/CT of Ec dhfr Tumors with [11C]TMP
(A) HCT116 tumors were xenografted subcutaneously (10 million cells) to the shoulders of nude mice. The tumors were grown for 10 days and imaged using small animal PET followed by CT. A representative animal is shown at imaging time point 85–90 min after ∼1 mCi of [11C]TMP injected i.v. (B) Quantification of mean SUV from in vivo experiment as in (A). Error bars, SD (n = 3). (C) Fold change of mean SUV with and without oral TMP block.
A biodistribution analysis was performed ex vivo 90 min after injection and subsequent imaging. Supporting the imaging data, Ec dhfr tumors showed over 3-fold increased signal compared to control (Figure 3). A representative animal pretreated with oral TMP was imaged 90 min after [11C]TMP injection and shows little specific uptake in the Ec dhfr tumor. To better model the adult human organ dose, a dosimetry experiment was performed. BALB/c mice were sacrificed at 2, 15, and 45 min. These data showed low retention of tracer (<2% ID/g) in the following tissues at 45 min: blood, heart, muscle, lung, pancreas, spleen, skin, brain, bone, and stomach (Figure S3). Estimated human dosimetry in a female patient using OLINDA/EXM software suggests that the kidneys and bladder are the most likely critical, dose-limiting organs (Figure S4).
Figure 3.

In Vivo Biodistribution of [11C]TMP
Biodistribution studies were completed 90 min after injection of [11C]TMP. Dissected tissues were analyzed with a gamma counter. Additional mice were pretreated with oral TMP (0.2 mg/mL final concentration) in the drinking water for 2 days prior to [11C]TMP injection. Error bars, SD (n = 3).
The primary route of metabolism was renal. Given that urinary tract infections are a major clinical use of TMP-SMX, a large amount of uptake from the kidneys was expected, ∼40% ID/g. [11C]TMP is excreted in the urine predominantly as the parent compound as measured by radio-thinlayer chromatography (Figure S5), and there is rapid accumulation of radiotracer in the bladder (Figure S6). The bladders of the animals appear distended by 45 min, as no urination occurred under anesthesia.
A smaller component of metabolism was the hepatobiliary system, with the liver uptake of 4.9% ID/g. Hepatobiliary excretion of the tracer in bile was supported by segmental areas of increased signal in the small bowel after GI tract explant (Figure S7). In fact, the signal in the small bowel appeared more concentrated than in the cecum, despite the fact that cecum contains the site of highest levels commensal bacterial colonization.17
Other areas of interest include the brain and thyroid. The uptake in the brain was low, measuring ∼0.2% ID/g, noting that earlier studies have shown TMP is capable of crossing the blood-brain barrier in rodents.14 The thyroid showed modest uptake, 3.2% ID/g, noting that animal studies have shown that chronic use of TMP-SMX was associated with thyroid dysregulation.18, 19
The minimum number and concentration of cells detectable by imaging in a particular tissue/volume is crucial information, especially for investigators interested whether adoptive cell therapies are reaching the target tumor. Thus, cell sensitivity experiments were performed to assess the minimum number of cells necessary for detection of radiotracer signal. Given the strong in vitro signal uptake, HEK293 cells carrying Ec dhfr transgene were diluted (3 M, 300 K, and 30 K) and injected in 150 μL of Matrigel matrix and imaged the next day. A representative mouse image is shown of the shoulder area (Figure 4A, 300 K), and ex vivo analysis by gamma counting supported imaging detection of 300 K cells in a small volume relative to muscle (p < 0.05, Figure 4B).
Figure 4.

Sensitivity of Detection of Transduced Ec dhfr Reporter Cells
(A) HEK293 dhfr cells were injected subcutaneously in Matrigel (150 μL) at concentrations of 3 × 106, 3 × 105, and 3 × 104 cells 24 hr prior to radiotracer administration and PET imaging. The Matrigel was harvested and measured on a gamma counter. Error bars, SEM (n = 3). There was a statistically significant difference between muscle control and 3 × 106 and 3 × 105 cell grafts (p < 0.05 Student’s t, two-tailed). (B) Representative images in three planes of the shoulder area containing 3 × 105Ec dhfr cells.
Discussion
We describe the synthesis, in vitro, and small animal testing of a clinically applicable PET radiotracer based on the synthetic, anti-folate antibiotic TMP. PET imaging provides volumetric localization and sensitivity, to enable monitoring of gene/cell therapy in humans.20 For applications such as stem cell or cancer immunotherapies in patients, clinicians are interested in questions such as: Are the cells getting to where they are supposed to go? How many cells are there? Are there sites of off-target accumulation? How long are the cells present in the desired location or in the tumor? These questions require tools that are sensitive, longitudinal, and non-immunogenic. To date, there is no single, facile reporter gene that allows monitoring transgenic cells in humans. In that light, Moroz et al.8 recently investigated several candidates for the best PET reporter genes, finding that meta-[18F]-fluorobenzylguanidine ([18F]MFBG) can detect human norepinephrine transporter (hNET) transduced cells down to 105 cells. hNET, however, has been hampered by widely variable transduction efficiencies and protein expression.21 HSV-tk has a reasonable sensitivity in a mouse model, detecting 106 cells, and has been applied in humans, but requires immune suppression to prevent immune clearance of cells containing the reporter gene.8, 22 [11C]TMP compares favorably to these other reporter genes. When imaging the next day after implantation, we were able to detect less than 1 million cells and potentially as few as 3 × 105 implanted cells. Current cell therapies typically implant greater than 107 cells, which implies that even if fewer than 10% of the cells reach the target tissue, [11C]TMP may be able to detect the appropriate homing of the cells.1 This sensitivity is expected to improve with longer-lived isotope derivatives of TMP, such as 18F derivatives that are currently under development, where a longer time from injection to imaging should allow continued background clearance of the tracer.
There are several advantages to using [11C]TMP for monitoring cell therapy. The synthetic route to [11C]TMP is two-steps and facile.13 The precursor is inexpensive and widely available. TMP as an antibiotic is often used clinically in combination with sulfamethoxazole and thus the toxicity profile is well established. As [11C]TMP is the isotopologue of the unlabeled antibiotic, it can be translated to the clinic rapidly through an institutional Radioactive Drug Research Committee (RDRC) pathway, noting that 18F derivatives may not have the same regulatory advantage.
[11C]TMP shows concentration-dependent specific cell uptake using an in vitro dot blot assay with recombinant protein and robust uptake in cells carrying YFP-Ec dhfr fusions. A 10-fold signal increase was seen in Ec dhfr compared to control cells, even after five half-lives of radioactive decay had occurred. The rapid wash conditions required during cell uptake experiments to remove unbound ligand and the short half-life inherent to 11C in vitro experiments made these experiments technically challenging and suggest that TMP radiotracers labeled with longer-lived isotopes may improve specific signal.
As expected, there was little [11C]TMP uptake in control cells in vitro. Co-treatment with cold TMP blocked specific uptake in Ec dhfr cells and co-treatment with inhibitory concentration of MTX showed no change in the fold-induction of uptake over the short incubation period and only minimal change in overall uptake in HEK293 control and Ec dhfr cells. This minimal change in overall uptake likely was due to a combination of cellular toxicity and competitive binding of Ec dhfr with a saturating dose of MTX (10 μM). No significant TMP binding of mammalian DHFR was observed. These experiments affirmed the known specificity of TMP for Ec dhfr over mammalian DHFR.23, 24
In a mouse xenograft model, there was rapid, specific radiotracer uptake in Ec dhfr containing HCT116 tumor cells highlighting that Ec dhfr/TMP radiotracers may be used as a PET reporter gene in vivo on a timescale compatible with 11C imaging. Tissues with very low background signal in dosimetry experiments included blood, heart, muscle, lung, spleen, skin, brain, bone, stomach, and pancreas, suggesting that homing to transgenic cells to these tissues would be potential areas for high contrast imaging. Competition with cold TMP showed a decrease in Ec dhfr tumor:control uptake ratio, again arguing for specificity of the tracer. The drop in absolute uptake of many tissues including the kidneys and thyroid with treatment of oral TMP could suggest increased urinary excretion of tracer and further testing will be performed to test this conclusion.
An additional advantage of [11C]TMP is that it appears to cross the blood-brain barrier in prior reports.14 This could be important for visualizing transgenic cells in areas of high background for other PET reporter proteins derived from nervous system tissues (e.g., hNET and D2R) or in cases where the small molecule probe does not cross the blood-brain barrier.21 Conversely, the abdominal compartment may be a sub-optimal region for [11C]TMP imaging as the tracer is cleared primarily through the renal excretion. In our mouse experiments, there is rapid bladder accumulation with an intravenous (i.v.) dose and low retention in the blood pool. Some of this prompt excretion may be related to the fact that mice have 8- to 10-fold circulating folate levels compared to humans.25 This excess of competing substrate may mildly affect Ec dhfr active site occupancy, which could affect the overall signal to noise, noting that inhibitors of dhfr enzymes bind to the enzyme 103 more strongly than dihydrofolate itself.16 Nonetheless, mice were maintained on low folate, antibiotic free diets for 2 weeks prior to experimentation to better approximate human folate levels. For adult human after a single oral dose of TMP, ∼50% of the tracer dose was in the urine by 24 hr and certainly scheduled hydration/urination would be part of a human protocol.26
There is a small amount of hepatobiliary metabolism of [11C]TMP that correlates with prior pharmacokinetic and metabolic analysis that showed TMP metabolism via O-demethylation.27 This appears to be minor pathway of TMP metabolism and of limited concern with short-live isotopes.
Prior to initiating these experiments, we were interested in whether the gastrointestinal tract contents, with high concentrations of bacteria (1011 CFU/mL), would act as a “molecular sink,” binding the majority of the [11C]TMP administered dose. At least with i.v. administration of [11C]TMP, however, there is relatively little uptake in bacteria in the large bowel (cecum/colon) on this short timescale. Rather, the signal in the gastrointestinal (GI) tract appears to be related to hepatobiliary excretion as seen by segmental, increased radiotracer signal in the small bowel compared to the large bowel in autoradiography experiments of explanted alimentary tracts. Taken together, both hepatobiliary metabolism and renal excretion of [11C]TMP may limit the abdominal compartment imaging, although some of this background signal may be mitigated by imaging timing.
An open question is whether using a bacterial protein in transgenic cells in human subjects will elicit an immune response as previously found with HSV-tk.7 Ec dhfr has several features that may engender less potential for immunogenicity. Ec dhfr is a small protein (18 kDa) and thus has fewer potential epitopes for antigen presentation compared to HSV-tk (43 kDa). Ec dhfr has 47% similarity to mammalian DHFR whereas HSV1-tk has 17% similarity to human cytosolic thymidine kinase (EMBOSS NEEDLE Pairwise Protein Alignment, Figure S8). Finally, humans live in a commensal state with GI bacteria, which may lead to tolerance of bacterial epitopes in comparison to viral epitopes. While immune clearance of these cells is possible and will be monitored closely during future first-in-human studies, a potential solution could be humanization of the Ec dhfr protein.
Additional active efforts in our lab include using TMP-based radiotracers to image pathologic bacteria. This use of [11C]TMP could be immediately clinically relevant especially in cases where bacterial infection cannot be discerned from bland inflammation or tumor based on anatomical imaging or other standard nuclear imaging techniques. Alternatively, [11C]TMP could be used to stage or assess response to therapy in cases of a chronic infection such as cystic fibrosis or osteomyelitis.
Conclusions
The radiosynthesis and in vivo application of [11C]TMP for PET reporter gene imaging is a straightforward solution for a quantitative, sensitive tool for imaging gene and cell therapies in humans. [11C]TMP may be rapidly translated to the clinic as the toxicity profile of the antibiotic is well known, and TMP radiotracers may have important impact on our understanding of gene and cell therapies for diverse pathologic processes.
Materials and Methods
Cell Culture and Transductions
HEK293 and HCT116 cells (American Type Cell Culture) were cultured in complete media: DMEM with 10% fetal bovine serum (Invitrogen), 2 mM glutamine, 100 U/mL penicillin, and 100 mg/mL streptomycin (all from GIBCO). Cells were maintained in a humidified incubator at 37°C. Yellow fluorescent protein (YFP)-Ec-dhfr cells (“dhfr” cells) were made by introducing a Ec-dhfr-YFP fusion gene cloned into pBMN Ec-dhfr-YFP (gift of the Wandless lab) used to generate amphotrophic retrovirus (gift of the Nolan lab). HEK293 and HCT116 cells were incubated with retrovirus and polybrene (6 μg/mL) for 4 hr at 37°C, passaged, and selected with YFP fluorescence-activated cell sorting (Becton Dickinson [BD]).
[11C]TMP Synthesis
Synthesis of 4-((2,4-Diaminopyrimidin-5-Yl)Methyl)-2,6-Dimethoxyphenol
4-((2,4-diaminopyrimidin-5-yl)methyl)-2,6-dimethoxyphenol (TMP-OH) was prepared similarly to a known method.13 Briefly, TMP (2.0 g, 6.88 mmol) was suspended in 48% aqueous hydrobromic acid (25.0 mL) and stirred for 20 min at 95°C. The reaction mixture was cooled down to room temperature, and the 50% (v/w) NaOH in water (5.94 mL) was added. The reaction mixture was then cooled to 4°C and the resulting crystals were filtered and rinsed with cold water. The crystals were dissolved in hot water (100 mL) and neutralized with 1 N NaOH solution and recrystallized at 4°C. The crystals were filtered, rinsed with cold water, and dried under high vacuum to give a pink solid (1.36 g, 71.6%). 1H NMR (360 MHz, DMSO-d6); 8.06 (-OH), 7.45 (s, 1H), 6.48 (s, 2H), 5.99 (s, 2H), 5.63 (s, 2H), 3.71 (s, 6H), and 3.41 (s, 2H).
Synthesis of 5-(3,5-Dimethoxy-4-([11C]Methoxy)Benzyl)Pyrimidine-2,4-Diamine
[11C]CO2 was produced by 14N(p,α)11C reaction using an IBA Cyclone 18 (Louvain-la-Neuve), [11C]CH3I was synthesized from this using a gas-phase module (GE Healthcare). [11C]CH3I was trapped in a mixture of TMP-OH (0.5 mg, 1.80 μmol) and 5 N NaOH aqueous solution (4 μL, 20 μmol) in DMF (200 μL) at room temperature. The reaction mixture was heated at 70°C for 5 min and diluted with high performance liquid chromatography (HPLC) mobile phase (1.8 mL, 12% EtOH in 0.01 M phosphate buffer, pH = 3.0). The solution was then injected onto a HPLC equipped with a semipreparative column (Phenomenex Gemini 5 μ, C18 110Å, New Column 250 × 10 mm) and eluted with HPLC mobile phase as above at a flow rate of 3 mL/min. The desired fraction eluted at 13–14 min was collected and used for biologic evaluation without concentration. For specific activity determination, an aliquot of [11C]TMP was injected onto an HPLC equipped with an analytical column (Agilent XDB-C18, 5 μ, 150 × 4.6 mm) and eluted with a 15% CH3CN:85% water with 0.1% TFA at a flow rate of 1 mL/min (tR = 5.1–5.2 min). Specific activity determinations were carried out by comparing the UV peak area (wavelength: 230 nm) of the desired radioactive peak with those of different concentrations of TMP by HPLC. An aliquot of [11C]TMP was coinjected with TMP into an HPLC system to confirm its identity.
In Vitro Assays
Dot Blot
Nitrocellulose membrane (Abcam) was cut to size and recombinant, bacterially produced Ec dhfr (gift of the Wandless lab) was dotted at 2 μL at various concentrations on to the membrane. The spots were dried and then blocked with 5% cold milk in TBS-T (20 mM Tris HCl, 150mM NaCl, pH 7.5, 0.05% Tween 20) and incubated with [11C]TMP (2 million counts per minute [CPM]) for 30 min. The blot was then washed with TBS ×2 and exposed to a phosphor plate (GE) and imaged on a Typhoon laser scanner (GE).
Microscopy
Ec-dhfr-YFP cells plated in a 6-well plate (1 × 105 cells/well) were incubated overnight at 37°C. Live cell imaging was performed with fluorescence microscope using a GFP/YFP filter and phase contrast (Zeiss).
Western Blot
The cells were lysed in radioimmunoprecipitation assay buffer (50 mM Tris, 150 mM sodium chloride, 1.0 mM EDTA, 1% Nonidet P40, and 0.25% SDS [pH 7.0]), supplemented with complete protease inhibitor cocktail (Roche) and phosphatase inhibitor cocktail 1 (Sigma Chemical). The cells were sonicated briefly, centrifuged at 13,000 × g for 20 min at 4°C and the supernatant collected. The protein concentration was determined using a Bio-Rad Dc protein assay kit (Bio-Rad Laboratories). Lysates containing 30 μg of protein were run on a 4%–20% acrylamide gel and transferred to a PVDF membrane using the Trans-Blot Turbo Transfer System (Bio-Rad Laboratories). The PVDF membrane was incubated with Odyssey blocking buffer (Li-Cor Biotechnology) for 1 hr at room temperature, then overnight with a mouse monoclonal antibody recognizing YFP (catalog #632381, Clontech) at a 1:1,000 dilution at 4°C, and finally with the secondary antibody, IRDye 680RD goat anti-mouse IgG (Li-Cor Biotechnology) at a 1:15,000 dilution. The same blot was incubated overnight with goat anti-GAPDH antibody (Santa Cruz) at a 1:300 dilution at 4°C and then with IRDye 680RD donkey anti-goat IgG secondary antibody (Li-Cor Biotechnology) at a 1:15,000 dilution. The signals were detected and quantified using the Odyssey CLx Infrared Imaging System (Li-Cor Biotechnology).
Cellular Uptake Studies
Ec dhfr or control cells were incubated with [11C]TMP (2 million CPM) in Opti-Mem (GIBCO) for 30 min at 37°C with and without cold TMP (10 μM, Sigma) or methotrexate (10 μM, Sigma). The cells were then washed in cold PBS and centrifuged to pellet the cells ×2. The cells were resuspended and assayed on a gamma counter (Perkin Elmer).
Mouse Models
Tumor Uptake
CD1 nu/nu mice (Charles River) received subcutaneous dorsal, shoulder injections of 8 × 106 HCT116 control or Ec dhfr cells. After 10 days growth, tumors were palpable and animals were anesthetized (2% isoflurane), given a tail vein injection of [11C]TMP (∼0.5–1 mCi/mouse) and placed on the warmed stage for small animal PET imaging. For competition experiments, mice were treated with TMP (0.2 mg/mL final concentration) in the drinking water for 2 days prior to injection. CT images were acquired with a micro CT (ImTek). Uptake was measure from elliptical region of interest (ROI) and mean standardized uptake value (SUV) was calculated using AMIDE software (Amide version 1.0.4) (http://www.amide.sourceforge.net).
Sensitivity Experiment
CD1 nu/nu mice were subcutaneously xenografted with 3 million, 300,000 and 30,000 HEK293 Ec dhfr cells and 3 million HEK293 control cells in 150 μL volume of Matrigel (Corning). The next day, animals were administered [11C]TMP IV (∼0.2–0.5 mCi/mouse) and imaged with CT prior to sacrifice and ex vivo analysis of the Matrigel tissues. Statistical analysis was performed with Prism (GraphPad) and statistical significance measure by (p < 0.05) using a paired Student’s t test. All animal studies were completed with institutional IACUC approval (protocol #80547).
Estimated Human Dosimetry
Biodistribution data from female BALB/c mice were used to estimate human dosimetry in an adult female human model that was predefined in OLINDA/EMX 1.1 (VU e-Innovations). Kinetic data from time points (2, 15, and 45 min) were fitted as percent-injected dose/organ over time. By fitting the kinetic data using %ID/organ we assume the [11C]TMP distribution would be relative to human and thus did not apply a scaling factor accounting for organ weight to subject total body weight between mouse and human.
Author Contributions
M.A.S. designed, performed research, and analyzed data. I.L. synthesized [11C]TMP. C.H., B.L., and C.Z. coordinated, performed research, and analyzed data. D.M. and R.H.M. designed the research and analyzed data. All authors wrote the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgments
We thank Joel Karp and Eric Blankemeyer of the UPenn Small Animal Imaging Facility, the Chris Contag and Tom Wandless labs (Stanford), Shihong Li and Hsiaoju Lee and the cyclotron staff, and Mehran Makvandi for reagents and helpful discussions. M.A.S. was supported by the RSNA Resident Research Award and the UPENN NIH T32 Radiology Research Training Grant (5T32EB004311-12). We received additional support from the Department of Energy Translational Radiochemistry Program (DE-SE0012476).
Footnotes
Supplemental Information includes eight figures and can be found with this article online at http://dx.doi.org/10.1016/j.ymthe.2016.10.018.
Contributor Information
Mark A. Sellmyer, Email: mark.sellmyer@uphs.upenn.edu.
Robert H. Mach, Email: rmach@mail.med.upenn.edu.
Supplemental Information
References
- 1.Kalos M., Levine B.L., Porter D.L., Katz S., Grupp S.A., Bagg A., June C.H. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klebanoff C.A., Rosenberg S.A., Restifo N.P. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat. Med. 2016;22:26–36. doi: 10.1038/nm.4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Waerzeggers Y., Monfared P., Viel T., Winkeler A., Voges J., Jacobs A.H. Methods to monitor gene therapy with molecular imaging. Methods. 2009;48:146–160. doi: 10.1016/j.ymeth.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 5.Collins S.A., Hiraoka K., Inagaki A., Kasahara N., Tangney M. PET imaging for gene & cell therapy. Curr. Gene Ther. 2012;12:20–32. doi: 10.2174/156652312799789271. [DOI] [PubMed] [Google Scholar]
- 6.Gambhir S.S. Molecular imaging of cancer with positron emission tomography. Nat. Rev. Cancer. 2002;2:683–693. doi: 10.1038/nrc882. [DOI] [PubMed] [Google Scholar]
- 7.Traversari C., Marktel S., Magnani Z., Mangia P., Russo V., Ciceri F., Bonini C., Bordignon C. The potential immunogenicity of the TK suicide gene does not prevent full clinical benefit associated with the use of TK-transduced donor lymphocytes in HSCT for hematologic malignancies. Blood. 2007;109:4708–4715. doi: 10.1182/blood-2006-04-015230. [DOI] [PubMed] [Google Scholar]
- 8.Moroz M.A., Zhang H., Lee J., Moroz E., Zurita J., Shenker L., Serganova I., Blasberg R., Ponomarev V. Comparative analysis of T cell imaging with human nuclear reporter genes. J. Nucl. Med. 2015;56:1055–1060. doi: 10.2967/jnumed.115.159855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prescher J.A., Contag C.H. Guided by the light: visualizing biomolecular processes in living animals with bioluminescence. Curr. Opin. Chem. Biol. 2010;14:80–89. doi: 10.1016/j.cbpa.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 10.Bushby S.R., Hitchings G.H. Trimethoprim, a sulphonamide potentiator. Br. Pharmacol. Chemother. 1968;33:72–90. doi: 10.1111/j.1476-5381.1968.tb00475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schnell J.R., Dyson H.J., Wright P.E. Structure, dynamics, and catalytic function of dihydrofolate reductase. Annu. Rev. Biophys. Biomol. Struct. 2004;33:119–140. doi: 10.1146/annurev.biophys.33.110502.133613. [DOI] [PubMed] [Google Scholar]
- 12.Baccanari D.P., Daluge S., King R.W. Inhibition of dihydrofolate reductase: effect of reduced nicotinamide adenine dinucleotide phosphate on the selectivity and affinity of diaminobenzylpyrimidines. Biochemistry. 1982;21:5068–5075. doi: 10.1021/bi00263a034. [DOI] [PubMed] [Google Scholar]
- 13.Calloway N.T., Choob M., Sanz A., Sheetz M.P., Miller L.W., Cornish V.W. Optimized fluorescent trimethoprim derivatives for in vivo protein labeling. ChemBioChem. 2007;8:767–774. doi: 10.1002/cbic.200600414. [DOI] [PubMed] [Google Scholar]
- 14.Iwamoto M., Björklund T., Lundberg C., Kirik D., Wandless T.J. A general chemical method to regulate protein stability in the mammalian central nervous system. Chem. Biol. 2010;17:981–988. doi: 10.1016/j.chembiol.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lewis W.S., Cody V., Galitsky N., Luft J.R., Pangborn W., Chunduru S.K., Spencer H.T., Appleman J.R., Blakley R.L. Methotrexate-resistant variants of human dihydrofolate reductase with substitutions of leucine 22. Kinetics, crystallography, and potential as selectable markers. J. Biol. Chem. 1995;270:5057–5064. doi: 10.1074/jbc.270.10.5057. [DOI] [PubMed] [Google Scholar]
- 16.Matthews D.A., Alden R.A., Bolin J.T., Freer S.T., Hamlin R., Xuong N., Kraut J., Poe M., Williams M., Hoogsteen K. Dihydrofolate reductase: x-ray structure of the binary complex with methotrexate. Science. 1977;197:452–455. doi: 10.1126/science.17920. [DOI] [PubMed] [Google Scholar]
- 17.Quigley E., Quera R. Small intestinal bacterial overgrowth: roles of antibiotics, prebiotics, and probiotics. Gastroenterology. 2006;130(2 Suppl 1):S78–S90. doi: 10.1053/j.gastro.2005.11.046. [DOI] [PubMed] [Google Scholar]
- 18.Hall I.A., Campbell K.L., Chambers M.D., Davis C.N. Effect of trimethoprim/sulfamethoxazole on thyroid function in dogs with pyoderma. J. Am. Vet. Med. Assoc. 1993;202:1959–1962. [PubMed] [Google Scholar]
- 19.Altholtz L.Y., La Perle K.M., Quimby F.W. Dose-dependant hypothyroidism in mice induced by commercial trimethoprim-sulfamethoxazole rodent feed. Comp. Med. 2006;56:395–401. [PubMed] [Google Scholar]
- 20.Weissleder R., Pittet M.J. Imaging in the era of molecular oncology. Nature. 2008;452:580–589. doi: 10.1038/nature06917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buursma A.R., Beerens A.M., de Vries E.F., van Waarde A., Rots M.G., Hospers G.A., Vaalburg W., Haisma H.J. The human norepinephrine transporter in combination with 11C-m-hydroxyephedrine as a reporter gene/reporter probe for PET of gene therapy. J. Nucl. Med. 2005;46:2068–2075. [PubMed] [Google Scholar]
- 22.Yaghoubi S.S., Jensen M.C., Satyamurthy N., Budhiraja S., Paik D., Czernin J., Gambhir S.S. Noninvasive detection of therapeutic cytolytic T cells with 18F-FHBG PET in a patient with glioma. Nat. Clin. Pract. Oncol. 2009;6:53–58. doi: 10.1038/ncponc1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bolin J.T., Filman D.J., Matthews D.A., Hamlin R.C., Kraut J. Crystal structures of Escherichia coli and Lactobacillus casei dihydrofolate reductase refined at 1.7 A resolution. I. General features and binding of methotrexate. J. Biol. Chem. 1982;257:13650–13662. [PubMed] [Google Scholar]
- 24.Matthews D.A., Bolin J.T., Burridge J.M., Filman D.J., Volz K.W., Kraut J. Dihydrofolate reductase. The stereochemistry of inhibitor selectivity. J. Biol. Chem. 1985;260:392–399. [PubMed] [Google Scholar]
- 25.Leamon C.P., Reddy J.A., Dorton R., Bloomfield A., Emsweller K., Parker N., Westrick E. Impact of high and low folate diets on tissue folate receptor levels and antitumor responses toward folate-drug conjugates. J. Pharmacol. Exp. Ther. 2008;327:918–925. doi: 10.1124/jpet.108.143206. [DOI] [PubMed] [Google Scholar]
- 26.Kremers P., Duvivier J., Heusghem C. Pharmacokinetic studies of co-trimoxazole in man after single and repeated doses. J. Clin. Pharmacol. 1974;14:112–117. doi: 10.1002/j.1552-4604.1974.tb02300.x. [DOI] [PubMed] [Google Scholar]
- 27.Mengelers M.J., Kleter G.A., Hoogenboom L.A., Kuiper H.A., Van Miert A.S. The biotransformation of sulfadimethoxine, sulfadimidine, sulfamethoxazole, trimethoprim and aditoprim by primary cultures of pig hepatocytes. J. Vet. Pharmacol. Ther. 1997;20:24–32. doi: 10.1046/j.1365-2885.1997.00048.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.