

Abstract
Inflammation, which is a highly regulated host response against danger signals, may be harmful if it is excessive and deregulated. Ideally, anti-inflammatory therapy should autonomously commence as soon as possible after the onset of inflammation, should be controllable by a physician, and should not systemically block beneficial immune response in the long term. We describe a genetically encoded anti-inflammatory mammalian cell device based on a modular engineered genetic circuit comprising a sensor, an amplifier, a “thresholder” to restrict activation of a positive-feedback loop, a combination of advanced clinically used biopharmaceutical proteins, and orthogonal regulatory elements that linked modules into the functional device. This genetic circuit was autonomously activated by inflammatory signals, including endogenous cecal ligation and puncture (CLP)-induced inflammation in mice and serum from a systemic juvenile idiopathic arthritis (sIJA) patient, and could be reset externally by a chemical signal. The microencapsulated anti-inflammatory device significantly reduced the pathology in dextran sodium sulfate (DSS)-induced acute murine colitis, demonstrating a synthetic immunological approach for autonomous anti-inflammatory therapy.
Keywords: anti-inflammatory therapy, gene circuit, mammalian cell engineering, synthetic biology
Graphical Abstract
Smole et al. describe an anti-inflammatory mammalian cell device in which inflammatory signals autonomously activate a genetic circuit to produce a therapeutic response. This can be reset externally by a chemical signal. The microencapsulated anti-inflammatory device was shown to ameliorate acute murine colitis.
Introduction
Inflammation is a complex widespread response of multi-cellular organisms that is activated by the innate immune system upon detection of danger signals emanating from pathogenic microorganisms or from a damaged tissue.1 Inflammation is generally beneficial for the organism because it alerts the immune system to eliminate the underlying cause and to restore homeostasis. However, deregulated and excessive inflammation is harmful and may become chronic due to positive feedback mediated by inflammatory cytokines. The immune response is one of the most complex biological subsystems with regulation involving various receptors, mediators, complex feedback mechanisms, and different effector molecules, which orchestrate the appropriate fine-tuning of the response to danger.2, 3 The rapid production of pro-inflammatory cytokines, such as tumor necrosis factor alpha (TNF-α), interleukin 1 beta (IL-1β), IL-6, and several others, is characteristic of autoimmune diseases, including inflammatory bowel disease (IBD), rheumatoid arthritis (RA), systemic juvenile idiopathic arthritis (sJIA), multiple sclerosis, psoriasis, and lupus erythematosus, as well as of autoinflammatory diseases, such as cryopyrin-associated periodic syndrome.4 The prevalence of inflammatory diseases associated with excessive and harmful inflammation is 5%–7%, even in societies with an advanced health care system.5
Non-steroidal anti-inflammatory drugs (NSAIDs) and corticosteroids are widely used and effective anti-inflammatory therapies, yet they can have adverse side effects. Currently, the most advanced clinically used therapies for inflammatory diseases are neutralizers of TNF-α (adalimumab, etanercept, and infliximab) and endogenous cytokine antagonists, such as anakinra (an IL-1β antagonist) that specifically and effectively inhibit the activity of pro-inflammatory cytokines.4 As inflammation is an essential component of the host defense system, long-term systemic suppression can cause adverse side effects, such as a decreased host immune response against infections and an increased risk of cancer development.4 Anti-inflammatory therapy that could match the responsiveness and tunability of the natural cellular response, but could be controlled externally, represents both a challenge and an opportunity for synthetic biology. The strategy according to the principles of synthetic biology is to construct biological systems that are orthogonal to the existing signaling components yet are able to read the inputs from the cellular processes or external signals and produce the physiologically relevant outputs. The modular construction of interacting components allows this type of systems to be adapted to the selected applications. Progress has already been made in this direction, such as engineering of the bacterium Lactococcus lactis to secrete IL-106 or anti-TNF-α nanobody,7 which has been used in situ to locally treat murine colitis. Escherichia coli that autonomously detect gut inflammation through nitric oxide sensing has been reported,8 suggesting the feasibility of more sophisticated inflammation management strategies.
Recently, synthetic biologists have demonstrated several innovative therapeutic strategies based on engineered mammalian cells designed to respond to a physiologically relevant signal in a highly predictable and controllable manner.9 Using microencapsulation technology, such engineered cells could be implanted into a host.10, 11 These cells can act as prosthetic devices, which do not require any host genome modification. Of particular interest are sensor-effector designs that can autonomously detect and treat various pathological conditions, such as a malfunction in urate homeostasis,12 diet-induced obesity,13 and diabetic ketoacidosis.14 Recently, an innovative designer cell device to treat experimental psoriasis by converting the disease-relevant inputs into the therapeutic output was presented.15
The therapeutic challenge in therapy of inflammatory diseases is to detect and suppress inflammatory flare-ups as soon as possible before they are amplified through positive-inflammatory-feedback loops and cause damage. Early intervention could clearly contribute to beneficial clinical outcomes in Crohn’s disease16, 17 or sIJA.18 As a safety and regulatory feature, implantable therapeutic cellular device aimed at treating inflammation should be amenable to external control and capability to rest into the standby state once the inflammation has been resolved. Additionally, autonomous activation of the device could also serve as a sensor of inflammation, reporting the previous inflammatory episode.
The inflammatory response is coordinated by highly complex regulatory networks, which consist of multiple, to some degree redundant inducers, sensors, mediators, and effectors.1 In this study, we constructed a gene regulatory network that captured the required functionalities of a fine-tuned therapeutic device aimed to provide support to the organism to cope with the dysregulated inflammatory response. The introduced regulatory elements comprise several interacting functional modules that form a functional genetic circuit, which can be integrated into and modulate the existing inflammatory response. The prototypic synthetic anti-inflammatory device autonomously detected inflammatory signals at physiologically relevant concentrations and responded by sustainable production of a combination of clinically used anti-inflammatory proteins that suppressed inflammation, namely an anti-hTNF-α antibody in combination with IL-10 or interleukin-1 receptor antagonist (IL-1RA). The anti-inflammatory device was activated upon encounter of inflammatory signals, including endogenous mouse inflammation induced by cecal ligation and puncture (CLP) as well as by a serum sample from sJIA patient, and reset by an external chemical inducer. The genetic device, implemented by implantation of alginate-poly-l-lysine-alginate (alginate-PLL-alginate) microencapsulated engineered cells into the peritoneal cavity of mice, reduced the pathological symptoms in dextran sodium sulfate (DSS)-induced acute murine colitis, demonstrating a novel route of designed immunological intervention.
Results
Design of the Functional Modules of the Synthetic Anti-inflammatory Gene Circuit
To design an efficient therapeutic anti-inflammatory device, we aimed to combine the following characteristics: (1) real-time monitoring of physiologically relevant markers of inflammation; (2) an ability to switch to a therapeutically active (ON) state in response to physiologically relevant concentrations of an inflammatory marker; (3) efficient suppression of inflammation via sustained production of anti-inflammatory effectors triggered by the activation of the device; (4) control mechanism for preventing pre-mature inactivation of the device by the neutralization of inflammation markers by effectors in the immediate vicinity of the therapeutic cells; (5) and an ability to reset the device to the standby state by external signal (e.g., small-molecule inducer) that could potentially also be used in clinical applications applied by a physician.
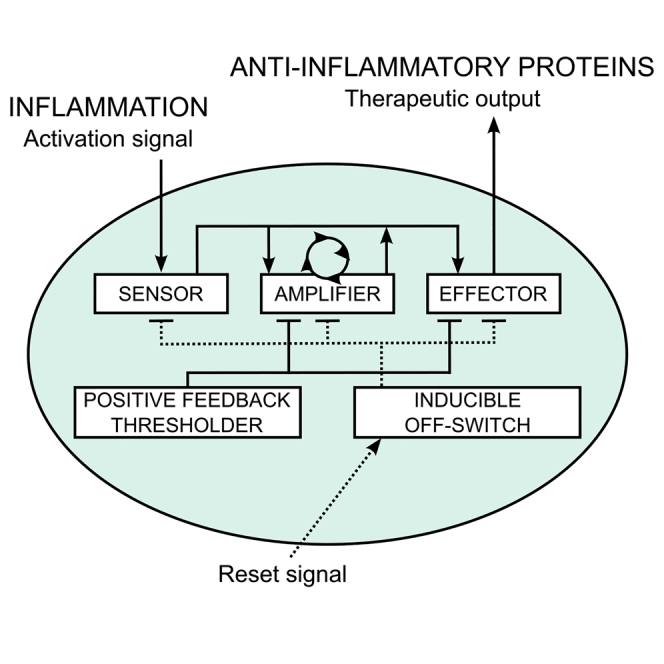
To detect and respond to an inflammatory signal, we developed a biomolecular circuit, which comprises three basic modules: a sensor, an amplifier, and an effector (Figure 1). These modules were interconnected via the well-characterized transcriptional activator Gal4-VP16 (GV16),19 which is orthogonal to mammalian intracellular signaling and transcriptional pathways. To demonstrate the concept of an anti-inflammatory device that could be used in patients with inflammatory diseases, we aimed to develop a system that could sense a common signal of inflammation. As a large fraction of signals from the danger-sensing receptors of the innate and adaptive immune system converge on the activation of the NF-κB transcription factor,20 the NF-κB-responsive sensor was chosen to monitor the inflammatory status of tissue as reported by cellular receptors. To build and validate the sensor, two different NF-κB-responsive elements were first examined. We utilized a validated combination of the κB DNA elements,21 which are specifically recognized by the NF-κB transcription factor, and used the luciferase reporter activity as a readout. Constructs pAS36 (PNF−κB1-PTAL-Luc, Table S1) and pAS37 (PNF-κB2-PTAL-Luc, Table S1) driving the respective PNF-κB-PTAL luciferase expression were introduced into human embryonic kidney 293 T (HEK293T) cells, and construct pAS37 was selected as the sensor design that produced the highest fold induction (FI) upon stimulation with IL-1β or TNF-α (Figure S1A). The ectopic expression of the receptors for IL-1β and TNF-α was not required for the activation in HEK293T cells (Figure S1A), indicating that the endogenous levels were sufficient, similarly as observed in other studies in HEK293 cells.22 Next, to decrease transcriptional leakage in the absence of stimulation, the PTAL minimal promoter (derived from the pISRE-Luc vector; Clontech) was replaced with the PMIN minimal promoter (derived from the pGL4.23[luc2/minP]; Promega) to build a pAS40 construct, which enabled PNF-κB2-PMIN-driven expression of luciferase (PNF-κB2-PMIN-Luc, Table S1). Apart from decreasing transcriptional leakage, this modification also increased the activation amplitude, which increased the FI in response to IL-1β stimulation from 25 to 150 (Figure S1B). Furthermore, the luciferase reporter gene from the pAS40 vector was replaced by a GV16 transcriptional activator to build pAS48 construct (PNF-κB2-PMIN-GV16, Table S1) with an aim of testing whether GV16 could act as a module-connecting mediator. IL-1β stimulation of the HEK293T cells transfected with pAS48 resulted in the activation of the co-transfected reporter plasmid pAS49 (UAS-PMIN-Luc, Table S1) that comprised five consecutive Gal4 binding sites (upstream activating sequence [UAS]), which enabled UAS-PMIN-driven luciferase expression (Figure 2A, left). To guide the experimental work by predicting the responsiveness of the device to stimuli and to determine the optimal transfection amounts, a deterministic model based on the ordinary differential equations (ODEs) was constructed using CellDesigner 4.4 software.23 The mathematical model that used the parameters derived from the experimentally determined values is represented by a constructed state transition diagram of the genetic circuit (Figure S2) and is explained in detail in Supplemental Materials and Methods. As predicted by the model (Figure 2A, right), in both stimulated and non-stimulated settings, the response increased with the increased amount of the transfected sensor. To visualize the activation of the sensor construct in the cells in situ, the mCitrine reporter gene was cloned in-frame with GV16 via the 2A peptide to build the construct pAS97 (TRE-PNF-κB2-PMIN-GV16-myc-2A-mCit, Table S1), which was monitored by confocal fluorescence microscopy. Activation was observed only in the IL-1β-stimulated cells transfected with the respective construct (Figure 2A, middle). Therefore, the sensor construct enabled us to repurpose the endogenous inflammation-characteristic NF-κB signaling for the activation of the anti-inflammatory device.
Figure 1.

Schematic Design of the Synthetic Anti-inflammatory Device
The anti-inflammatory device comprises three basic modules (a sensor, an amplifier, and an effector), which are connected through the transcriptional activator Gal4-VP16 (GV16) and two supporting modules (an inducible OFF-switch and a thresholder). The latter allows additional control of the system performance.
Figure 2.

Characterization of the Functional Modules of the Synthetic Anti-inflammatory Gene Circuit
(A) Validation of the sensor construct (pAS48; PNF-κB2-PMIN-GV16) in HEK293T cells. Left: Dual luciferase test. Middle: confocal fluorescence microscopy (mCherry represents a seeding/transfection control). Right: model simulation of the stimulated and non-stimulated sensor-reporter pairs with increasing sensor amounts 24 hr after stimulation. (B) Validation of the amplifier construct (pAS53; UAS-PMIN-GV16) in HEK293T cells. Left: Dual luciferase test. Middle: confocal fluorescence microscopy (mCherry represents a seeding/transfection control). The amplifier autoactivates at higher amounts of transfected construct (cGV16− conditions and a confocal fluorescence image). Additionally, it amplifies an effect of constitutively expressed GV16 as observed in cGV16+ conditions and from FI calculated from the relative light units (RLU) for each amount of the transfected amplifier according the formula [cGV16+A+/cGV16+A−], where A− represents constitutive expression of GV16 from the pSGVP in the absence of the amplifier (A(0ng) in the RLU graph). Right: model simulation of the amplifier-reporter pairs with increasing amplifier amounts after 24 hr. The effect of cGV16 was examined by setting the particle number of GV16 at t = 0 to 1 × 106 for the cGV16+ simulation. (C) Functional circuit, composed of all the basic elements, except for the thresholder, tested in HEK293T cells. Left: dual luciferase test, reporter pAS51; TRE-UAS-PMIN-Luc. Right: model simulation and parameter scan. The graph and heat maps depict the level of the device activation in stimulated and non-stimulated setting with increasing sensor and amplifier amounts after 24 hr. The amount of each respective construct was increased from 0 to 10 ng, in 0.1-ng increments. Fold induction was calculated by dividing the effector particle number after 24 hr of stimulation with the corresponding effector particle number of non-stimulated circuit after 24 hr for each simulation. S, sensor; A, amplifier; FI, fold induction. Error bars indicate SD (n = 4).
Once the activation of the device was optimized, we then aimed to achieve sustained high-level production of anti-inflammatory proteins. This production should be persistent even after the input signal from the sensor, delivered by the inflammatory cytokines in the close proximity of the sensing cells, had decreased. The sustainability is important to prevent pre-mature shutdown of the production of anti-inflammatory therapeutics that neutralize the inflammatory cytokines (i.e., the input signal) in the immediate vicinity of the device. The sustainability of the system activation was achieved by incorporation of the positive-feedback amplifier module involving the transcriptional activator GV16, which activates its own transcription by binding to the UAS located upstream of the PMIN (Figure 1). The mathematical model predicted that the amplifier switches to an activated state once a certain level of GV16 has been achieved due to the positive-feedback loop (Figure 2B, right). To test the functionality of the amplifier construct, HEK293T cells were transfected with increasing amounts of the pAS53 construct (UAS-PMIN-GV16, Table S1). Based on the mathematical model predictions, we reasoned that, when the leakage surpasses the certain level, auto-activation would be observed. By monitoring pAS49 reporter activity, this turned out to be in the interval between 1 and 10 ng of pAS53 per well, which is in accordance with the mathematical model. We have also demonstrated that the amplifier augmented the low-level signal of the constitutively expressed GV16 (plasmid pSGVP) up to 4-fold in the relevant range where the amplifier itself was not yet auto-active (Figure 2B, left). The auto-activation of the amplifier-2A-tagBFP construct was also observed directly by confocal fluorescence microscopy (pAS98; TRE-UAS-PMIN-GV16-myc-2A-tagBFP, Table S1) (Figure 2B, middle). Notably, the 2A fusion of mCitrine or tagBFP did not affect the activity of the respective constructs (Figure S1C). Therefore, by including a positive-feedback amplifier module, our device gained the ability to sustain high-level synthesis of therapeutic anti-inflammatory proteins.
To enable the external control over the activity of the device, an inducible OFF-switch, implemented as the rtTR-KRAB repressor,24 was introduced. This switch reset the device by external chemical signal and shut down production of the anti-inflammatory effectors when they were no longer required. The orthogonal repressor binds to the Tet-binding sites (Tet-responsive element [TRE]) and silences gene expression only in the presence of doxycycline (Dox). The repression effect of KRAB domain spreads several kilobases upstream and downstream of its binding site.25 Thus, we positioned the TRE into all three modules upstream of the other regulatory elements to avoid disrupting the existing transcriptional architecture, which could potentially impair the function of the fine-tuned constructs. The constitutively expressed rtTR-KRAB (pAS58; PCMV-rtTR-NLS-KRAB, Table S1) repressed the reporter construct pAS51 carrying the TRE-UAS-PMIN-driven luciferase expression unit (TRE-UAS-PMIN-Luc, Table S1), which was activated by the GV16 (plasmid pSGVP19). Efficient and dose-dependent repression was achieved, whereas in the absence of external reset signal the rtTR-KRAB (pAS58) alone did not significantly influence the system activation (Figure S1D, left). Similarly, we reset both the activated sensor and the amplifier circuit (Figure S1D, middle and right), suggesting a successful implementation of the inducible OFF-switch function.
After testing and optimizing all the modules, they were incorporated into a functional device. Based on the mathematical model of the genetic network, we predicted higher leakage of the system at the increased amounts of the transfected sensor and amplifier constructs (Figure 2C, middle and right). Based on those predictions, the ratio of the sensor construct pAS60 (TRE-PNF-κB2-PMIN-GV16-myc, Table S1) encoding TRE-PNF-κB2-PMIN-driven expression of GV16-myc and the amplifier construct pAS62 (pAS62; TRE-UAS-PMIN-GV16-myc, Table S1) encoding TRE-UAS-PMIN-driven GV16-myc expression together with the inducible OFF-switch pAS58 and reporter pAS51 was fine-tuned (Figure 2C, left). The lowest amounts of the sensor and the amplifier constructs were used in all further experiments. As a result, we observed a robust system induction triggered by IL-1β, which was efficiently repressed by the addition of Dox (Figure 2C, left).
Introduction of the “Thresholder” and Sustained System Activity by the Amplifier
It is important to restrict activation of the positive-feedback loop until the signal is sufficiently strong. To decrease the transcriptional leakage of the system and increase the FI, a thresholder module was introduced. The thresholder concept is based on competition between GV16 and the constitutively expressed Gal4 that lacks the VP16 activation domain, implemented as the construct pAS67 (PHSV-TK-G-myc, Table S1; schematic representation in Figure 1), for binding to the UAS operator in the amplifier and effector constructs, which acts as a threshold for the system activation. Using model simulations, we predicted that the constitutively expressed thresholder, which provides the constant amount of the UAS binding domain, should prevent activation of the system until the signal from the sensor surpasses the selected level, defined by the amount of the expressed thresholder (Figure 3A, upper right and bottom). According to the model simulations, the thresholder decreased the unwanted background activity while retaining high amplification (Figure 3A, upper right). The mathematical simulations represented by the heat maps demonstrated the effect of increasing thresholder amounts on the level of the device activation with rising leaky transcription rates (kl) (Figure 3A, bottom). Experimental testing of the thresholder demonstrated its function in agreement with the simulations, decreasing leaky transcription and consequently increasing the FI (Figure 3A, upper left and middle). Importantly, the activated state remained high even at thresholder amounts that strongly decreased the leakage of the unstimulated state. This made it possible to tune the thresholder amount that gave the highest FI of approximately 60 (Figure 3A, upper left and middle). To our knowledge, this is a novel approach for decreasing transcriptional leakage in mammalian cells, which could be applicable to other systems employing positive-feedback regulation.
Figure 3.

Introduction of the Thresholder and the Effect of the Amplifier
(A) Left: transcriptional leakage of the system was efficiently reduced by a thresholder (pAS67; PHSV-TK-G-myc) tested in HEK293T cells. Right: model simulation and parameter scan. The graph depicts the level of the device activation in stimulated and non-stimulated setting with the increasing thresholder. Bottom panels: heat maps represent the effect of increasing thresholder amounts on the level of the device activation with rising leaky transcription rates (kl) after 24 hr. (B) Anti-inflammatory device-engineered HEK293T cells (reporter pAS72; TRE-UAS-PMIN-SEAP) were stimulated with hIL-1β for 24 hr, and then the input signal was removed by exchanging the medium. SEAP production was quantified in the culture supernatants every 24 hr along with a medium exchange to observe the kinetics of the system activation. S, sensor; A, amplifier; R, reporter; FI, fold induction. Error bars indicate SD (n = 4).
Furthermore, we tested additional characteristics of the final circuit when both the amplifier and the thresholder constructs were present. To evaluate the contribution of the amplifier to the amplitude and kinetics of the response, the system was stimulated for 24 hr (or 4 hr, Figure S3A). The medium was then replaced to remove the inducer. The amplifier sustained the response, which remained increased as much as 5-fold after 72 hr, compared to the decreased response of the system without the amplifier (Figure 3B). Thus, the amplifier enabled sustained system activation, which is important to avoid pre-mature decrease of anti-inflammatory therapeutics production. Importantly, at the same time, the fine-tuned strength of the positive-feedback loop enabled us to retain concentration-dependent system activation supported by the sensor construct (shown later in the text).
Additionally, the response of the pAS97 and pAS98 constructs in the final device was monitored directly. As shown in Figure S3B, they were both activated only when an input signal was present, and they remained silent if Dox was also added. The amplifier construct should, in principle, switch to an ON state only after the sensor has been activated and the threshold level of Gal4 has been outcompeted by GV16 induced by the sensor. Thus, we verified whether the activated sensor and amplifier constructs were present in the same cell. This was demonstrated by colocalization of the fluorescently tagged activated sensor and amplifier (Figure S3C).
After the successful construction of the system, its activation capacity was investigated. Low amounts (Table S2) of transfected sensor and amplifier constructs in the fine-tuned system yielded a comparable output from the reporter pAS51 as triggered by the equal amount of the constitutive CMV-driven reporter (Figure S1C).
Activation of the Anti-Inflammatory Device Ex Vivo and In Vivo
Next, we investigated the responsiveness of the anti-inflammatory device to physiologically relevant inflammatory signals. As described in the previous sections, we fine-tuned the strength of the positive-feedback loop to retain concentration-dependent system activation. First, we stimulated the engineered cells with increasing amounts of IL-1β and TNF-α, and observed a gradual increase in activation (Figure 4A). Inflammatory cytokine (IL-1β, TNF-α, and IL-6) levels in healthy humans range between undetectable amounts to approximately 10 pg/mL,26, 27 whereas they are strongly elevated in patients with active inflammatory disease. For example, in RA patients, levels of as much as 109 pg/mL of TNF-α and 184 pg/mL of IL-6 were detected.28 Because IL-1β has a short half-live in vivo,29, 30 its secretion is often investigated indirectly by stimulating cultured peripheral blood mononuclear cells (PBMCs). It was shown that serum from sIJA patients induced secretion of an approximately 100 pg/mL of IL-1β in healthy PBMCs.31 As our aim was to design a system that could, in principle, function in humans, it was important that the anti-inflammatory device-engineered cells responded at physiologically relevant concentrations. Notably, the system was far less responsive to mouse cytokines, as human receptors are not activated by mouse cytokines as potently as by human cytokines (Figure 4A).
Figure 4.

Activation of the Anti-inflammatory Device In Vitro and In Vivo
(A) The responsiveness of the system to recombinant human and mouse interleukin-1β (IL-1β) and tumor necrosis factor α (TNF-α) was tested in the anti-inflammatory device-engineered HEK293T cells by a dual luciferase test (reporter pAS51; TRE-UAS-PMIN-Luc) after a 24-hr activation period. Error bars indicate SD (n = 4). (B) The responsiveness of the system to pooled healthy human serum (n = 4) and the serum of a patient with systemic juvenile idiopathic arthritis (sJIA). Error bars indicate SD (n = 4). (C) Activation of the anti-inflammatory device-engineered HEK293T cells ex vivo by a peritoneal lavage fluid of mice, injected i.p. with hIL-1β (300 ng/mice) or hTNF-α (250 ng/mice). The peritoneal lavage fluid was collected at different time points. Error bars indicate SD (n = 4). (D) Activation of the anti-inflammatory device-engineered HEK293T cells ex vivo by endogenous mouse cytokines in a peritoneal lavage fluid of mice that were subjected to the cecal ligation and puncture (CLP). The peritoneal lavage fluid was collected 24 hr after CLP procedure. Error bars indicate SD (n = 4). (E) The responsiveness of the system in vivo. The anti-inflammatory device-engineered HEK293T cells (reporter pAS72; TRE-UAS-PMIN-SEAP) were microencapsulated and implanted i.p. An inflammatory signal was induced i.p. with an injection of hIL-1β (300 ng/mouse) or hTNF-α (250 ng/mouse) 1 and 24 hr post-implantation. Serum SEAP levels were profiled after 48 hr. Error bars indicate SD (n = 5). (F) The responsiveness of the implanted anti-inflammatory device to endogenous CLP-induced inflammation in mice. CLP procedure was performed 24 hr before microcapsules implantation, and SEAP levels were measured 48 hr post-implantation. Y, mass concentration; FI, fold induction; Ctrl. m., control microcapsules; Control, control mice. Error bars indicate SD (n = 5).
We further demonstrated the physiological relevance of the anti-inflammatory device by demonstrating that the serum of healthy human individuals did not activate the anti-inflammatory device, whereas the 1:1-diluted human serum from a sIJA patient activated the system approximately 3.5-fold (Figure 4B).
We also observed high ex vivo system activation using peritoneal lavage fluid collected 1 hr after an injection of human IL-1β and TNF-α (intraperitoneally [i.p.]) in mice (Figure 4C). The activation, however, strongly decreased when using peritoneal lavage fluid collected 6 hr after the injection and was undetectable when stimulating with the one, collected after 24 hr (Figure 4C), that highlights the short half-life of the injected cytokines (Figure S4A). To demonstrate activation of the anti-inflammatory device by endogenous inflammation in mice, CLP procedure, which is frequently utilized as an animal model of sepsis and is characterized by high level of inflammatory cytokines (Figure S4B), was performed.32 Peritoneal lavage fluid collected 24 hr after the CLP procedure demonstrated ex vivo activation of the designed device transfected in HEK293T cells (Figure 4D).
To test the functionality of the implemented anti-inflammatory device in vivo, the engineered cells were microencapsulated in alginate-PLL-alginate beads and were implanted into the peritoneal cavity of mice. An intracellular luciferase reporter, present in the pAS51, was replaced by a secretory alkaline phosphatase (SEAP) to construct the pAS72 reporter plasmid (TRE-UAS-PMIN-SEAP, Table S1). A significant increase in serum SEAP levels was observed 48 hr after i.p. injection of human IL-1β or TNF-α, whereas the device remained silent in the absence of cytokines (Figure 4E). A similar increase in the SEAP activity was detected in the serum after 24 hr and in the peritoneal lavage fluid after 48 hr (Figure S4C). The trend of in vivo studies was similar to that observed in parallel in vitro experiments (Figure S4D, left). As an internal control of the system, a construct for a secretory Renilla luciferase under the constitutive promoter, SR-Luc (pAS75; PCMV-SR-Luc, Table S1), was designed and co-transfected along with the anti-inflammatory device. As seen in Figure S4D, right, the SR-Luc was comparable among the groups, indicating similar amounts of implanted microcapsules and similar microencapsulated cell viability. In a similar vein to ex vivo experiments, we have also demonstrated that the CLP procedure in mice resulted in elevated inflammatory cytokines, measured in the peritoneal lavage fluid and serum (Figure S4B), and led to an autonomous activation of the implanted anti-inflammatory device by the endogenous inflammation in vivo (Figure 4F).
These results therefore demonstrated that the microencapsulated anti-inflammatory device was able to respond reliably to an inflammatory signal in a complex biological environment when implanted into the peritoneal cavity of mice.
Design and Biological Activity of the Composite Anti-Inflammatory Therapeutic Proteins
Upon activation, a functional anti-inflammatory device should produce a therapeutically relevant output. For this purpose, we designed an advanced effector construct based on clinically used anti-inflammatory biopharmaceuticals, anti-human TNF-α antibody (anti-hTNF-α Ab with CDR from the adalimumab33), human IL-1β receptor antagonist (hIL-1RA), and mouse interleukin 10 (mIL-10). To produce a stoichiometric amount of the heavy and light antibody chain, the effector construct was designed as a single mRNA transcript comprising coding sequences for three polypeptide chains, an anti-hTNF-α antibody heavy chain, an anti-hTNF-α antibody light chain, and hIL-1RA or mIL-10. The polypeptide-coding chains were linked through the coding sequence for the 2A peptide, which interrupts the translation of the polypeptide chain, similar as described previously34 (Figure 5A). These composite anti-inflammatory therapeutics were integrated in the final effector constructs pAS116 (TRE-UAS-PMIN-anti-hTNF-α Ab-hIL-1RA) and pAS134 (TRE-UAS-PMIN-anti-hTNF-α Ab-mIL-10, Table S1). The constitutive and inducible production of the anti-hTNF-α antibody and hIL-1RA separately, or coupled in the final effector construct, was validated by western blot analysis (Figure S5A). The ability and efficiency of the antibody molecules to traverse the membrane of the microcapsules was demonstrated (Figures S5B–S5D). Production of hIL-1RA, anti-hTNF-α, and mIL-10 from the pAS116 or pAS134-transfected microencapsulated cells was quantified by an ELISA. We determined the production of hIL-1RA, anti-hTNF-α antibody, and mIL-10 in the stimulated system (Figure 5B; see Figure S5E for SR-Luc). The unstimulated and the stimulated and subsequently Dox-reset system did not produce substantial amounts of anti-inflammatory effectors (Figure 5B; see Figure S5E for SR-Luc). Similar results were obtained in vitro with an inducible anti-hTNF-α (pAS113; TRE-UAS-PMIN-anti-hTNF-α Ab, Table S1), mIL-10 in the setting with or without the effector construct pAS134, and constitutive hIL-1RA (pAS35; Pcmv-hIL-1RA-myc, Table S1) alone (Figure S5F).
Figure 5.

Design and Biological Activity of Composite Anti-inflammatory Therapeutics
(A) Schematic representation of the double effector construct. The construct was designed as a single mRNA transcript comprising coding sequences for the anti-hTNF-α antibody heavy chain, anti-hTNF-α antibody light chain, and mouse IL-10 (mIL-10) or human interleukin-1 receptor antagonist (hIL-1RA), linked through the coding sequence for the 2A peptide. Each coding sequence contains its own signal peptide (SP) and an additional furin cleavage site to truncate the length of 2A cleavage residues. After the protein translation, self-processing, and furin and signal peptide cleavage, the mature anti-inflammatory effectors were assembled, resulting in a mixture of proteins that counteracted inflammation. (B and C) In vitro (B) and in vivo (C) expression of the anti-inflammatory therapeutics from the effector constructs indicated in the figure. The anti-inflammatory device-engineered HEK293T cells were microencapsulated, and 700 μL of suspension was either incubated in vitro in 10 mL of media or implanted i.p. in mice. In vitro, the anti-inflammatory device was stimulated with a combination of interleukin-1β (IL-1β) (10 ng/mL) and tumor necrosis factor α (TNF-α) (10 ng/mL) or, in addition, subsequently shut down by doxycycline (Dox) (1 μg/mL). For in vivo expression, the mice were injected i.p. with a combination of 500 ng of hIL-1β and 250 ng of hTNF-α per mouse 1 hr after implantation. Serum and peritoneal lavage fluid were collected 48 or 72 hr post-implantation as indicated in the figure. The anti-hTNF-α antibody, mIL-10, and hIL-1RA concentrations were determined by an ELISA test. (D) The responsiveness of the implanted anti-inflammatory device to endogenous CLP-induced inflammation in mice. CLP procedure was performed 3 hr before microcapsules implantation, and SEAP levels were measured 72 hr post-implantation. (E) The biological activity of hIL-1RA and the anti-hTNF-α antibody was determined in vitro by competitive inhibition of hIL-1β and hTNF-α signaling via the NF-κB promoter (reporter pAS40; PNF-κB2-PMIN-Luc). We stimulated cells with 20 ng/mL hIL-1β or hTNF-α for 24 hr, to which we added (simultaneously or with 30-min preincubation) the titrated supernatant, containing anti-inflammatory proteins. SN, supernatant; Ctrl. m., control microcapsules; M−, without microcapsules; M+, with microcapsules. Error bars indicate SD (n = 4).
The in vivo production of the effectors from the microencapsulated engineered cells implanted into the peritoneal cavity of mice was demonstrated. We detected 1 ng/mL of hIL-1RA and 150 ng/mL of anti-hTNF-α in the serum or 7 ng/mL of hIL-1RA and 30 ng/mL of anti-hTNF-α in the peritoneal lavage fluid 48 hr post-implantation only when the mice with implanted device were stimulated with human IL-1β and TNF-α. Mouse IL-10 was detected in the peritoneal lavage fluid at 200 pg/mL 72 hr post-implantation (Figure 5C; see Figure S5E for SR-Luc). Similar results were obtained for the constitutive in vivo production of hIL-RA (Figure S5G). Similarly as described above (Figure 4F), we have shown autonomous in vivo activation of the device by the CLP procedure, detected by anti-inflammatory therapeutics 72 hr post-implantation (Figure 5D). We speculate that the different concentration and distribution of therapeutic proteins between serum and peritoneal lavage fluid are due to the two factors. First, a longer serum half-life of human IgG1 antibody (approximately 3 days35) enables wide distribution of anti-hTNF-α and its accumulation in the serum, compared to the shorter-lived hIL-1RA and mIL-10 (serum half-life of an approximately 30 min36, 37 and 2 hr,38 respectively), which results in the highest concentration in the vicinity of the implanted microcapsules. Second, the peritoneal lavage fluid-collecting procedure (see Materials and Methods) dilutes the local concentration of therapeutic proteins. In this way, the concentration of more evenly distributed anti-hTNF-α is higher in the serum than in the diluted peritoneal lavage fluid, whereas the concentration of hIL-1RA and mIL-10 is higher in peritoneal lavage fluid even after dilution, because their short circulating half-life compromises their accumulation in the serum.
The biological activity of hIL-1RA and anti-hTNF-α, produced in the HEK293T cells, was demonstrated in vitro by the inhibition of hIL-1β or hTNF-α signaling, respectively. Approximately 3-fold molar excess was required for 50% inhibition for both effectors, which was similar to the recombinant proteins used as controls (Figure 5E). Altogether, in this set of experiments, we were able to successfully construct and test composite anti-inflammatory therapeutic proteins.
Resetting of the Anti-Inflammatory Device In Vitro and In Vivo
Next, we demonstrated in vitro that, once activated, the device could be reset to the resting state by the addition of Dox after 24 hr. Complementary, when the device was initially reset by Dox, it could be reactivated by an inflammatory signal (Figures 6A and S6A). Furthermore, we wanted to recapitulate these results in vivo. First, we stimulated the implanted device for 1 hr to activate the circuit and then injected Dox, which prevented full system activation, as measured by SEAP release (Figure 6B; see Figure S6B for SR-Luc). Next, we wanted to demonstrate in vivo reset of the fully activated system by injecting Dox 24 hr after activation (thin arrow in Figure 6C) and measuring the anti-inflammatory therapeutics concentrations (pAS134; TRE-UAS-PMIN-anti-hTNF-α Ab-mIL-10, Table S1). Considering a long half-life of human IgG1 antibody (approximately 3 days35) and likewise of the SEAP (approximately 1 day39) in vivo in mice and taking in account the kinetics of the reset switch shown in Figure 6A, there was no difference in the anti-hTNF-α antibody serum concentrations 48 hr after Dox injection, because the fully activated device secreted anti-hTNF-α antibody that accumulated in the circulation during that period (Figure 6C). Therefore, the experiment was prolonged and the device was additionally reset 72 hr after the first activation (thick arrow in Figure 6C). In this case, the device was activated simultaneously with the reset. This was done to ensure the full activity of the circuit receiving signals from the sensor and the amplifier constructs in the ON group throughout the time course of the experiment and to show the ability to keep the system shut down even in the presence of the inflammatory signal (i.e., the dominant effect of the OFF-switch) in the OFF group. One hundred twenty hours after the implantation, the activity of the initially fully activated system dropped to around 20%, demonstrating an efficient reset in vivo (Figures 6C and S6C). Additionally, when mIL-10, which has a short serum half-life (approximately 2 hr38), was monitored, we demonstrated that one injection of Dox 24 hr after the system activation (thin arrow in Figure 6D) decreased the activity of fully activated system to around 20% after 48 hr (Figures 6D and S6D). Finally, we have shown in vivo that, if the system was initially reset by Dox injection 1 hr after implantation, it could be reactivated by a single application of human IL-1β and TNF-α 3 days after, as measured by the presence of anti-hTNF-α antibody in the peritoneal lavage fluid 120 hr after implantation (Figure 6E). However, the in vivo reactivation was low by means of capacity, presumably because of the transient nature of the system as shown in Figure S7A and possibly also due to the residual Dox in the bloodstream (half-life of an approximately 3 hr in mice40).
Figure 6.

Reversibility of the Synthetic Anti-inflammatory Device In Vitro and In Vivo
(A) Reversibility of the synthetic anti-inflammatory device in vitro. Left: shutdown of the system. The anti-inflammatory device-engineered HEK293T cells (reporter pAS72; TRE-UAS-PMIN-SEAP) were stimulated with hIL-1β (1 ng/mL) for 24 hr and then shut down by the addition of the doxycycline (Dox) (1 μg/mL) by exchanging the medium (indicated by the arrows). Right: rebooting of the initially shut down system. The anti-inflammatory device-engineered HEK293T cells (reporter pAS72; TRE-UAS-PMIN-SEAP) were shut down by Dox (1 μg/mL) for 24 hr and then reactivated by the addition of hIL-1β (1 ng/ml) during medium exchange (indicated by the arrows). Error bars indicate SD (n = 4). (B) In vivo restraint of the full system activation. The anti-inflammatory device-engineered HEK293T cells (reporter pAS72; TRE-UAS-PMIN-SEAP) were microencapsulated and implanted i.p. One hour after implantation, the mice were injected i.p. with a combination of 300 ng of hIL-1β and 250 ng of hTNF-α per mouse. After 1 hr and then every 24 hr, the device was shut down by an i.v. Dox injection (20 mg/kg). Serum was collected daily, and SEAP levels were measured. (C and D) In vivo shut down of the system. The anti-inflammatory device-engineered HEK293T cells (effector pAS134; TRE-UAS-PMIN-anti-hTNF-α Ab-mIL-10) were microencapsulated, implanted i.p., and fully activated by injection of a combination of 300 ng of hIL-1β and 250 ng of hTNF-α per mouse. After 24 hr (thin arrow), the device was shut down by an i.v. Dox injection (20 mg/kg). (C) The device was additionally activated and shut down after 72 hr (thick arrow) to reach the relevant time span needed for an observation of the anti-hTNF-α antibody kinetics. Serum was collected at different time points, and (C) anti-hTNF-α antibody or (D) mIL-10 levels were measured. (E) Reactivation of the anti-inflammatory device in vivo. The anti-inflammatory device-engineered HEK293T cells (effector pAS134; TRE-UAS-PMIN-anti-hTNF-α Ab-mIL-10) were microencapsulated and shut down by i.v. Dox injection (20 mg/kg) 1 hr after implantation. After 72 hr, the system was activated by a single application of 300 ng of hIL-1β and 250 ng of hTNF-α per mouse. Implanted anti-inflammatory device alone represented the control group. Anti-hTNF-α antibody concentration was profiled in the peritoneal lavage fluid 120 hr after implantation. Error bars indicate SD (n = 4).
Application of the Anti-inflammatory Device In Vivo in the Acute Murine Colitis
The anti-inflammatory device was designed and optimized to function in humans. Here, we tested the robustness of the system and the efficiency of the composite anti-inflammatory therapeutics also in an animal disease model. We tested the in vivo physiological functionality of the implanted device in the amelioration of acute (8-day) DSS-induced murine colitis. Although we have shown activation of the anti-inflammatory device by endogenous mouse cytokines in the CLP model (Figures 4F and 5D), the DSS-induced murine colitis did not result in the sufficient increase of the endogenous systemic inflammatory cytokines to activate the system, as substantial amounts were detected only in the supernatants of colon biopsy samples (Figure S7B) and the receptors of human HEK293T cells are far more responsive to the human than mouse inflammatory cytokines as shown in Figure 4A. To mimic an inflammatory flare and to reach an optimal effect in a DSS-induced murine colitis, we stimulated the microencapsulated anti-inflammatory device by injecting human inflammatory cytokines. As a therapeutic output, a combination of anti-hTNF-α, which also inhibits mTNF-α,33 and mIL-10 was used, because both anti-inflammatory therapeutics have previously shown efficiency in a mouse colitis model.6, 7 When the microencapsulated anti-inflammatory device was implanted on the fifth day of the experimental model (i.e., 5 days after the starting of the DSS exposure), a significantly reduced colon shortening was observed, when mice were sacrificed on the eighth day, in comparison to the control without microcapsules or the implants containing the engineered cells without the effector module (Figure 7A). The microencapsulated anti-inflammatory device clearly improved the intestinal morphology as observed at the end of the in vivo experiment, compared to that of the controls (Figure 7B), and significantly reduced the daily monitored disease activity index, defined by the stool form and stool blood (Figure 7C). Furthermore, the anti-inflammatory device improved the colon histopathology (Figure 7E) and consequently the histological score (Figure 7F), which was evaluated on the eighth day upon sacrifice of the mice. Colon from animals with the implanted anti-inflammatory device showed less infiltration of lymphocytes and neutrophil granulocytes, less crypt damage, and no formation of ulcers. In control animals, foci of hydropic degeneration were also observed in contrast to mice with implanted active device. When the anti-inflammatory device was implanted on the third day after DSS exposure, activated, and simultaneously restrained by Dox, a significant difference in the amount of body mass lost was measured on the fifth day (2 days after the implantation of the device), compared to the device that remained activated (Figure 7D). However, at later time points, the device was not able to reverse the body mass loss, presumably because of the severity of the damage to the gastrointestinal tract caused by DSS and possibly also because of the decrease of the circuit’s therapeutic capacity at later time points due to the transient nature of the system (Figure S7A).
Figure 7.

Anti-inflammatory Device Demonstrates Efficacy by Reducing the Symptoms in the Acute Murine Colitis
The anti-inflammatory device-engineered HEK293T cells (double effector pAS134; TRE-UAS-PMIN-anti-hTNF-α Ab-mIL-10) were microencapsulated and implanted i.p. into mice with DSS-induced acute murine colitis on the fifth day (except for D) of the experimental model (i.e., 5 days after exposing mice to DSS). One hour after implantation, the device was activated by a combined inflammatory signal (an i.p. injection containing a combination of 300 ng of hIL-1β and 250 ng of hTNF-α per mouse). (A) The anti-inflammatory device blocked colon shortening induced by DSS (the mean ± SD is shown) and (B) improved the intestinal morphology as validated at the end of the in vivo experiment; (C) reduced daily monitored disease activity index, defined by the combined scores of stool consistency and stool blood (two-tailed Student’s t test assuming unequal variances; mean ± SEM is shown); (D) temporarily reduced daily monitored body mass loss (each data point represents the body mass measurement of one mouse); and, as measured upon the sacrifice of the mice, (E) improved colon histopathology, and (F) the histological score (Mann-Whitney U test; mean ± SEM is shown). Without m., without microcapsules.
Discussion
Several innovative concepts for a variety of medical applications have already been introduced employing the principles of synthetic biology.41 One particularly promising strategy is the modification of mammalian cells by designed genetic therapeutic networks that autonomously sense pathological conditions (i.e., metabolites or other signature molecules) and respond by a therapeutic output.9 When implanted into the host organism, such engineered cells act as therapeutic prosthetic devices. In this way, synthetic biology could contribute to the next generation of cell-based therapies for important medical problems, representing a new pillar of pharmacotherapy.42
Inflammatory processes are central to many widespread diseases such as IBD, which alone affects more than 2 million people in Europe.43 As pro-inflammatory cytokines are the key pathophysiological components in the inflammatory disease manifestation,44 biological drugs that interfere with those mediators represent an effective therapy.45
The anti-inflammatory device developed in this study enabled a prompt detection of inflammatory signals requiring only 4 hr of stimulation (Figure S3A) and the initiation of an in vivo localized therapeutic response designed to be strengthened by the amplifier device. In addition to its inflammatory signal-induced rapid response, a major advantage of the therapeutic device compared to established biological therapies, such as TNF-α and other inflammatory cytokine neutralizers, is the avoidance of long-term systemic blockage of beneficial inflammatory reactions. Upon future developments of the therapeutic systems, the main medical conditions that might benefit from the anti-inflammatory device are chronic inflammatory diseases or medical conditions with high risk of acute inflammatory flare-ups.
We demonstrated that the sensor module enabled dose-dependent activation of the device at physiologically relevant concentrations of inflammatory mediators both in vitro and in vivo in mice (Figures 4 and 5). Similarly as in other studies,13 the dynamic range of the genetic circuit in the in vivo conditions did not fully recapitulate the in vitro setting, but nevertheless we have demonstrated that the device enabled a reasonable FI with a relatively low leakage when measuring SEAP as an output (Figures 4E and 4F). When therapeutic proteins were profiled in the serum or peritoneal lavage fluid of mice, a better dynamic range was observed, which was attributed to the lower background (Figures 5C and 5D). Additionally, ex vivo activation by human serum from a sIJA patient was demonstrated (Figure 4B). sJIA is an auto-inflammatory disease affecting 250,000 children in the United States alone.31 Anakinra has been previously shown to have a beneficial effect on the disease outcome, when administered early in the disease progression before arthritis has become well established.18 That is exactly what the described anti-inflammatory device enables and anakinra as a therapeutic output was also validated in this study (Figures 5B, 5C, and 5E). The activation of the system by the human serum from sIJA patient was lower than in the in vitro titration experiment. This is in part due to the dilution of the serum used in this experiment, but also due to a very short half-life of hIL-1β in the human serum.29, 30 Considering these limitations, a 3.5-fold induction is probably an underestimation of what the systems’ response would be in a hypothetical in vivo setting, where the circulating hIL-1β would fuel the activity of the implanted device. Nevertheless, it remains to be investigated, whether this level of activation would suffice for the therapeutic efficacy in the in vivo setting, which clearly depends on the particular inflammatory disease being targeted. Although the anti-inflammatory device was optimized for a human setting, activation by endogenous mouse cytokines was shown in the CLP model of sepsis in vivo (Figures 4F and 5D).
The notion and application of a threshold for regulation has already been established in DNA nanotechnology, where in vitro DNA strand displacement enabled a construction of neural networks by linear threshold circuits.46 The novelty of our designed circuit is introduction of the thresholder, which decreases the transcriptional leakage in the mammalian cell system. This was achieved by a competition between transcriptionally active and inactive proteins for the same DNA binding site (Figure 3A). This principle contributes to the synthetic biology toolbox for mammalian cell engineering and could be applied in genetic circuits that are prone to a transcriptional leakage. It has been demonstrated by others47, 48 that repeated DNA sequences containing transcription factor binding sites that are not coupled to system activation can act as decoys. In this way, by sequestering transcriptional activator, decoy binding sites can influence the properties of the circuit by increasing its ultra-sensitivity. On the other hand, when designing complex gene circuits by joining several modules, decoy DNA binding sites can apply a load to a system and negatively influence circuit function, which was alleviated by a designed load driver device.49 However in this study, we did not test how the thresholder affected ultra-sensitivity of the system, because our primary goal was to decrease the transcriptional leakage of the circuit. Upon introduction of the thresholder, our circuit retained graded response, which depended on the concentration of the input inflammatory signals (Figure 4A).
The activated device produced sufficient amounts of biologically active anti-inflammatory therapeutic proteins in vitro (Figure 5B) and in vivo (Figures 5C and 5D) to impair the excessive inflammation and its pathological effects. Sustained production of effector proteins was achieved by incorporation of the positive-feedback amplifier module (Figure 3B), bypassing device inactivation by sequestration of the inflammatory signals in the immediate vicinity of the implant. The production of therapeutic proteins with this device can be terminated by the administration of a clinically licensed chemical mediator Dox. Dox, acting via a repressor module, resets the therapeutic device into the standby state as demonstrated in vitro (Figures 2C and 6A) and in vivo (Figures 6B–6D). At the same time, the integrated OFF-switch does not prevent the recurrent system activation as shown in vitro (Figure 6A) and in vivo (Figure 6E).
The designed synthetic strategies against inflammation have already been reported and included orally administered L. lactis secreting IL-106 or anti-TNF-α nanobody7 and adenoviral IL-10 gene therapy,50 which constitutively produce anti-inflammatory proteins. These approaches demonstrated efficacy in the murine colitis models. The production capacity of the anti-inflammatory device, which was activated autonomously upon encountering inflammatory signals, was similar to these studies. As an example, we demonstrated the in vivo efficacy of the microencapsulated anti-inflammatory device in a DSS-induced acute murine colitis model evaluated by several physiological parameters (Figure 7).
From an engineering perspective, the anti-inflammatory device in the present study can be considered a gene regulatory circuit, in which the active, therapeutic protein-producing state is triggered by an inflammatory signal and the OFF signal is provided by an external chemical signal. This setup allows for a rapid autonomous activation and the external control of the reset (e.g., when a physician decides that the therapeutic production of anti-inflammatory proteins is no longer required).
Recently, E. coli was engineered to detect and record the presence of anhydrotetracycline in the mammalian gut, serving as a living diagnostic device.51 Detection of in vivo anti-inflammatory device activation (Figures 4E and 4F), representing an inflammatory episode, could serve as a diagnostic reporter of an ongoing inflammatory process or as a log of past inflammatory events, without having to frequently sample the body fluids of an organism.
The modular design of the device enables adaptation of a combination of effector proteins for the particular pathology and for an optimal effect. Changing the sensor module allows alternative endogenous signals to be detected. The latter ability of the device was demonstrated by introduction of the STAT3 responsive element, which enabled IL-6 detection (Figure S4E). Combining two or more input signals by an AND or similar logic could increase the specificity of the response to different combinations of inflammatory mediators. As this type of device would be used for patients diagnosed with chronic inflammatory diseases, we speculate that the activation of the device mediated by the NF-κB signal, activated also by other stimuli, such as bacterial or viral infection, should not cause a problem as also infection-triggered inflammation can aggravate or even initiate the disease.52, 53, 54 Furthermore, the ability to switch off the production of anti-inflammatory therapeutics by applying a chemical signal would allow the anti-inflammatory action to be bypassed, if required.
Although the capacity of the system decreased with time due to the transient nature of the system, the device was still active also 6 days after transfection in vitro; however, the productivity decreased at later time points (Figure S7A). The underlying cause is probably a well-documented phenomena of the intracellular plasmid degradation, spontaneous loss, and dilution during cell divisions in transiently transfected cells.55, 56 We have demonstrated that the sensor and the amplifier constructs were present and activated in the same cell 48 hr after the transfection (Figure S3C). However, as expected, the plasmid copy number per cell dropped within 5 days after transfection, but nevertheless, the amplifier construct, which was introduced at the lowest amounts, and the inducible OFF-switch representing a safety mechanism, were still present at that time (Figure S7C). Importantly, the initial amounts of the inducible OFF-switch were much higher than the amounts of the sensor and amplifier, which are the drivers of the device activation, ensuring that the safety switch is functional for the duration of the device activity.
Although the reliability and robustness of the anti-inflammatory device were demonstrated in vitro and in vivo, it should be noted that, in this configuration, the device is a prototype that needs further refinements for clinical applications in human therapy. In particular, long-term stable cell lines would need to be established by genome integration of the device, either using autologous or validated safe cell lines.
Particularly for the translation of the system from mice to a human setting, the capacity to produce anti-inflammatory proteins and pharmacodynamics analysis of the local concentration in the affected organs will have to be taken into account for therapeutic applications. The results indicated that the production levels of the anti-inflammatory effectors were sufficient to suppress inflammatory reactions in mice (Figure 7) and that the engineered microencapsulated cells remained viable in the in vitro culture (Figure S7D) and, more importantly, in the peritoneal cavity of mice (Figure S7E).
Alginate microencapsulation has already been tested in clinical trials,57, 58 for example, microencapsulated human islets transplanted into a type 1 diabetic patient aided insulin independence for 9 months.59 Besides alginate microencapsulation, there are alternative cell implantation technologies, such as biocompatible hollow fibers, which may enable an easy removal of the implanted device.60 Advancing safety and efficacy of cell implantation technologies may foster the translation of the designed synthetic biology-inspired prosthetic devices into the clinic.
In conclusion, the concept of an autonomous anti-inflammatory device may provide an alternative to anti-inflammatory therapy and the emerging field of synthetic immunology61 may significantly contribute to the development of novel therapeutic solutions for important medical problems.
Materials and Methods
Plasmid Construction
The genetic constructs used and designed in this study are listed and explained in detail (including their architecture, construction methods, and annotated sequences of relevant elements) in Table S1. Key plasmids include the following: the sensor construct pAS60, encoding the TRE-PNF-κB2-PMIN-driven GV16-myc expression unit (TRE-PNF-κB2-PMIN-GV16-myc); the amplifier construct pAS62, encoding the TRE-UAS-PMIN-driven GV16-myc expression unit (TRE-UAS-PMIN-GV16-myc); the double effector construct pAS134, encoding the TRE-UAS-PMIN-driven anti-hTNF-α antibody-mIL-10-myc double effector expression unit (TRE-UAS-PMIN-anti-hTNF-α Ab-mIL-10; DE2); the positive-feedback thresholder construct pAS67, encoding the constitutive HSV-TK promoter-driven Gal4-myc expression unit (PHSV-TK-G-myc); the inducible OFF-switch construct pAS58, encoding constitutive CMV-promoter-driven rtTR-NLS-KRAB expression (PCMV-rtTR-NLS-KRAB); the luciferase reporter construct pAS51, encoding the TRE-UAS-PMIN-driven luciferase expression unit (TRE-UAS-PMIN-Luc); and the SEAP reporter construct pAS72, encoding the TRE-UAS-PMIN-driven SEAP expression unit (TRE-UAS-PMIN-SEAP).
Cell Culture and Transfection
The HEK293T cell line (American Type Culture Collection) was cultured in DMEM+GlutaMAX-I medium (Invitrogen) (supplemented with 10% fetal bovine serum) (BioWhittaker) at 37°C in a 5% CO2 humidified atmosphere. At 50%–70% confluence, cells were transfected with a mixture of DNA and JetPEI (Polyplus-transfection) according to the manufacturer’s instructions.
Inducer Molecules
For in vitro experiments, doxycycline hyclate (Sigma-Aldrich; D9891) was dissolved in MQ at a concentration of 0.1 mg/mL, sterile filtered, and used at a final concentration of 1 μg/mL. For in vivo experiments, doxycycline was dissolved in PBS at 5 mg/mL (pH 7.3), sterile filtered, and used at a final concentration of 20 mg/kg. Human IL-1β and human TNF-α (R&D Systems) were used at a final concentration of 1 ng/mL in the in vitro experiments and at 250 ng/mouse in the in vivo experiments, unless stated otherwise.
Transfection and Dual Luciferase Assay
The HEK293T cells (2 × 104 cells per well) were seeded into CoStar White 96-well plates (Corning). At a confluence of 50%–70%, cells were transfected with a mixture of DNA and JetPEI (Polyplus-transfection) according to the manufacturer’s instructions. Detailed information about the transfected plasmids and their amount in a specific experiment are provided in Table S2. After 24 hr, cells were activated as stated in the specific experiment. After 24 or 48 hr, cells were harvested and lysed with 30 μL of 1× passive lysis buffer (Promega). Firefly luciferase and Renilla luciferase expression were measured using a dual luciferase assay (Promega) and an Orion II microplate reader (Berthold Technologies). The relative luciferase activity was calculated by normalizing each sample’s firefly luciferase activity to the constitutive Renilla luciferase activity determined in the same sample.
Stimulation with Human Serum
Human serum was collected from four healthy individuals and from a patient with a sJIA. Pooled serum samples (n = 4) from healthy individuals were tested along with the serum from a patient in a 1:1 dilution for the anti-inflammatory device activation according to the protocol described in Transfection and Dual Luciferase Assay (for information about the transfected plasmids, see Table S2). Written informed consent for obtaining blood samples from healthy controls and from a patient was obtained. Principles of the Declaration of Helsinki were followed, and the Slovenian National Medical Ethics Committee approved the study (decree number 29_2_13).
SEAP Detection
Human placental SEAP levels were determined in cell culture supernatants using a QUANTI-Blue assay (InvivoGen) according to the manufacturer’s instructions. Briefly, 30 μL of supernatant was added to 200 μL of pre-warmed (37°C), sterile-filtered QUANTI-Blue reagent solution. After 15 min, the absorbance was measured at 630 nm using a Synergy Mx automated microplate reader (BioTec). The SEAP levels in blood samples and peritoneal lavage fluid of the mice were quantified using a chemiluminescence-based assay (Great EscAPe SEAP Chemiluminescence Kit 2.0; Clontech) according to the manufacturer’s instructions, with a few adaptations. Briefly, 5 μL of serum or 15 μL of peritoneal lavage fluid was mixed with 95 or 85 μL of a 1× dilution buffer, respectively, and incubated for 30 min at 65°C to heat inactivate endogenous phosphatases. After cooling to room temperature (22°C), 100 μL of substrate solution was added. After 10 min, the samples were centrifuged (2 min, 20,000 × g), and 100 μL of the supernatant was transferred to CoStar White 96-well plates (Corning). Thirty minutes after the addition of the substrate solution, the luminescence was measured with an Orion II microplate reader (Berthold Technologies). As an internal control, the activity of the constitutive secretory Renilla luciferase SR-Luc (pFLAG-CMV3-R-Luc; pAS75, Table S1) was measured in 50 μL of the same sample.
Microencapsulation
The cells were microencapsulated in 400-μm alginate-PLL-alginate microcapsules (approximately 200 cells/microcapsule) using the Encapsulator Biotech (Büchi), according to the manufacturer’s protocol and as previously described.62 The following settings were applied: a 200 μM single nozzle, a 20-mL syringe with a flow of 19 mL/min, a vibration frequency of 1,200 Hz, an electrostatic dispersion of 1,300 V, and a stirrer speed of 200 rpm. We used 1.4% ultrapure alginate (PRONOVA UP LVG; Novamatrix) and 0.04% poly-l-lysine hydrobromide (molecular weight [mol wt], 15.000-30.000 PLL; Sigma-Aldrich; P7890). The microencapsulated cells were used for in vitro or in vivo experiments.
Confocal Fluorescence Microscopy
To directly observe the system’s performance in vitro, HEK293T cells were seeded onto eight-well microscopic chambers (Ibidi) at a density of 5 × 104 cells per well. Cells were transfected with the constructs pAS97 (to observe the sensor’s activity) and/or pAS98 (to observe the amplifier’s activity), pAS72, pAS67, pAS58, pAS75, and pmCherry-C1 (Table S1). Twenty-four hours after transfection, the system was stimulated with human IL-1β (R&D Systems) at a final concentration of 1 ng/mL. Four hours after stimulation, the medium was removed and the cells were washed three times using fresh medium to remove input signal. After 24 hr, the responsiveness of the system was visualized, and microscopic images were acquired using a Leica TCS SP5 inverted laser-scanning microscope on a Leica DMI 6000 CS module equipped with a HCX PL Fluotar L (20×; numerical aperture, 0.4) (Leica Microsystems).
Quantification and Biological Activity of Anti-inflammatory Proteins
Supernatants from cell cultures or in vivo samples (the preparation of the serum and peritoneal lavage fluid was as described in Animal Models) were collected, and protein levels were measured with a sandwich ELISA (eBioscience). hIL-1RA was detected with a human IL-1RA Platinum ELISA (eBioscience; BMS2080), anti-hTNF-α antibody was detected with a human IgG1 Platinum ELISA (eBioscience; BMS2092TWO), and mouse IL-10 was detected with a mouse IL-10 ELISA Ready-SET-Go! (2nd Generation) (eBioscience; 88-7105-22). The biological activity of the hIL-1RA and anti-hTNF-α antibody was determined by competitive inhibition of hIL-1β and hTNF-α signaling through the NF-κB promoter. First, hIL-1RA and anti-hTNF-α antibody were produced in supernatants of HEK293T cells, transfected with constitutive promoter-driven expression form pAS35 and pAS114 (Table S1), respectively. We transfected the reporter cell line HEK293T with pAS40 and phRL-TK (Table S1) to observe NF-κB-driven luciferase activity. The cells were stimulated with 20 ng/mL hIL-1β or hTNF-α for 24 hr, to which we added (simultaneously or with 30-min preincubation) the titrated supernatant, containing anti-inflammatory proteins to observe the molar ratio-dependent decrease in luciferase activity. Recombinant human IL-1RA (Sigma-Aldrich; SRP3084) and anti-hTNF-α-hIgG1 (InvivoGen; htnfa-mab1) were used as controls.
Animal Models
For the all in vivo experiments, 8- to 10-week-old C57BL/6JOlaHsd mice of mixed sexes (evenly distributed between the groups) were used. The numbers of mice are stated in the figure captions.
The microcapsules were produced as described in Microencapsulation. A 1:1 suspension of microcapsules with MOPS buffer (10 mM MOPS, 0.85% NaCl) was prepared and 700 μL were injected i.p. into the mice using a 20G needle (10,000 microcapsules per mouse, 200 cells per microcapsule), as described previously.62
To determine the responsiveness of the synthetic anti-inflammatory device to an inflammatory signal in vivo (for information on the transfected plasmids, see Table S2), we injected mice i.p. with 300 ng of hIL-1β per mouse or 250 ng of hTNF-α per mouse 1 and 24 hr after implantation. Serum was collected using Multivette 600 (Sarstedt; 3,000 rpm, 30 min) 24 and 48 hr later, and peritoneal lavage fluid was collected after 48 hr (5 mL of PBS). We tested the peritoneal lavage fluid for system activation ex vivo.
To demonstrate activation of the anti-inflammatory device by endogenous mouse cytokines, we performed CLP model following the described method.32, 63 In Figure 4A, we collected peritoneal lavage fluid 24 hr after CLP procedure; in Figure 4F, we performed CLP 1 day before, and in Figure 5D, 3 hr before microcapsules implantation.
For the in vivo restraint (Figure 6B) (see Table S2 for information on the transfected plasmids used in the study), the mice were first injected i.p. with a combination of 300 ng of hIL-1β and 250 ng of hTNF-α per mouse 1 hr after implantation. After 1 hr and every 24 hr, 20 mg/kg of Dox was injected intravenously (i.v.).
For the in vivo shut down (Figures 6C and 6D) (see Table S2 for information on the transfected plasmids used in the study), the mice were first injected i.p. with a combination of 300 ng of hIL-1β and 250 ng of hTNF-α per mouse 1 hr after implantation. After 24 hr (Figures 6C and 6D) and in addition after 72 hr (Figure 6C), 20 mg/kg of Dox was injected i.v. In the case of Figure 6C, at 72-hr time point, the mice in ON and OFF groups were injected i.p. also with a combination of 300 ng of hIL-1β and 250 ng of hTNF-α per mouse simultaneously with Dox injection. Serum was collected, and anti-hTNF-α and mIL-10 concentrations were profiled. Control values were subtracted from the ON and OFF state, and results were represented as a percentage of a fully activated system (raw data are presented in Figures S6C and S6D).
For the in vivo reactivation (Figure 6E) (see Table S2 for information on the transfected plasmids used in the study), 20 mg/kg of Dox was injected i.v. into mice 1 hr after implantation. After 72 hr, the system was activated by a single application of 300 ng of hIL-1β and 250 ng of hTNF-α per mouse, and peritoneal lavage fluid was collected 120 hr after implantation.
To produce therapeutic anti-inflammatory proteins in vivo, the mice were injected i.p. with a combination of 500 ng of hIL-1β and 250 ng of hTNF-α per mouse 1 hr after implantation. After 48 hr, serum and peritoneal lavage fluid were collected.
The application of the anti-inflammatory device in vivo was then demonstrated in an acute murine colitis model.64 The mice were given either water or 2% DSS (TdB Consultancy, DB001-31) for 8 days as previously described, and on the fifth day (Figure 7, except for D) of this time-course experimental model, the microcapsules containing anti-inflammatory device-engineered cells were implanted i.p. The control groups without microcapsules received 700 μL of MOPS buffer. One hour after implantation and then daily, the mice were injected i.p. with a combination of 300 ng of hIL-1β and 250 ng of hTNF-α per mouse. Weight, stool blood, and stool consistency were monitored daily. Disease activity index (DAI) was determined by combining the stool blood and stool consistency scores. Each score was determined as follows: stool blood, 0–2 (0, no blood; 1, hemoocult bleeding; 2, gross bleeding); and stool consistency, 0–2 (0, normal stool; 1, soft stool; 2, diarrhea). Upon the sacrifice of the mice, the colon length was measured from the body of the caecum to the distal part of the rectum. Histological samples of the sacrificed mice were fixated overnight in 10% neutral buffered formalin (Sigma-Aldrich; HT501128) and then embedded in paraffin (Leica; Paraplast; 39601006). The paraffin blocks were cut 7 μm thick with a rotation microtome RM 2245 (Leica; 1492245UL01) and stained with hematoxylin and eosin (Sigma-Aldrich; MHS32; HT110332). The histological score was determined as previously described: formation of ulcus, 0–4; epithelium morphology, 0–4; immune cell infiltration, 0–4; and lymph follicles, 0–4.65 Alternatively, the microcapsules were implanted on the third day after DSS exposure (Figure 7D). In this case, the control group comprised full anti-inflammatory system that was initially activated by injecting the mice with a combination of 300 ng of hIL-1β and 250 ng of hTNF-α per mouse, and the activation of the circuit was simultaneously restrained by a daily i.v. Dox injection (20 mg/kg). All of the animal experiments were performed according to the directives of the EU 2010/63, approved by the Ministry of Agriculture, Forestry, and Foods, Republic of Slovenia (no. U34401-55/2013/6).
Mathematical Model and Simulations
The mathematical model and simulations are explained in detail in Supplemental Mathematical Model. Briefly, the mathematical model was generated using CellDesigner 4.4 software23 by first constructing a state transition diagram and describing the transitions with ODEs according to mass action kinetics. The initial quantities of genetic constructs were approximated by estimating the efficiency of transient transfection. The input signal for the modeled genetic device was the NF-κB particle number, and the output signal was the effector protein particle number. The simulations were run with the CellDesigner 4.4 software using an integrated SBML ODE Solver.66
Statistics
Representative experiments are shown. All values are represented as the mean ± SD, except for in Figures 7C and 7F, where the mean ± SEM is shown. The data were analyzed with two-tailed Student’s t test assuming unequal variances, except in Figure 7F, where a Mann-Whitney U test was used. p values are shown for each comparison of interest.
Author Contributions
R.J. conceived and supervised the study. A.S. and R.J. designed the experiments, with the help of D.L., U.B., and S.H. A.S., D.L., and U.B. performed the experimental work. A.S., D.L., U.B., S.H., and R.J. analyzed and discussed the results. D.L. and S.H., with the assistance of A.S., performed the in vivo experimental work. U.B. designed the mathematical model and performed the simulations. A.S. and R.J. wrote the manuscript, with help from D.L., U.B., and S.H. A.S., D.L., U.B., S.H., and R.J. discussed and commented on the manuscript before submission.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgments
We thank the Slovenian Research Agency (grants P4-0176 and J1-6740) and EN-FIST Centre of Excellence for their financial support. We thank Luka Smole for help with introducing microencapsulation technology in our laboratory. We thank Dr. Mojca Benčina and Dr. Jelka Pohar for help with the confocal fluorescence microscopy. We thank Lucija Kadunc for help with preparing the implant membrane hollow fibers. We thank Professor Dr. Martin Fussenegger (Institute of Biotechnology, Swiss Federal Institute of Technology), Dr. Rok Gaber, Dr. Jelka Pohar, Dr. Slavko Čeru, and Jan Lonzarić for fruitful discussions. We thank Karen Butina for suggestions with the statistical analysis and Robert Bremšak, Darija Oven, and Irena Škraba for technical support. We thank Vesna Mrak for maintenance of the mouse colony. We thank Professor Dr. Tadej Avčin, MD (University Children’s Hospital, University Medical Centre Ljubljana, Department of Allergology, Rheumatology and Clinical Immunology; Faculty of Medicine, University of Ljubljana), and Professor Dr. Uroš Potočnik (Faculty of Medicine, Centre for Human Molecular Genetics and Pharmacogenomics, University of Maribor; Faculty for Chemistry and Chemical Engineering, Laboratory for Biochemistry, Molecular Biology and Genomics, University of Maribor) for providing human blood samples.
Footnotes
Supplemental Information includes Supplemental Materials and Methods, seven figures, five tables, and can be found with this article online at http://dx.doi.org/10.1016/j.ymthe.2016.10.005.
Supplemental Information
References
- 1.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 2.Dempsey P.W., Vaidya S.A., Cheng G. The art of war: innate and adaptive immune responses. Cell. Mol. Life Sci. 2003;60:2604–2621. doi: 10.1007/s00018-003-3180-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clark R., Kupper T. Old meets new: the interaction between innate and adaptive immunity. J. Invest. Dermatol. 2005;125:629–637. doi: 10.1111/j.0022-202X.2005.23856.x. [DOI] [PubMed] [Google Scholar]
- 4.Dinarello C.A. Anti-inflammatory agents: present and future. Cell. 2010;140:935–950. doi: 10.1016/j.cell.2010.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.El-Gabalawy H., Guenther L.C., Bernstein C.N. Epidemiology of immune-mediated inflammatory diseases: incidence, prevalence, natural history, and comorbidities. J. Rheumatol. Suppl. 2010;85:2–10. doi: 10.3899/jrheum.091461. [DOI] [PubMed] [Google Scholar]
- 6.Steidler L., Hans W., Schotte L., Neirynck S., Obermeier F., Falk W., Fiers W., Remaut E. Treatment of murine colitis by Lactococcus lactis secreting interleukin-10. Science. 2000;289:1352–1355. doi: 10.1126/science.289.5483.1352. [DOI] [PubMed] [Google Scholar]
- 7.Vandenbroucke K., de Haard H., Beirnaert E., Dreier T., Lauwereys M., Huyck L., Van Huysse J., Demetter P., Steidler L., Remaut E. Orally administered L. lactis secreting an anti-TNF Nanobody demonstrate efficacy in chronic colitis. Mucosal Immunol. 2010;3:49–56. doi: 10.1038/mi.2009.116. [DOI] [PubMed] [Google Scholar]
- 8.Archer E.J., Robinson A.B., Süel G.M. Engineered E. coli that detect and respond to gut inflammation through nitric oxide sensing. ACS Synth. Biol. 2012;1:451–457. doi: 10.1021/sb3000595. [DOI] [PubMed] [Google Scholar]
- 9.Ye H., Aubel D., Fussenegger M. Synthetic mammalian gene circuits for biomedical applications. Curr. Opin. Chem. Biol. 2013;17:910–917. doi: 10.1016/j.cbpa.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 10.Ausländer S., Wieland M., Fussenegger M. Smart medication through combination of synthetic biology and cell microencapsulation. Metab. Eng. 2012;14:252–260. doi: 10.1016/j.ymben.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 11.Murua A., Portero A., Orive G., Hernández R.M., de Castro M., Pedraz J.L. Cell microencapsulation technology: towards clinical application. J. Control. Release. 2008;132:76–83. doi: 10.1016/j.jconrel.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 12.Kemmer C., Gitzinger M., Daoud-El Baba M., Djonov V., Stelling J., Fussenegger M. Self-sufficient control of urate homeostasis in mice by a synthetic circuit. Nat. Biotechnol. 2010;28:355–360. doi: 10.1038/nbt.1617. [DOI] [PubMed] [Google Scholar]
- 13.Rössger K., Charpin-El-Hamri G., Fussenegger M. A closed-loop synthetic gene circuit for the treatment of diet-induced obesity in mice. Nat. Commun. 2013;4:2825. doi: 10.1038/ncomms3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ausländer D., Ausländer S., Charpin-El Hamri G., Sedlmayer F., Müller M., Frey O., Hierlemann A., Stelling J., Fussenegger M. A synthetic multifunctional mammalian pH sensor and CO2 transgene-control device. Mol. Cell. 2014;55:397–408. doi: 10.1016/j.molcel.2014.06.007. [DOI] [PubMed] [Google Scholar]
- 15.Schukur L., Geering B., Charpin-El Hamri G., Fussenegger M. Implantable synthetic cytokine converter cells with AND-gate logic treat experimental psoriasis. Sci. Transl. Med. 2015;7:318ra201. doi: 10.1126/scitranslmed.aac4964. [DOI] [PubMed] [Google Scholar]
- 16.Miyoshi J., Hisamatsu T., Matsuoka K., Naganuma M., Maruyama Y., Yoneno K., Mori K., Kiyohara H., Nanki K., Okamoto S. Early intervention with adalimumab may contribute to favorable clinical efficacy in patients with Crohn’s disease. Digestion. 2014;90:130–136. doi: 10.1159/000365783. [DOI] [PubMed] [Google Scholar]
- 17.Fascì Spurio F., Aratari A., Margagnoni G., Doddato M.T., Papi C. Early treatment in Crohn’s disease: do we have enough evidence to reverse the therapeutic pyramid? J. Gastrointestin. Liver Dis. 2012;21:67–73. [PubMed] [Google Scholar]
- 18.Nigrovic P.A., Mannion M., Prince F.H.M., Zeft A., Rabinovich C.E., van Rossum M.A.J., Cortis E., Pardeo M., Miettunen P.M., Janow G. Anakinra as first-line disease-modifying therapy in systemic juvenile idiopathic arthritis: report of forty-six patients from an international multicenter series. Arthritis Rheum. 2011;63:545–555. doi: 10.1002/art.30128. [DOI] [PubMed] [Google Scholar]
- 19.Sadowski I., Ma J., Triezenberg S., Ptashne M. GAL4-VP16 is an unusually potent transcriptional activator. Nature. 1988;335:563–564. doi: 10.1038/335563a0. [DOI] [PubMed] [Google Scholar]
- 20.Ruland J. Return to homeostasis: downregulation of NF-κB responses. Nat. Immunol. 2011;12:709–714. doi: 10.1038/ni.2055. [DOI] [PubMed] [Google Scholar]
- 21.Chen F.E., Huang D.B., Chen Y.Q., Ghosh G. Crystal structure of p50/p65 heterodimer of transcription factor NF-kappaB bound to DNA. Nature. 1998;391:410–413. doi: 10.1038/34956. [DOI] [PubMed] [Google Scholar]
- 22.Hellweg C.E., Arenz A., Bogner S., Schmitz C., Baumstark-Khan C. Activation of nuclear factor κ B by different agents: influence of culture conditions in a cell-based assay. Ann. N Y Acad. Sci. 2006;1091:191–204. doi: 10.1196/annals.1378.066. [DOI] [PubMed] [Google Scholar]
- 23.Funahashi A., Morohashi M., Kitano H., Tanimura N. CellDesigner: a process diagram editor for gene-regulatory and biochemical networks. Biosilico. 2003;1:159–162. [Google Scholar]
- 24.Szulc J., Wiznerowicz M., Sauvain M.O., Trono D., Aebischer P. A versatile tool for conditional gene expression and knockdown. Nat. Methods. 2006;3:109–116. doi: 10.1038/nmeth846. [DOI] [PubMed] [Google Scholar]
- 25.Groner A.C., Meylan S., Ciuffi A., Zangger N., Ambrosini G., Dénervaud N., Bucher P., Trono D. KRAB-zinc finger proteins and KAP1 can mediate long-range transcriptional repression through heterochromatin spreading. PLoS Genet. 2010;6:e1000869. doi: 10.1371/journal.pgen.1000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kleiner G., Marcuzzi A., Zanin V., Monasta L., Zauli G. Cytokine levels in the serum of healthy subjects. Mediators Inflamm. 2013;2013:434010. doi: 10.1155/2013/434010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lyke K.E., Burges R., Cissoko Y., Sangare L., Dao M., Diarra I., Kone A., Harley R., Plowe C.V., Doumbo O.K., Sztein M.B. Serum levels of the proinflammatory cytokines interleukin-1 beta (IL-1beta), IL-6, IL-8, IL-10, tumor necrosis factor alpha, and IL-12(p70) in Malian children with severe Plasmodium falciparum malaria and matched uncomplicated malaria or healthy controls. Infect. Immun. 2004;72:5630–5637. doi: 10.1128/IAI.72.10.5630-5637.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manicourt D.H., Triki R., Fukuda K., Devogelaer J.-P., Nagant de Deuxchaisnes C., Thonar E.J. Levels of circulating tumor necrosis factor α and interleukin-6 in patients with rheumatoid arthritis. Relationship to serum levels of hyaluronan and antigenic keratan sulfate. Arthritis Rheum. 1993;36:490–499. doi: 10.1002/art.1780360409. [DOI] [PubMed] [Google Scholar]
- 29.Lopez-Castejon G., Brough D. Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev. 2011;22:189–195. doi: 10.1016/j.cytogfr.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kudo S., Mizuno K., Hirai Y., Shimizu T. Clearance and tissue distribution of recombinant human interleukin 1 beta in rats. Cancer Res. 1990;50:5751–5755. [PubMed] [Google Scholar]
- 31.Pascual V., Allantaz F., Arce E., Punaro M., Banchereau J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J. Exp. Med. 2005;201:1479–1486. doi: 10.1084/jem.20050473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dejager L., Pinheiro I., Dejonckheere E., Libert C. Cecal ligation and puncture: the gold standard model for polymicrobial sepsis? Trends Microbiol. 2011;19:198–208. doi: 10.1016/j.tim.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 33.Abbott Biotechnology, Ltd. July 2000. Human antibodies that bind human TNFα. U.S. patent 6,090,382.
- 34.Fang J., Qian J.-J., Yi S., Harding T.C., Tu G.H., VanRoey M., Jooss K. Stable antibody expression at therapeutic levels using the 2A peptide. Nat. Biotechnol. 2005;23:584–590. doi: 10.1038/nbt1087. [DOI] [PubMed] [Google Scholar]
- 35.Dall’Acqua W.F., Woods R.M., Ward E.S., Palaszynski S.R., Patel N.K., Brewah Y.A., Wu H., Kiener P.A., Langermann S. Increasing the affinity of a human IgG1 for the neonatal Fc receptor: biological consequences. J. Immunol. 2002;169:5171–5180. doi: 10.4049/jimmunol.169.9.5171. [DOI] [PubMed] [Google Scholar]
- 36.Liu M., Huang Y., Hu L., Liu G., Hu X., Liu D., Yang X. Selective delivery of interleukine-1 receptor antagonist to inflamed joint by albumin fusion. BMC Biotechnol. 2012;12:68. doi: 10.1186/1472-6750-12-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petrasek J., Bala S., Csak T., Lippai D., Kodys K., Menashy V., Barrieau M., Min S.Y., Kurt-Jones E.A., Szabo G. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J. Clin. Invest. 2012;122:3476–3489. doi: 10.1172/JCI60777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li L., Elliott J.F., Mosmann T.R. IL-10 inhibits cytokine production, vascular leakage, and swelling during T helper 1 cell-induced delayed-type hypersensitivity. J. Immunol. 1994;153:3967–3978. [PubMed] [Google Scholar]
- 39.Salucci V., Scarito A., Aurisicchio L., Lamartina S., Nicolaus G., Giampaoli S., Gonzalez-Paz O., Toniatti C., Bujard H., Hillen W. Tight control of gene expression by a helper-dependent adenovirus vector carrying the rtTA2(s)-M2 tetracycline transactivator and repressor system. Gene Ther. 2002;9:1415–1421. doi: 10.1038/sj.gt.3301813. [DOI] [PubMed] [Google Scholar]
- 40.Böcker R., Estler C.J., Maywald M., Weber D. Comparison of distribution of doxycycline in mice after oral and intravenous application measured by a high-performance liquid chromatographic method. Arzneimittelforschung. 1981;31:2116–2117. [PubMed] [Google Scholar]
- 41.Ruder W.C., Lu T., Collins J.J. Synthetic biology moving into the clinic. Science. 2011;333:1248–1252. doi: 10.1126/science.1206843. [DOI] [PubMed] [Google Scholar]
- 42.Fischbach M.A., Bluestone J.A., Lim W.A. Cell-based therapeutics: the next pillar of medicine. Sci. Transl. Med. 2013;5:179ps7. doi: 10.1126/scitranslmed.3005568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Molodecky N.A., Soon I.S., Rabi D.M., Ghali W.A., Ferris M., Chernoff G., Benchimol E.I., Panaccione R., Ghosh S., Barkema H.W., Kaplan G.G. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142:46–54.e42. doi: 10.1053/j.gastro.2011.10.001. quiz e30. [DOI] [PubMed] [Google Scholar]
- 44.Strober W., Fuss I.J. Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology. 2011;140:1756–1767. doi: 10.1053/j.gastro.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ford A.C., Sandborn W.J., Khan K.J., Hanauer S.B., Talley N.J., Moayyedi P. Efficacy of biological therapies in inflammatory bowel disease: systematic review and meta-analysis. Am. J. Gastroenterol. 2011;106:644–659. doi: 10.1038/ajg.2011.73. quiz 660. [DOI] [PubMed] [Google Scholar]
- 46.Qian L., Winfree E., Bruck J. Neural network computation with DNA strand displacement cascades. Nature. 2011;475:368–372. doi: 10.1038/nature10262. [DOI] [PubMed] [Google Scholar]
- 47.Lee T.-H.H., Maheshri N. A regulatory role for repeated decoy transcription factor binding sites in target gene expression. Mol. Syst. Biol. 2012;8:576. doi: 10.1038/msb.2012.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lu M.S., Mauser J.F., Prehoda K.E. Ultrasensitive synthetic protein regulatory networks using mixed decoys. ACS Synth Biol. 2013;16:65–72. doi: 10.1021/sb200010w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mishra D., Rivera P.M., Lin A., Del Vecchio D., Weiss R. A load driver device for engineering modularity in biological networks. Nat. Biotechnol. 2014;32:1268–1275. doi: 10.1038/nbt.3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sasaki M., Mathis J.M., Jennings M.H., Jordan P., Wang Y., Ando T., Joh T., Alexander J.S. Reversal of experimental colitis disease activity in mice following administration of an adenoviral IL-10 vector. J. Inflamm. (Lond.) 2005;2:13. doi: 10.1186/1476-9255-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kotula J.W., Kerns S.J., Shaket L.A., Siraj L., Collins J.J., Way J.C., Silver P.A. Programmable bacteria detect and record an environmental signal in the mammalian gut. Proc. Natl. Acad. Sci. USA. 2014;111:4838–4843. doi: 10.1073/pnas.1321321111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Navaneethan U., Venkatesh P.G.K., Shen B. Clostridium difficile infection and inflammatory bowel disease: understanding the evolving relationship. World J. Gastroenterol. 2010;16:4892–4904. doi: 10.3748/wjg.v16.i39.4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shih D.Q., Targan S.R. Immunopathogenesis of inflammatory bowel disease. World J. Gastroenterol. 2008;14:390–400. doi: 10.3748/wjg.14.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mann E.A., Saeed S.A. Gastrointestinal infection as a trigger for inflammatory bowel disease. Curr. Opin. Gastroenterol. 2012;28:24–29. doi: 10.1097/MOG.0b013e32834c453e. [DOI] [PubMed] [Google Scholar]
- 55.Kichler A., Leborgne C., Coeytaux E., Danos O. Polyethylenimine-mediated gene delivery: a mechanistic study. J. Gene Med. 2001;3:135–144. doi: 10.1002/jgm.173. [DOI] [PubMed] [Google Scholar]
- 56.Oh Y.-K., Suh D., Kim J.M., Choi H.-G., Shin K., Ko J.J. Polyethylenimine-mediated cellular uptake, nucleus trafficking and expression of cytokine plasmid DNA. Gene Ther. 2002;9:1627–1632. doi: 10.1038/sj.gt.3301735. [DOI] [PubMed] [Google Scholar]
- 57.Jacobs-Tulleneers-Thevissen D., Chintinne M., Ling Z., Gillard P., Schoonjans L., Delvaux G., Strand B.L., Gorus F., Keymeulen B., Pipeleers D., Beta Cell Therapy Consortium EU-FP7 Sustained function of alginate-encapsulated human islet cell implants in the peritoneal cavity of mice leading to a pilot study in a type 1 diabetic patient. Diabetologia. 2013;56:1605–1614. doi: 10.1007/s00125-013-2906-0. [DOI] [PubMed] [Google Scholar]
- 58.Buder B., Alexander M., Krishnan R., Chapman D.W., Lakey J.R. Encapsulated islet transplantation: strategies and clinical trials. Immune Netw. 2013;13:235–239. doi: 10.4110/in.2013.13.6.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Soon-Shiong P., Heintz R.E., Merideth N., Yao Q.X., Yao Z., Zheng T., Murphy M., Moloney M.K., Schmehl M., Harris M. Insulin independence in a type 1 diabetic patient after encapsulated islet transplantation. Lancet. 1994;343:950–951. doi: 10.1016/s0140-6736(94)90067-1. [DOI] [PubMed] [Google Scholar]
- 60.Krishnan R., Alexander M., Robles L., Foster C.E., 3rd, Lakey J.R.T. Islet and stem cell encapsulation for clinical transplantation. Rev. Diabet. Stud. 2014;11:84–101. doi: 10.1900/RDS.2014.11.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Geering B., Fussenegger M. Synthetic immunology: modulating the human immune system. Trends Biotechnol. 2015;33:65–79. doi: 10.1016/j.tibtech.2014.10.006. [DOI] [PubMed] [Google Scholar]
- 62.Weber W., Fux C., Daoud-el Baba M., Keller B., Weber C.C., Kramer B.P., Heinzen C., Aubel D., Bailey J.E., Fussenegger M. Macrolide-based transgene control in mammalian cells and mice. Nat. Biotechnol. 2002;20:901–907. doi: 10.1038/nbt731. [DOI] [PubMed] [Google Scholar]
- 63.Rittirsch D., Huber-Lang M.S., Flierl M.A., Ward P.A. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat. Protoc. 2009;4:31–36. doi: 10.1038/nprot.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wirtz S., Neufert C., Weigmann B., Neurath M.F. Chemically induced mouse models of intestinal inflammation. Nat. Protoc. 2007;2:541–546. doi: 10.1038/nprot.2007.41. [DOI] [PubMed] [Google Scholar]
- 65.Kojouharoff G., Hans W., Obermeier F., Männel D.N., Andus T., Schölmerich J., Gross V., Falk W. Neutralization of tumour necrosis factor (TNF) but not of IL-1 reduces inflammation in chronic dextran sulphate sodium-induced colitis in mice. Clin. Exp. Immunol. 1997;107:353–358. doi: 10.1111/j.1365-2249.1997.291-ce1184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Machné R., Finney A., Müller S., Lu J., Widder S., Flamm C. The SBML ODE Solver Library: a native API for symbolic and fast numerical analysis of reaction networks. Bioinformatics. 2006;22:1406–1407. doi: 10.1093/bioinformatics/btl086. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.