

Abstract
Lithium promotes acute poststroke neuronal survival, which includes mechanisms that are not limited to GSK3β inhibition. However, whether lithium induces long-term neuroprotection and enhanced brain remodeling is unclear. Therefore, mice were exposed to transient middle cerebral artery occlusion and lithium (1 mg/kg bolus followed by 2 mg/kg/day over up to 7 days) was intraperitoneally administered starting 0–9 h after reperfusion onset. Delivery of lithium no later than 6 h reduced infarct volume on day 2 and decreased brain edema, leukocyte infiltration, and microglial activation, as shown by histochemistry and flow cytometry. Lithium-induced neuroprotection persisted throughout the observation period of 56 days and was associated with enhanced neurological recovery. Poststroke angioneurogenesis and axonal plasticity were also enhanced by lithium. On the molecular level, lithium increased miR-124 expression, reduced RE1-silencing transcription factor abundance, and decreased protein deubiquitination in cultivated cortical neurons exposed to oxygen–glucose deprivation and in brains of mice exposed to cerebral ischemia. Notably, this effect was not mimicked by pharmacological GSK3β inhibition. This study for the first time provides efficacy data for lithium in the postacute ischemic phase, reporting a novel mechanism of action, i.e. increased miR-124 expression facilitating REST degradation by which lithium promotes postischemic neuroplasticity and angiogenesis.
Keywords: Cerebral ischemia, neuroregeneration, lithium, miRNA, stroke, RE1-silencing transcription factor
Introduction
For more than 60 years, the mood stabilizer lithium has successfully been used for treatment of bipolar disorders.1 In recent years, however, evidence suggested that lithium also exerts neuroprotective effects in experimental models of ischemic stroke, traumatic brain injury, and neurodegenerative diseases,2–6 which subsequently gave rise to clinical trials. Whereas lithium failed to improve outcome in patients suffering from amyotrophic lateral sclerosis,7 a first investigation on a small study group provided evidence that lithium might improve outcome in stroke patients.7,8
In view of these translational bench-to-bedside efforts, it is surprising that the majority of studies used prophylactic, i.e. preischemic delivery strategies for lithium.9–11 In fact, only two studies evaluated effects of postischemic lithium delivery in experimental stroke models. Whereas one study administered lithium immediately after unilateral global hypoxia-ischemia in neonatal rats, another study evaluated effects of lithium delivery 3 h after transient focal cerebral ischemia in rats.10,12 Observation periods in these studies were short. In the latter study, effects of lithium were evaluated only over 7 days. These studies did not allow for assessment of effects of lithium on postacute brain remodeling and plasticity.
The molecular pathways altered by lithium are diverse and include but are not limited to inhibition of the GSK3β pathway.13 Further pathways include prevention of excitotoxicity, upregulation of anti-apoptotic Bcl-2 and brain-derived-neurotrophic factor (BDNF) as well as induction of prosurvival phosphatidylinositol 3-kinase (PI3K)/Akt, all of which promote neuronal survival.14–18 Moreover, lithium stimulates endogenous neurogenesis and promotes neuronal differentiation under physiological conditions12,19–23 and in models of global cerebral ischemia.12,21
Neuronal differentiation is critically controlled by the RE1-silencing transcription factor (REST, also known as NRSF). In non-neuronal cells, REST abundance is high leading to repression of brain microRNA-124 (miR-124), whereas REST abundance is low in cells committed to neuronal fate.24,25 REST abundance is controlled by the ubiquitin-proteasome-pathway26 and has been shown to increase upon global27 and focal27,28 cerebral ischemia. Using a mouse model of focal cerebral ischemia in which miR-124 has intracerebrally been administered, we have recently demonstrated a novel negative loop from miR-124 to REST via the deubiquitinating enzyme Usp14 resulting in neuroprotection, neurogenesis, and angiogenesis.28
While regulation of REST abundance by lithium has never been described under conditions of ischemia, previous studies suggested a lithium-induced downregulation of REST in neural progenitor cells (NPCs) under nonischemic conditions in cell culture.29,30 In the present study, we aimed to show whether or not lithium’s neuroprotective properties persist in the postacute ischemic phase, and whether they are associated with sustained neurological recovery and brain plasticity. Considering the role of the miR-124/REST loop in controlling postischemic neurogenesis and angiogenesis, we evaluated the role of miR-124 and REST in lithium-induced brain remodeling.
Materials and methods
Experimental design
Experiments were performed according to EU guidelines for the care and use of laboratory animals following the ARRIVE guidelines and were approved by local government authorities (LANUV and Bezirksregierung Braunschweig). Both experimenters and analysts were blinded to study groups, and animals were strictly randomized to groups throughout the study. Time points of animal sacrifice were 2, 4, and 56 days after stroke induction. Animals were intraperitoneally (i.p.) treated with normal saline (control group) or lithium chloride (Sigma-Aldrich, Germany; subsequently addressed as lithium only). Lithium dosage was chosen according to previous reports10,12 with small modifications. Thus, a dose of 1 mmol/kg was i.p. administered during the first injection, followed by daily doses of 2 mmol/kg from day 1 until animal sacrifice (animals randomized to 2 or 4 days survival) or day 7 poststroke (animals randomized to 56 days survival). In order to assess the therapeutic time window of lithium, the first in vivo study protocol included different time points of first lithium injections, i.e. immediately after reperfusion onset (“0 h”), 3, 6, or 9 h after induction of cerebral ischemia. The treatment paradigm is summarized in Supplementary Information Figure 1. In some experiments, 20% dimethyl sulfoxide (DMSO) in normal saline or the GSK3β inhibitor SB216763 (2 mg/kg; Sigma-Aldrich, Germany), dissolved in 20% DMSO in normal saline, was i.p. delivered at the onset of reperfusion. In accordance with a previously published protocol,31 these animals were sacrificed on day 4 poststroke.
Induction of focal cerebral ischemia
Transient focal cerebral ischemia was induced in male C57BL6 mice (Charles River, Germany) as previously described.32 Under anesthesia with isofluran (1.5%), O2 (30%), and N2O (68.5%), the left common carotid artery was isolated and a silicon-coated nylon monofilament with a tip diameter of 180 µm (Doccol, USA) was inserted. The monofilament was gently moved toward the left middle cerebral artery (MCA) and stayed in place for 45 min under constant laser Doppler flow (LDF) control. After monofilament removal, the wounds were carefully sutured and LDF was monitored for an additional 15 min to ensure reperfusion.
Analysis of postischemic cell proliferation, neurogenesis, and angiogenesis
For labeling of proliferating cells, mice received daily i.p. bromodeoxyuridine (BrdU; 50 mg/kg; Sigma-Aldrich, Germany) injections on days 8–56 poststroke. The delayed delivery of BrdU injections was chosen in order to avoid staining of proliferating microglia during the acute and subacute stage of the stroke. Animals were transcardially perfused with 4% paraformaldehyde in 0.1 M phosphate-buffered saline (PBS). Brains were removed and frozen. Cryostat sections of 20 µm thick were obtained, which were used for quantitative analysis of cell proliferation and differentiation that were evaluated in peri-infarct tissue defining four regions of interests (ROI) at 0.14 mm anterior, 2.5–3.25 mm ventral, and 1.5–2.25 mm lateral to bregma, as previously described.32 These ROI were essentially used for all immunohistochemical analyses in this study (see Supplementary Information Figure 2). Quantitative analysis was done using a Zeiss (Germany) fluorescence microscope equipped with an Apotome.
Analysis of postischemic axonal plasticity
Axonal plasticity was analyzed using the anterograde tract tracer biotinylated dextran amine (BDA; Molecular Probes, USA), which was stereotactically infused over 5 min into the contralateral cortex 0.5 mm rostral to the bregma, 2.5 mm lateral to the midline, and 1.5 mm below the brain surface on day 42 poststroke.32,33 After infusion, the infusion needle was kept in situ for additional 5 min before removal. Animals were sacrificed on day 56 poststroke, and brains were used for immunohistochemical analysis of midline-crossing BDA-labeled fibers innervating the peri-infarct cortex, that were detected by 3,3′-diaminobenzidine (DAB) staining. Axonal density was evaluated in eight sections of each mouse by analyzing six fields per section divided by total mean densities of all mice. Data are always given as percentage of proportional areas.
Analysis of postischemic neurological recovery
Neurological recovery was evaluated over as long as 56 days poststroke using the rota rod, tight rope, corner turn, and the balance beam tests, as previously described by our group.34 Mice were trained 2 days before stroke induction. In the rota rod test, the time until the animal dropped an accelerating rotating rod (4–40 rpm) was recorded (maximal testing time 300 s). In the tight rope test the animal’s ability to cross a tight rope was evaluated using a validated score from 0 (min) to 20 (max). In the corner turn test, the mouse was placed into an apparatus consisting of two vertical boards forming an angle of 30°. When placed into the corner, a healthy animal randomly leaves the corner to either side, whereas an ischemic mouse preferentially leaves the corner to the nonimpaired body side (i.e. the left side). The laterality index (number of left turns/10) was calculated after ten trials per test day. A healthy animal typically reaches a score of “0.5,” whereas a severely impaired animal reaches a score of up to “1.” The balance beam test consists of a long beam with constantly reduced width, which was elevated from the ground. Animals had to reach the platform at the end of the beam, and the time, until they reached the platform was measured (maximum testing time 60 s). With exception of the corner turn test, which was performed 10 times per day, all tests were performed twice on occasion of each day, and means were calculated for both tests that were used for further data analysis.
Preparation of cultured neurons and oxygen–glucose deprivation
Cortical neurons were prepared as previously described.28 Cells were seeded on glass cover slips at a density of 125,000/cm2 and incubated under standard cell culture conditions in a humidified atmosphere at 5% CO2. Oxygen–glucose deprivation (OGD) was induced by transferring cells to Sterofundin medium (Braun, Germany) containing 1 mM mannitol and incubated at 37℃ in a hypoxic chamber (1% O2, 5% CO2, remainder N2) for 45 min with subsequent re-incubation under standard cell culture conditions for 24 h. Treatment with lithium (1 mM) was done 24 h before OGD and repeated at the beginning of reoxygenation. For some assays, cells were treated with the GSK3β inhibitor SB216763 (1 µM solved in 20% DMSO; Sigma-Aldrich, Germany) during reoxygenation at the end of OGD as previously described.31 Cell viability was assessed using a Live/Dead Viability/Cytotoxicity kit (Cambrex, Germany).
Evaluation of brain injury
Infarct volume and brain edema were analyzed by 2,3,5-triphenyltetrazolium chloride (TTC) staining on 2 mm-thick brain slices on day 4 poststroke using image J software.28 DNA-fragmented cells were evaluated by TUNEL staining on day 2 poststroke. For this purpose, 20 µm brain sections were incubated with proteinase K (7 min, 37℃), followed by exposure to terminal desoxynucleotidyl transferase (TDT) mix, containing 12.5 mg/ml TDT and 25 mg/ml biotinylated dUTP TdT and enzymes according to the manufacturer’s manual (Roche, Germany). After repeated washing steps, sections were stained with a streptavidin-Alexa488-conjugated secondary antibody (2 h, room temperature; Abcam, Germany).
Immunohistochemical analyses
Brain sections were labeled with the following primary antibodies that were used alone or in combination with each other: rat anti-BrdU (1:50; Abcam), mouse anti-BrdU (1:400; Roche Diagnostics, Switzerland), goat anti-doublecortin (anti-Dcx, 1:50; Santa Cruz Biotechnology, Germany), mouse anti-NeuN (1:200; Millipore, Germany), and rat anti-CD31 (1:200; BD Biosciences, USA). After repeated washing steps, sections were incubated for 1 h at room temperature with secondary antibodies that included goat anti-mouse Cy-3 (1:400; Dianova, Germany), goat anti-rat Alexa 594 (1:400; Dianova), donkey anti-goat Alexa 488 (1:250; Invitrogen, Germany), goat anti-mouse Alexa 488 (1:100; Jackson ImmunoResearch, Germany), and goat anti-rat Alexa 488 (1:250; Invitrogen). For analysis of activated microglia, sections were labeled with biotinylated anti-Ib4 antibody (1:25, Vector, USA) that was detected by Alexa488-conjugated streptavidine (1:50, Invitrogen).
Analysis of brain leukocyte infiltration by flow cytometry
Absolute leukocyte numbers were determined in ischemic hemispheres by flow cytometry on day 4 poststroke using Percoll gradient as described by our group before.32,35,36 Briefly, ischemic left hemispheres were mechanically homogenized in lysis buffer (collagenase type XI (125 U/ml), hyaluronidase (60 U/ml), and collagenase (450 U/ml) in Ca2+/Mg2+ supplemented PBS). Cells were incubated with a rat anti-CD45 antibody (BioLegend, Germany). Only the total amount of CD45+high leukocytes were counted as a means to calculate total leukocyte numbers.
Measurement of oxidative stress
Oxidative stress was measured on day 2 poststroke as previously described.28 Briefly, left ischemic hemispheres were homogenized, and thiobarbituric acid (TBA) reactive substances (TBARS) giving rise to a chromogenic compound during peroxidation were photometrically measured.
Quantitative reverse transcriptase polymerase chain reaction for miR-124 expression analysis
MiR-124 was evaluated in cell lysates 24 h after ODG or brain lysates 4 days after MCA occlusion following previously published protocols.37,38 Total RNA was extracted using the miR-Vana RNA kit (Life Technologies, Germany), of which 10 ng was used for further sample processing. MiR124 expression was measured using the TaqMan® MicroRNA Assay (Life Technologies). Data were evaluated as fold expression change using the comparative Ct (ΔΔCt) method with U6 as endogenous control.
Western blot analysis of REST abundance
REST abundance was examined by Western blotting in cell lysates 24 h after ODG or brain lysates 4 days after MCA occlusion.28 Briefly, cells or tissue samples were complemented with lysis buffer (50 mmol/l Tris, pH 8.0, 150 mmol/l NaCl, 1% Triton X-100, and protease inhibitors), homogenized, and centrifuged. After SDS-PAGE and transferal of proteins onto PVDF membranes, membranes were incubated with a rabbit antibody directed against amino acids 1-290 of the REST molecule (Santa Cruz Biotechnology, Germany) or goat anti-actin antibody (Millipore, Germany). Membranes were incubated with peroxidase-coupled secondary antibody (Santa Cruz Biotechnology) and exposed to ECL-Hyperfilm (Amersham, Freiburg, Germany).
Determination of deubiquitination activity
Deubiquitination activity was measured as previously described28 using 50 μM of ubiquitin-7-amino-4-methylcoumarin (AMC; Enzo Life Sciences, Germany) as reaction substrate in cell lysates 24 h after ODG or brain lysates 4 days after MCA occlusion. Cells or brain samples were complemented with lysis buffer containing 100 mM Tris-HCl, 145 mM NaCl, 10 mM EDTA, and 0.5% Triton X-100 at pH 7.3. Thereafter, lysates were incubated with reaction buffer that consisted of 50 mM Tris-HCl (pH 7.5), 1 mM EDTA, 1 mM ATP (Sigma-Aldrich), 5 mM MgCl2, 1 mM DTT, and 1 mg/ml ovalbumin (Sigma-Aldrich). Protease activities were given as arbitrary fluorescence units. Protein contents were measured using the Bradford assay.
Statistical analysis
Statistical analysis was performed using Student’s t tests (comparison between two groups) or one-way analysis of variance (ANOVA) followed by Tukey’s post hoc tests (comparison between multiple groups). Data were presented as means ± standard deviations (SDs). p values < 0.05 were regarded to indicate statistical significance.
Results
Lithium induces long-term neuroprotection associated with functional neurological recovery when delivered up to 6 h after reperfusion
Previous studies using postischemic delivery strategies of lithium are scarce.10,12 focusing either on neonatal models of unilateral global hypoxia–ischemia with immediate postischemic lithium delivery or on models of focal cerebral ischemia with lithium delivery within 3 h poststroke and observation periods of 7 days only. We therefore analyzed the therapeutic window of lithium’s neuroprotective properties using TTC stainings, demonstrating that lithium reduced infarct volume on day 4 poststroke when delivered up to 6 h after reperfusion (Figure 1). In view of these findings, we systematically administered lithium at 6 h poststroke in all subsequent studies.
Figure 1.
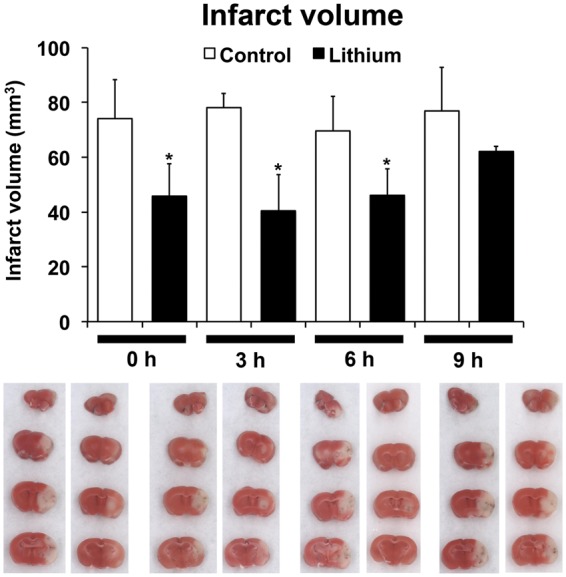
Lithium protects against focal cerebral ischemia when delivered up to 6 h poststroke. Mice were exposed to 45 min of intraluminal middle cerebral artery (MCA) occlusion, intraperitoneally treated with normal saline (control) or lithium (1 mg/kg bolus, followed by 2 mg/kg/day) starting at the indicated time-points and sacrificed after 4 days. Infarct volume was determined by triphenyltetrazolium chloride (TTC) staining (n = 6 mice per group). Representative TTC stainings for each condition are shown in the same order as in the graph. *Significantly different from control with p:0.026.
Lithium reduced the density of TUNEL positive, i.e. DNA-fragmented cells in the ischemic striatum 2 days poststroke (Figure 2(a)), which is in line with the aforementioned reduction of infarct volume on day 4. Likewise, brain edema was significantly reduced 4 days poststroke after treatment with lithium (Figure 2(c)). Of note, lithium-induced neuroprotection was not transient, but persisted over as long as 56 days, as revealed by an increased density of surviving NeuN+ neurons in the peri-infarct striatum (Figure 2(d)). Structural neuroprotection induced by lithium was associated with reduced motor coordination impairment in the rota rod, tight rope, corner turn, and balance beam tests (Figure 3).
Figure 2.

Lithium induces long-term neuroprotection that persists in the postacute stroke phase. Mice were exposed to 45 min of intraluminal MCA occlusion and intraperitoneally treated with normal saline (control) or lithium (1 mg/kg bolus, followed by 2 mg/kg/day) starting at 6 h after reperfusion over up to 7 days. (a) Analysis of TUNEL positive cells in the ischemic striatum after 2 days, (b) TTC-analysis for depicture of infarct volume on day 4 taken from Figure 1, (c) analysis of brain edema using TTC from (b), and (d) analysis of long-term neuronal survival in the ischemic striatum using NeuN immunohistochemistry after 56 days (n = 12–13 mice per group). Representative TUNEL stainings and NeuN immunohistochemistries are shown. Scale bars: 50 µm. *Significantly different from controls with p:0.012 (a), p:0.037 (b), p:0.021 (c), and p:0.017 (d).
Figure 3.

Lithium induces sustained postischemic neurological recovery. Mice were exposed to 45 min of intraluminal MCA occlusion and intraperitoneally treated with normal saline (control) or lithium (1 mg/kg bolus, followed by 2 mg/kg/day) starting at 6 h after reperfusion over up to 7 days. Neurological recovery was assessed using (a) rota rod, (b) tight rope, (c) corner turn, and (d) balance beam tests (n = 12–13 mice per group). *Significantly different from controls with p:0.009–0.021 (a), p:0.015–0.043 (b), p:0.024–0.032 (c), and p:0.014–0.046 (d).
Lithium reduces postischemic inflammation and ameliorates oxidative stress
Brain inflammation and oxidative stress are key components contributing to ischemic injury development.39 We therefore analyzed microglial activation and leukocyte infiltration in the ischemic brain 2 days poststroke by Ib4 immunohistochemistry and CD45 flow cytometry, furthermore evaluating oxidative stress by TBARS formation. Lithium reduced the density of reactive Ib4+ microglia (Figure 4(a)), decreased the number of CD45+high leukocytes (Figure 4(b)), and diminished oxidative stress (Figure 4(c)) in the ischemic brain tissue.
Figure 4.

Lithium reduces postischemic brain inflammation and oxidative stress. Mice were exposed to 45 min of intraluminal MCA occlusion, intraperitoneally treated with normal saline (control) or lithium (1 mg/kg bolus, followed by 2 mg/kg/day) starting at 6 h after reperfusion and sacrificed after 2 days. (a) Reactive microglia in the ischemic striatum, assessed by Ib4 immunohistochemistry, (b) total number of CD45+high leukocytes in the ischemic hemisphere, examined by flow cytometry, and (c) analysis of oxidative stress in the ischemic hemisphere, evaluated by thiobarbituric acid reactive substances (TBARS) formation (n = 7 mice per group). Representative Ib4 immunohistochemistries are shown. Scale bars: 40 µm. *Significantly different from control with p:0.011 (a), p:0.020 (b), and p:0.042 (c).
Lithium stimulates postischemic neurogenesis, angiogenesis, and axonal plasticity
Based on observations that lithium promotes neurogenesis in models of global cerebral ischemia,12,21 we next analyzed whether sustained neuroprotection was associated with increased brain plasticity (also depicted in Supplementary Information Figure 3). Delivery of lithium increased the density of BrdU+, i.e. proliferating cells in the peri-infarct tissue at 56 days poststroke (Figure 5(a)). Differentiation analysis revealed increased expression of the neuronal markers Dcx and NeuN in BrdU+ cells of lithium treated mice (Figure 5(b) and (c)), indicating that lithium enhanced neurogenesis. Of note, one has to keep in mind that BrdU labeling does not exclusively indicate neurogenesis but cell proliferation per se and also DNA repair,40 albeit the likelihood of these events are limited due to the BrdU labeling protocol chosen.
Figure 5.
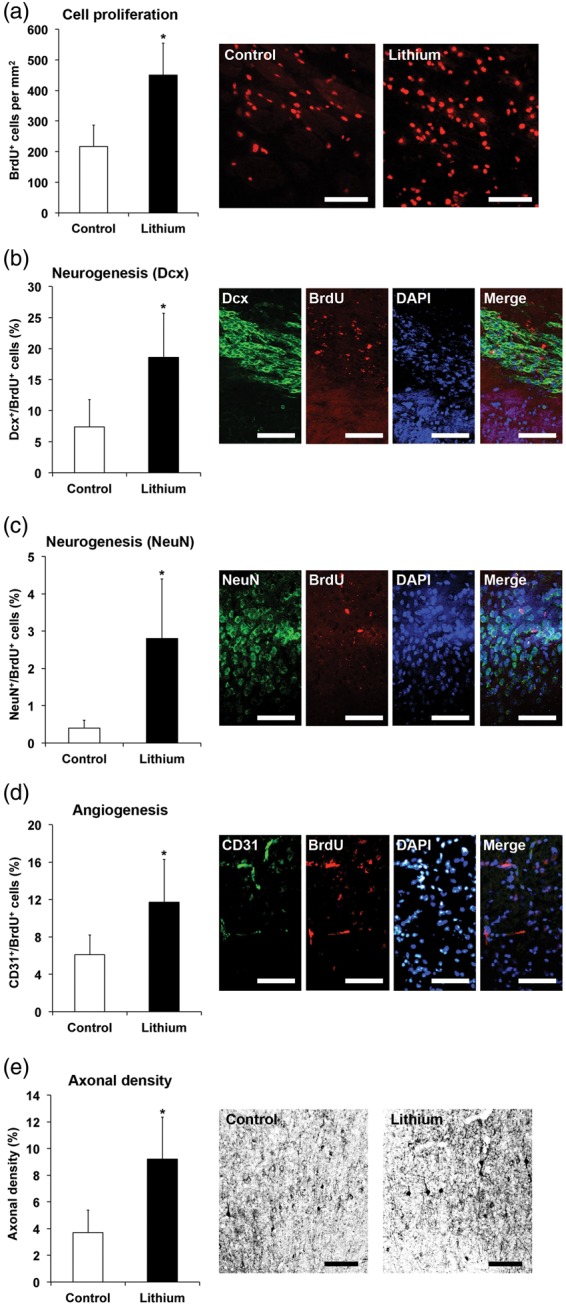
Lithium stimulates peri-infarct cell proliferation, neurogenesis, angiogenesis, and axonal plasticity. Mice were exposed to 45 min of intraluminal MCA occlusion, intraperitoneally treated with normal saline (control) or lithium (1 mg/kg bolus, followed by 2 mg/kg/day) starting at 6 h after reperfusion over up to 7 days and sacrificed after 56 days. (a) Assessment of postischemic cell proliferation by bromodeoxyuridine (BrdU) incorporation in the ischemic striatum, (b–d) differentiation analysis of BrdU+ cells in the ischemic striatum based on co-expression of neuronal markers Dcx (in b) and NeuN (in c) (indicative of new-born neurons, i.e. neurogenesis) or endothelial marker CD31 (in d) (indicative of new-born endothelium, i.e. angiogenesis), as well as (e) determination of axonal density in the peri-infarct cortex after contralesional injection of the anterograde tract tracer biotinylated dextran amine (BDA) (n = 12–13 per group). Data for axonal densities are given as percentage of proportional areas as indicated in the materials and methods section. *Significantly different from controls with p:0.029 (a), p:0.005 (b), p:0.013 (c), p:0.045 (d), and p:0.008 (e). Scale bars: 50 µm.
Considering that neurogenesis is closely associated with angiogenesis in the ischemic brain,41 we further analyzed the expression of the endothelial marker CD31 in BrdU+ cells. In these studies, an increased co-localization of BrdU and CD31 was noticed in CM-treated mice, suggesting that lithium stimulated angiogenesis (Figure 5(d)).
To evaluate whether lithium increased axonal plasticity, we also analyzed the density of terminal fibers in the peri-infarct cerebral cortex by means of anterograde tract-tracing using BDA. Delivery of lithium increased the density of axons in the peri-infarct cortex originating from the contralesional motor cortex at 56 days poststroke (Figure 5(e)). Thus, lithium had profound effects on brain plasticity that were likely to contribute to neurological recovery.
Lithium increases miR-124 expression and decreases REST abundance after OGD
Although robust evidence suggests that lithium similar to other mood stabilizers regulates miRNA expression in vitro,42,43 there have hitherto been no data suggesting poststroke regulation of miR-124 by lithium. As a proof of concept, we therefore analyzed in cultivated cortical neurons exposed to OGD whether miR-124 expression was influenced by lithium. As expected, lithium protected neurons against cell death 24 h after OGD induction (Figure 6(a)). Neuroprotection by lithium was associated with increased miR-124 expression (Figure 6(b)). Of note, delivery of a selective GSK3β inhibitor, SB216763, did not induce miR-124 expression (Figure 6(b)), albeit SB216763 conferred neuroprotection in cortical neurons (data not shown). In line with the increased miR-124 expression, REST abundance in cortical neurons was significantly reduced by lithium (Figure 6(c)). Since REST abundance is controlled by the ubiquitin-proteasome pathway28 we also measured protein deubiquitination in cortical neurons exposed to OGD. These studies showed that lithium reduced deubiquitination activity (Figure 6(d)).
Figure 6.

Lithium increases miR-124 expression, reduces REST abundance, and decreases protein deubiquitination in cortical neurons exposed to oxygen–glucose deprivation (OGD). Cultivated cortical neurons were exposed to OGD for 45 min and subsequently recultivated under standard cell culture conditions for 24 h. Cells were exposed to normal saline (control) or lithium (1 mM dissolved in normal saline) starting 24 h before OGD, while the GSK3β inhibitor SB216763 (1 µM dissolved in DMSO) was added at the end of OGD. (a) Cell death, evaluated using a Live/Dead Viability/Cytotoxicity kit, (b) miR-124 expression, assessed by RT-PCR, (c) REST abundance, analyzed by Western blots (using actin as housekeeping protein for controlling protein loading), and (d) protein deubiquitination, measured by ubiquitin-AMC (always n = 4 independent experiments or samples). Note that the GSK3β inhibitor SB216763 similar to its solvent DMSO did not affect miR-124 expression in (b), indicating that miR-124 induction by lithium was independent of lithium’s GSK3β inhibitory activity. *Significantly different from controls with p:0.047 (a), p:0.034 (b), p:0.036 (c), and p:0.002–0.027 (d).
Lithium increases miR-124 expression and decreases REST abundance after focal cerebral ischemia
In light of these in vitro studies, we further evaluated lithium’s effects on miR-124 expression, REST abundance, and protein deubiquitination after focal cerebral ischemia in mice. Similar to OGD in vitro, lithium elevated miR-124 expression, reduced REST abundance, and decreased protein deubiquitination in ischemic brain tissue 4 days poststroke (Figure 7(a) to (c)). Again, the GSK3β inhibitor SB216763 did not influence miR-124 expression (Figure 7(a)), although SB216763 reduced ischemic injury (data not shown). Hence, lithium stimulates miR-124 expression in vitro after OGD and in vivo after stroke, reducing REST abundance most likely due to increased proteasomal degradation.
Figure 7.

Lithium increases miR-124 expression, reduces REST abundance and decreases protein deubiquination after focal cerebral ischemia in mice. Mice were exposed to 45 min of intraluminal MCA occlusion, intraperitoneally treated with normal saline (control) or lithium (1 mg/kg bolus, followed by 2 mg/kg/day) starting at 6 h after reperfusion and sacrificed after 4 days. In some studies, the GSK3β inhibitor SB216763 (2 mg/kg dissolved in DMSO) was intraperitoneally administered at the onset of reperfusion. (a) miR-124 expression, assessed by RT-PCR, (b) REST abundance, analyzed by Western blots (using actin as housekeeping protein for controlling protein loading), and (c) protein deubiquitination, measured by ubiquitin-AMC, in ischemic brain tissue (n = 7 mice per group). Note that the GSK3β inhibitor SB216763 similar to its solvent DMSO did not affect miR-124 expression in (a), indicating that miR-124 induction by lithium was independent of lithium’s GSK3β inhibitory activity. *Significantly different from controls with p:0.016 (a), p:0.029 (b), p:0.018–0.042 (c).
Discussion
In the present study we show that postischemic delivery of lithium induces long-term neuroprotection after focal cerebral ischemia in mice, when administered up to 6 h after reperfusion onset. The survival-promoting effects of lithium persisted in the postacute stroke phase, up to 56 days poststroke, and were associated with enhanced neurological recovery in a battery of motor-coordination tests. Delivery of lithium increased neurogenesis, angiogenesis, and axonal plasticity in the peri-infarct brain tissue. On the molecular level, lithium increased the expression of miR-124, reduced the abundance of REST, and decreased protein deubiquitination in peri-infarct brain tissue. Our data suggest that the regulation of the miR-124 and REST pathways contributed to lithium-induced postischemic brain remodeling via mechanisms involving protein degradation via the proteasome.
Only two studies so far evaluated effects of postischemic lithium delivery in experimental models resembling stroke. Whereas one study administered lithium immediately after unilateral global hypoxia-ischemia in rats, another study evaluated effects of lithium delivery 3 h after transient focal cerebral ischemia in rats.10,12 The present study expands these data to mice, demonstrating that lithium protects against stroke, when delivered up to 6 h after reperfusion onset. Based on these findings, lithium might offer a promising add-on treatment next to thrombolysis. In earlier studies with postischemic delivery of lithium, observation periods were short. In the study by Ren et al.,10 postischemic recovery was evaluated over up to 7 days. These studies did not allow for assessment of long-term effects of lithium on postacute brain remodeling and plasticity. By evaluating animals over up to 56 days, we now show that the recovery-promoting effects of lithium are sustainable and that they are associated with profound brain remodeling and plasticity.
Indeed, by means of BrdU incorporation studies and anterograde tract tracing experiments using BDA we show that lithium enhances postischemic cell proliferation, neurogenesis, angiogenesis, and axonal plasticity in the peri-infarct brain tissue in the postacute stroke phase. Promotion of neurogenesis by lithium has previously been demonstrated under nonischemic conditions19,20 and in models of global cerebral ischemia.12,21 While Li et al. observed increased postischemic cell proliferation and elevated neuronal differentiation in the dentate gyrus after lithium delivery in a rat model of unilateral neonatal hypoxia-ischemia, Yan et al. reported increased cell proliferation without enhanced neuronal differentiation after prophylactic lithium delivery in an adult rat model of global cerebral ischemia. Differences in animal models (neonatal vs. adult rats/unilateral vs. bilateral global cerebral ischemia) and observation periods (3 vs. 7 weeks) may explain diverging findings in these earlier studies.
In the present study, lithium increased neurogenesis in the peri-infarct brain tissue over up to 56 days. Overall neuronal differentiation rates were low, suggesting that new-born cells did not replace lost neural tissue to a relevant extent. Rather, new-born cells may have acted in a paracrine way, releasing trophic factors into the brain tissue that stimulated remodeling of the ischemic brain parenchyma.32,44–48 Interestingly, both angiogenesis and axonal plasticity were also enhanced by lithium in the peri-infarct brain tissue, again supporting the hypothesis that neurogenesis, angiogenesis, and axonal plasticity are tightly linked in the ischemic brain.41
The mechanisms via which lithium promoted neurogenesis, angiogenesis, and axonal plasticity are still unknown. Previous work has shown that brain miR-124, which is negatively regulated by REST, is intimately involved in neuronal differentiation.49–54 Upon conditions of global and focal cerebral ischemia, REST abundance in the brain was increased and contributed to brain injury.27,28 Previous studies of our group identified a hitherto unknown mechanism by which miR-124 inhibits the postischemic increase of REST abundance.28 Although some papers described a regulation of REST or miRNAs by mood stabilizers such as lithium under nonischemic conditions,29,30,42,43 the regulation of miR-124 and REST by lithium is new.
That miR-124 is increased after focal cerebral ischemia55 and protects against ischemic injury28 has previously been shown by others and ourselves. Of note, the lithium-induced elevation of miR-124 expression is likely to be independent of the GSK3β inhibition properties of lithium.13 Thus, the delivery of the GSK3β inhibitor SB216763 did not mimic lithium’s effects on miR-124 expression, neither in vitro nor in vivo. In view of earlier findings,28 we hypothesize that the elevation of miR-124 expression may have been responsible for the degradation of REST. As previously reported for miR-124 delivery,28 the increased miR-124 expression after lithium delivery was associated with reduced deubiquitination activity. Following miR-124 delivery, the inhibition of deubiquitination activity was evoked by the inhibition of the deubiquitinating enzyme Usp14.28 It facilitated proteasomal REST degradation.28 Yet, one has to keep in mind that the lithium-regulated signaling pathways observed in the present study might not exclusively be causally connected with each other. Such a conclusion would have needed further experiments which were beyond the scope of the present work.
The regulation of miR-124 and REST by lithium further expands previous evidence that miR-124 and REST might represent promising targets, which might successfully be modulated by neurorestorative drugs. In light of the recently conducted clinical trial on the therapeutic benefit of lithium in stroke patients,8,56 the effects of lithium on postacute ischemic brain remodeling and plasticity will deserve attention in future experimental and preclinical studies. Lithium represents a cheap pharmacological compound with substantial clinical experience even after prolonged delivery in mood disorders. Nevertheless, lithium also possesses a narrow therapeutic window which limits its clinical use. As such, studies using lithium analogues with better side effect profiles as well as different lithium salt compounds are currently under investigation with respect to their therapeutic potential against mood disorders.57 Corresponding studies in the stroke field, however, do not yet exist. Therefore, additional studies using either lithium or analogues will have to rule out dose-responses and will have to test whether or not lithium and its analogues are similarly effective in aged animals with vascular risk factors, thus better reflecting the actual clinical situation with stroke patients.
Supplementary Material
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The present study was supported by the German Research Council (DFG; #HE3173/2-2 and #HE3173/3-1 to DMH) and TUBITAK (#2221 to TRD).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
Doeppner: concept and design of the study, performance of research, writing of the manuscript, financial support. Kaltwasser: performance of research. Sanchez-Mendoza: performance of research. Caglayan: performance of research. Hermann: writing of the manuscript and financial support. Bähr: financial support.
Supplementary material
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data.
References
- 1.Manji HK, Lenox RH. Lithium: A molecular transducer of mood-stabilization in the treatment of bipolar disorder. Neuropsychopharmacology 1998; 19: 161–166. [DOI] [PubMed] [Google Scholar]
- 2.Yu F, Wang Z, Tchantchou F, et al. Lithium ameliorates neurodegeneration, suppresses neuroinflammation, and improves behavioral performance in a mouse model of traumatic brain injury. J Neurotrauma 2012; 29: 362–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kang K, Kim YJ, Kim YH, et al. Lithium pretreatment reduces brain injury after intracerebral hemorrhage in rats. Neurol res 2012; 34: 447–454. [DOI] [PubMed] [Google Scholar]
- 4.Wang ZF, Fessler EB, Chuang DM. Beneficial effects of mood stabilizers lithium, valproate and lamotrigine in experimental stroke models. Acta pharmacol Sin 2011; 32: 1433–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu F, Wang Z, Tanaka M, et al. Posttrauma cotreatment with lithium and valproate: Reduction of lesion volume, attenuation of blood-brain barrier disruption, and improvement in motor coordination in mice with traumatic brain injury. J Neurosurg 2013; 119: 766–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiu CT, Chuang DM. Molecular actions and therapeutic potential of lithium in preclinical and clinical studies of CNS disorders. Pharmacol Ther 2010; 128: 281–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aggarwal SP, Zinman L, Simpson E, et al. Safety and efficacy of lithium in combination with riluzole for treatment of amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled trial. Lancet Neurol 2010; 9: 481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mohammadianinejad SE, Majdinasab N, Sajedi SA, et al. The effect of lithium in post-stroke motor recovery: A double-blind, placebo-controlled, randomized clinical trial. Clin Neuropharmacol 2014; 37: 73–78. [DOI] [PubMed] [Google Scholar]
- 9.Nonaka S, Chuang DM. Neuroprotective effects of chronic lithium on focal cerebral ischemia in rats. Neuroreport 1998; 9: 2081–2084. [DOI] [PubMed] [Google Scholar]
- 10.Ren M, Senatorov VV, Chen RW, et al. Postinsult treatment with lithium reduces brain damage and facilitates neurological recovery in a rat ischemia/reperfusion model. Proc Natl Acad SciUSA 2003; 100: 6210–6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu J, Culman J, Blume A, et al. Chronic treatment with a low dose of lithium protects the brain against ischemic injury by reducing apoptotic death. Stroke 2003; 34: 1287–1292. [DOI] [PubMed] [Google Scholar]
- 12.Li H, Li Q, Du X, et al. Lithium-mediated long-term neuroprotection in neonatal rat hypoxia-ischemia is associated with antiinflammatory effects and enhanced proliferation and survival of neural stem/progenitor cells. J Cereb Blood Flow Metab 2011; 31: 2106–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chuang DM, Wang Z, Chiu CT. GSK-3 as a target for lithium-induced neuroprotection against excitotoxicity in neuronal cultures and animal models of ischemic stroke. Front Mol Neurosci 2011; 4: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bian Q, Shi T, Chuang DM, et al. Lithium reduces ischemia-induced hippocampal CA1 damage and behavioral deficits in gerbils. Brain Res 2007; 1184: 270–276. [DOI] [PubMed] [Google Scholar]
- 15.Chuang DM. The antiapoptotic actions of mood stabilizers: Molecular mechanisms and therapeutic potentials. Ann N Y Acad Sci 2005; 1053: 195–204. [DOI] [PubMed] [Google Scholar]
- 16.Gold AB, Herrmann N, Lanctot KL. Lithium and its neuroprotective and neurotrophic effects: Potential treatment for post-ischemic stroke sequelae. Curr Drug Targets 2011; 12: 243–255. [DOI] [PubMed] [Google Scholar]
- 17.Sasaki T, Han F, Shioda N, et al. Lithium-induced activation of Akt and CaM kinase II contributes to its neuroprotective action in a rat microsphere embolism model. Brain Res 2006; 1108: 98–106. [DOI] [PubMed] [Google Scholar]
- 18.Hashimoto R, Takei N, Shimazu K, et al. Lithium induces brain-derived neurotrophic factor and activates TrkB in rodent cortical neurons: An essential step for neuroprotection against glutamate excitotoxicity. Neuropharmacology 2002; 43: 1173–1179. [DOI] [PubMed] [Google Scholar]
- 19.Chen G, Rajkowska G, Du F, et al. Enhancement of hippocampal neurogenesis by lithium. J Neurochem 2000; 75: 1729–1734. [DOI] [PubMed] [Google Scholar]
- 20.Kim JS, Chang MY, Yu IT, et al. Lithium selectively increases neuronal differentiation of hippocampal neural progenitor cells both in vitro and in vivo. J Neurochem 2004; 89: 324–336. [DOI] [PubMed] [Google Scholar]
- 21.Yan XB, Hou HL, Wu LM, et al. Lithium regulates hippocampal neurogenesis by ERK pathway and facilitates recovery of spatial learning and memory in rats after transient global cerebral ischemia. Neuropharmacology 2007; 53: 487–495. [DOI] [PubMed] [Google Scholar]
- 22.Young W. Review of lithium effects on brain and blood. Cell Transplant 2009; 18: 951–975. [DOI] [PubMed] [Google Scholar]
- 23.Zigova T, Willing AE, Tedesco EM, et al. Lithium chloride induces the expression of tyrosine hydroxylase in hNT neurons. Exp Neurol 1999; 157: 251–258. [DOI] [PubMed] [Google Scholar]
- 24.Westbrook TF, Hu G, Ang XL, et al. SCFbeta-TRCP controls oncogenic transformation and neural differentiation through REST degradation. Nature 2008; 452: 370–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Conaco C, Otto S, Han JJ, et al. Reciprocal actions of REST and a microRNA promote neuronal identity. Proc Natl Acad SciU S A 2006; 103: 2422–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang Z, Wu Q, Guryanova OA, et al. Deubiquitylase HAUSP stabilizes REST and promotes maintenance of neural progenitor cells. Nat Cell Biol 2011; 13: 142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Calderone A, Jover T, Noh KM, et al. Ischemic insults derepress the gene silencer REST in neurons destined to die. J Neurosci 2003; 23(6): 2112–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doeppner TR, Doehring M, Bretschneider E, et al. MicroRNA-124 protects against focal cerebral ischemia via mechanisms involving Usp14-dependent REST degradation. Acta Neuropathol 2013; 126: 251–265. [DOI] [PubMed] [Google Scholar]
- 29.Warburton A, Savage AL, Myers P, et al. Molecular signatures of mood stabilisers highlight the role of the transcription factor REST/NRSF. J Affect Disord 2014; 172C: 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishii T, Hashimoto E, Ukai W, et al. Lithium-induced suppression of transcription repressor NRSF/REST: Effects on the dysfunction of neuronal differentiation by ethanol. Eur J Pharmacol 2008; 593: 36–43. [DOI] [PubMed] [Google Scholar]
- 31.Valerio A, Bertolotti P, Delbarba A, et al. Glycogen synthase kinase-3 inhibition reduces ischemic cerebral damage, restores impaired mitochondrial biogenesis and prevents ROS production. J Neurochem 2011; 116: 1148–1159. [DOI] [PubMed] [Google Scholar]
- 32.Doeppner TR, Kaltwasser B, Teli MK, et al. Effects of acute versus post-acute systemic delivery of neural progenitor cells on neurological recovery and brain remodeling after focal cerebral ischemia in mice. Cell Death Dis 2014; 5: e1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan T, Chopp M, Ye X, et al. Niaspan increases axonal remodeling after stroke in type 1 diabetes rats. Neurobiol Dis 2012; 46: 157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Doeppner TR, Kaltwasser B, Bahr M, et al. Effects of neural progenitor cells on post-stroke neurological impairment-a detailed and comprehensive analysis of behavioral tests. Front Cell Neurosci 2014; 8: 338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doeppner TR, Herz J, Gorgens A, et al. Extracellular vesicles improve post-stroke neuroregeneration and prevent postischemic immunosuppression. Stem Cells Transl Med 2015; 4: 1131–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Herz J, Hagen SI, Bergmuller E, et al. Exacerbation of ischemic brain injury in hypercholesterolemic mice is associated with pronounced changes in peripheral and cerebral immune responses. Neurobiol Dis 2014; 62: 456–468. [DOI] [PubMed] [Google Scholar]
- 37.Silber J, Hashizume R, Felix T, et al. Expression of miR-124 inhibits growth of medulloblastoma cells. Neuro Oncol 2013; 15: 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Silber J, Lim DA, Petritsch C, et al. miR-124 and miR-137 inhibit proliferation of glioblastoma multiforme cells and induce differentiation of brain tumor stem cells. BMC Med 2008; 6: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Macrez R, Ali C, Toutirais O, et al. Stroke and the immune system: From pathophysiology to new therapeutic strategies. Lancet Neurol 2011; 10: 471–480. [DOI] [PubMed] [Google Scholar]
- 40.Taupin P. BrdU immunohistochemistry for studying adult neurogenesis: Paradigms, pitfalls, limitations, and validation. Brain Res Rev 2007; 53: 198–214. [DOI] [PubMed] [Google Scholar]
- 41.Hermann DM, Chopp M. Promoting brain remodelling and plasticity for stroke recovery: Therapeutic promise and potential pitfalls of clinical translation. Lancet Neurol 2012; 11: 369–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou R, Yuan P, Wang Y, et al. Evidence for selective microRNAs and their effectors as common long-term targets for the actions of mood stabilizers. Neuropsychopharmacology 2009; 34: 1395–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hunsberger JG, Fessler EB, Chibane FL, et al. Mood stabilizer-regulated miRNAs in neuropsychiatric and neurodegenerative diseases: Identifying associations and functions. Am J Transl Res 2013; 5: 450–464. [PMC free article] [PubMed] [Google Scholar]
- 44.Bacigaluppi M, Pluchino S, Martino G, et al. Neural stem/precursor cells for the treatment of ischemic stroke. J Neurol sci 2008; 265: 73–77. [DOI] [PubMed] [Google Scholar]
- 45.Bacigaluppi M, Pluchino S, Peruzzotti Jametti L, et al. Delayed post-ischaemic neuroprotection following systemic neural stem cell transplantation involves multiple mechanisms. Brain 2009; 132: 2239–2251. [DOI] [PubMed] [Google Scholar]
- 46.Martino G, Bacigaluppi M, Peruzzotti-Jametti L. Therapeutic stem cell plasticity orchestrates tissue plasticity. Brain 2011; 134: 1585–1587. [DOI] [PubMed] [Google Scholar]
- 47.Martino G, Pluchino S, Bonfanti L, et al. Brain regeneration in physiology and pathology: The immune signature driving therapeutic plasticity of neural stem cells. Physiol Rev 2011; 91: 12811304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Doeppner TR, Ewert TA, Tonges L, et al. Transduction of neural precursor cells with TAT-heat shock protein 70 chaperone: Therapeutic potential against ischemic stroke after intrastriatal and systemic transplantation. Stem Cells (Dayton, Ohio) 2012; 30: 1297–1310. [DOI] [PubMed] [Google Scholar]
- 49.Mishima T, Mizuguchi Y, Kawahigashi Y, et al. RT-PCR-based analysis of microRNA (miR-1 and -124) expression in mouse CNS. Brain Res 2007; 1131: 37–43. [DOI] [PubMed] [Google Scholar]
- 50.Liu K, Liu Y, Mo W, et al. MiR-124 regulates early neurogenesis in the optic vesicle and forebrain, targeting NeuroD1. Nucleic Acids Res 2011; 39: 2869–2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Pietri Tonelli D, Pulvers JN, Haffner C, et al. miRNAs are essential for survival and differentiation of newborn neurons but not for expansion of neural progenitors during early neurogenesis in the mouse embryonic neocortex. Development 2008; 135: 3911–3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Papagiannakopoulos T, Kosik KS. MicroRNA-124: Micromanager of neurogenesis. Cell Stem Cell 2009; 4: 375–376. [DOI] [PubMed] [Google Scholar]
- 53.Cheng LC, Pastrana E, Tavazoie M, et al. miR-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nat Neurosci 2009; 12: 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu JY, Chung KH, Deo M, et al. MicroRNA miR-124 regulates neurite outgrowth during neuronal differentiation. Exp Cell Res 2008; 314: 2618–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sun Y, Gui H, Li Q, et al. MicroRNA-124 protects neurons against apoptosis in cerebral ischemic stroke. CNS Neurosci Ther 2013; 19: 813–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weng H, Shen C, Hirokawa G, et al. Plasma miR-124 as a biomarker for cerebral infarction. Biomed Res 2011; 32: 135–141. [DOI] [PubMed] [Google Scholar]
- 57.Smith AJ, Kim SH, Tan J, et al. Plasma and brain pharmacokinetics of previously unexplored lithium salts. RSC Adv 2014; 4: 12362–12365. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.