

Abstract
Insulin resistance contributes to the high prevalence of type 2 diabetes mellitus, leading to cardiac anomalies. Emerging evidence depicts a pivotal role for mitochondrial injury in oxidative metabolism and insulin resistance. Mitochondrial aldehyde dehydrogenase (ALDH2) is one of metabolic enzymes detoxifying aldehydes although its role in insulin resistance remains elusive. This study was designed to evaluate the impact of ALDH2 overexpression on insulin resistance-induced myocardial damage and mechanisms involved with a focus on autophagy. Wild-type (WT) and transgenic mice overexpressing ALDH2 were fed sucrose or starch diet for 8 weeks and cardiac function and intracellular Ca2+ handling were assessed using echocardiographic and IonOptix systems. Western blot analysis was used to evaluate Akt, heme oxygenase-1 (HO-1), PGC-1α and Sirt-3. Our data revealed that sucrose intake provoked insulin resistance and compromised fractional shortening, cardiomyocyte function and intracellular Ca2+ handling (p < 0.05) along with unaltered cardiomyocyte size (p > 0.05), mitochondrial injury (elevated ROS generation, suppressed NAD+ and aconitase activity, p < 0.05 for all), the effect of which was ablated by ALDH2. In vitro incubation of the ALDH2 activator Alda-1, the Sirt3 activator oroxylin A and the histone acetyltransferase inhibitor CPTH2 rescued insulin resistance-induced changes in aconitase activity and cardiomyocyte function (p < 0.05). Inhibiting Sirt3 deacetylase using 5-amino-2-(4-aminophenyl) benzoxazole negated Alda-1-induced cardioprotective effects. Taken together, our data suggest that ALDH2 serves as an indispensable cardioprotective factor against insulin resistance-induced cardiomyopathy with a mechanism possibly associated with facilitation of the Sirt3-dependent PGC-1α deacetylation.
Keywords: sucrose, insulin resistance, ALDH2, Sirt3, PGC-1, Pathology Section
INTRODUCTION
Epidemiological studies estimate that the global prevalence of diabetes mellitus was 285 million among adults in 2010, and this number is anticipated to reach an alarming 439 million by 2030 [1, 2]. Among all contributing factors for diabetes, compromised insulin sensitivity is perhaps most crucial leading to the consequence of an overstimulation of pancreatic secretion of insulin and dampened glucose and fatty acid metabolism [3, 4]. Insulin resistance is associated with accumulation of reactive oxygen species (ROS), oxidative stress, onset and development of cardiac dysfunction possibly related to peroxidation of lipids and proteins, mitochondrial damage, and changes in excitation contraction coupling proteins [5]. Earlier work from our group and others has depicted that sucrose diet intake triggers whole body insulin resistance and cardiac dysfunction [6-8], which is in line with the clinical manifestations of cardiac dysfunction and increased mortality among patients with insulin resistance and type 2 diabetes [9]. Framingham Heart Study also correlates soft drink consumption with an increased incidence of metabolic risk, suggesting a close tie between carbonhydrate intake and metabolic diseases [10]. Nonetheless, neither the precise pathological mechanism nor effective clinical management is available for cardiac anomalies associated with sucrose diet-induced insulin resistance.
Recent evidence has indicated a key role for mitochondrial aldehyde dehydrogenase (ALDH2) in a wide array of heart diseases including ischemic heart disease [11-13], dilated cardiomyopathy [14], alcoholic cardiomyopathy [15, 16], diabetic cardiomyopathy [17, 18], and cardiac aging [19]. In addition, ALDH2 is reported to inhibit inflammatory response and regulate atherosclerotic plaque formation [20], which affects cardiac function indirectly. Up-to-date, the main cellular machineries behind ALDH2-offered cardiac benefits are perceived to be mediated through detoxification of toxic reactive aldehydes including 4-hydroxy-2-nonenal (4-HNE) and regulation of autophagy [11, 13, 15, 21-24]. Recent report also suggested a role for ALDH2 in the regulation of Sirt3 activation [25]. Sirt3 is one of sirtuin family proteins belonging to class III histone deacetylase (HDAC) localized predominantly in mitochondria. Sirt3 may serve as a mitochondrial stress sensor for the modulation of mitochondrial proteins governing metabolism [26, 27]. Given the important role for ALDH2 in cardiovascular diseases [17, 19, 21, 22, 28] and the high prevalence of cardiac dysfunction in insulin resistance [6-8], this study was designed to evaluate the impact of ALDH2 in cardiac function in the sucrose diet feeding-induced insulin resistance. In an effort to examine the possible mechanism of action involved in ALDH2-offered response to insulin resistance-associated cardiomyopathy, if any. Mitochondrial integrity was examined using aconitase activity, mitochondrial permeation pore opening (NAD+ levels) and mitochondrial biogenesis cofactor peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) as well as posttranslational modification of PGC-1α. Emerging evidence has revealed a likely role for the transcriptional coactivator PGC-1α in mitochondrial biosynthesis where reduced PGC-1α activity is associated with multiple glycolytic metabolisms in the heart [27, 29]. We also examined the levels of heme oxygenase-1 (HO-1), which is best known for its ability to regulate mitochondrial function and cardiac homeostasis in myocardial pathologies [30].
RESULTS
Echocardiographic properties of WT and ALDH2 mice challenged with sucrose
Neither sucrose feeding nor ALDH2 overexpression, or both, significantly affected body weight, heart weight and size (heart weight normalized to body weight), organ (liver and kidney) weight, heart rate, LV wall thickness, and LVEDD (p > 0.05 vs. starch group for all these indices). Sucrose diet feeding greatly enhanced LV ESD and suppressed fractional shortening (p < 0.05 vs. starch group for both indices), the effects of which were obliterated by ALDH2 transgene (p < 0.05 vs. sucrose group for both indices, Table 1). These data suggest that ALDH2 is capable of alleviating sucrose diet-induced myocardial dysfunction.
Table 1. General characteristics of WT and ALDH2 mice fed starch or sucrose diet.
| WT-Starch | WT-Sucrose | ALDH2-Starch | ALDH2-Sucrose | |
|---|---|---|---|---|
| Body Weight (g) | 28.8 ± 0.6 | 30.9 ± 1.1 | 30.1 ± 0.8 | 31.2 ± 0.8 |
| Heart Weight (mg) | 130 ± 3 | 141 ± 3 | 127 ± 4 | 138 ± 4 |
| Heart/Body Weight (mg/g) | 4.53 ± 0.15 | 4.57 ± 0.13 | 4.24 ± 0.10 | 4.44 ± 0.05 |
| Liver Weight (g) | 1.19 ± 0.04 | 1.24 ± 0.04 | 1.21 ± 0.04 | 1.25 ± 0.05 |
| Kidney Weight (g) | 0.35 ± 0.02 | 0.37 ± 0.01 | 0.34 ± 0.01 | 0.34 ± 0.01 |
| Heart Rate (bpm) | 473 ± 12 | 467 ± 7 | 476 ± 12 | 474 ± 16 |
| LV Wall Thickness (mm) | 0.96 ± 0.08 | 0.95 ± 0.05 | 0.89 ± 0.08 | 0.93 ± 0.07 |
| LVEDD (mm) | 2.33 ± 0.16 | 2.49 ± 0.11 | 2.26 ± 0.13 | 2.25 ± 0.13 |
| LVESD (mm) | 1.04 ± 0.12 | 1.55 ± 0.19* | 0.91 ± 0.11 | 0.95 ± 0.10# |
| Fractional Shortening (%) | 54.6 ± 1.5 | 43.0 ± 2.1* | 56.6 ± 1.5 | 53.3 ± 3.1# |
LV, left ventricular; EDD, end-diastolic diameter; ESD, end-systolic diameter, Mean ± SEM, n = 6-7 mice per group,
p < 0.05 vs. WT-Starch group,
p < 0.05 vs. WT-Sucrose group.
Effect of sucrose diet on energy expenditure and glucose tolerance in mice
Impact of insulin resistance and ALDH2 transgene on global metabolism was examined using the open-circuit indirect calorimetry. Our data revealed that diet-induced insulin resistance increased O2 consumption (VO2) (Figure 1A), CO2 production (Figure 1B) and respiratory exchange ratio (RER; Figure 1C & 1D) compared with starch group (p < 0.05 vs. starch group for all indices), favoring carbohydrate oxidation as a preferential energy source. Although ALDH2 transgene itself did not affect O2 consumption, CO2 production and RER (p > 0.05), it partially although significantly attenuated sucrose feeding-induced increase in RER (p < 0.05 vs. sucrose group) but not O2 consumption and CO2 production (p > 0.05 vs. sucrose group, Figure 1A-1D). Neither sucrose diet intake nor ALDH2 transgene, or both, significantly affacted total activity or heat production (data not shown). IPGTT test confirmed global insulin resistance following 8 weeks of sucrose diet feeding. Baseline glucose levels were unchanged by sucrose diet intake (p > 0.05 vs. starch group) although fasting blood glucose levels rose significantly at 30 and 60 min (p < 0.05 vs. starch group, Figure 1E), consistent with increased area under the curve (AUC) (p = 0.003 vs. starch group, Figure 1F) in sucrose diet group. ALDH2 transgene did not alter glucose tolerance in the starch-fed mice, although it partially but significantly attenuated sucrose feeding-induced increase in AUC (p = 0.024 vs. sucrose group, Figure 1F).
Figure 1.

Effect of 8 weeks of sucrose diet (starch as control diet) intake on (A) oxygen consumption (VO2), (B) carbon dioxide production (VCO2), (C) real-time RER; (D) Pooled RER, (E) IPGTT curve and (F) area underneath the IPGTT curve in WT and ALDH2 transgenic mice. Mean ± SEM, n = 6- 8 mice per group, *p < 0.05 vs. WT-Starch group, #p < 0.05 vs. WT-Sucrose group.
Effect of sucrose diet intake and ALDH2 on cardiomyocyte cross-sectional area
Data from H&E staining revealed that neither sucrose diet nor ALDH2 overexpression, or both, affected cardiomyocyte cross-sectional area (p > 0.05 among all 4 groups, Figure 2A-2B), suggesting minimal impact of sucrose diet intake-induced insulin resistance or ALDH2 transgene on cardiac remodeling.
Figure 2. Effect of 8 weeks of sucrose diet (starch as control diet) intake on histological property in WT and ALDH2 transgenic mice.
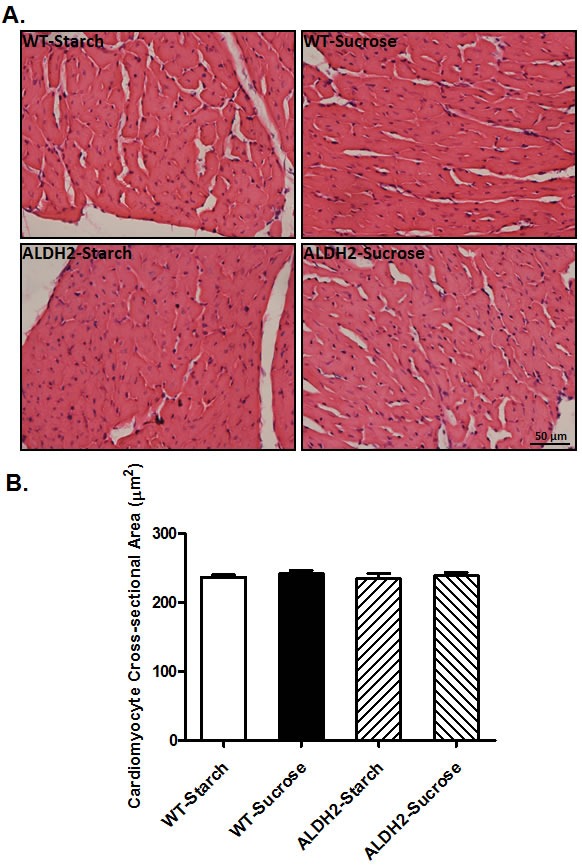
(A) Representative H&E staining photomicrographs (original magnification 400×) of myocardial sections from respective groups; and (B) Pooled data of cardiomyocyte cross-sectional area. Mean ± SEM, n = 10-15 cells per mouse heart image from 5 mice per group, p > 0.05 among all groups.
Effect of sucrose diet intake and ALDH2 on cardiomyocyte contractile function and intracellular Ca2+handling
Neither sucrose diet nor ALDH2 overtly affected phenotype of cardiomyocytes (data not shown) or resting cell length in cardiomyocytes. Sucrose diet feeding-induced insulin resistance resulted in a significant decrease in peak shortening (PS) and maximal velocities of shortening/ relengthening (± dL/dt) as well as prolonged TR90 (p < 0.05 vs. starch group for all these indices) without affecting TPS (p > 0.05 vs. starch group). Although ALDH2 transgene itself did not overtly affect these mechanical parameters (p > 0.05 vs. starch group), it significantly attenuated or abrogated (p < 0.05 vs. sucrose group) sucrose diet-induced mechanical changes (Figure 3). Data presented in Figure 4 displayed that sucrose diet significantly suppressed baseline and peak fura-2 fluorescence intensity (FFI), and electrically-stimulated rise in fura-2 fluorescence intensity (ΔFFI) as well as prolonging intracellular Ca2+ clearance (p < 0.001 vs. starch group for all these indices). Although ALDH2 itself did not affect these intracellular Ca2+ parameters (p > 0.05), it restored sucrose diet-induced changes in peak FFI, ΔFFI and intracellular Ca2+ decay rate (p < 0.001 vs. sucrose group).
Figure 3. Effect of 8 weeks of sucrose diet (starch as control diet) intake on cardiomyocyte contractile function in WT and ALDH2 transgenic mice.

(A) Resting cell length; B) peak shortening; (C) maximal velocity of shortening (+ dL/dt); (D) maximal velocity of relengthening (-dL/dt); (E), Time-to-peak shortening (TPS); and (F) Time-to-90% relengthening (TR90). Mean ± SEM, n = 70 - 74 cells from 3 mice per group, *p < 0.05 vs. WT-Starch group, # p < 0.05 vs. WT-Sucrose group.
Figure 4. Effect of 8 weeks of sucrose diet (starch as control diet) intake on intracellular Ca2+ homeostasis in cardiomyocytes from WT and ALDH2 transgenic mice.
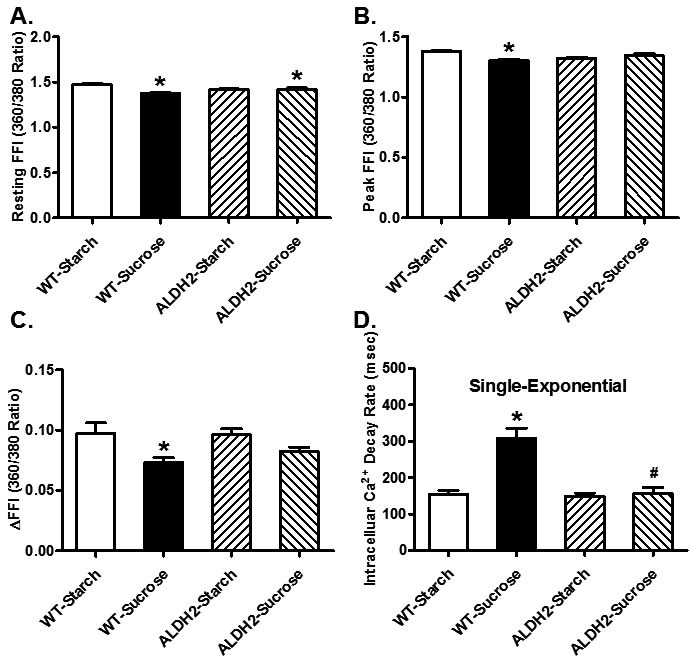
(A) Baseline fura-2 fluorescence intensity (FFI); (B) peak FFI; (C) change in FFI in response to electrical stimuli (ΔFFI) and (D) single exponential intracellular Ca2+ decay rate. Mean ± SEM, n = 60 cells from 3 mice per group, *p < 0.05 vs. WT-Starch group, # p < 0.05 vs. WT-Sucrose group.
Effect of sucrose diet and ALDH2 feeding on total and phosphorylated Akt, HO-1, PGC1α, Sirt3 in mice
Our further analysis revealed that sucrose diet intake significantly suppressed Akt phosphorylation, as well as downregulated levels of HO-1, PGC-1α and Sirt3 (p < 0.05 vs. starch group for all these indices) without any overt effect on pan Akt levels. ALDH2 transgene itself failed to alter the levels of phosphorylated Akt, HO-1, PGC-1α and Sirt3 (p > 0.05 vs. starch group) although it significantly attenuated or abrogated (p < 0.05 vs. sucrose group) sucrose diet-induced changes in these protein markers (Figure 5).
Figure 5.
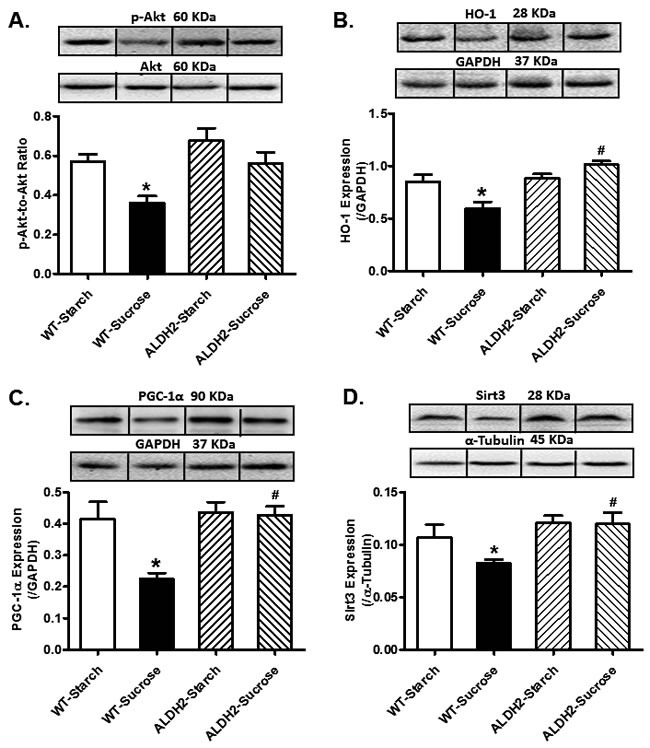
Effect of 8 weeks of sucrose diet (starch as control diet) intake on (A) phosphorylated levels of Akt; (B) protein level of HO-1, (C) protein level of PGC-1α; and (D) protein level of Sirt3 in WT and ALDH2 mice. Insets: Representative gel blots depicting expression of pan and phosphorylated Akt, HO-1, PGC-1α and Sirt3 using specific antibodies. GAPDH or α-tubulin was used as loading control; Mean ± SEM, n = 5 - 6 mice per group; *p < 0.05 vs. WT-Starch group, # p < 0.05 vs. WT-Sucrose group.
Effect of sucrose diet and ALDH2 on ROS production, aconitase activity and NAD+ activity
Assessment of mitochondrial function revealed that insulin resistance significantly decreased mitochondrial aconitase and NAD+ levels, suggesting mitochondrial injury. Aconitase is an iron sulfur enzyme located in citric acid cycle, and the mitochondrial aconitase activity is closely associated with oxidative stress related mitochondrial damage [21]. Although ALDH2 itself did not affect the activities of aconitase or NAD+ (p > 0.05 vs. starch group), it rectified insulin resistance-induced decrease in aconitase and NAD+ activities (p < 0.001 vs. sucrose group, Figure 6A-6B). Cardiomyocytes from WT and ALDH2 mice with or without sucrose diet intake were stained with DCF fluorescence dye prior to the assessment of tissue ROS levels. Our data revealed that insulin resistance overtly enhanced ROS generation (p < 0.001 vs. starch group), the effect of which was mitigated by ALDH2 (p < 0.001 vs. sucrose group). ALDH2 itself did not affect ROS production (p > 0.05, Figure 6C).
Figure 6.
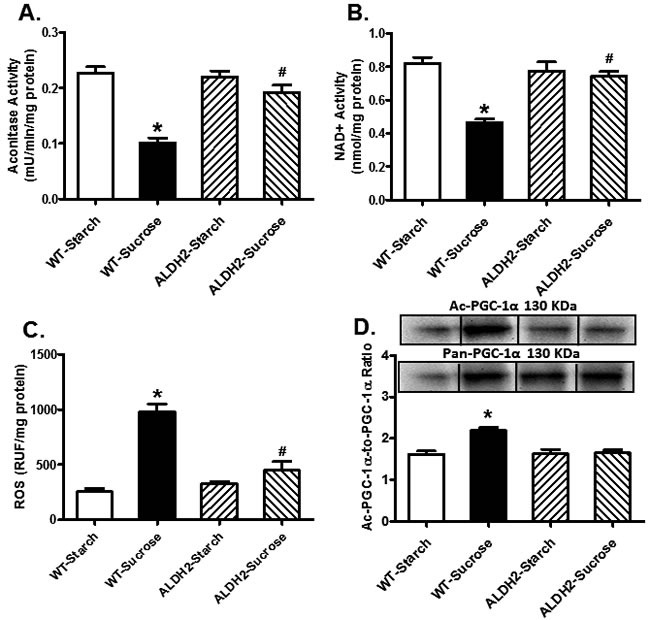
Effect of 8 weeks of sucrose diet (starch as control diet) intake on (A) aconitase activity; (B) NAD+ levels; (C) ROS accumulation; and (D) acetylated PGC-1α.protein levels in WT and ALDH2 mice. Insets: Representative gel blots depicting expression of pan and acetylated PGC-1α using specific antibodies. GAPDH was used as loading control; Mean ± SEM, n = 5 - 6 mice per group; *p < 0.05 vs. WT-Starch group, # p < 0.05 vs. WT-Sucrose group.
Role of PGC1α acetylation in insulin resistance and ALDH2-induced cardiac responses
Given that Sirt3 is a crucial deacetylase regulating activity of PGC-1α [31], acetylated levels of PGC-1α were evaluated. Data shown in Figure 6D indicated insulin resistance promoted PGC-1α acetylation (p = 0.0213 vs. starch group), the effect of which was significantly attenuated by ALDH2 (p = 0.0213 vs. sucrose group) without any effect by ALDH2 transgene itself. To further discern a potential role of posttranslational modification of PGC-1α in ALDH2 transgene-offered protective effect against insulin resistance-induced cardiomyocyte anomalies, isolated murine cardiomyocytes from WT mice were pretreated with 25 ng/ml insulin for 2 hrs to establish insulin resistance in vitro [32, 33]. Assessment of aconitase activity and cardiomyocyte function revealed significantly decreased aconitase activity and cardiomyocyte function as manifested by reduced PS, ± dL/dt and prolonged TR90 in insulin resistant group (p < 0.05 vs. control group), in a manner reminiscent to in vivo insulin resistance elicited by sucrose diet feeding. Consistent with in vivo observation, co-incubation of the ALDH2 activator Alda-1 alleviated high insulin-induced mitochondrial and cardiomyocyte contractile anomalies (p < 0.05 vs. control group), the effect of which was cancelled off by Sirt3 inhibitor AAPBO (which promotes acetylation). The Sirt3 activator Oroxylin A and the histone acetyltransferase inhibitor CPTH2 obliterated (p < 0.05 vs. high insulin group) or overtly dampened insulin resistance-induced cardiomyocyte dysfunction and mitochondrial injury (aconitase activity) without eliciting any effects themselves. Moreover, the Sirt3 inhibitor AAPBO failed to produce additive effect to insulin resistance-induced cardiomyocyte contractile and aconitase anomalies (p > 0.05 between insulin and insulin-AAPBO groups for all indices, Figure 7).
Figure 7.
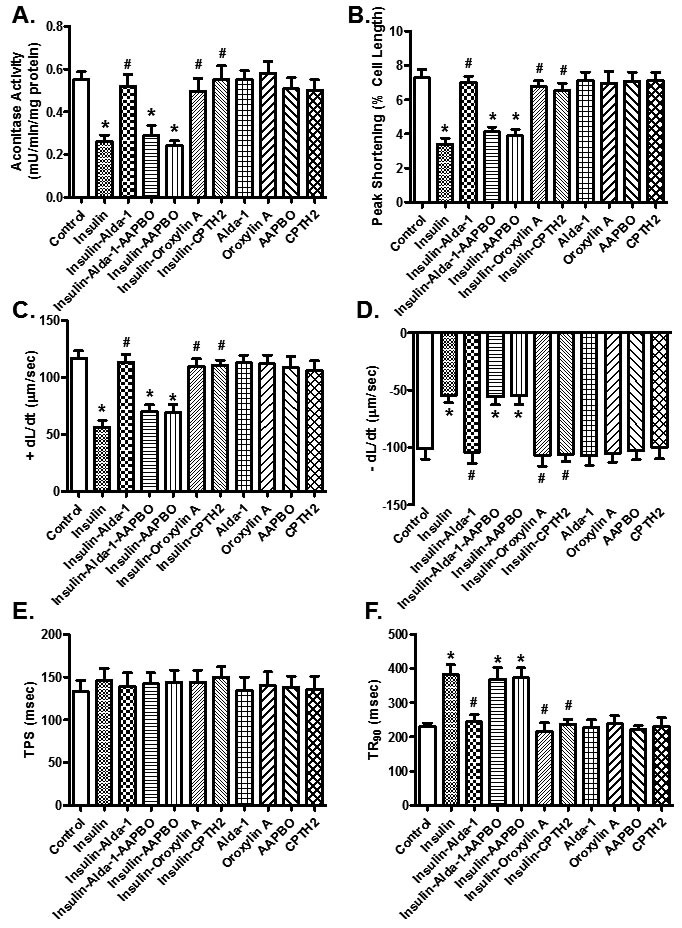
Assessment of aconitase activity and contractile function in cardiomyocytes cultured in vitro with high insulin (25 ng/ml, as a model for insulin resistance) for 2 hrs in the presence or absence of the ALDH2 activator Alda-1 (20 μM), the Sirt3 activator, Oroxylin A (100 μM) or the histone acetyltransferase inhibitor CPTH2 (200 μM). A cohort of Alda-1-treated high insulin cultured cardiomyocytes was co-incubated with the Sirt3 inhibitor 5-amino-2-(4-aminophenyl) benzoxazole (AAPBO, 100 μM) at the same time. (A) aconitase activity; (B) peak shortening, (C) maximal velocity of shortening (+ dL/dt), (D) maximal velocity of relengthening (-dL/dt); (E) time-to-peak shortening (TPS); and (F) time-to-90% relengthening (TR90). Mean ± SEM, n = 55 - 60 cells from 3 mice per group, *p < 0.05vs.control group, # p < 0.05 vs. insulin resistance group.
DISCUSSION
The salient findings of our study indicated that ALDH2 exerts a protective effect against insulin resistance-induced cardiac contractile dysfunction through preservation of mitochondrial integrity and Sirt3 function. Our findings indicated that sucrose diet intake-induced changes in myocardial contractile and intracellular Ca2+ properties in the absence of cardiac remodeling are closely associated with dampened phosphorylation of Akt and mitochondrial function. ALDH2 is likely to offer its protection by reversing insulin resistance-induced changes in mitochondrial integrity, Sirt3, and PGC-1α (pan/acetylated form). Furthermore, the possible involvement of Sirt3-mediated deacetylation of PGC-1α, in insulin resistance- and ALDH2-induced cardiac contractile and mitochondrial responses was substantiated by the pharmacological manipulation of Sirt3 acetylation and HAT. ALDH2-offered protection was recapitulated by the use of its agonist Alda-1, supporting the therapeutic potential of this enzyme in hyperinsulinemia. Interestingly, Alda-1-elicited beneficial effects against high insulin-induced cardiac dysfunction were effectively abolished by the specific Sirt3 deacetylation inhibitor AAPBO. Likewise, high insulin-elicited mitochondrial and functional anomalies were effectively reversed by the Sirt3 activator, Oroxylin A and the HAC inhibitor CPTH2. These findings support a pivotal role for the Sirt3 deacetylation in insulin resistance- and/or ALDH2-induced regulation of mitochondrial and contractile function in the heart. Taken together, these findings revealed that ALDH2 protects against insulin resistance-induced cardiac defect likely through a Sirt3-dependent deacetylation of PGC-1α.
The sucrose enriched dietary model offers an important means for cellular changes in the heart during early stages of development of type 2 diabetes and metabolic syndrome [6]. Data from the open-circuit calorimetry suggests presence of metabolic derangement following sucrose diet intake for 8 weeks. Balanced energy intake and expenditure is essential to a healthy energy homeostasis [34]. Our results did not notice any significant difference in total activity or heat production among all mouse groups, not favoring a role for energy expenditure in sucrose diet-induced metabolic and cardiac derangement. RER denotes the ratio between CO2 production and O2 consumption, suggesting the energetic substrate which is being oxidized. A higher RER ratio indicates increased oxidation of carbohydrate in reference to oxidation of fatty acid, whereas a lower RER denotes an increased oxidation of fat over carbohydrate [35]. In our hands, the RER value round 0.8 in the starch-fed mice suggests energy source from both fatty acid oxidation and carbohydrate oxidation [36]. With the high sucrose diet (68% of energy provided from sucrose which offers more carbohydrate relative to starch diet [37]), RER was significantly elevated to around 1 compared with the starch group, suggesting a more predominant role for carbohydrates as the energy source. Interestingly, ALDH2 transgene partially reversed sucrose diet-induced changes in RER, suggesting a role for the mitochondrial enzyme in governance of energetic substrate oxidation. However, ALDH2 transgene cannot fully alleviate sucrose diet-induced high carbohydrate oxidation as the predominant energy source.
Unfavorable changes in myocardial function are seen in sucrose-fed mice characterized by compromised contractility, prolonged diastolic duration intracellular Ca2+ mishandling [6, 37]. In our hands, sucrose diet-induced insulin resistance triggered myocardial contractile defects including enlarged LVESD, reduced fractional shortening, peak shortening, ± dL/dt and prolonged TR90 in the absence of overt change in cardiomyocyte cross-sectional area. These findings are somewhat in agreement with the previous notion of myopathic changes in chronic alcoholism-, insulin resistant- and diabetic cardiomyopathy [4, 6, 9, 15, 17, 18, 37-39]. Our data revealed interrupted intracellular Ca2+ homeostasis in response to sucrose diet intake manifested as depressed baseline, peak and electrically-stimulated rise in intracellular Ca2+, as well as prolonged intracellular Ca2+ decay, denoting an essential role of intracellular Ca2+ dysregulation in insulin resistance-induced cardiomyopathy. Perhaps the most intriguing observation from our study is that overexpression of ALDH2 alleviated insulin resistance-induced myocardial injury and mitochondrial defect. Data from our current study revealed that ALDH2 counteracts insulin resistance-induced cardiomyopathy (myocardial, cardiomyocyte function and intracellular Ca2+ handling) possibly in association with regulation of Sirt3 and mitochondrial integrity. Although ALDH2 transgene itself did not affect intracellular Ca2+ homeostasis, it unveiled a dampened release of intracellular Ca2+ and decrease in intracellular Ca2+ clearance in pre-diabetic myopathic changes. These findings suggest that ALDH2 plays a role in insulin resistance challenge-disturbed cardiac homeostasis. In accordance with cardiac dysfunction, comprised mitochondrial integrity was noted under insulin resistance (decreased aconitase levels, HO-1 levels, and NAD+ activity and elevated production of ROS). Through degradation of heme and generation of cytoprotective byproducts, HO-1 helps to maintain mitochondrial quality control by disengaging the positive feedback between oxidative stress and mitochondrial injury [40]. Consistent with its effect on cardiac contractile and intracellular Ca2+ properties in the face of sucrose diet intake, ALDH2 alleviated insulin resistance-induced ROS accumulation, as well as loss in HO-1 levels, aconitase activity and NAD+ levels. This was further supported by the findings from in vitro studies using pharmacological regulation of Sirt3 and ALDH2. In particular, Sirt3 activator effectively mitigated high insulin-induced cardiomyocyte and mitochondrial injury whereas the Sirt3 inhibitor cancelled off Alda-1-induced beneficial effect against high insulin.
In the pre-diabetic insulin resistance state, the accumulation of ROS is evident in hearts. ALDH2 is known to lower ROS levels in humans and animals [21] although the mitochondrial enzyme itself may not directly work as an antioxidant against ROS production. Interestingly, Sirt3 is well recognized as critical inhibitor to ROS. To elucidate possible mechanisms of action behind ALDH2 on sucrose diet-induced cardiac derangement, levels of Sirt3 and PGC-1α were examined with western blotting. Pre-diabetes are known to downregulate PGC-1α protein levels [41], thus we evaluated the effects of sucrose diet intake on Sirt3 and PGC-1α protein levels. In addition, since Sirt3 is the most important deacetylase that contributes to deacetylation of PGC-1α [31], we evaluated the acetylated lysine levels of this mitochondrial biogenesis marker that modulates mitochondrial dynamics and oxidative stress. Our immunoprecipitation data further revealed that insulin resistance promoted PGC-1 acetylation, the effect of which may be reversed by ALDH2. These data revealed that cardiac dysfunction under sucrose diet-induced insulin resistance is probably the result of decreased nuclear genes encoding mitochondrial proteins as a result of PGC-1α acetylation [42]. Our in vitro insulin resistant model confirmed that the benefit of Alda-1 may be nullified by the Sirt-3 inhibitor 5-amino-2-(4-aminophenyl)benzoxazole. In addition, direct activation of Sirt3 mimicked the Alda-1-offered protection against high insulin-induced cardiomyocyte contractile function and mitochondrial integrity. In our hands, acetylation of PGC-1α was noted in insulin resistant mouse hearts, which may contribute to its activity and thus mitochondrial function. Downregulated or acetylated PGC1α has been shown in failing hearts in numerous rodent models [43, 44]. Given that PGC-1α acetylation can be induced through activation of GCN5 acetyl transferase [45, 46], CPTH2 was employed to specifically inhibit HAT which modulates the GCN5 network [46]. As expected, CPTH2 offered beneficial effect against high insulin-induced cardiomyocyte contractile and mitochondrial injuries.
Experimental limitations: Although our current study has provided several lines of causal relationship among cardiac mechanical function, insulin resistance and PGC-1α acetylation, these results should not offer conclusive answers to the precise pathogenesis of cardiac defect in human insulin resistance. Use of more clinically relevant models of insulin resistance (such as defects in insulin receptor or post-receptor signaling components) should help to better understand the role of PGC-1α and its post-translational modification in the progression of insulin resistance-induced cardiomyopathy. Moreover, other post-translational modification modalities should not be discounted for regulation of mitochondrial biogenesis at this time.
In summary, findings from our present study have provided rather convincing evidence that ALDH2 rescues against cardiac anomalies including mechanical defect, intracellular Ca2+ dysregulation and mitochondrial injury through Sirt3-mediated deacetylation of mitochondrial biogenesis cofactor PGC-1α. Our data further revealed alleviated ROS production with ALDH2 overexpression under insulin resistance. A schematic diagram is provided here to summarize the proposed mechanism of action behind ALDH2-offered protection against insulin resistance-induced cardiac anomalies (Figure 8). Further study is warranted to unveil the mechanism behind ALDH2-mediated Sirt3 deacetylation in a more clinically relevant setting of insulin resistance-induced cardiomyopathy.
Figure 8.

Schematic diagram depicting proposed mechanism for ALDH2-offered protection against insulin resistance-induced cardiac dysfunction. It is proposed that downregulation of acetylation of the mitochondrial biogenesis cofactor PGC-1α in response to insulin resistance challenge imposes suppression of mitochondrial function, en route to ROS production, intracellular Ca2+ derangement, and myocardial dysfunction. ALDH2 prevents insulin resistance-induced loss of PGC-1α function through Sirt3-mediated deacetylation to preserve mitochondrial integrity and cardiac function. Arrowheads refer to promoting subsequent response. ALDH2: mitochondrial aldehyde dehydrogenase; ROS: reactive oxygen species; Ac: acetylation.
MATERIALS AND METHODS
Experimental animals, model of insulin resistance and intraperitoneal glucose tolerance tests (IPGTT)
Experimental procedures used here were approved by the Zhongshan Hospital Fudan University and University of Wyoming Institutional Animal Use and Care Committees. Insulin resistance was induced in Friend virus B (FVB) wild-type (WT) and ALDH2 transgenic mice. In brief, 3-month-old adult male mice were randomly divided into two groups to receive either starch diet or a sucrose diet (68% of energy from sucrose; Research Diets, D11725) for 8 weeks [6]. On the last day (23:00 hours) of diet treatment, food was removed and all mice were fasted for 12 hrs prior to IPGTT test. Mice were challenged with glucose (2 g/kg, ip) and blood was drawn immediately before and 30, 60, 90, and 120 min after glucose injection. Fasting blood glucose levels were assessed using an Accu-Chek glucose analyzer (Roche Diagnostics, Indianapolis, IN).
Metabolic measurements
After 8 weeks of dietary treatment, mice were housed individually in metabolic cages, acclimated for 24 hrs to minimize the novelty effect, and then monitored for 24 hrs in 12-hour light/dark cycle by indirect open-circuit calorimetry (Oxymax System; Columbus Instruments, Columbus, OH) [47, 48]. Metabolic examination was performed over the course of 3 days while respective diet was maintained. Calibration of the calorimeter was performed at the beginning of each measurement day. Oxygen consumption and carbon dioxide production were measured by volume every 10 minutes. Respiratory exchange ratio (RER) was calculated as the ratio of VCO2/VO2. Energy expenditure was calculated as (3.815 + 1.232 × RER) × VO2 and normalized to body weight [49].
Histological examination
Following anesthesia, hearts were excised and immediately placed in 10% neutral-buffered formalin at room temperature for 24 hrs after a brief rinse with PBS. The specimen were embedded in paraffin, cut in 5-μm sections and stained with hematoxylin and eosin (H&E). Cardiomyocyte cross-sectional areas were calculated on a digital microscope (x400) using the Image J (version1.34S) software [50].
Echocardiographic assessment
Cardiac geometry and contractile function were evaluated in anesthetized (ketamine 80 mg/kg and xylazine 12 mg/kg, i.p.) mice using a 2-dimensional (2-D) guided M-mode echocardiography (Phillips Sonos 5500) equipped with a 15-6 MHz linear transducer (Phillips Medical Systems, Andover, MD). Adequate depth of anesthesia was monitored using toe reflex. The heart was imaged in the 2-D mode in the parasternal long-axis view with a depth setting of 2 cm. The M-mode cursor was positioned perpendicular to interventricular septum and posterior wall of left ventricle (LV) at the level of papillary muscles from the 2-D mode. The sweep speed was 100 mm/sec for the M-mode. Diastolic wall thickness, left ventricular (LV) end diastolic dimension (EDD) and LV end systolic dimension (ESD) were measured. All measurements were done from leading edge to leading edge in accordance with the Guidelines of the American Society of Echocardiography. LV fractional shortening was calculated as [(EDD-ESD)/EDD] × 100 [17].
Cardiomyocyte isolation and cell mechanics
Hearts were removed rapidly from mice sedated with ketamine (80 mg/kg, ip) and xylazine (12 mg/kg, ip) and perfused with Krebs-Henseleit bicarbonate (KHB) solution consisting of (in mM) 118 NaCl, 4.7 KCl, 1.2 MgSO4, 1.2 KH2PO4, 25 NaHCO3, 10 HEPES, and 11.1 glucose. Hearts were digested with Liberase BlendzymeTH (Roche Diagnostics) for 15 min. After removal and mincing of the left ventricle, Ca2+ was added back to a final concentration of 1.25 mM. Cardiomyocytes with no spontaneous contractions and clear edges were used for shortening and Ca2+ cycling experiments. The IonOptix soft-edge system (IonOptix, Milton, MA) was employed to assess the mechanical properties of isolated myocytes. Myocytes were mounted on the stage of an Olympus IX-70 microscope in contractile buffer containing (in mM) 131 NaCl, 4 KCl, 1CaCl2,1 MgCl2, 10 glucose, and 10 HEPES. Myocytes were stimulated at0.5 Hz with cell shortening and relengthening evaluated using the following indices: peak shortening (PS), time to peak shortening (TPS), time to 90% relengthening (TR90), and maximal velocities of shortening/relengthening (± dL/dt) [51].
Intracellular Ca2+ transients
A cohort of myocytes was loaded with fura-2/AM (0.5 μM) for 10 min, and fluorescence intensity was recorded with a dual-excitation fluorescence photomultiplier system (IonOptix). Myocytes were placed onto an Olympus IX-70 inverted microscope and imaged through a Fluor 40 oil objective. Cells were exposed to light emitted by a 75W lamp and passed through either a 360 or a 380 nm filter, while being stimulated to contract at 0.5 Hz. Fluorescence emissions were detected between 480 and 520 nm, and qualitative change in fura-2 fluorescence intensity (FFI) was inferred from the FFI ratio at the two wavelengths (360/380). Fluorescence decay time was measured as an indication of the intracellular Ca2+ clearing rate. Single exponential curve fit was used to calculate the intracellular Ca2+ decay constant [52].
Intracellular fluorescence measurement of ROS
Intracellular ROS were measured by changes in fluorescence intensity resulting from intracellular probe oxidation [53]. Cardiomyocytes were loaded with 5-(6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA, 1 μM, Molecular Probes, Eugene, OR) for 30 min at 37°C. Fluorescence intensity was measured at an excitation wavelength of 480 nm and an emission wavelength of 530 nm [53].
In vitro insulin resistant model
Insulin was added to isolated cardiomyocytes to induce insulin resistance at a level of 25 ng/ml for 2 hrs [32, 33]. To assess the effect of ALDH2 activation and Sirt3 on insulin resistance-induced change in cardiomyocyte function, freshly isolated murine cardiomyocytes from WT mice were pretreated with the ALDH2 activator, Alda-1 (20 μM) [54], the Sirt3 activator, Oroxylin A (100 μM) [55], the Sirt3 inhibitor, 5-amino-2-(4-aminophenyl) benzoxazole (AAPBO, 100 μM) [56] or the histone acetyltransferase inhibitor CPTH2 (200 μM) [57] for 2 hrs prior to assessment of cardiomyocyte mechanical function.
Aconitase activity
Mitochondria prepared from whole heart homogenate were resuspended in 0.2 mM sodium citrate. After determination of protein content, aconitase activity assay (Aconitase activity assay kit, Aconitase-340 assay™, Oxisresearch, Portland, OR) was performed according to manufacturer instructions with minor modifications. Briefly, mitochondrial sample (50 μl) was mixed in a 96-well plate with 50 μl trisodium citrate (substrate) in Tris-HCl pH 7.4,50 μl isocitrate dehydrogenase (enzyme) in Tris-HCl, and 50 μl NADP in Tris-HCl. After incubating for 15 min at 37°c with 50 rpm shaking, the absorbance was dynamically recorded at340 nm every min for 5 min with a spectramax 190 microplate spectrophotometer. During theassay, citrate is isomerized by aconitase into isocitrate and eventually α-ketoglutarate. Theaconitase-340 assay™ measures NADPH formation, a product of the oxidation of isocitrate to α-ketoglutarate. Tris-HCl buffer (pH 7.4) served as a blank. All results were normalized to respective protein content [58].
Determination of NAD+
NAD+ was extracted from frozen ventricular tissues using perchloric acid. For these determinations, 30 mg of fresh frozen tissue was powdered in a mortar under liquid nitrogen and thoroughly mixed with 150 μl 0.6 M perchloric acid. The mixture was then homogenized, neutralized with 150 μl 3 M potassium hydroxide. NAD+ concentrations were determined fluorometrically in dilutions of the supernatant sample using alcohol dehydrogenase (Sigma-Aldrich, St. Louis, MO). Excitation was at 339 nm, and emission wavelength was at 460 nm in a spectrofluorimeter (Spectra MaxGeminiXS, Sunnyvale, CA) [58].
Western blot analysis
Heart tissue was homogenized in a RIPA lysis buffer and centrifuged at 4°C for 20 min at 12,000xg. Supernatants were removed and protein concentration was measured using the Bradford assay. About 50 μg of equivalent protein samples were separated on a 7%, 10%, or 15% SDS-PAGE gel in a mini gel apparatus (Mini-PROTEAN II;Bio-Rad). Membranes were blocked with 5% milk in Tris-buffered saline/Tween 20 and incubated in anti-Sirt3, anti-PGC1α, anti-Akt, anti-phospho-Akt (pAkt), anti-HO-1, anti-GAPDH and anti-α-tubulin (Cell Signaling Technology, Beverly, MA) antibodies at the 1:1,000 dilution factor overnight at 4°C. Membranes were rinsed and incubated with horseradish peroxidase conjugated secondary antibodies and exposed using enzymatic chemiluminescence. The intensity of immunoblot bands was detected with a Bio-Rad Calibrated Densitometer (Model: GS-800). Images obtained by Western blot were analyzed using an ImageJ software (NIH) to quantify gel densities [51].
Immunoprecipitation
Co-IP assay was performed following the protocol of the Co-IP kit (Pierce). Briefly, 50 μg of purified IR and PGC-1 antibodies were immobilized with coupling resin. Protein extracts (500 μg) were incubated with antibody-coupled resin gently end-over-end mixing for 2 hours at room temperature. The resin was washed, and the protein complexes bound to the antibody were eluted with 50 μl of elution buffer. The eluted protein was boiled and separated by 10% SDS-PAGE, transferred to a nitrocellulose membrane, and incubated with anti-Acetylated Lysine antibody. Antibody binding was detected using the enhanced chemiluminescence. The film was scanned and the intensity of immunoblotting bands was detected with a Bio-Rad Calibrated Densitometer (model GS-800) [38].
Statistical analysis
Data were Mean ± SEM. Statistical comparison was performed by a 1-way ANOVA followed by the Tukey post hoc test. Significance was set at p < 0.05.
Footnotes
CONFLICTS OF INTEREST
None of the authors declare any conflict of interest associated with this work.
GRANT SUPPORT
This work was supported in part by the National Natural Science Foundation of China (NSFC 81522004, 81370195). Portion of this work was presented in the 2013 American Diabetes Association Scientific Session in Chicago, IL.
REFERENCES
- 1.Zhang P, Zhang X, Brown J, Vistisen D, Sicree R, Shaw J, Nichols G. Global healthcare expenditure on diabetes for 2010 and 2030. Diabetes research and clinical practice. 2010;87(3):293–301. doi: 10.1016/j.diabres.2010.01.026. [DOI] [PubMed] [Google Scholar]
- 2.Santos JM, Tewari S, Benite-Ribeiro SA. The effect of exercise on epigenetic modifications of PGC1: The impact on type 2 diabetes. Medical hypotheses. 2014;82(6):748–753. doi: 10.1016/j.mehy.2014.03.018. [DOI] [PubMed] [Google Scholar]
- 3.Dhalla NS, Takeda N, Rodriguez-Leyva D, Elimban V. Mechanisms of subcellular remodeling in heart failure due to diabetes. Heart failure reviews. 2014;19(1):87–99. doi: 10.1007/s10741-013-9385-8. [DOI] [PubMed] [Google Scholar]
- 4.Jia G, DeMarco VG, Sowers JR. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nature reviews Endocrinology. 2016;12(3):144–153. doi: 10.1038/nrendo.2015.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol. 2011;301(6):H2181–2190. doi: 10.1152/ajpheart.00554.2011. [DOI] [PubMed] [Google Scholar]
- 6.Roe ND, He EY, Wu Z, Ren J. Folic acid reverses nitric oxide synthase uncoupling and prevents cardiac dysfunction in insulin resistance: role of Ca2+/calmodulin-activated protein kinase II. Free radical biology & medicine. 2013;65:234–243. doi: 10.1016/j.freeradbiomed.2013.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dong F, Kandadi MR, Ren J, Sreejayan N. Chromium (D-phenylalanine)3 supplementation alters glucose disposal, insulin signaling, and glucose transporter-4 membrane translocation in insulin-resistant mice. The Journal of nutrition. 2008;138(10):1846–1851. doi: 10.1093/jn/138.10.1846. [DOI] [PubMed] [Google Scholar]
- 8.Vasanji Z, Cantor EJ, Juric D, Moyen M, Netticadan T. Alterations in cardiac contractile performance and sarcoplasmic reticulum function in sucrose-fed rats is associated with insulin resistance. American journal of physiology Cell physiology. 2006;291(4):C772–780. doi: 10.1152/ajpcell.00086.2005. [DOI] [PubMed] [Google Scholar]
- 9.Rezende PC, Rahmi RM, Uchida AH, da Costa LM, Scudeler TL, Garzillo CL, Lima EG, Segre CA, Girardi P, Takiuti M, Silva MF, Hueb W, Ramires JA, Kalil Filho R. Type 2 diabetes mellitus and myocardial ischemic preconditioning in symptomatic coronary artery disease patients. Cardiovascular diabetology. 2015;14:66. doi: 10.1186/s12933-015-0228-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dhingra R, Sullivan L, Jacques PF, Wang TJ, Fox CS, Meigs JB, D'Agostino RB, Gaziano JM, Vasan RS. Soft drink consumption and risk of developing cardiometabolic risk factors and the metabolic syndrome in middle-aged adults in the community. Circulation. 2007;116(5):480–488. doi: 10.1161/CIRCULATIONAHA.107.689935. [DOI] [PubMed] [Google Scholar]
- 11.Pang JJ, Barton LA, Chen YG, Ren J. Mitochondrial aldehyde dehydrogenase in myocardial ischemia-reperfusion injury: from bench to bedside. Sheng li xue bao. 2015;67(6):535–544. [PubMed] [Google Scholar]
- 12.Ebert AD, Kodo K, Liang P, Wu H, Huber BC, Riegler J, Churko J, Lee J, de Almeida P, Lan F, Diecke S, Burridge PW, Gold JD, Mochly-Rosen D, Wu JC. Characterization of the molecular mechanisms underlying increased ischemic damage in the aldehyde dehydrogenase 2 genetic polymorphism using a human induced pluripotent stem cell model system. Science translational medicine. 2014;6(255):255ra130. doi: 10.1126/scitranslmed.3009027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma H, Guo R, Yu L, Zhang Y, Ren J. Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: role of autophagy paradox and toxic aldehyde. Eur Heart J. 2011;32(8):1025–1038. doi: 10.1093/eurheartj/ehq253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun A, Cheng Y, Zhang Y, Zhang Q, Wang S, Tian S, Zou Y, Hu K, Ren J, Ge J. Aldehyde dehydrogenase 2 ameliorates doxorubicin-induced myocardial dysfunction through detoxification of 4-HNE and suppression of autophagy. Journal of molecular and cellular cardiology. 2014;71:92–104. doi: 10.1016/j.yjmcc.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y, Ren J. ALDH2 in alcoholic heart diseases: molecular mechanism and clinical implications. Pharmacol Ther. 2011;132(1):86–95. doi: 10.1016/j.pharmthera.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doser TA, Turdi S, Thomas DP, Epstein PN, Li SY, Ren J. Transgenic overexpression of aldehyde dehydrogenase-2 rescues chronic alcohol intake-induced myocardial hypertrophy and contractile dysfunction. Circulation. 2009;119(14):1941–1949. doi: 10.1161/CIRCULATIONAHA.108.823799. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Zhang Y, Babcock SA, Hu N, Maris JR, Wang H, Ren J. Mitochondrial aldehyde dehydrogenase (ALDH2) protects against streptozotocin-induced diabetic cardiomyopathy: role of GSK3beta and mitochondrial function. BMC medicine. 2012;10:40. doi: 10.1186/1741-7015-10-40. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.Guo Y, Yu W, Sun D, Wang J, Li C, Zhang R, Babcock SA, Li Y, Liu M, Ma M, Shen M, Zeng C, Li N, He W, Zou Q, Zhang Y, et al. A novel protective mechanism for mitochondrial aldehyde dehydrogenase (ALDH2) in type i diabetes-induced cardiac dysfunction: role of AMPK-regulated autophagy. Biochimica et biophysica acta. 2015;1852(2):319–331. doi: 10.1016/j.bbadis.2014.05.017. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Mi SL, Hu N, Doser TA, Sun A, Ge J, Ren J. Mitochondrial aldehyde dehydrogenase 2 accentuates aging-induced cardiac remodeling and contractile dysfunction: role of AMPK, Sirt1, and mitochondrial function. Free radical biology & medicine. 2014;71:208–220. doi: 10.1016/j.freeradbiomed.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pan C, Xing JH, Zhang C, Zhang YM, Zhang LT, Wei SJ, Zhang MX, Wang XP, Yuan QH, Xue L, Wang JL, Cui ZQ, Zhang Y, Xu F, Chen YG. Aldehyde dehydrogenase 2 inhibits inflammatory response and regulates atherosclerotic plaque. Oncotarget. 2016;7(24):35562–35576. doi: 10.18632/oncotarget.9384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen CH, Ferreira JC, Gross ER, Mochly-Rosen D. Targeting aldehyde dehydrogenase 2: new therapeutic opportunities. Physiological reviews. 2014;94(1):1–34. doi: 10.1152/physrev.00017.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang B, Zhang Y, La Cour KH, Richmond KL, Wang XM, Ren J. Mitochondrial aldehyde dehydrogenase obliterates endoplasmic reticulum stress-induced cardiac contractile dysfunction via correction of autophagy. Biochim Biophys Acta. 2013;1832(4):574–584. doi: 10.1016/j.bbadis.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo R, Xu X, Babcock SA, Zhang Y, Ren J. Aldehyde dedydrogenase-2 plays a beneficial role in ameliorating chronic alcohol-induced hepatic steatosis and inflammation through regulation of autophagy. Journal of hepatology. 2015;62(3):647–656. doi: 10.1016/j.jhep.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.Mali VR, Ning R, Chen J, Yang XP, Xu J, Palaniyandi SS. Impairment of aldehyde dehydrogenase-2 by 4-hydroxy-2-nonenal adduct formation and cardiomyocyte hypertrophy in mice fed a high-fat diet and injected with low-dose streptozotocin. Exp Biol Med (Maywood) 2014;239(5):610–618. doi: 10.1177/1535370213520109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xue L, Xu F, Meng L, Wei S, Wang J, Hao P, Bian Y, Zhang Y, Chen Y. Acetylation-dependent regulation of mitochondrial ALDH2 activation by SIRT3 mediates acute ethanol-induced eNOS activation. FEBS letters. 2012;586(2):137–142. doi: 10.1016/j.febslet.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 26.Pillai VB, Sundaresan NR, Jeevanandam V, Gupta MP. Mitochondrial SIRT3 and heart disease. Cardiovascular research. 2010;88(2):250–256. doi: 10.1093/cvr/cvq250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsushima S, Sadoshima J. The role of sirtuins in cardiac disease. American journal of physiology Heart and circulatory physiology. 2015;309(9):H1375–1389. doi: 10.1152/ajpheart.00053.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y, Ren J. Autophagy in ALDH2-elicited cardioprotection against ischemic heart disease: slayer or savior? Autophagy. 2010;6(8):1212–1213. doi: 10.4161/auto.6.8.13652. [DOI] [PubMed] [Google Scholar]
- 29.Ping Z, Zhang LF, Cui YJ, Chang YM, Jiang CW, Meng ZZ, Xu P, Liu HY, Wang DY, Cao XB. The Protective Effects of Salidroside from Exhaustive Exercise-Induced Heart Injury by Enhancing the PGC-1 alpha -NRF1/NRF2 Pathway and Mitochondrial Respiratory Function in Rats. Oxidative medicine and cellular longevity. 2015;2015:876825. doi: 10.1155/2015/876825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Otterbein LE, Foresti R, Motterlini R. Heme Oxygenase-1 and Carbon Monoxide in the Heart: The Balancing Act Between Danger Signaling and Pro-Survival. Circulation research. 2016;118(12):1940–1959. doi: 10.1161/CIRCRESAHA.116.306588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klaman LD, Boss O, Peroni OD, Kim JK, Martino JL, Zabolotny JM, Moghal N, Lubkin M, Kim YB, Sharpe AH, Stricker-Krongrad A, Shulman GI, Neel BG, Kahn BB. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Molecular and cellular biology. 2000;20(15):5479–5489. doi: 10.1128/mcb.20.15.5479-5489.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown JD, Naples SP, Booth FW. Effects of voluntary running on oxygen consumption, RQ, and energy expenditure during primary prevention of diet-induced obesity in C57BL/6N mice. Journal of applied physiology. 2012;113(3):473–478. doi: 10.1152/japplphysiol.00668.2011. [DOI] [PubMed] [Google Scholar]
- 33.Panzhinskiy E, Hua Y, Lapchak PA, Topchiy E, Lehmann TE, Ren J, Nair S. Novel curcumin derivative CNB-001 mitigates obesity-associated insulin resistance. The Journal of pharmacology and experimental therapeutics. 2014;349(2):248–257. doi: 10.1124/jpet.113.208728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu N, Han X, Lane EK, Gao F, Zhang Y, Ren J. Cardiac-specific overexpression of metallothionein rescues against cigarette smoking exposure-induced myocardial contractile and mitochondrial damage. PloS one. 2013;8(2):e57151. doi: 10.1371/journal.pone.0057151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu N, Yang L, Dong M, Ren J, Zhang Y. Deficiency in adiponectin exaggerates cigarette smoking exposure-induced cardiac contractile dysfunction: Role of autophagy. Pharmacological research. 2015;100:175–189. doi: 10.1016/j.phrs.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 36.Turdi S, Ge W, Hu N, Bradley KM, Wang X, Ren J. Interaction between maternal and postnatal high fat diet leads to a greater risk of myocardial dysfunction in offspring via enhanced lipotoxicity, IRS-1 serine phosphorylation and mitochondrial defects. Journal of molecular and cellular cardiology. 2013;55:117–129. doi: 10.1016/j.yjmcc.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Y, Li L, Hua Y, Nunn JM, Dong F, Yanagisawa M, Ren J. Cardiac-specific knockout of ET(A) receptor mitigates low ambient temperature-induced cardiac hypertrophy and contractile dysfunction. Journal of molecular cell biology. 2012;4(2):97–107. doi: 10.1093/jmcb/mjs002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marshall S, Olefsky JM. Effects of insulin incubation on insulin binding, glucose transport, and insulin degradation by isolated rat adipocytes. Evidence for hormone-induced desensitization at the receptor and postreceptor level. The Journal of clinical investigation. 1980;66(4):763–772. doi: 10.1172/JCI109914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goodner CJ, Sweet IR, Harrison HC., Jr Rapid reduction and return of surface insulin receptors after exposure to brief pulses of insulin in perifused rat hepatocytes. Diabetes. 1988;37(10):1316–1323. doi: 10.2337/diab.37.10.1316. [DOI] [PubMed] [Google Scholar]
- 40.Xu X, Hueckstaedt LK, Ren J. Deficiency of insulin-like growth factor 1 attenuates aging-induced changes in hepatic function: role of autophagy. Journal of hepatology. 2013;59(2):308–317. doi: 10.1016/j.jhep.2013.03.037. [DOI] [PubMed] [Google Scholar]
- 41.Wei L, Zhou Y, Dai Q, Qiao C, Zhao L, Hui H, Lu N, Guo QL. Oroxylin A induces dissociation of hexokinase II from the mitochondria and inhibits glycolysis by SIRT3-mediated deacetylation of cyclophilin D in breast carcinoma. Cell death & disease. 2013;4:e601. doi: 10.1038/cddis.2013.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salo HS, Laitinen T, Poso A, Jarho E, Lahtela-Kakkonen M. Identification of novel SIRT3 inhibitor scaffolds by virtual screening. Bioorganic & medicinal chemistry letters. 2013;23(10):2990–2995. doi: 10.1016/j.bmcl.2013.03.033. [DOI] [PubMed] [Google Scholar]
- 43.Yin YW, Jin HJ, Zhao W, Gao B, Fang J, Wei J, Zhang DD, Zhang J, Fang D. The Histone Acetyltransferase GCN5 Expression Is Elevated and Regulated by c-Myc and E2F1 Transcription Factors in Human Colon Cancer. Gene expression. 2015;16(4):187–196. doi: 10.3727/105221615X14399878166230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu N, Dong M, Ren J. Hydrogen sulfide alleviates cardiac contractile dysfunction in an Akt2-knockout murine model of insulin resistance: role of mitochondrial injury and apoptosis. American journal of physiology Regulatory, integrative and comparative physiology. 2014;306(10):R761–771. doi: 10.1152/ajpregu.00327.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Y, Yuan M, Bradley KM, Dong F, Anversa P, Ren J. Insulin-like growth factor 1 alleviates high-fat diet-induced myocardial contractile dysfunction: role of insulin signaling and mitochondrial function. Hypertension. 2012;59(3):680–693. doi: 10.1161/HYPERTENSIONAHA.111.181867. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 46.Lombard DB, Zwaans BM. SIRT3: as simple as it seems? Gerontology. 2014;60(1):56–64. doi: 10.1159/000354382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Galgani J, Ravussin E. Energy metabolism, fuel selection and body weight regulation. Int J Obes (Lond) 2008;32(Suppl 7):S109–119. doi: 10.1038/ijo.2008.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ferrannini E. The theoretical bases of indirect calorimetry: a review. Metabolism. 1988;37(3):287–301. doi: 10.1016/0026-0495(88)90110-2. [DOI] [PubMed] [Google Scholar]
- 49.Oosterman JE, Foppen E, van der Spek R, Fliers E, Kalsbeek A, la Fleur SE. Timing of fat and liquid sugar intake alters substrate oxidation and food efficiency in male Wistar rats. Chronobiology international. 2015;32(2):289–298. doi: 10.3109/07420528.2014.971177. [DOI] [PubMed] [Google Scholar]
- 50.Davidoff AJ, Mason MM, Davidson MB, Carmody MW, Hintz KK, Wold LE, Podolin DA, Ren J. Sucrose-induced cardiomyocyte dysfunction is both preventable and reversible with clinically relevant treatments. American journal of physiology Endocrinology and metabolism. 2004;286(5):E718–724. doi: 10.1152/ajpendo.00358.2003. [DOI] [PubMed] [Google Scholar]
- 51.Li SY, Gilbert SA, Li Q, Ren J. Aldehyde dehydrogenase-2 (ALDH2) ameliorates chronic alcohol ingestion-induced myocardial insulin resistance and endoplasmic reticulum stress. Journal of molecular and cellular cardiology. 2009;47(2):247–255. doi: 10.1016/j.yjmcc.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hull TD, Boddu R, Guo L, Tisher CC, Traylor AM, Patel B, Joseph R, Prabhu SD, Suliman HB, Piantadosi CA, Agarwal A, George JF. Heme oxygenase-1 regulates mitochondrial quality control in the heart. JCI insight. 2016;1(2):e85817. doi: 10.1172/jci.insight.85817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Park YM, Kanaley JA, Zidon TM, Welly RJ, Scroggins RJ, Britton SL, Koch LG, Thyfault JP, Booth FW, Padilla J, Vieira-Potter VJ. Ovariectomized High Fit Rats Are Protected against Diet-Induced Insulin Resistance. Medicine and science in sports and exercise. 2016. [DOI] [PMC free article] [PubMed]
- 54.Sihag S, Cresci S, Li AY, Sucharov CC, Lehman JJ. PGC-1alpha and ERRalpha target gene downregulation is a signature of the failing human heart. Journal of molecular and cellular cardiology. 2009;46(2):201–212. doi: 10.1016/j.yjmcc.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Patten IS, Arany Z. PGC-1 coactivators in the cardiovascular system. Trends in endocrinology and metabolism. 2012;23(2):90–97. doi: 10.1016/j.tem.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 56.Papanicolaou KN, O'Rourke B, Foster DB. Metabolism leaves its mark on the powerhouse: recent progress in post-translational modifications of lysine in mitochondria. Frontiers in physiology. 2014;5:301. doi: 10.3389/fphys.2014.00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tian X, Zhao F, Cheng Z, Zhou M, Zhi X, Li J, Hu K. GCN5 acetyltransferase inhibits PGC1alpha-induced hepatitis B virus biosynthesis. Virologica Sinica. 2013;28(4):216–222. doi: 10.1007/s12250-013-3344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tavares CD, Sharabi K, Dominy JE, Lee Y, Isasa M, Orozco JM, Jedrychowski MP, Kamenecka TM, Griffin PR, Gygi SP, Puigserver P. The Methionine Transamination Pathway Controls Hepatic Glucose Metabolism through Regulation of the GCN5 Acetyl Transferase and the PGC-1alpha Transcriptional Coactivator. The Journal of biological chemistry. 2016. [DOI] [PMC free article] [PubMed]