

Abstract
Genetic polymorphisms are associated with breast cancer risk. Clinical and epidemiological observations suggest that clinical characteristics of breast cancer, such as estrogen receptor or HER2 status, are also influenced by hereditary factors. To identify genetic variants associated with pathological characteristics of breast cancer patients, a Genome Wide Association Study was performed in a cohort of 9365 women from the French nationwide SIGNAL/PHARE studies (NCT00381901/RECF1098). Strong association between the FGFR2 locus and ER status of breast cancer patients was observed (ER-positive n=6211, ER-negative n=2516; rs3135718 OR=1.34 p=5.46×10−12). This association was limited to patients with HER2-negative tumors (ER-positive n=4267, ER-negative n=1185; rs3135724 OR=1.85 p=1.16×10−11). The FGFR2 locus is known to be associated with breast cancer risk. This study provides sound evidence for an association between variants in the FGFR2 locus and ER status among breast cancer patients, particularly among patients with HER2-negative disease. This refinement of the association between FGFR2 variants and ER-status to HER2-negative disease provides novel insight to potential biological and clinical influence of genetic polymorphisms on breast tumors.
Keywords: breast, estrogen receptor, HER2, association, GWAS
INTRODUCTION
Since the completion of the Human Genome Project, the Genome Wide Association Scan (GWAS) has become the tool of choice for the detection of associations between disease risk and common genetic variation. The first breast cancer risk variants identified in the GWAS era were in the FGFR2 locus [1,2].
Further analyses, mainly in case-control and prospective cohorts, have reinforced this association as well as identified over 90 additional breast cancer risk loci [3]. GWAS studies with cases selected based on the estrogen receptor (ER) status of their tumors, and control subjects not affected by breast cancer, have shown divergent associations between ER+ and ER- tumors. In these analyses, variants in FGFR2 are more strongly associated with ER+ disease [4–14], as opposed to ER- disease, when comparing cases to healthy controls. Few single studies, however, have sufficient detail or sample size to carry out case-only analyses to further explore the relationship between genetic variants and disease characteristics, particularly with respect to amplification of the HER2 gene. Therefore analyses by subtype are often secondary, based on findings of the primary analyses of overall breast cancer risk. Furthermore, these studies are now carried out in large consortia with the potential for heterogeneity in definitions of various case characteristics, particularly ER and HER2 status.
For example, Broeks et al. [13] examined the association between low penetrance breast cancer loci and specific breast tumor subtypes in the context of the Breast Cancer Association Consortium (BCAC). rs2981582 in the FGFR2 locus was significantly associated with ER+/PR+/HER2- breast cancer (ncases=7201, p = 2.2 × 10−29), less so with ER+/PR+/HER2+ cases (ncases=996, p=5.5×10−4), and no association was observed with triple negative breast cancer (ncases=1480, p=0.841) or ER-/PR-/HER2+ breast cancer (ncases=627, p=0.396). A case-only comparison of HER2 status was carried out within ER+/PR+ and ER-/PR- groups, and neither showed any association (p=0.23 and 0.15, respectively).
In the present study, a case-only GWAS approach was used to study differences in the distribution of variants between breast cancer cases in a large, multi-center study with centralized data collection and handling, the SIGNAL/PHARE case-cohorts (NCT00381901/RECF1098).
RESULTS
Genotype data was generated from 9365 SIGNAL/PHARE participants. All subjects had greater than 95% genotyping success rate. 26 pairs of individuals were identified with Identity by State (IBS) > 30%, with the subject having the most complete genotype data from each pair retained for analyses. 551 further individuals were excluded from the present study due to PCA analyses. Finally, 61 subjects with missing clinical data were excluded. A total of 8727 patients including 2516 patients with ER- breast cancer were analyzed. Furthermore, 5452 patients had HER2-negative breast cancer, of which 1185 were ER-.
The search for variants associated with ER status showed only one region with a highly significant association, corresponding to FGFR2 (best p-value for rs3135718 p-value=6.0×10−12, Figure 1 and Supplementary Figure S1). Restricting our analyses to HER2-negative cases found that associations between variants at the FGFR2 locus remained significant at the genome-wide level (best p-value for rs3135724 = 5.2×10−11, Figure 2). Among HER2-positive tumors, the lowest p-value in the FGFR2 locus for the association with ER status was found for rs2981578 (p = 3.3×10−4 Table 1). The four variants in Table 1 were chosen to highlight the difference in associations between HER2+ and HER2− patients. Despite the smaller sample size among HER2-positive cases, this study has nearly 100% power to detect a per-allele OR = 1.8 as observed among the HER2-negative tumors, and greater than 80% power to detect a per-allele OR ≈ 1.3. The observed direction of the association was consistent with observations in prior case-control studies, with for example the C allele of rs3135718 being more frequently reported among women with ER+ tumors.
Figure 1. Manhattan plot of associations between SNPs and ER status overall.
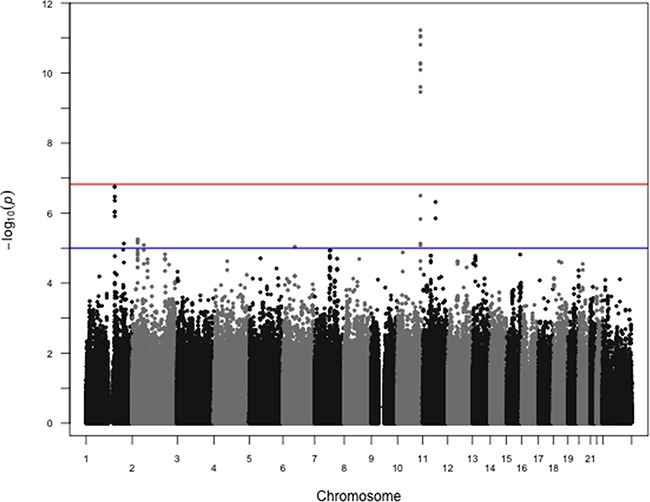
P-values from logistic regression comparing estrogen receptor positive cases to estrogen receptor negative cases, controlling for age at diagnosis and first two principal components, are shown. rs3135718 on chromosome 10 at the FGFR2 locus shows the strongest association. 914144 SNPs were included in these analyses, with 6211 ER+ and 2516 ER- cases. The red horizontal line corresponds to the empirical significance threshold of 1.48×10−7, while the blue horizontal line corresponds to an arbitrary level of 1.0×10−5. The inflation factor (λ) for these analyses is 1.02.
Figure 2. Manhattan plot of associations between SNPs and ER status restricted to HER2− cases.
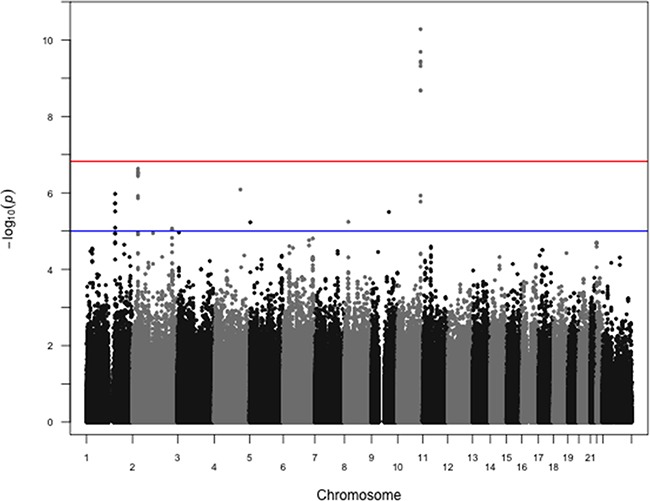
P-values from logistic regression comparing estrogen receptor positive cases to estrogen receptor negative cases restricted to HER2− cases, controlling for age at diagnosis and first two principal components, are shown. rs2981578 on chromosome 10 at the FGFR2 locus shows the strongest association. The same 914144 SNPs were included in these analyses, with 4267 HER2-/ER+ and 1185 HER2-/ER- cases. The red horizontal line corresponds to the empirical significance threshold of 1.48×10−7, while the blue horizontal line corresponds to an arbitrary level of 1.0×10−5. The inflation factor (λ) for these analyses is 1.02.
Table 1. Selected variants at the FGFR2 locus and ER status among breast cancer cases.
| Overall | HER2+ | HER2− | |||||
|---|---|---|---|---|---|---|---|
| SNP | I/G* (Rsq, Quality) | OR (95% CI) | p | OR (95% CI) | p | OR (95% CI) | p |
| rs3135718 | I (0.64, 0.89) | 1.33 (1.23 - 1.45 | 6.0×10-12 | 1.19 (1.04 - 1.35) | 7.9×10-3 | 1.47 (1.30 - 1.64) | 2.0×10-10 |
| rs3135724 | I (0.41, 0.84) | 1.51 (1.33 - 1.69) | 8.1×10-11 | 1.18 (0.97 - 1.41) | 9.3×10-2 | 1.79 (1.49 - 2.13) | 5.2×10-11 |
| rs2981578 | G (NA, NA) | 1.24 (1.16-1.32) | 3.5×10-10 | 1.20 (1.09-1.33) | 3.3×10-4 | 1.26 (1.14-1.38) | 1.7×10-6 |
| rs2981579 | G (NA, NA) | 1.25 (1.16 - 1.33) | 5.5×10-11 | 1.15 (1.03 - 1.27) | 9.2×10-3 | 1.33 (1.20 - 1.47) | 2.1×10-9 |
Imputed (I) or genotyped (G). Values reported from MACH output
As mentioned previously, variants in the FGFR2 locus were the first identified via GWAS with respect to breast cancer risk. The most recent fine-mapping effort of the FGFR2 locus explored functional variants, and identified three separate independent sets of correlated highly associated variants (ICHAVs [18]). In the present analyses restricted to HER2-negative tumors, rs3135724 was the SNP with the strongest association for ER status. These data included rs2981579 and rs2981578, from ICHAVs 1 and 3 respectively (Table 1). Unfortunately, rs45631563 from ICHAV 2 was not included, and no SNPs showed significant linkage disequilibrium with this marker in the current 1000 genomes data (http://1000genomes.org accessed July 8, 2015). Therefore additional analyses were carried out including rs3135724, rs2981579, and rs2981578 in the same logistic regression model. In our analyses of HER2− breast cancer, we found no evidence for independent association between these variants and tumor ER status (data not shown).
DISCUSSION
The identification of variants associated with specific molecular subtypes of breast cancer was a primary aim of the prospective SIGNAL/PHARE cohort. In this high-powered GWAS performed in a case-cohort of breast cancer patients with detailed clinical data, further information with respect to variants in the FGFR2 locus and their influence on breast cancer were provided, particularly regarding tumor ER status. In addition, the association between variants in FGFR2 and ER status in breast cancer was stronger among patients with HER2− tumors. While not including an independent validation set is a drawback of our analyses, the large sample size allowed us to have sufficient power to fully define this association, and the p-values obtained were well below empirical estimations of significance thresholds (1.48×10−7) as well as the generic GWAS significance threshold of 5×10−8.
Our hypothesis is that genetic variants that are associated with molecular subtypes will provide novel insights regarding disease etiology, and may lead to further developments regarding disease prevention and treatment. As our main focus was the construction of a clinical cohort, we have focused on collecting information with respect to histo-pathology and treatments, and patient follow-up. Therefore, we have not collected detailed information regarding epidemiological data such as body-mass index, reproductive history and menopausal status, or family history/BRCA mutations. The participants have been given a self-administered questionnaire with some of these variables, but as this questionnaire was administered after cancer diagnosis, we have chosen to not exploit these data at this time.
We have focused on the FGFR2 locus, which showed the strongest association with ER status, particularly among HER- breast cancer patients. There is growing evidence that genetic variants may be more strongly associated with specific breast cancer subtypes. For the most part, these analyses are extensions of current prospective cohort and case-control analyses. For example, recent analyses by Michailidou et al. [3] included stratification by estrogen receptor status for the 77 variants included in their polygenic risk score. A number of these variants showed differential associations with respect to estrogen receptor status. However as the authors state in their discussion, the number of estrogen receptor negative cases made accurately determining risk estimates difficult for this cancer subtype. Future analyses in our case-cohort will investigate other variants previously shown to influence breast cancer subtype.
A potential limitation of our study is the use of an internal imputation process, as opposed to imputing to the commonly used 1000 Genomes data or the Michigan Imputation Server. As highlighted in the Methods this was our original study design prior to the availability of these resources. We have continued with this approach in order to avoid any potential population differences with respect to linkage disequilibrium between our population of French breast cancer cases and the populations that provided data for publicly available resources. This approach leads to a lower number of variants on the absolute scale, meaning that we may be unable to detect any additional variants not captured through genotyping with the Illumina Omni5, which captures over 80% of common variants among Caucasian populations, and strict quality filtering of data (See Methods section).
For aspects of response to treatment, SIGNAL/PHARE has not yet accrued enough follow-up to fully explore the implication of variants on patient's outcome. This will be of course an obvious next step of our analyses, particularly as pertains to response to hormone therapy and FGFR2 variants in ER+/HER- breast cancer patients.
In conclusion, we further refine the influence of variants in the FGFR2 locus with respect to molecular characteristics of breast tumors, in that they are more strongly associated with estrogen receptor status among cancers without amplification of the HER2 gene.
METHODS
PHARE was a randomized phase 3 clinical trial comparing 6- and 12-month trastuzumab adjuvant exposure [15], which included a subset of 1,430 HER2-positive breast cancer cases with DNA available for GWAS analyses. SIGNAL was a prospective cohort specifically designed for GWAS analyses of 8,406 early breast cancer patients, enrolled at the time of the adjuvant chemotherapy from June 2009 to December 2013. The combined data set, the PHARE/SIGNAL study, included 9,365 breast cancer patients. Clinical and pathological characteristics were prospectively collected using standardized forms, and centralized at the French National Cancer Institute (INCa). For both studies, patients provided blood samples that were centralized at the Centre d'Etude du Polymorphisme Humain (CEPH) in Paris, France, for DNA extraction using standard protocols. Genotyping was carried out at the Centre National de Génotypage (CNG) in Evry, France.
The original study plan called for a two-staged genotyping strategy using only study participants. This approach aimed at reducing the potential that population structure in French breast cancer cases would influence imputation, while maximizing the proportion of the genome covered. Briefly, all cases were genotyped using the Illumina HumanCore Exome chip set, composed of over 264000 variants for a “GWAS Backbone” and over 244000 “exome-centered” variants. Variants were filtered based on completion rates (<95% SNP success, N = 8122), departure from Hardy-Weinberg Equilibrium (HWE p<0.001, N = 20357), and low minor allele frequency (MAF<0.001, N=200628). Principal Components Analysis (PCA) and k-means were then used to characterize the ancestry of the participants and only the main cluster of European individuals was included in the present analysis, to reduce risk of population stratification (See Supplementary Figure S2). A random subset of 1449 individuals from the main “European” cluster was selected for genotyping using the Illumina Omni5 chip set, composed of over 4M variants (See Supplementary Figure S2). Complete (SNP success = 100%, N=2049173) Omni5 data were then filtered using similar cutoffs as the HumanCore Exome data, specifically HWE (p<0.001, N=91018) were then used to impute missing genotypes from the remaining subjects genotyped using the HumanCore Exome array. SNPs with imputation quality score < 30% were excluded from analyses (N=783416), and finally variants with a MAF < 0.01 were excluded (N=82847). A total of 914144 SNPs were included in the GWAS analyses. Standard GWAS logistic regressions were carried out using the ProbABEL package [16]. Age at diagnosis and the first two principal components were included in regression analyses.
Genome-wide significance levels were estimated using the effective number of tests based on linkage disequilibrium between all markers used in our population through the SimpleM function in R [17]. The number of effective markers is estimated at 345906, corresponding to a Bonferroni-corrected p-value threshold of 1.48×10−7.
SUPPLEMENTARY MATERIALS FIGURES
Acknowledgments
The authors would like to thank the patients who took part in the study as well as medical staff for their cooperation. We also would like to thank Nicolas Thammavong for his contribution in clinical data management, Alexia Renoud for her contribution in clinical data management and descriptive statistics analyses, and the CEPH Biological Resource Center and CNG genotyping staff for technical assistance.
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare.
GRANT SUPPORT
PHARE and SIGNAL are academic trials sponsored by the French National Cancer Institute (INCa). David G. Cox receives support from the Ligue Contre le Cancer, Comité de l'Ain and the association Amis de l'Université de Lyon 1
REFERENCES
- 1.Hunter D.J, Kraft P, Jacobs K.B, Cox D.G, Yeager M, Hankinson S.E, Wacholder S, Want Z, Welch R, Hutchinson A, et al. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat. Genet. 2007;39:870–874. doi: 10.1038/ng2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Easton D.F, Pooley K.A, Dunning A.M, Pharoah P.D.P, Thompson D, Ballinger D.G, Stuewing J.P, Morrison J, Field H, Luben R, et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447:1087–1093. doi: 10.1038/nature05887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Michailidou K, Beesley J, Lindstrom S, Canisius S, Dennis J, Lush M.J, Maranian M.J, Bolla M.K, Wang Q, Shah M, et al. Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat. Genet. 2015;47:373–380. doi: 10.1038/ng.3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siddiq A, Couch F.J, Chen G.K, Lindström S, Eccles D, Millikan R.C, Michailidou K, Stram D.O, Beckmann L, Rhie K.S, et al. A meta-analysis of genome-wide association studies of breast cancer identifies two novel susceptibility loci at 6q14 and 20q11. Hum. Mol. Genet. 2012;21:5373–5384. doi: 10.1093/hmg/dds381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haiman C.A, Chen G.K, Vachon C.M, Canzian F, Dunning A, Millikan R.C, Wang X, Ademuyiwa F, Ahmed S, Ambrosone C.B, et al. A common variant at the TERT-CLPTM1L locus is associated with estrogen receptor-negative breast cancer. Nat. Genet. 2011;43:1210–1214. doi: 10.1038/ng.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Figueroa J.D, Garcia-Closas M, Humphries M, Platte R, Hopper J.L, Southey M.C, Apicella C, Hammet F, Schmidt M.K, Broeks A, et al. Associations of common variants at 1p11.2 and 14q24.1 (RAD51L1) with breast cancer risk and heterogeneity by tumor subtype: findings from the Breast Cancer Association Consortium. Hum. Mol. Genet. 2011;20:4693–4706. doi: 10.1093/hmg/ddr368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stevens K.N, Fredericksen Z, Vachon C.M, Wang X, Margolin S, Lindblom A, Nevanlinna H, Breco D, Aittomäki K, Blomqvist C, et al. 19p13.1 is a triple-negativespecific breast cancer susceptibility locus. Cancer Res. 2012;72:1795–1803. doi: 10.1158/0008-5472.CAN-11-3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garcia-Closas M, Couch F.J, Lindstrom S, Michailidou K, Schmidt M.K, Brook M.N, Orr N, Rhie S.K, Riboli E, Feigelson H.S, et al. Genome-wide association studies identify four ER negative-specific breast cancer risk loci. Nat. Genet. 2013;45:392–398. doi: 10.1038/ng.2561. 398e1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lambrechts D, Truong T, Justenhoven C, Humphreys M.K, Wang J, Hopper J.L, Dite G.S, Apicella C, Southey M.C, Schmidt M.K, et al. 11q13 is a susceptibility locus for hormone receptor positive breast cancer. Hum. Mutat. 2012;33:1123–1132. doi: 10.1002/humu.22089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Purrington K.S, Slager S, Eccles D, Yannoukakos D, Fasching P.A, Miron P, Carpenter J, Chang-Claude J, Martin N.G, Montgomery G.W, et al. Genome-wide association study identifies 25 known breast cancer susceptibility loci as risk factors for triple-negative breast cancer. Carcinogenesis. 2014;35:1012–1019. doi: 10.1093/carcin/bgt404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Warren H, Dudbridge F, Fletcher O, Orr N, Johnson N, Hopper J.L, Apicella C, Southey M.C, Mahmoodi M, Schmidt M.K, et al. 9q31.2-rs865686 as a susceptibility locus for estrogen receptor-positive breast cancer: evidence from the Breast Cancer Association Consortium. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 2012;21:1783–1791. doi: 10.1158/1055-9965.EPI-12-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stevens K.N, Vachon C.M, Lee A.M, Slager S, Lesnick T, Olswold C, Fasching P.A, Miron P, Eccles D, Carpenter J.E, et al. Common breast cancer susceptibility loci are associated with triple-negative breast cancer. Cancer Res. 2011;71:6240–6249. doi: 10.1158/0008-5472.CAN-11-1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Broeks A, Schmidt M.K, Sherman M.E, Couch F.J, Hopper H.L, Dite G.S, Apicella C, Smith L.D, Hammet F, Southey M.C, et al. Low penetrance breast cancer susceptibility loci are associated with specific breast tumor subtypes: findings from the Breast Cancer Association Consortium. Hum. Mol. Genet. 2011;20:3289–3303. doi: 10.1093/hmg/ddr228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cen Y.-L, Qi M.-L, Li H.-G, Su Y, Chen L.-J, Lin Y, Chen W.-Q, Xie X.-M, Tang L.-Y, Ren Z.-F. Associations of polymorphisms in the genes of FGFR2, FGF1, and RBFOX2 with breast cancer risk by estrogen/progesterone receptor status. Mol. Carcinog. 2013;52(Suppl 1):E52–59. doi: 10.1002/mc.21979. [DOI] [PubMed] [Google Scholar]
- 15.Pivot X, Romieu G, Debled M, Pierga J.-Y, Kerbrat P, Bachelot T, Lortholary A, Espié A, Fumoleau P, Serin D, et al. 6 months versus 12 months of adjuvant trastuzumab for patients with HER2-positive early breast cancer (PHARE): a randomised phase 3 trial. Lancet Oncol. 2013;14:741–748. doi: 10.1016/S1470-2045(13)70225-0. [DOI] [PubMed] [Google Scholar]
- 16.Aulchenko Y.S, Struchalin M.V, van Duijn C.M. ProbABEL package for genome-wide association analysis of imputed data. BMC Bioinformatics. 2010;11:134. doi: 10.1186/1471-2105-11-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goa X, Becker L.C, Becker D.M, Starmer J.D, Province M.A. Avoiding the high Bonferroni penalty in genome-wide association studies. Genet. Epidemiol. 2010;341:100, 105. doi: 10.1002/gepi.20430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meyer K.B, O'Reilly M, Michailidou K, Saskia C, Edwards S.L, French J.D, Prathalingham R, Dennis J, Bolla M.K, Wang Q, et al. Fine-scale mapping of the FGFR2 breast cancer risk locus: putative functional variants differentially bind FOXA1 and E2F1. Am. J. Hum. Genet. 2013;93:1046–1060. doi: 10.1016/j.ajhg.2013.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.