

Abstract
The palms are a family of tropical origin and one of the main constituents of the ecosystems of these regions around the world. The two main species of palm represent different challenges: coconut (Cocos nucifera L.) is a source of multiple goods and services in tropical communities, while oil palm (Elaeis guineensis Jacq) is the main protagonist of the oil market. In this study, we present a workflow that exploits the comparative genomics between a target species (coconut) and a reference species (oil palm) to improve the transcriptomic data, providing a proteome useful to answer functional or evolutionary questions. This workflow reduces redundancy and fragmentation, two inherent problems of transcriptomic data, while preserving the functional representation of the target species. Our approach was validated in Arabidopsis thaliana using Arabidopsis lyrata and Capsella rubella as references species. This analysis showed the high sensitivity and specificity of our strategy, relatively independent of the reference proteome. The workflow increased the length of proteins products in A. thaliana by 13%, allowing, often, to recover 100% of the protein sequence length. In addition redundancy was reduced by a factor greater than 3. In coconut, the approach generated 29,366 proteins, 1,246 of these proteins deriving from new contigs obtained with the BRANCH software. The coconut proteome presented a functional profile similar to that observed in rice and an important number of metabolic pathways related to secondary metabolism. The new sequences found with BRANCH software were enriched in functions related to biotic stress. Our strategy can be used as a complementary step to de novo transcriptome assembly to get a representative proteome of a target species. The results of the current analysis are available on the website PalmComparomics (http://palm-comparomics.southgreen.fr/).
Introduction
Palms are a plant family represented by more than 2,500 species in 184 genera distributed in tropical and subtropical regions of America, Africa and Asia [1]. As indicated by Couvreur et al. [1], evolution of palms is closely linked to rain tropical forest history. Several species of this family are keystone species, i.e their size and abundance play a major role in shaping their ecosystem.
The most popular species of palms is coconut (Cocos nucifera L). This species is the main source of income of multiple tropical communities and it played a role unequaled in the colonization of the pacific islands [2]. Coconut production in the coming decades faces several challenges to plant breeders as a result of climate change and market demand for increasing yield and diversification of products. Genetic and genomic knowledge in this species is still minimal; to date NCBI database reports only 1,008 ESTs sequences and 3,903 proteins [3] and several transcriptomes [4–6]. On the contrary, oil palm (Elaeis guineensis L.) receives greater attention because of its high productivity and profitability, which made palm oil the most common oil on the world market [7]. The interest in oil palm led to the sequencing of the genome of a palm species for the first time [7]. Genomic resources have also been developed for other palms such as the date palm [8] offering the opportunity to resolve fundamental questions of palms biology and opening new routes to the genetic improvement of these species [8–9]. For example, was the whole genome duplication experienced by Elaeis guineensis [10] shared by coconut and did it affect gene families diversification?. Improving the coconut genomic resources quality would help the understanding of the evolutionary history of this species compared to other palms and consider possible routes for a future coconut plant breeding.
In this context, translational genomics, i.e translating genomic knowledge from a model plant to a closely related orphan or non-model plant [11] is a mean to deepen the understanding of coconut biology by using advances on the oil palm research. Here, we propose a bioinformatics methodology that improves coconut transcriptome data by using an oil palm proteome. The methodology responds to several of the limitations of the transcriptomic data, by exploiting the sequence similarity between species, and offers as product a coconut proteome that can be used for different types of analysis such as phylogenetics, evolution of gene families, diversity, among others.
Transcriptomic data obtained by the new generation sequencing technologies (NGS) are a powerful and rapid mean to obtain valuable information on the genetic composition and expression of a given genome [12]. However, given the complex task of assembling the coding sequences without a reference genome, de novo transcriptomes undergo several drawbacks [13]. Wang et al, identified two types of errors that can occur in the clustering of expressed sequence tags (EST). Type I error occurs when ESTs of the same gene are not grouped into the same cluster, while type II error appears when ESTs of different genes are grouped in the same cluster. The type I error involves the fragmentation of genetic sequences, whereas type II represents chimeric sequences [13]. Transcriptomic data are also difficult to assemble because of a redundancy problem: it is not obvious to separate alternative transcripts of a gene, alleles, and gene copies of a multigenic family. These factors prevent obtaining a correct proteome that can be used in downstream analysis. A recent study showed that Trinity, a software dedicated to de novo assembly, has a high resilience to chimerism, while this software and some others with high performance (CLCscaf-S, SOAPtrans-S) have a significant rate of fragmentation as a result of the difficulty of assembling coding sequences with either low or high levels of expression [14].
Some studies have explored translational genomics to improve de novo transcriptome in non-model organisms. In order to identify the allelic variation present in the transcriptome of 14 different genotypes of Lolium perenne, a methodology based on orthology was developed [15]. This strategy called Orthology Guided Assembly (OGA) identifies the transcripts that can be orthologous to genes of a group of reference species and reduces redundancy by using CAP3 software. Another approach with CAP3 software is the scaffolding translation mapping (STM) method that maps reads and transcripts to the proteome of a reference species [16]. CAP3 is considered to be a conservative tool, demanding a high similarity between sequences [17]. This feature can prevent this tool to capture all the redundancy in de novo contigs or perform the scaffolding when the degree of overlap between the fragments is not long enough to be identified by this program. Thus CAP3 may not be completely adequate to reduce contig redundancy and their scaffolding.
In order to overcome these hindrances, we developed a methodology that reduces the fragmentation and redundancy of de novo transcriptomes by providing a workflow to build a reference proteome in a neglected species (coconut). The main characteristics of this workflow are: i) Identify new contig sequences with BRANCH software [18], aligning reads and de novo transcriptome to a closely related well-studied organism (oil palm). ii) Translate a large pool of polypeptide sequences from transcriptome with FrameDP software [19]. iii) Process the alignments of these polypeptides to an oil palm proteome (Fig 1).
Fig 1. Methodology and results obtained on coconut.

(A) Principal steps of the methodology to recover a target proteome, using transcriptomic data and a reference proteome from a related better studied organism. (B). Number of sequences generated on coconut by our methodology starting from four coconut transcriptomes. Sequences in red represent the number of new sequences found by BRANCH. Sequences in orange represent de novo transcriptomes assembled by Trinity. Protein products (PP) generated at step 5 (in dark red) combine information from both categories of sequences.
We validated our methodology by reversing the logic of model species and reassembling a well-known proteome from RNA-Seq data. Arabidopsis thaliana is a model species and has been the first plant genome to be sequenced [20]. Genomic resources are also available on related species such as Arabidopsis lyrata and Capsella rubella [21, 22]. These species have diverged around 12 and 23 M.Y.A respectively from Arabidopsis thaliana [23]. In order to evaluate the performance of our methodology and the influence of the reference species on the results, we assembled the Arabidopsis thaliana proteome from transcriptomic data, using Capsella rubella and Arabidopsis lyrata proteomes as references. This analysis, applied to a well-known genome, indicated that the workflow can produce a proteome with a high precision and respond significantly to the problems of redundancy and fragmentation.
This validation allowed us to apply our methodology to coconut and to produce an improved proteome, made available as a comparative genome hub in reference to oil palm pseudochromosomes. We finally assessed the functional representation of the coconut proteome, product of the workflow.
Material & methods
Workflow used to improve a target transcriptome by using a reference genome
Our methodology, starting from short nucleotide sequence reads generated by New Generation Sequencing technologies (NGS) of a transcriptome, involves five steps summarized in Fig 1A.
Step 1. de novo assembly of a target transcriptome
NGS transcriptomic studies are generally represented by paired-end reads. The target paired-end reads were trimmed using Trimmomatic V033 [24]. Transcriptomes were assembled with the software Trinity [25].
We used the BRANCH software as a first strategy to improve de novo transcriptome by translational genomics. BRANCH can identify novel contigs, extend incomplete contigs and join fragmented ones by using partial or related genomic sequences [18]. In the current analysis we used BRANCH with default parameters to find new contigs by using the reads and target contigs assembled at step 1 and the mRNA of the reference species as a related genomic sequence.
Step 2. Contigs translation into polypeptide sequences
The target transcriptomes and the new sequences found by BRANCH software were pooled. Identical sequences (full-length and fragments) were removed with cd-hit-est V4.6.4 software [26], retaining the longest sequence as the representative of the redundant group. These transcripts were translated into polypeptide sequences with FrameDP V1.2.2 software [19] and default parameters, using UniProtKB viridiplantae proteins [27] and relevant reference databases [28–30].
Step 3. Alignment of polypeptides to reference proteins
The target polypeptides of the previous step were aligned to the proteins of the reference species using BLASTP [31]. The alignments were processed to recover high-scoring segment pair (HSP) which do not overlap over more than 1 amino acids (aa). The alignments were subjected to the following algorithm to identify potential chimeric sequences:
All HSPs with a at least of 50% identity between the target polypeptides and the proteins of the reference species were identified.
If at least two disjoint regions of the target polypeptide aligned to two different proteins of the reference species, and none of these alignments covered more than 70% of the target polypeptide, this sequence was tagged as chimeric.
Step 4. Selection of the target polypeptides best hit in the reference proteome
The selection of the best hit was based on two parameters of quality of alignment: the identity and the coverage of target polypeptides. It was more stringent for small sequences to ensure accurate results in the last step. The best hit choice involved three steps:
An alignment was retained if the identity was at least 80% and the coverage of the target polypeptide was at least 70% (with a coverage of the reference protein below 40%) or 60% (with a coverage of the reference protein equal or above 40%).
The list of alignments was ordered according to the coverage of target polypeptide, the identity of alignment and coverage of the reference protein in that order of importance.
The best alignment of a target polypeptide was considered as the one at the top of the ordered list.
Step 5. Identification of redundant polypeptides and scaffolding
This step aimed to reduce redundancy and join target polypeptides into a single protein product (PP) by using the homology to the reference proteome. This step was done by processing the best hits to the reference proteome through the following steps:
All target polypeptides having the same reference protein as best hit were identified.
The non-redundant target polypeptides were identified, i.e. the proteins which had the best coverage of a region of the reference protein. All others target polypeptides that had a lower coverage of this region of the reference protein were labeled as redundant.
The non-redundant target polypeptides were sorted according to their first position of alignment to the reference protein.
If there was only one non-redundant polypeptide it was declared as the protein product. If there was more than one non-redundant polypeptides, the positions of alignment of two consecutive target polypeptides were compared. If the positions were overlapping, the rightmost aa in the alignments shared by the two target polypeptides was identified and this served as a position of cut and join for the two sequences. Otherwise, the target polypeptides were joined with n times”X” where n represents the distance between the alignments of these polypeptides.
Steps 4 and 5 and the identification of potential chimeric sequences were executed by a series of scripts that can be obtained at http://palm-comparomics-pj.southgreen.fr/Download.html/Scaff.RRed.tar.gz. A more detailed explanation of the use of this script is found in S1 File.
Precision assessment using A. thaliana
Precision was evaluated by two parameters: sensitivity and specificity, assessed by comparing the proteins obtained with the workflow in A. thaliana to an “expected group” of proteins of this species.
The “expected group” of A. thaliana proteins were the sequences in TAIR database [32], which met two conditions, i) there is evidence that these proteins are present in the polypeptides derived from contigs and ii) these proteins have significant homology to the A. lyrata or C. rubella proteomes. We counted as evidence of the presence of a protein of A. thaliana database in the polypeptide sequences produced at step 2, the fact that polypeptides were significantly aligned (i.e. identity > 60, target coverage > 70) to A. thaliana proteins with BLASTP software. To meet the second condition, the proteins of C. rubella/A. lyrata were aligned to proteins of A. thaliana that met the first condition, these alignments were filtered (identity > 60, target coverage > 70), and A. thaliana proteins retained were considered the “expected group”.
After applying the workflow, we aligned the resulting A. thaliana protein product (PP) with BLASTP software to the “expected group” of proteins defining three categories:
True Positive (TP): A A. thaliana PP has a significant alignment to a protein of the “expected group”.
False Positive (FP): A A. thaliana PP does not have a significant alignment to a protein of the “expected group”.
False Negative (FN): A protein of the expected group does not have a significant alignment to a A. thaliana PP.
An alignment was considered significant if the identity was at least 60% and the coverage of the protein from the “expected group” was minimum 70%. The categories enabled to calculate the measures of sensitivity and specificity.
The sensitivity is the ability of the methodology to recover the proteins of the expected group and was calculated by:
The specificity decreases when the methodology suggests proteins that are not present in the expected group. This measure was calculated by:
The behavior of these measures was studied by varying the parameters of identity and coverage in the alignments of step 4 of the workflow. There are three alignment parameters: the first represents the identity, the second is the coverage for target polypeptides covering more than 40% of the protein of expected group (reference) and the last is the coverage of target polypeptides covering 40% or less of the protein of the expected group. These parameters varied from 0/0/0 to 80/75/85.
Impact of scaffolding step in length, coverage and identity of the A. thaliana proteins
The increase in length of A. thaliana PPs was assessed by analyzing the coverage and the identity of alignments of polypeptides before and after step 5 to A. thaliana proteins TAIR 10 when A. lyrata was used as reference species.
Functional annotation in coconut and comparative analysis of metabolic pathways
Gene Ontologies: The gene ontology terms of coconut polypeptides before and after step 5 were determined using PRIAM software [33]. The distribution of proteins in the categories of level II in the three graphs (cellular component, molecular function, biological process) was compared between coconut proteins and Oryza sativa proteins [27] with WEGO software [34].
The enrichment analysis of coconut PPs which consist exclusively of polypeptides derived from new contigs (contigs founded by BRANCH software not identical at the sequence level to the contigs assembled by Trinity) was carried out with GOstats packages version 2.38.0 [35] in R [36]. A concise visualization of the results was performed with the software Revigo [37].
Biosynthetic pathways: The nomenclature of KEGG encyclopedia of coconut polypeptides before and after step 5 was inferred by InterproScan software [38]. The metabolic pathways of coconut polypeptides was identified with Pathway Tools [39] and compared to O. sativa pathways [40].
Transcriptome representation on a comparative genome hub
A website called PalmComparomics http://palm-comparomics.southgreen.fr/ was developed as a representation of the current analysis. This site is based on in-house Apache tools and tools of the Generic Model Organism Database (GMOD) project [41]. For instance, coconut scaffolded polypeptide matches along oil palm genome are viewable using JBrowse [42]. We exploited the facilities of Bio::DB::SeqFeature::Store schema to create a MySQL database with the annotations of coconut polypeptides from gff3 files. The website also included the CocoCyc Pathway/Genome Database (PGDB) containing the predicted metabolic pathways.
Data
Workflow application in coconut
The methodology was applied to coconut transcriptomic data recovered from NCBI [4–5]. The first dataset corresponds to a transcriptomic analysis of endosperm, embryo and leaf of a dwarf variety (SRX518095, SRX534380 and SRX534428) [4], while the second represents transcriptome data from a mix of tissues from Hainan tall cultivar (SRX198908) [5]. The new contigs were identified by using mRNAs of oil palm recovered from the NCBI Reference Sequence Database (RefSeq), annotation release 100 [28]. The translation into polypeptide sequences of coconut contigs was performed using as reference database the proteins of Elaeis guineensis [28], uniprotKB viridiplantae [27], Phoenix dactylifera [29] and Musa acuminata [30]. Finally, Elaeis guineensis proteome (RefSeq 100) [28] was used in step 5 of the workflow to produce a coconut proteome.
Sensitivity and specificity test
We applied the workflow to three transcriptomic datasets of A. thaliana recovered from NCBI: SRR1640366, SRR1640370, SRR2895761 [43, 14]. The proteins of “expected group” of A. thaliana was derived from TAIR10 database [32]. The Arabidopsis lyrata [21] and Capsella rubella [22] proteomes were recovered from phytozome v. 11 [44].
Functional annotation
The gene ontology terms of Oryza sativa v 7.0 were recovered from UniprotKB [27]. The biosynthetic pathways of Oryza sativa were recovered from PlantCyc [40].
A complete description of the software and the parameters used in each of the steps of the workflow is described in the document S1 File. This document also presents how you can obtain the main results of our analysis of the website PalmComparomics (http://palm-comparomics.southgreen.fr/).
Data used in the present paper can be retrieved at the following sites:
Transcriptomic data at NCBI site (https://www.ncbi.nlm.nih.gov/)
Cocos nucifera: datasets SRR1273180, SRR1273070 SRR606452, SRR606452
Arabidopsis Thaliana: datasets SRR1640366, SRR1640370, SRR2895761
mRNA and proteins: see links provided in refs 28, 29 and 30
Other data available at the following sites
Uniprot gene ontology rice: ftp://ftp.ebi.ac.uk/pub/databases/GO/goa/proteomes/23240.O_sativa_indica.goa
Uniprot viridiplantae: http://www.uniprot.org/taxonomy/33090
Phytozome v11.0: https://phytozome.jgi.doe.gov/pz/portal.html
PlantCyc sites: ftp://ftp.plantcyc.org/Pathways/Data_dumps/PMN10_June2015/oryzacyc_pathways.20150608
Data produced during the present work can be downloaded at http://palm-comparomics.southgreen.fr/Download.html or at https://github.com/armerovillanueva/PalmComparomics
Results
Our methodology offers a high precision when generating a new proteome
Before applying our methodology to coconut, we tested its precision on a well studied species: Arabidopsis thaliana. In this study several proteomes were obtained by varying the filtering parameters in step 4 using Arabidopsis lyrata and Capsella rubella as reference species. These proteomes were aligned to an expected group of proteins in A. thaliana and sensitivity and specificity were calculated.
The expected group for A. thaliana, i.e. proteins with significant alignments to polypeptides derived from contigs and presenting homology to A. lyrata/C.rubella respectively, consisted of 22,015 proteins with A. lyrata and 20,200 proteins with C. rubella.
The methodology described in Fig 1A was applied to three groups of transcriptomic data of A. thaliana (S1 Table). A pool of 121,076 contigs were obtained from de novo assembly, BRANCH and elimination of redundancy of identical sequences (full-length and fragments) (step 1, Fig 1A). We found a translation to polypeptide sequence for 101,688 contigs with FrameDP software (step 2, Fig 1A). These polypeptides were aligned and filtered to different thresholds of identity and coverage to A. lyrata/C. rubella proteomes (step 3 and 4, Fig 1A). Finally, elimination of redundancy and scaffolding for each of these groups of polypeptides was performed according to the last step of workflow (step 5, Fig 1A).
We tested several filtering parameter sets (see below and S1 Table) and develop here the results for the 80/60/70 parameter set. At the end of step 4, we recovered 77,541 and 70,803 A. thaliana polypeptides when A. lyrata and C. rubella were used respectively as references. The numbers of final protein products (PP) after step 5 were 19,943 (A. lyrata) and 18,615 (C. rubella) resulting in a reduction of redundancy by factors of 3.88 and 3.80 respectively.
When varying the filtering parameters, the sensitivity and specificity varied in a similar way between the two species used as reference (Fig 2). In both analyses, it was evident that an initial filtration of the alignments performed at step 4 is required to improve the specificity. This trend was especially notable in C. rubella where a first filtration increased the specificity from 0.75 to 0.93. After this initial point, there was a slight increase of specificity and a stable behavior of sensitivity as filtration parameters became more stringents until the parameters 80/60/70 (Fig 2). In this combination, sensitivity reached 88% and specificity was higher than 96%. When using more stringent filtering parameters, sensitivity decreased slightly, as a consequence of an increase of false negatives (S1 Table, Fig 2).
Fig 2. Analysis of sensitivity and specificity in Arabidopsis thaliana.

Curves of sensitivity (triangles) and specificity (stars) in Arabidopsis thaliana for different filtering parameters (step 4) using Arabidopsis lyrata (in red) and Capsella rubella (in blue) as reference species. The filtering parameters consist of three digits. The first represents the identity, the second is the coverage for target polypeptides covering more than 40% of the protein of expected group (reference), and the last is the coverage of target polypeptides covering 40% or less of the protein of the expected group.
We compared the number of true positives found with the two reference species in filtering parameters (80/60/70). 16,569 polypeptides were shared by the two analyses, 2,801 were specific to A. lyrata analysis and 1,281 to C. rubella.
The methodology improves the polypeptide sequences derived from de novo transcriptome
We assessed the performance of the methodology to recover the full-length sequence of a protein ensuring the amino acid identity. The polypeptide sequences of step 4 and 5 of the methodology were compared in terms of length, coverage and identity to the corresponding proteins of A. thaliana in TAIR10.
The comparison between the ranges of protein lengths between step 4 and 5 of workflow showed that step 5 reduced the percentage of short polypeptides while increasing the percentage of the large polypeptides, approaching protein length distribution of A. thaliana TAIR10 (Fig 3). Before step 5, 36.2% of the polypeptide sequences had a length lower than 200 aa, while after step 5 this percentage dropped to 21.3%. In all other ranges of length, the percentage of sequences was increased after step 5, concentrating 78% of sequences between 200 to 2,000 aa. In the sequences with lengths greater than or equal to 2000 aa, the percentage was doubled after step 5 (0.18–0.36%), approaching to that observed in A. thaliana (0.43%). Finally, the average length of the polypeptide sequences was increased from 339 to 429 aa.
Fig 3. Increase on the length of A. thaliana PPs.

Length distribution of polypeptides in step 4 (in orange) and 5 (in green) and distribution of Arabidopsis thaliana TAIR10 proteome (in blue). Protein lengths are indicated as discrete length ranges, from 0–200 to >2000 aa.
The identity of alignments to proteins from A. thaliana TAIR10 before and after step 5 remained unchanged (99%), while the average coverage of protein in database was increased from 0.87 to 0.90. To describe the impact of the methodology on the coverage, the distribution of this trait was divided into ranges, and amount of polypeptides that changed their coverage range after the scaffolding of step 5 was calculated (S2 Table). 53.0% (10,745) polypeptides before scaffolding had already a 100% coverage to proteins in TAIR10. Among the 9,106 remaining polypeptides, 26.4% (2,425) moved to a higher range of coverage after scaffolding. The polypeptides that were improved had an average increase of 18% in coverage, i.e. an average gain of 145 aa in length. The percentage of polypeptides with 100% coverage was increased by 7.4% (1,481). Only 1.2% of proteins moved to a lower range.
The methodology largely eliminates chimeric sequences
We evaluated the number of chimeric sequences at the main steps of the methodology. We identified 1,272 A. thaliana polypeptides sequences that are rated as chimeric sequences when aligned to the proteome reference of A. lyrata (step 3). In order to an exact evaluation of the amount of potential chimeric sequences the polypeptides were also aligned to the proteome reference of A. thaliana. As a result, the amount of chimeric sequences was reduced to 745, i.e. 0.61% of all polypeptides. Moreover, only 77 of these 1,272 sequences were retained in step 4 after filtration (80/60/70). In the step 5, 54 of these potential chimeric sequences were declared redundant, 5 were cut and joined to other sequences in the scaffolding process and the other 18 sequences represent complete PPs.
On the other hand, the scaffolding process could create new chimeric sequences by binding polypeptides belonging to different proteins. For this reason we evaluated the amount of PPs that could be considered as potential chimeras. When PPs were aligned to A. lyrata proteome, 47 potential chimeric sequences were identified. When PPs were aligned to the A. thaliana proteome, the amount of chimeric sequences was 142, representing 0.71% of all PPs.
The methodology provides an improved Cocos nucifera proteome
We applied the methodology in order to obtain a proteome of coconut using oil palm as model species.
In order to obtain a better representation of the coconut proteome, we worked with two different transcriptomes [4, 5]. The contigs obtained with Trinity and BRANCH from the four different groups (20,847 new/264,349 de novo dwarf embryo, 29,122/185,242 dwarf endosperm, 27,625/88,707 dwarf leaf and 23,283/89,133 tall tissue mix) were pooled and identical sequences (full-length and fragments) were eliminated, yielding 591,511 sequences (Fig 1B, step 1). Of these contigs, 14% (86,356) were identified by BRANCH software. 74% (439,300) of contigs were translated into polypeptides (step 2) sequences and 48% (285,975) of these had significant alignments to oil palm proteins (step 3 and 4). At step 5, these polypeptides sequences aligning to oil palm proteins were integrated into 29,366 coconut protein products (PPs). Each of these polypeptides represented on average 9,74 sequences of step 4 and 24% (7,058) of them were improved by scaffolding.
35% polypeptides (153,325) were lost between step 2 and step 3 because they had no significant hits in oil palm proteome used in this analysis. Among them, 7,716 had significant alignments (identity > = 80%, coverage > = 70%) to proteins in TrEMBL database. The remaining 145,609 polypeptide sequences had an average length of 106 aa in comparison to a length of 336 aa in polypeptides that had significant alignments to oil palm or other species. These polypeptides without significant alignments may represent orphan genes or artifacts of de novo transcriptome due to the presence of genomic DNA in the sample and/or non-coding RNA.
We quantified the degree of chimerism in coconut sequences. Among all the polypeptides that could be derived from the transcripts (step 3), 6,332 were identified as potential chimeric sequences (1.44%). Only 153 of these potential chimeric polypeptides were retained in step 4. 130 of these potential chimeric sequences were declared redundant, 7 intervened in the scaffolding process and 16 represented full PPs sequences. Finally, the alignment of coconut PPs to oil palm proteome revealed the existence of 23 PPs that could be considered chimeric.
The methodology identifies more coconut contigs
In this section we consider a new contig as a sequence that has been found by the software BRANCH and is not identical at the level of nucleotide composition to contigs obtained with Trinity. These new sequences represent contigs with evidence in RNAseq data and that were assembled using the oil palm mRNA in step 1 of the methodology.
In step 4 there were 79,021 polypeptide sequences derived from new contigs. 14,258 of these sequences were used in step 5 to obtain coconut PPs, while the remaining polypeptides obtained from the new contigs were identified as redundant. 11,196 of these 14,258 polypeptides were involved in the scaffolding of 4,059 coconut PPs, and 3,062 represented each a complete coconut PP sequence. This represented an average gain of 122 aa in the corresponding polypeptides.
Our workflow produced 1,246 coconut PPs that were completely new sequences, i.e these sequences would not have been recovered from the polypeptides derived from de novo assembly contigs with Trinity. Usually, these polypeptides represented only part of the reference proteins: when they were aligned to the corresponding proteins of reference species, the average coverage was significantly lower (0.60) than the coverage of all the proteome product of the workflow (0.89).
The methodology allows the identification of full-length protein products
An example of scaffolding of coconut polypeptides is depicted in Fig 4. The oil palm protein used as a reference is coded as XP_010917798.1 [28]. This protein is homologous to sequences known as ECERIFERUM 26-like, enzymes involved in epicuticular wax biosynthesis and pollen fertility [45]. In Fig 4A, the gene coding XP_010917798.1 is presented in its genomic context, with the representation of alignments of coconut polypeptides in step 4 and after step 5.
Fig 4. Scaffolding of coconut polypeptides homologous to oil palm ECERIFERUM 26-like protein.
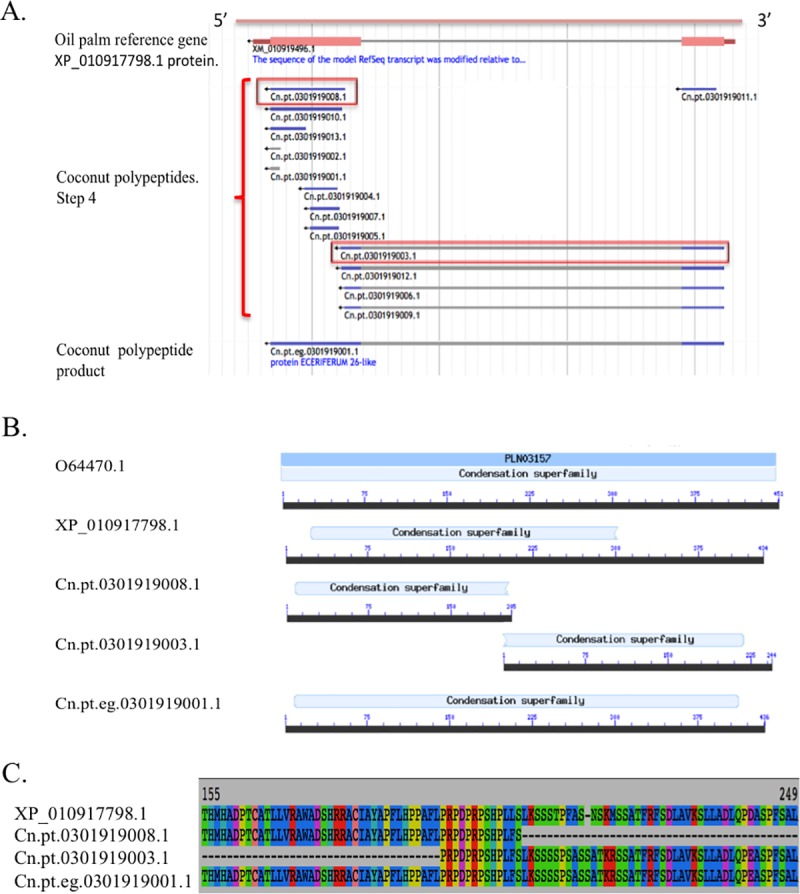
(A) Snapshot of oil palm protein, coconut polypeptides and coconut PP in ‘Palmcomparomics’ Jbrowse. Polypeptides within red squares were used for scaffolding of coconut PP. (B) Functional domain identified in coconut sequences and oil palm protein. The sequence O64470.1 of Arabidopsis thaliana is representative of the condensation superfamily. (C) Alignment between oil palm protein, coconut polypeptides and coconut PP around the overlap region.
In this example, several redundant coconut polypeptides sequences overlapped to two sequences (Cn.pt.0301919003.1 and Cn.pt.0301919008.1, Fig 4A) which were scaffolded and, together, represented the entire homologous coconut PP. The protein domains were identified with CDD software [46] and are represented in Fig 4B. Polypeptides of oil palm and coconut shared a ‘condensation’ domain in their sequences. This domain catalyses a condensation reaction to form peptide bonds in nonribosomal peptide biosynthesis [46]. The scaffolding step accurately joined the two coconut polypeptides (Fig 4C), thereby obtaining the complete condensation domain after scaffolding, despite the fact that the quality of the oil palm sequences does not allow to recover the whole domain in oil palm.
The comparison of coconut proteome to public data shows the ability of our methodology to reduce transcript fragmentation
We compared the proteome obtained in the current analyses with the polypeptides derived of two published transcriptomes [4, 5]. 57,175 transcripts recovered from Fan et al [5] and 153,852 from Huang et al [4] were translated into 37,743 and 76,553 polypeptides (66% and 49% of the transcripts respectively). The percentage of translated transcripts in our methodology was 66%.
These polypeptides datasets were aligned to rice proteome. The polypeptides of step IV have the highest amount of best-hits in rice (19,470), followed by Coconut PPs derived from our methodology (13,237), compared to 11,018 in Huang et al [5] and 9,535 in Fan et al [4]. The average identity for all datasets was similar, at around 66%. The S3 Fig shows the distributions of the coverage of rice proteins for the different datasets. While the distribution is approximately flat for polypeptides obtained from Huang et al, the other three distributions were characterized by a large peak at high values of coverage of rice proteins (>90%). The distribution of PPs (step 5) presents the highest concentration of sequences with good coverage, followed by the polypeptides of step 4, which in turn have a slightly better distribution than that observed in Fan et al.
The methodology allows an accurate functional representation of coconut transcriptome
We annotated the coconut PPs by identifying the gene ontology (GO) terms, including the 1,246 sequences that were never identified before (see above). The resulting GO terms at level II were compared to the functional annotation of Oryza sativa (the model species of monocots).
At least one GO term was assigned to 186,185 (42%) of coconut polypeptides in step 4, to 19,628 (67%) coconut PPs and to 22,674 (53%) O. sativa proteins. S1 Fig reveals the similarity between the distribution of polypeptides in the GO categories before and after the last step of workflow, indicating that the elimination of redundancy does not alter the functional representation of this species. Similarly, the distribution of coconut PP in gene ontology categories was similar to the distribution of O. sativa. The categories of biological processes with a high percentage of genes in rice and coconut were related to the basic metabolism necessary to cell survival (S1 Fig).
“Cellular processes” is one of the major categories in the coconut PPs and O. sativa proteome (GO:0009987). This category includes the processes involved in reproduction, growth and cell death. Another category with an important representation in the proteomes of these species is “metabolic process” (GO:0008152). This category represents the metabolic processes of biosynthesis and degradation of primary and secondary metabolites. Biological processes related to cell cycle regulation (GO:0043473) or the regulation of transcription activity and phosphorylases (GO:0065007) also have an important representation in rice and coconut sequences. Among the molecular functions with an important representation, we found “binding activity” (GO:0005488) and “catalytic activity” (GO:0003824), the latter covering a broad spectrum of enzymes such as transferases, transposase, ligases, lyases etc.
Nevertheless, the distribution of proteins in the gene ontology categories revealed interesting differences between coconut and rice, this comparison suggests a significant degree of specialization of rice proteins. For example, no coconut PP was found for the category “viral process” (GO:0016032), i.e. a multi-organism process in which a virus is a participant (S1 Fig). There are eight rice proteins belonging to this category, including CYP701A8, an enzyme involved in phytoalexins biosynthesis [47]. We aligned these proteins to the coconut PPs. The best hit in coconut for rice protein CYP701A8 was Cn.pt.eg.0300260001.1 (Id = 58%, Cv = 96%). Cn.pt.eg.0300260001.1 was aligned to the non-redundant proteins NCBI database with smartblast [31]. The best alignment was obtained for ALF95896.1, a coconut mRNA derived polypeptide (Id = 99%, Cv = 99%). Putting aside palms, the best alignment for coconut PP was NP_197962.1 (Id = 63%, Cv = 87%), a ent-kaurene oxidase from A. thaliana involved in gibberellin biosynthesis [48].
New sequences have functions related to biotic stress
It was possible to identify at least one Gene Ontology term in 748 of the 1,246 proteins derived exclusively from new contigs (S3 Table). This group of sequences was enriched in four main biological processes: response to biotic stimulus, protein phosphorylation, organelle organization and glucosamine-containing compound metabolism. In several of these groups the genes formed clusters on the chromosomes.
Response to biotic stimulus is especially important in coconut, which is subject a variety of lethal diseases [6, 49]. In the group of polypeptides involved in responses to biotic stimulus (GO:0009607) and defense response (GO:000695) 14 coconut PPs were identified as homologous to oil palm proteins, annotated as pathogenesis-related 1-like protein and MLO-related protein (Mildew Locus O) [50]. Five of the nine proteins of oil palm annotated as pathogenesis-related 1-like proteins formed a cluster on chromosome 14. Five coconut polypeptides were derived from oil palm protein annotated as MLO-related protein and had significant alignments to various A. thaliana proteins belonging to the MLO gene family that acting as susceptibility factors towards fungi causing the powdery mildew (S3 Table) [50].
“Protein amino acid phosphorylation” (GO:0006468) is a diverse group of 72 serine/threonine kinases. We identified 10 coconut PPs derived from oil palm references bearing the annotation “G-type lectin-like receptor S-serine/ threonine-protein kinase”. In oil palm, 7 of these proteins are part of two clusters, one on chromosome 1 and the other one on chromosome 2. Another important group of phosphorylases found in the new sequences are cysteine-rich protein receptor-like kinases. Four of these proteins also form a cluster on chromosome 2 of oil palm. Other kind of phosphorylases cover a wide spectrum of functions, e.g. Leucine-rich repeat receptor kinases (LRR-RKS) [51] and strubbelig receptor family (S3 Table) [52].
An important group of 25 coconut PPs had similarity to oil palm proteins with functions related to organelle organization (GO:0006996). Some are involved in the assembly of mitochondria (mitochondrial fission proteins) and peroxisomes biogenesis. Other proteins do a negative regulation of these morphogenetic processes by preventing polymerization of actin filaments or by promoting depolymerization of this molecule (F-actin-capping protein). Among these proteins, some also have a negative control over the organization of the cytoskeleton and mitosis (mitotic spindle checkpoint protein MAD2-like protein). Finally, some of these new proteins are histones or NAP1-related protein 2-like with functions related to chromatin modification [27].
A small group of three coconut proteins with glycoside hydrolase functions were related to chitinase of oil palm (GO:0006040). The Pfam annotation indicated that two of these proteins belong to the family hydrolase Glycoside 19, involved in plant defense against fungi and other pathogens. The other protein belongs to the family hydrolase Glycoside 18 (S3 Table) [53].
The coconut proteome has a large number of specialized metabolism pathways
The presence of the major classes of biosynthetic pathways of coconut polypeptides was assessed, and this result was compared with the pathways identified in O. sativa (S2 Fig). The coconut proteome presents 567 metabolic pathways, of which 453 (80%) belong to the “biosynthesis” class, while rice has 415 of this type representing 85% of total metabolic pathways in this species. S2 Fig presents the quantity of pathways subclasses in each species that are shared or exclusive to one species, in the main classes of biosynthetic pathways. In most subclasses, pathways shared by the two species are the most numerous. This was the case for carbohydrate biosynthesis, fatty acid and lipid biosynthesis, nucleosides and nucleotides biosynthesis (S2 Fig). A different trend was represented by secondary metabolites biosynthetic class, where shared and specific pathways were represented by a similar number. At the other end of the spectrum, biosynthetic pathways of amines and polyamines were all species specific.
PalmComparomics
In order to facilitate the access to genomic information in coconut, we developed the web site “PalmComparomics” (http://palm-comparomics.southgreen.fr/) that contains the main results of the current analysis. This web site, hosted by the Southgreen bioinformatic platform, offers several tools for data querying and visual analyses. PalmComparomics has a browser where oil palm genes and the mapping of coconut polypeptides to oil palm pseudo chromosomes are represented. Data can be retrieved for specific terms of gene ontology, Pfam domains, metabolism pathways, Interpro domains or sequence homology. PalmComparomics provides a BLAST tool that allows the user to align sequences against the coconut polypeptides and contigs. The polypeptides and proteins products of our methodology can be downloaded in FASTA format. It is also possible to retrieve a GFF3 file with gene ontology annotations, and Interpro or Pfam domains of coconut polypeptides.
Discussion
Fragmentation, redundancy and chimerism
The methodology developed in this work corrected transcripts fragmentation to a large extent. In the methodology validation made in A. thaliana, a substantial percentage of polypeptides with shorter sequences than expected were improved, and this gain in length resulted in recovering the entire sequence of the polypeptides in many cases. In coconut, a quarter of all PPs of the workflow were obtained by scaffolding. The relevance of the scaffolding process was reflected in the comparison of our coconut proteome to polypeptides obtained from Fan et al, [5] and Huang et al, [4] transcriptomes. In fact, the last step of the methodology increased the frequency of polypeptides that have high coverage of the rice proteins. The scaffolding could recover the full functional domain of a protein, although it was initially divided into polypeptides fragments, and regardless of the completion of the protein used as reference. This plasticity of the methodology to resolve the correct union of the fragments in most cases was the result of our scaffolding strategy on polypeptide sequences.
Our strategy differs from previous approaches using CAP3 like OGA or STM by using the protein of reference species as a guide for the scaffolding based on the alignment positions of target polypeptides. It is more flexible than these approaches because using reference proteins avoids the need to determine the similarity and coverage between the polypeptides to be joined.
These strengths of the methodology respond to the potential drawbacks of the fragmentation in downstream analysis, such as the reliable assessment of homology [54] and errors in multiple alignments [55].
Our methodology applied to coconut generated a comprehensive group of protein products that are representative of polypeptides derived from a large number of contigs aligning to the proteome of a reference species. Reduction of redundancy is an important factor in functional and evolutionary analysis. Ono et al.[56] showed that reducing redundancy in transcriptomes improves detection of differentially expressed genes. Redundancy could be a problem in identifying orthologs and paralogs, defining multiples alignments and constructing distance matrix between sequences [57]. This new coconut proteome could therefore lead the way to more in-depth genomic evolution analysis in the palm group.
The intensity of fragmentation and redundancy is related to the species under study. Reducing redundancy and scaffolding in A. thaliana analyses was clearly lower than in coconut. Although this could result from the quality of A. thaliana genome annotation or the large number of contigs assembled in coconut, it may also suggest some impact of biological factors explaining these results. Below we discuss some of these biological factors.
Our methodology is heavily shielded against chimeric sequences. Our analysis showed that, in the first steps, the percentage of chimeric sequences can mount beyond 1% of the total proteins that are obtained from the translation of the transcripts. However, using appropriate alignment quality parameters allows filtering out most chimeric sequences. Additionally, step V functions as another filtering point for the chimeric sequences. These could be declared redundant, or they could be cut in the process of scaffolding eliminating chimerism. As a result, only a small proportion of the final PPs are chimeric sequences, that users should analyze further attentively, since many of these apparent chimerisms may represent whole proteins that have additional sequence segments with minor alignments against other reference proteins.
Filtration issues in comparative analysis
The analysis of sensitivity and specificity showed the dependence of these variables in relation to the filtering parameters (identity and coverage) used in step 4 of workflow. The selection of filtering parameters determines the similarity between the sequences of target species and species reference. Relaxed parameters may result in chimeric sequences at the scaffolding step, producing background noise (increasing the number of false positives) and decreasing slightly the specificity of the outcome. Conversely, very stringent parameters reduce slightly the sensitivity by unnecessarily eliminating correct protein product sequences (increasing the number of false negatives).
The precision analysis performed in A. thaliana suggests that a combination of parameters (80/60/70) represents a good compromise in both references species (A. lyrata, C. rubella). It acts as a turning point, where the gain in specificity resulting from more stringent parameters no longer compensates the loss of sensitivity. We used these parameters to present the final results in A. thaliana and in coconut.
Choosing the right reference species
The choice of a reference species involves the assessment of several factors. Perhaps the most influential of these factors is the availability of good quality genomic resources in closely related species. Although the rate of publication of genomes increase at a high-speed [58], there are still major taxonomic groups in which there is a low amount of published genomes, likes palms. Another important aspect is the quality of the structural annotation of genomes In plants, some species like Arabidopsis thaliana have benefited from great attention in manual curation and experimental support of gene function [58].
Another factor that should be taken in consideration is the evolutionary divergence between the target species and the potential reference species. Two aspects come into play. The first one is the divergence time between the species under analyses. The second one is the rate of evolution in the considered species or taxon, which is determined by life history traits such as organism size, metabolism rate, the life cycle and the ecological niche [59]. In A. thaliana, the divergence time may be the main explanation to the observed differences in the number of expected proteins, protein products and true positives obtained with A. lyrata compared to C. rubella.
Conversely, coconut and oil palm diverged around 60 M.Y.A [60]. However, a very good genome conservation has been found among palm species as shown by the redundancy reduction and scaffolding. Genome sequencing of oil palm showed that 94.4% of the genes of this species are homologous to genes of Phoenix dactylifera supporting our observations [7]. This genome conservation may result from a longer generation time compared to Arabidopsis [61].
Functional representation of coconut proteome
The comparison of the distribution of gene ontology terms among coconut polypeptides before and after step 5 of the workflow showed that the later reduces redundancy without loss of functional representation. This functional profile is characterized by a large number of proteins involved in cellular and metabolic processes with catalytic and binding activities.
Our functional profile for the coconut PPs is also similar to the distribution of transcriptomes published by Fan et al [5] and Huang et al [4]. As indicated by Huang et al, this profile is shared by other species of monocots like banana and pineapple [4]. It also applies to the recently published transcriptome of Nypa fruticans [9], the only monocot adapted to mangrove.
While there is a great similarity between the GO distribution of coconut and rice proteins, there are also differences that may indicate a lineage specific processes. We evidenced one of these differences with CYP701A8, a rice protein that acquired a new role in the biosynthesis of phytoalexins, proteins implicated in resistance to biotic stress [47]. Although it was possible to identify sequences with significant similarity to CYP701A8 in the coconut proteome, this coconut sequences presented a higher degree of similarity to dicots proteins with functions in the gibberellins biosynthetic pathway. These results would need confirmation because our proteome is not necessarily complete. Therefore, the absence of evidence of a protein sharing the same function as CYP701A8 is not evidence of its absence.
The similarity of proteomes of coconut and rice was also confirmed through the comparison of the respective biosynthetic pathways. These species have an important number of pathways in common. However, the data did not show pathways of amines and polyamines shared by these species. This result could reflect true biological differences, but it has to be viewed with caution because it may be the product of transcriptomic bias and/or the limitations of the automatic annotation by pathway tools.
Interestingly, coconut has a high proportion of pathways involved in specialized metabolism that are not shared with rice. Secondary metabolites are important in the interaction of the plant with its environment [62]. In a comparative analysis of the pathways through the major taxonomic groups of plants, Chae et al., [63] showed that angiosperms are enriched in biochemical reactions involved in specialized metabolism. Some of the gene families involved in specialized metabolism appeared to be under selection and derived from local duplications [63]. Our results could motivate further investigation on the structure and evolution of secondary metabolism pathways in coconut and other palms.
Finally, we found that the new sequences identified with BRANCH are enriched in functions involved in biotic stress and organelles morphogenesis. These sequences are fragments with low coverage of complete sequence of mRNA, suggesting that these fragments are part of mRNA degradation product. This should be investigated further because many sequences with roles in biotic stress were identified as being over-expressed in coconut phytoplasma infection [6]. This is the case for pathogenesis-related proteins, MLO family and chitinases. Functions involved in the morphogenesis of peroxisomes and mitochondria could also be related to this coconut infection, considering the role of these organelles in the metabolism of reactive-oxygen species and response to stress [64]. Our current work shows that oil palm genes that are homologous to coconut diseases resistances genes are grouped in genomic clusters. This observation suggests active evolutionary mechanisms for local duplication of these genes.
Limits of the comparative analysis
There are other factors to be taken into account for the application of the methodology. The methodology cannot recover proteins that are absents in the reference species, but are present in the target species.
In A. thaliana analysis, C. rubella allowed to identify 1,281 PPs that were not initially revealed when A. lyrata was the reference species. This result suggests a complementary approach that would consist in using more than one reference species. For coconut, a complementary reference species could be Phoenix dactylifera, which genome has been also sequenced [8]. This could prove useful if the main goal is to have an exhaustive protein set however, there is a risk to introduce false positives.
Other proteins that can be recovered from the polypeptides derived from contigs are the orphan genes or “taxonomically restricted genes” i.e. genes uniques to the species or with a narrow phylogenetic distribution. The average percentage of orphan genes in a species are between 5 and 15% in plants [65]. These proteins may be retrieved from the initial set of sequences as those which do not match to the reference proteome, but present a “canonic” coding sequence feature. They must be further characterized by BLAST for highly conserved short stretches on a large set of sequenced genomes [66] or by InterproScan. The sequences, initially identified by these bioinformatic methods as candidate orphan genes, could be verified by transcriptomic or proteomic studies. The ratio of non-synonymous vs. synonymous substitutions of the coding sequences could be an additional signal of a selection pressure acting on those genes [67].
Finally, one last point to consider in the results of the methodology is the allelic diversity. In our study we used different transcriptomes, originating from separate genetic resources. The combination of transcripts from several studies allows a better coverage of protein sequence product and complementarity that may exist between different studies at the tissue level. The protein product identified by our methodology could be used as a reference to explore sequence diversity at the next step. In order to take allelic diversity into account, one approach would be to align the sequences that have been identified as redundant to the reference protein and determine sequence polymorphism at the nucleotide level, with potential implications as genetic markers or impact on the three-dimensional structure of proteins and their functions.
Conclusion
We developed a simple and effective method to improve protein sequences which can be obtained from a transcriptome by translational genomics. This methodology is an additional step to de novo assembly and solves the problem of fragmentation and redundancy of contigs. A more complete protein sequence enables reliable phylogenetic inference of the evolution of gene families. This analysis represents a first step before the expected publication of genomic sequence of coconut, offering a proteome that would help improving the structural genome annotation.
Supporting information
The table shows the number of contigs (QC) or polypeptides (QP) obtained at each step of the workflow, as well as the sensitivity and specificity analysis in Arabidopsis thaliana using Arabidopsis lyrata and Capsella rubella as reference species. Results are indicated for several filtering parameters, indicated as three digits: the first represents the identity, the second the coverage for target polypeptides covering more than 40% of the protein of expected group, and the last is the coverage of polypeptides covering 40% or less of the protein of the expected group. The filtering parameters retained for coconut are highlighted in yellow.
(XLSX)
The table represents the amount of polypeptides that change or maintain the coverage rank (BLASTP alignment) of the respective proteins of Arabidopsis thaliana of TAIR10, between step 4 and 5 of the methodology using Arabidopsis lyrata as a reference species.
(XLSX)
The table provides original data for new coconut PPs (derived exclusively from contigs found by BRANCH software). Unique identifiers and coordinates on the oil palm pseudochromosomes allows to retrieve the sequences in our ‘palmcomparomics’ database. GO terms, Pfam, E.C numbers and annotation for the corresponding oil palm protein are indicated.
(XLSX)
The figure presents the distribution of rice proteins (in red), coconut polypeptides from step 4 (in grey), and protein products (PPs) from step 5 (in orange) in Gene Ontology terms for three main GO categories (cellular component, molecular function, biological process).
(TIF)
The figure indicates the number of subclasses in the main classes of biosynthetic pathways that are shared (in blue) or specific to coconut (in orange) or rice (in green)
(TIF)
The frequency distribution of four sets of coconut polypeptides are plotted according to their coverage of rice proteins.
(TIF)
(A). Visualization of the alignment of the polypeptides of step 4 and Cn.pt.eg.Un05781001.1 coconut PP in relation to the XP_010910661.1 oil palm protein in Jbrowse. (B). Functional domains of coconut polypeptides, PP and the protein reference of oil palm.
(TIF)
Detailed description of the parameters of the software used in the workflow and of the data produced by our research.
(DOCX)
Acknowledgments
We thank Mr Gaëtan Droc and Mr Bertrand Pitollat, from Cirad, UMR AGAP, for providing guidance during the database construction.
Data Availability
Data used in the present paper can be retrieved at the following sites: Transcriptomic data at NCBI site (https://www.ncbi.nlm.nih.gov/); Cocos nucifera: datasets SRR1273180, SRR1273070, SRR606452, SRR606452; Arabidopsis Thaliana: datasets SRR1640366, SRR1640370, SRR2895761; mRNA and proteins: see links provided in refs 28, 29 and 30. Other data available at the following sites: Uniprot gene ontology rice: ftp://ftp.ebi.ac.uk/pub/databases/GO/goa/proteomes/23240.O_sativa_indica.goa, Uniprot viridiplantae: http://www.uniprot.org/taxonomy/33090, Phytozome v11.0: https://phytozome.jgi.doe.gov/pz/portal.html, PlantCyc sites: ftp://ftp.plantcyc.org/Pathways/Data_dumps/PMN10_June2015/oryzacyc_pathways.20150608. Data produced during the present work can be downloaded at http://palm-comparomics.southgreen.fr/Download.html or at https://github.com/armerovillanueva/PalmComparomics.
Funding Statement
The research works were funded by Montpellier SupAgro through a graduate research allocation to Alix Armero, and by CIRAD through a specific financial support. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Couvreur TL, Baker WJ. Tropical rain forest evolution: palms as a model group. BMC Biology. 2013;11(1):1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gunn BF, Baudouin L, Olsen KM. Independent Origins of Cultivated Coconut (Cocos nucifera L.) in the Old World Tropics. PLoS ONE. 2011. June 22;6(6):e21143 10.1371/journal.pone.0021143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cocos nucifera—GQuery: Global Cross-database NCBI search—NCBI [Internet]. [cited 2016 Aug 2]. Available from http://www.ncbi.nlm.nih.gov/gquery/?term=Cocos+nucifera
- 4.Huang Y-Y, Lee C-P, Fu JL, Chang BC-H, Matzke AJM, Matzke M. De Novo Transcriptome Sequence Assembly from Coconut Leaves and Seeds with a Focus on Factors Involved in RNA-Directed DNA Methylation. G3: Genes|Genomes|Genetics. 2014. November;4(11):2147–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fan H, Xiao Y, Yang Y, Xia W, Mason AS, Xia Z, et al. RNA-Seq Analysis of Cocos nucifera: Transcriptome Sequencing and De Novo Assembly for Subsequent Functional Genomics Approaches. PLoS ONE. 2013. March 29;8(3):e59997 10.1371/journal.pone.0059997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nejat N, Cahill DM, Vadamalai G, Ziemann M, Rookes J, Naderali N. Transcriptomics-based analysis using RNA-Seq of the coconut (Cocos nucifera) leaf in response to yellow decline phytoplasma infection. Molecular Genetics and Genomics Mol Genet Genomics. 2015;290(5):1899–910. 10.1007/s00438-015-1046-2 [DOI] [PubMed] [Google Scholar]
- 7.Singh R, Ong-Abdullah M, Low E-TL, Manaf MAA, Rosli R, Nookiah R, et al. Oil palm genome sequence reveals divergence of interfertile species in Old and New worlds. Nature. 2013. August 15;500(7462):335–9. 10.1038/nature12309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Al-Mssallem IS, Hu S, Zhang X, Lin Q, Liu W, Tan J, et al. Genome sequence of the date palm Phoenix dactylifera L. Nature Communications. 2013. August 6;4:2274 10.1038/ncomms3274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He Z, Zhang Z, Guo W, Zhang Y, Zhou R, Shi S. De Novo Assembly of Coding Sequences of the Mangrove Palm (Nypa fruticans) Using RNA-Seq and Discovery of Whole-Genome Duplications in the Ancestor of Palms. PLoS ONE. 2015. December 18;10(12):e0145385 10.1371/journal.pone.0145385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiao Y, Li J, Tang H, Paterson AH. Integrated Syntenic and Phylogenomic Analyses Reveal an Ancient Genome Duplication in Monocots. The Plant Cell. 2014;26(7):2792–802. 10.1105/tpc.114.127597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kang YJ, Lee T, Lee J, Shim S, Jeong H, Satyawan D, et al. Translational genomics for plant breeding with the genome sequence explosion. Plant Biotechnology Journal. 2016;14(4):1057–69. 10.1111/pbi.12449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marguerat S, Bähler J. RNA-seq: from technology to biology. Cellular and Molecular Life Sciences. 2010. February;67(4):569–79. 10.1007/s00018-009-0180-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang J-PZ, Lindsay BG, Leebens-Mack J, Cui L, Wall K, Miller WC, et al. EST clustering error evaluation and correction. Bioinformatics. 2004. November 22;20(17):2973–84. 10.1093/bioinformatics/bth342 [DOI] [PubMed] [Google Scholar]
- 14.Honaas LA, Wafula EK, Wickett NJ, Der JP, Zhang Y, Edger PP, et al. Selecting Superior De Novo Transcriptome Assemblies: Lessons Learned by Leveraging the Best Plant Genome. PLoS ONE. 2016. January 5;11(1):e0146062 10.1371/journal.pone.0146062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruttink T, Sterck L, Rohde A, Bendixen C, Rouzé P, Asp T, et al. Orthology Guided Assembly in highly heterozygous crops: creating a reference transcriptome to uncover genetic diversity in Lolium perenne. Plant Biotechnology Journal. 2013;11(5):605–17. 10.1111/pbi.12051 [DOI] [PubMed] [Google Scholar]
- 16.Surget-Groba Y, Montoya-Burgos JI. Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Research. 2010. October 1;20(10):1432–40. 10.1101/gr.103846.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mundry M, Bornberg-Bauer E, Sammeth M, Feulner PGD. Evaluating Characteristics of De Novo Assembly Software on 454 Transcriptome Data: A Simulation Approach. PLoS ONE. 2012. February 27;7(2):e31410 10.1371/journal.pone.0031410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bao E, Jiang T, Girke T. BRANCH: boosting RNA-Seq assemblies with partial or related genomic sequences. Bioinformatics. 2013;29(10):1250–9. 10.1093/bioinformatics/btt127 [DOI] [PubMed] [Google Scholar]
- 19.Gouzy J, Carrere S, Schiex T. FrameDP: sensitive peptide detection on noisy matured sequences. Bioinformatics. 2009;25(5):670–1. 10.1093/bioinformatics/btp024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000. December 14;408(6814):796–815. 10.1038/35048692 [DOI] [PubMed] [Google Scholar]
- 21.Hu TT, Pattyn P, Bakker EG, Cao J, Cheng J-F, Clark RM, et al. The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nature Genetics Nat Genet. 2011October;43(5):476–81. 10.1038/ng.807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Slotte T, Hazzouri KM, Agren JA, Koenig D, Maumus F, Guo Y-L, et al. The Capsella rubella genome and the genomic consequences of rapid mating system evolution. Nat Genet. 2013. July;45(7):831–5. 10.1038/ng.2669 [DOI] [PubMed] [Google Scholar]
- 23.Murat F, Louis A, Maumus F, Armero A, Cooke R, Quesneville H, et al. Understanding Brassicaceae evolution through ancestral genome reconstruction. Genome Biology. 2015;16(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014. January;30(15):2114–20. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol Nature Biotechnology. 2011;29(7):644–52. 10.1038/nbt.1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li W, Jaroszewski L, Godzik A. Clustering of highly homologous sequences to reduce the size of large protein databases. Bioinformatics. 2001;17(3):282–3. [DOI] [PubMed] [Google Scholar]
- 27.Consortium TU. UniProt: a hub for protein information. Nucleic Acids Research. 2015;43(D1):D204–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.National Center for Biotechnology Information [Internet]. National Center for Biotechnology Information. U.S. National Library of Medicine; [cited 2016Jun16]. Available from: ftp://ftp.ncbi.nih.gov/genomes/Elaeis_guineensis/
- 29.National Center for Biotechnology Information [Internet]. National Center for Biotechnology Information. U.S. National Library of Medicine; [cited 2016Jun16]. Available from: ftp://ftp.ncbi.nih.gov/genomes/Phoenix_dactylifera/
- 30.National Center for Biotechnology Information [Internet]. National Center for Biotechnology Information. U.S. National Library of Medicine; [cited 2016Jun16]. Available from: ftp://ftp.ncbi.nih.gov/genomes/Musa_acuminata/
- 31.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of Molecular Biology. 1990. October 5;215(3):403–10. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- 32.Berardini TZ, Reiser L, Li D, Mezheritsky Y, Muller R, Strait E, et al. The arabidopsis information resource: Making and mining the “gold standard” annotated reference plant genome. genesis. 2015;53(8):474–85. 10.1002/dvg.22877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Claudel-Renard C. Enzyme-specific profiles for genome annotation: PRIAM. Nucleic Acids Research. 2003;31(22):6633–9. 10.1093/nar/gkg847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Research. 2006;34(suppl 2):W293–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beißbarth T, Speed TP. GOstat: find statistically overrepresented Gene Ontologies within a group of genes. Bioinformatics. 2004;20(9):1464–5. 10.1093/bioinformatics/bth088 [DOI] [PubMed] [Google Scholar]
- 36.R Development Core Team (2008). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0, URL http://www.R-project.org.
- 37.Supek F, Bošnjak M, Škunca N, Šmuc T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE. 2011. July 18;6(7):e21800 10.1371/journal.pone.0021800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hunter S, Jones P, Mitchell A, Apweiler R, Attwood TK, Bateman A, et al. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Research. 2012;40(D1):D306–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karp PD, Latendresse M, Paley SM, Krummenacker M, Ong QD, Billington R, et al. Pathway Tools version 19.0 update: software for pathway/genome informatics and systems biology. Briefings in Bioinformatics [Internet]. 2015. October 10; Available from: http://bib.oxfordjournals.org/content/early/2015/09/25/bib.bbv079.abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mueller LA, Zhang P, Rhee SY. AraCyc: A Biochemical Pathway Database for Arabidopsis. Plant Physiology. 2003. June 1;132(2):453–60. 10.1104/pp.102.017236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Droc G, Larivière D, Guignon V, Yahiaoui N, This D, Garsmeur O, et al. The Banana Genome Hub. Database [Internet]. 2013 Jan 1;2013. Available from: http://database.oxfordjournals.org/content/2013/bat035.abstract [DOI] [PMC free article] [PubMed]
- 42.Skinner ME, Uzilov AV, Stein LD, Mungall CJ, Holmes IH. JBrowse: A next-generation genome browser. Genome Research. 2009. September 1;19(9):1630–8. 10.1101/gr.094607.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu H, Tian C, Yu Y, Jiao Y. Transcriptome Survey of the Contribution of Alternative Splicing to Proteome Diversity in Arabidopsis thaliana. Molecular Plant. 2016;9(5):749–52. 10.1016/j.molp.2015.12.018 [DOI] [PubMed] [Google Scholar]
- 44.Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Research. 2012;40(D1):D1178–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bourdenx B, Bernard A, Domergue F, Pascal S, Léger A, Roby D, et al. Overexpression of Arabidopsis ECERIFERUM1 Promotes Wax Very-Long-Chain Alkane Biosynthesis and Influences Plant Response to Biotic and Abiotic Stresses. Plant Physiology. 2011. May 1;156(1):29–45. 10.1104/pp.111.172320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marchler-Bauer A, Anderson JB, Cherukuri PF, DeWeese-Scott C, Geer LY, Gwadz M, et al. CDD: a Conserved Domain Database for protein classification. Nucleic Acids Research. 2005. January 1;33(suppl 1):D192–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Q, Hillwig ML, Wu Y, Peters RJ. CYP701A8: A Rice ent-Kaurene Oxidase Paralog Diverted to More Specialized Diterpenoid Metabolism. Plant Physiology. 2012. March 1;158(3):1418–25. 10.1104/pp.111.187518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morrone D, Chen X, Coates RM, Peters RJ. Characterization of the kaurene oxidase CYP701A3, a multifunctional cytochrome P450 from gibberellin biosynthesis. Biochem J. 2010. October 11;431(3):337 10.1042/BJ20100597 [DOI] [PubMed] [Google Scholar]
- 49.Magalhães JAS, De Moraes Neto A. H. A., Miguens FC. Nematodes of Rhynchophorus palmarum, L. (Coleoptera: Curculionidae), vector of the Red Ring disease in coconut plantations from the north of the Rio de Janeiro State. Parasitology Research Parasitol Res. 2008;102(6):1281–7. 10.1007/s00436-008-0906-7 [DOI] [PubMed] [Google Scholar]
- 50.Iovieno P, Andolfo G, Schiavulli A, Catalano D, Ricciardi L, Frusciante L, et al. Structure, evolution and functional inference on the Mildew Locus O (MLO) gene family in three cultivated Cucurbitaceae spp. BMC Genomics. 2015;16:1112 10.1186/s12864-015-2325-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Torii KU. Leucine-Rich Repeat Receptor Kinases in Plants: Structure, Function, and Signal Transduction Pathways In: International Review of Cytology [Internet]. Academic Press; 2004. p. 1–46. Available from: http://www.sciencedirect.com/science/article/pii/S0074769604340015 [DOI] [PubMed] [Google Scholar]
- 52.Eyüboglu B, Pfister K, Haberer G, Chevalier D, Fuchs A, Mayer KF, et al. Molecular characterisation of the STRUBBELIG-RECEPTOR FAMILY of genes encoding putative leucine-rich repeat receptor-like kinases in Arabidopsis thaliana. BMC Plant Biol BMC Plant Biology. 2007;7(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Research. 2016. January 4;44(D1):D279–85. 10.1093/nar/gkv1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de la Torre JE, Egan MG, Katari MS, Brenner ED, Stevenson DW, Coruzzi GM, et al. ESTimating plant phylogeny: lessons from partitioning. BMC Evolutionary Biology. 2006;6(1):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thompson JD, Linard B, Lecompte O, Poch O. A Comprehensive Benchmark Study of Multiple Sequence Alignment Methods: Current Challenges and Future Perspectives. PLoS ONE. 2011. March 31;6(3):e18093 10.1371/journal.pone.0018093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ono H, Ishii K, Kozaki T, Ogiwara I, Kanekatsu M, Yamada T. Removal of redundant contigs from de novo RNA-Seq assemblies via homology search improves accurate detection of differentially expressed genes. BMC Genomics. 2015;16:1031 10.1186/s12864-015-2247-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang Y, Smith SA. Optimizing de novo assembly of short-read RNA-seq data for phylogenomics. BMC Genomics. 2013;14(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Michael TP, VanBuren R. Progress, challenges and the future of crop genomes. Current Opinion in Plant Biology. 2015. April;24:71–81. 10.1016/j.pbi.2015.02.002 [DOI] [PubMed] [Google Scholar]
- 59.Bromham L. Why do species vary in their rate of molecular evolution? Biol Lett. 2009. May 11;5(3):401 10.1098/rsbl.2009.0136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baker WJ, Norup MV, Clarkson JJ, Couvreur TLP, Dowe JL, Lewis CE, et al. Phylogenetic relationships among arecoid palms (Arecaceae: Arecoideae). Annals of Botany. 2011. December 1;108(8):1417–32. 10.1093/aob/mcr020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tomlinson PB. The uniqueness of palms. Botan J Linn Soc Botanical Journal of the Linnean Society. 2006;151(1):5–14. [Google Scholar]
- 62.Shang Y, Ma Y, Zhou Y, Zhang H, Duan L, Chen H, et al. Biosynthesis, regulation, and domestication of bitterness in cucumber. Science. 2014. November 27;346(6213):1084 10.1126/science.1259215 [DOI] [PubMed] [Google Scholar]
- 63.Chae L, Kim T, Nilo-Poyanco R, Rhee SY. Genomic Signatures of Specialized Metabolism in Plants. Science. 2014. May 1;344(6183):510 10.1126/science.1252076 [DOI] [PubMed] [Google Scholar]
- 64.Bonekamp NA, Völkl A, Fahimi HD, Schrader M. Reactive oxygen species and peroxisomes: Struggling for balance. BioFactors. 2009;35(4):346–55. 10.1002/biof.48 [DOI] [PubMed] [Google Scholar]
- 65.Arendsee ZW, Li L, Wurtele ES. Coming of age: orphan genes in plants. Trends in Plant Science. 2014. November;19(11):698–708. 10.1016/j.tplants.2014.07.003 [DOI] [PubMed] [Google Scholar]
- 66.Tautz D, Domazet-Lošo T. The evolutionary origin of orphan genes. Nat Rev Genet. 2011. October;12(10):692–702. 10.1038/nrg3053 [DOI] [PubMed] [Google Scholar]
- 67.Prabh N, Rödelsperger C. Are orphan genes protein-coding, prediction artifacts, or non-coding RNAs? BMC Bioinformatics. 2016;17(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The table shows the number of contigs (QC) or polypeptides (QP) obtained at each step of the workflow, as well as the sensitivity and specificity analysis in Arabidopsis thaliana using Arabidopsis lyrata and Capsella rubella as reference species. Results are indicated for several filtering parameters, indicated as three digits: the first represents the identity, the second the coverage for target polypeptides covering more than 40% of the protein of expected group, and the last is the coverage of polypeptides covering 40% or less of the protein of the expected group. The filtering parameters retained for coconut are highlighted in yellow.
(XLSX)
The table represents the amount of polypeptides that change or maintain the coverage rank (BLASTP alignment) of the respective proteins of Arabidopsis thaliana of TAIR10, between step 4 and 5 of the methodology using Arabidopsis lyrata as a reference species.
(XLSX)
The table provides original data for new coconut PPs (derived exclusively from contigs found by BRANCH software). Unique identifiers and coordinates on the oil palm pseudochromosomes allows to retrieve the sequences in our ‘palmcomparomics’ database. GO terms, Pfam, E.C numbers and annotation for the corresponding oil palm protein are indicated.
(XLSX)
The figure presents the distribution of rice proteins (in red), coconut polypeptides from step 4 (in grey), and protein products (PPs) from step 5 (in orange) in Gene Ontology terms for three main GO categories (cellular component, molecular function, biological process).
(TIF)
The figure indicates the number of subclasses in the main classes of biosynthetic pathways that are shared (in blue) or specific to coconut (in orange) or rice (in green)
(TIF)
The frequency distribution of four sets of coconut polypeptides are plotted according to their coverage of rice proteins.
(TIF)
(A). Visualization of the alignment of the polypeptides of step 4 and Cn.pt.eg.Un05781001.1 coconut PP in relation to the XP_010910661.1 oil palm protein in Jbrowse. (B). Functional domains of coconut polypeptides, PP and the protein reference of oil palm.
(TIF)
Detailed description of the parameters of the software used in the workflow and of the data produced by our research.
(DOCX)
Data Availability Statement
Data used in the present paper can be retrieved at the following sites: Transcriptomic data at NCBI site (https://www.ncbi.nlm.nih.gov/); Cocos nucifera: datasets SRR1273180, SRR1273070, SRR606452, SRR606452; Arabidopsis Thaliana: datasets SRR1640366, SRR1640370, SRR2895761; mRNA and proteins: see links provided in refs 28, 29 and 30. Other data available at the following sites: Uniprot gene ontology rice: ftp://ftp.ebi.ac.uk/pub/databases/GO/goa/proteomes/23240.O_sativa_indica.goa, Uniprot viridiplantae: http://www.uniprot.org/taxonomy/33090, Phytozome v11.0: https://phytozome.jgi.doe.gov/pz/portal.html, PlantCyc sites: ftp://ftp.plantcyc.org/Pathways/Data_dumps/PMN10_June2015/oryzacyc_pathways.20150608. Data produced during the present work can be downloaded at http://palm-comparomics.southgreen.fr/Download.html or at https://github.com/armerovillanueva/PalmComparomics.