

Abstract
Objective
—Increasing evidence suggests that contractile dysfunction in smooth muscle cells (SMCs) plays a critical role in aortic biomechanical dysfunction and aortic aneurysm and dissection (AAD) development. However, the mechanisms underlying SMC contractile dysfunction in sporadic AAD are poorly understood. In this study, we examined the role of the NLRP3–caspase-1 inflammasome, a key inflammatory cascade, in SMC contractile dysfunction in AAD.
Approach and Results
—We observed significant SMC contractile protein degradation in aortas from patients with sporadic thoracic AAD. The contractile protein degradation was associated with activation of the NLRP3–caspase-1 inflammasome cascade. In SMCs, caspase-1 bound and directly cleaved and degraded contractile proteins, leading to contractile dysfunction. Furthermore, Nlrp3 or caspase-1 deficiency in mice significantly reduced angiotensin II–induced contractile protein degradation, biomechanical dysfunction, and AAD formation in both thoracic and abdominal aortas. Finally, blocking this cascade with the inflammasome inhibitor, glyburide (an antidiabetic medication), reduced angiotensin II–induced AAD formation.
Conclusions
—Inflammasome-caspase-1–mediated degradation of SMC contractile proteins may contribute to aortic biomechanical dysfunction and AAD development. This cascade may be a therapeutic target in AAD formation. Additionally, glyburide may have protective effects against AAD development.
Keywords: aneurysm, contractility, cardiovascular diseases, vessels
Graphical abstract
Aortic aneurysms and dissections (AAD) are common interrelated cardiovascular disorders that cause more than 10,000 deaths in the United States each year and are a leading cause of death in people 55 years of age or older.1 The structural integrity of the aortic wall depends on the homeostasis of vascular smooth muscle cells (SMCs) and the extracellular matrix (ECM).
A growing body of evidence has suggested that SMC contractile dysfunction2–6 plays a critical role in AAD development. Specific genetic defects have been identified as the underlying cause of SMC contractile dysfunction in hereditary aortic disorders.2–5 Genomic copy number variants that disrupt cell contractile function have been identified in sporadic thoracic aortic diseases,6 which account for 80% of AAD.7 However, the molecular mechanisms responsible for SMC contractile dysfunction in sporadic aortic disease remain largely unknown.
The NLRP3 (nucleotide oligomerization domain–like receptor family, pyrin domain containing 3) inflammasome functions as a molecular platform for mediating cellular responses to stress. In response to danger signals, the canonical NLRP3 inflammasome complex (NLRP3-ASC [apoptosis-associated speck-like protein containing a caspase recruitment domain]-caspase-1) triggers cell injury and dysfunction in a caspase-1–dependent manner.8 Gain-of-function mutations in the NLRP3 gene result in several inflammatory diseases.9 In addition, the NLRP3–caspase-1 pathway has been implicated in the pathogenesis of several chronic disorders including type 2 diabetes10,11 and atherosclerosis.12 Genetic variability involving one of the components of the NLRP3–caspase-1 inflammasome complex—the caspase recruitment domain-containing protein 8 (CARD8 SNP rs2043211)—has been associated with altered susceptibility to abdominal aortic aneurysm (AAA) formation.13 These findings raise the question of whether enhanced NLRP3–caspase-1 inflammasome activation can trigger SMC injury and dysfunction leading to AAD development.
Given the critical role of this cascade in tissue inflammation and destruction, we conducted this study to examine the hypothesis that the NLRP3–caspase-1 inflammasome is activated in aortic SMCs in patients with sporadic AAD and that its activation is important in SMC dysfunction and aortic disease development. Our findings suggest that the NLRP3-caspase-1 inflammasome cascade promotes the degradation of contractile proteins in SMCs that causes aortic contractile dysfunction, leading to AAD development.
Material and Methods
Materials and Methods are available in the online-only data supplement.
Results
Smooth Muscle Contractile Proteins are Degraded in Human Sporadic Thoracic AAD Tissues
To determine the potential cause of SMC dysfunction, we examined the structural integrity of the SMC-specific contractile proteins, myosin heavy chain and tropomyosin, in patient aortic tissues. We observed a significant amount of degradation products of myosin heavy chain and tropomyosin in the aortic wall of patients with ascending thoracic aortic aneurysm (aTAA) (Figure 1A), descending thoracic aortic aneurysm (dTAA), and descending thoracic aortic dissection (dTAD) (Figure 1B). These findings clearly show increased protein degradation of contractile proteins in diseased tissues from patients with sporadic thoracic AAD.
Figure 1.
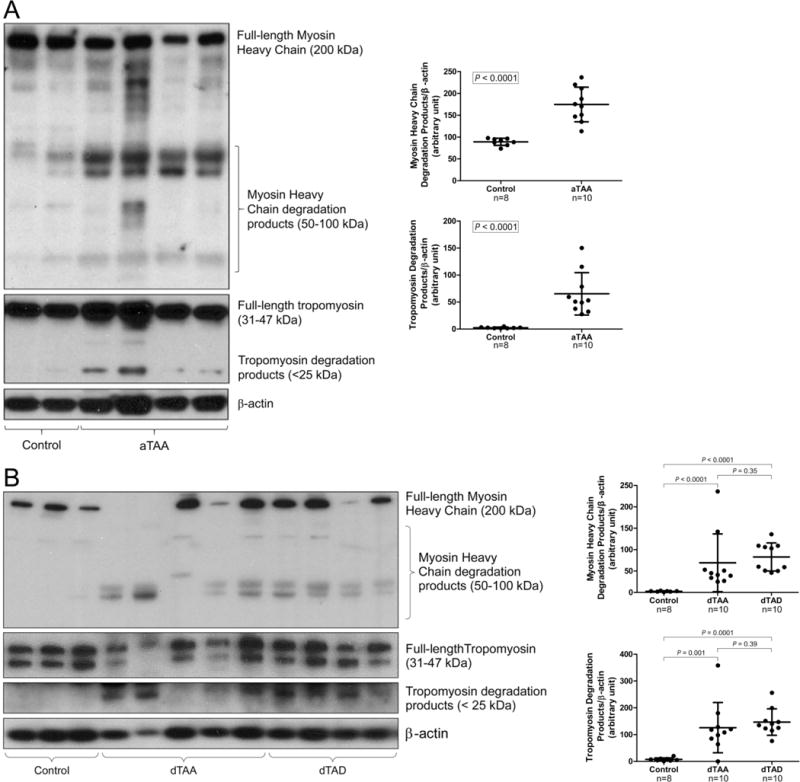
Significant smooth muscle contractile protein degradation in human thoracic aortic aneurysm and dissection (AAD) tissues. A, Ascending aortic tissues from patients with ascending thoracic aortic aneurysm without dissection (aTAA, n=10) and from organ donors (ascending control, n=8) were examined. Representative western blot images and quantification studies show significant degradation products of myosin heavy chain and tropomyosin in aTAA. B, Descending aortic tissues from patients with descending thoracic aortic aneurysm without dissection (dTAA, n=10), patients with descending thoracic aortic dissection with aneurysm (dTAD, n=10), and organ donors (descending control, n=8) were examined. Representative western blot images and quantification studies show significant degradation products of myosin heavy chain and tropomyosin in dTAA and dTAD.
NLRP3–caspase-1 Inflammasome is Activated in Human Sporadic Thoracic Tissues
To identify potential pathways responsible for the degradation of contractile proteins in sporadic AAD, we examined the activation of the NLRP3–caspase-1 inflammasome cascade in aortic diseases in the same set of patient tissues described above. We found that protein levels of NLRP3, ASC, procaspase-1, and active caspase-1 (p10 and p20) were increased in ascending (Figure 2A) and descending (Figure 2B) sections of diseased aortas when compared with levels in similar segments of control aortas. Immunostaining analysis further indicated that NLRP3 and caspase-1 protein levels were increased in the media and adventitia of diseased aortas (Figure 2C, particularly in CD68+ macrophages and SM22-α+ SMCs (Figure 2D). Together, these findings indicate that the NLRP3–caspase-1 inflammasome cascade is activated in both SMCs and macrophages, and the activation of the cascade is associated with degradation of myosin heavy chain and tropomyosin in SMCs in the aortic wall of patients with sporadic thoracic AAD.
Figure 2.
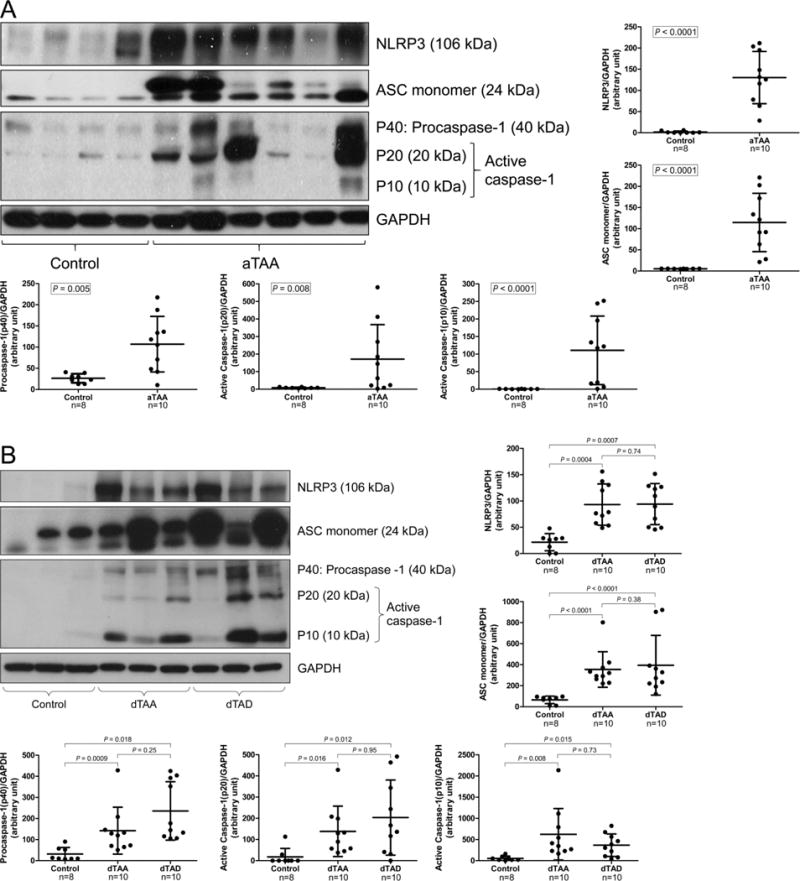
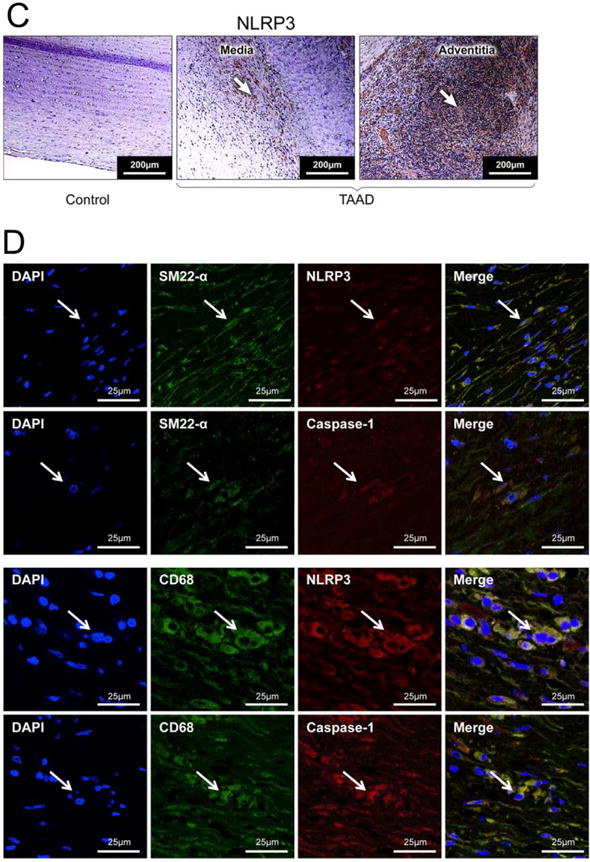
Significant activation of NLRP3–caspase-1 inflammasome in human thoracic aortic aneurysm and dissection (AAD) tissues. A, Representative western blot images and quantification studies show higher levels of NLRP3, ASC, procaspase-1, and active caspase-1 in aortic tissues from patients with ascending thoracic aortic aneurysm without dissection (aTAA, n=10) than in aortic tissues from ascending aorta controls (ascending control, n=8). B, Representative western blot images and quantification studies show higher levels of NLRP3, ASC, procaspase-1, and active caspase-1 in aortic tissues from patients with descending thoracic aortic aneurysm without dissection (dTAA, n=10) and descending thoracic aortic dissection with aneurysm (dTAD, n=10) than in aortic tissues from descending aorta controls (descending control, n=8). C, Representative immunostaining shows abundant NLRP3 in the media and adventitia of TAAD tissue as compared with control tissue. D, Representative immunostaining(dTAA, n=10,dTAD, n=10) shows the expression of NLRP3 and caspase-1 in SM22-α+ smooth muscle cells and CD68+ macrophages in TAAD tissues. The white arrows indicate positive staining.
Caspase-1 Degrades SMC Contractile Proteins Leading to SMC Contractile Dysfunction
We then examined whether the NLRP3–caspase-1 inflammasome cascade plays a role in contractile protein degradation. Palmitic acid (PA) mimics in cells the metabolic stress created by a high-fat diet (HFD) in challenged mice. We therefore exposed human thoracic aortic SMCs to PA (0.3 mM) as a trigger in these studies. PA consistently increased the levels of NLRP3, pro-caspase-1, and active caspase-1 in SMCs (Figure 3A). PA also induced the cleavage and degradation of tropomyosin and myosin heavy chain in SMCs (Figure 3B). The siRNA-mediated reduction of the inflammasome components partially prevented the cleavage/degradation of tropomyosin and myosin heavy chain in PA-challenged SMCs, suggesting the involvement of this cascade in contractile protein degradation. Because the NLRP3–caspase-1 inflammasome cascade executes its activity mainly through the caspase-1–mediated cleavage of target proteins, we determined whether this cascade directly targets and cleaves contractile proteins. Immunoprecipitation studies showed that caspase-1 directly bound to tropomyosin and myosin heavy chain in cultured SMCs, and the binding was induced by PA treatment (Figure 3C). Caspase-1 also bound to tropomyosin and myosin heavy chain in aortic tissues from sporadic TAAD patients (Figure 3D). In addition, recombinant caspase-1 directly cleaved tropomyosin and myosin heavy chain (Figure 3E), and the cleavage was reduced by the caspase-1 inhibitor Z-VAD-FMK. Finally, our SMC contractility assay showed that stimulation with PA significantly suppressed the ability of SMCs to contract in a collagen matrix, an effect that was partially reversed by the sequential siRNA-mediated knockdown of the NLRP3–caspase-1 inflammasome cascade (Figure 3F). Together, our findings suggest that PA induces activation of the NLRP3–caspase-1 inflammasome components in SMCs and that, through caspase-1, the NLRP3–caspase-1 inflammasome cascade degrades contractile proteins, leading to SMC contractile dysfunction.
Figure 3.

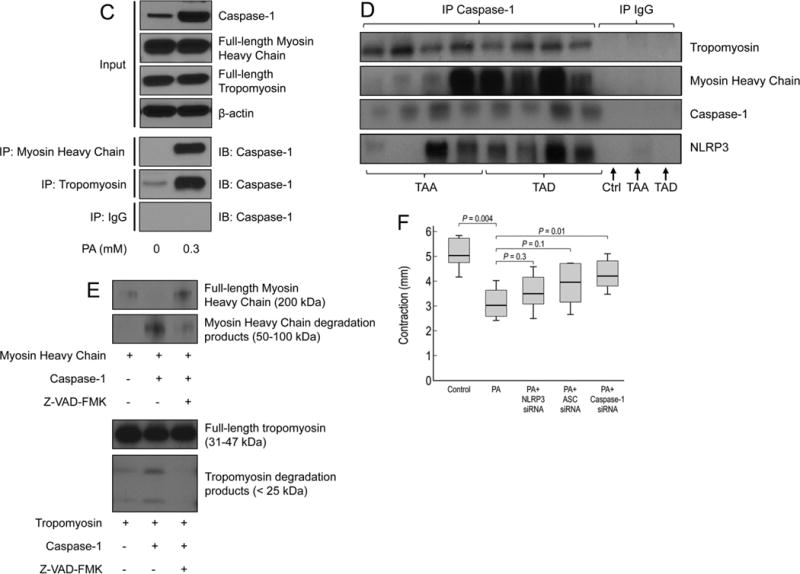
Caspase-1 degrades smooth muscle cell (SMC) contractile proteins leading to SMC contractile dysfunction. A, Representative western blot images and quantification studies show that palmitic acid (PA) increased the expression of NLRP3 and caspase1 in human aortic SMCs (n>3 in independent experiments). B, PA induced the cleavage/degradation of tropomyosin and myosin heavy chain (MHC). The cleavage was reduced by treatment with NLRP3 siRNA, ASC siRNA, or caspase-1 siRNA. C, Co-immunoprecipitation studies show that caspase-1 bound to myosin heavy chain and tropomyosin in SMCs (n>3 in independent experiments) and D, in diseased aortic tissues. E, Cleavage assay results show that recombinant caspase-1 directly cleaved and degraded recombinant tropomyosin and myosin heavy chain. The cleavage was reduced by the caspase-1 inhibitor Z-VAD-FMK (n>3 in independent experiments). F, SMCs were treated and polymerized in collagen matrix; their ability to contract in collagen matrix was examined by determining the reduction in collagen diameter (from 3 independent experiments). PA treatment significantly decreased SMC contraction, and this effect was partially prevented by treatment with NLRP3 siRNA, ASC siRNA, or caspase-1 siRNA.
NLRP3–caspase-1 Inflammasome Cascade Contributes to Aortic Contractile Protein Degradation and Biomechanical Dysfunction in Mice
Using a sporadic AAD mouse model, we examined whether the inflammasome plays a role in aortic biomechanical dysfunction. In this model, challenging C57BL/6 WT mice with a HFD and angiotensin II (AngII) infusion14 induced aortic enlargement (Figure IA in the online-only Data Supplement), AAD formation (Figure IB in the online-only Data Supplement), and aortic rupture (Figure IC in the online-only Data Supplement), which were associated with activation of the NLRP3–caspase-1 inflammasome complex (Figure ID in the online-only Data Supplement).
We then evaluated aortic function. Advanced aortic disease with significant aortic destruction and fibrotic remodeling can result in aortic stiffness and biomechanical dysfunction. To eliminate these effects, we studied the biomechanical function of descending thoracic aortas with mild disease (without coexisting dissection and aneurysm). Aortic diameters were slightly larger in challenged than in unchallenged WT mice; however, the diameters were similar among the challenged groups with different genetic backgrounds. The density of alpha -adrenergic receptors was also similar among the challenged groups (Figure II in the online-only Data Supplement).
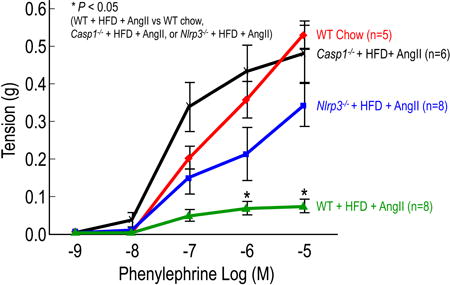
Wire myograph analysis of descending thoracic aortic rings showed that the contractile response to phenylephrine was significantly reduced in challenged WT mice when compared with that in unchallenged mice (Figure 4A). However, the contractile response to phenylephrine was preserved in challenged NLRP3-deficient (Nlrp3−/−) mice and caspase-1–deficient (Casp1−/−) mice, when compared with that of challenged WT mice (Figure 4A). Similarly, pressure myograph analysis of descending thoracic aortic rings showed that the diameter changes per increase in pressure were greater in challenged WT mice than in unchallenged WT mice, indicating loss of elastic fiber integrity and/or aortic contractile ability (Figure 4B). Compared with challenged WT mice, challenged Nlrp3−/− and Casp1−/− mice had reduced pressure-induced diameter enlargement (Figure 4B), suggesting preserved aortic function in these mice.
Figure 4.
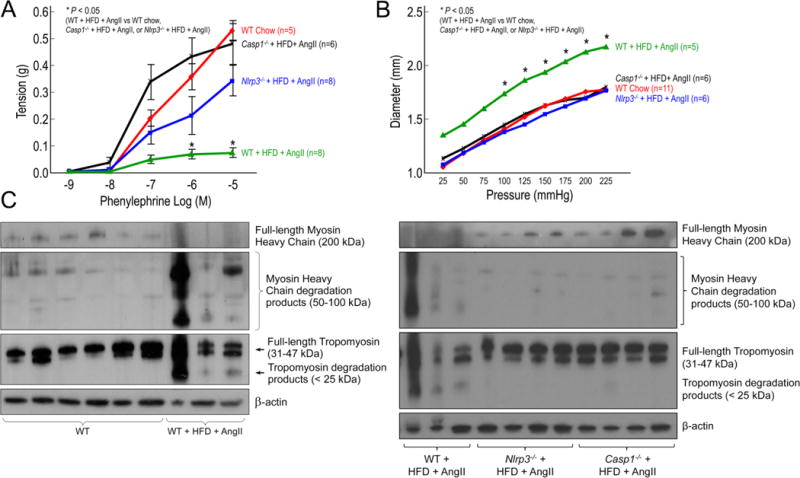
Deficiency in NLRP3–caspase-1 inflammasome reduces challenge-induced aortic contractile protein degradation and biomechanical dysfunction in mice. Wild type (WT), Nlrp3−/−, and Casp1−/− mice were unchallenged or challenged with AngII infusion and a high-fat diet (HFD). A, Wire myograph analysis of descending thoracic aortic rings shows a significantly reduced contractile response to phenylephrine in challenged WT mice when compared with that in unchallenged WT mice (WT chow). In contrast, aortic rings from challenged Nlrp3−/− and Casp1−/− mice exhibit partial preservation of contractile ability. B, Pressure myograph analysis of descending thoracic aortic segments shows greater diameters with increment increases in pressure in challenged WT mice than in unchallenged WT mice (WT chow). In contrast, aortic segments from challenged Nlrp3−/− and Casp1−/− mice exhibit diameters comparable to those in unchallenged WT mice. C, Western blot analysis of aortic protein lysates shows increased degradation of tropomyosin and myosin heavy chain in challenged WT mice when compared with unchallenged WT mice, and preserved full-length tropomyosin and myosin heavy chain in challenged Nlrp3−/− and Casp1−/− mice when compared with challenged WT mice. *P<0.05, WT+HFD+AngII vs. WT chow, Nlrp3−/−+HFD+AngII, or Casp1−/−+HFD+AngII.
We examined whether the inflammasome cascade contributes to contractile protein degradation during AAD development in mice. Compared with unchallenged WT mice, challenged WT mice showed increased degradation of myosin heavy chain and tropomyosin (Figure 4C). Importantly, aortas from challenged Nlrp3−/− and Casp1−/− mice showed reduced degradation of and preserved levels of full-length myosin heavy chain and tropomyosin when compared with aortas from challenged WT mice (Figure 4C), suggesting a critical role of the NLRP3–caspase-1 inflammasome cascade in contractile protein degradation. These findings suggest that the NLRP3–caspase-1 inflammasome cascade is involved in contractile protein degradation, which may contribute to aortic biomechanical failure and AAD formation.
The NLRP3–caspase-1 Inflammasome Cascade Contributes to Aortic Destruction and AAD Formation
We determined the role of the NLRP3–caspase-1 inflammasome cascade in AAD formation in mice challenged with a HFD and AngII infusion. Routine blood pressure monitoring in challenged mice showed significant increases in blood pressure after AngII infusion, but there were no differences in blood pressure between groups (data not shown). Nlrp3−/− mice and Casp1−/− mice showed markedly preserved gross aortic architecture (Figure 5A) and significantly reduced aortic enlargement (Figure 5B) and AAD incidence in various aortic segments (Figure 5C) when compared with challenged WT mice. Moreover, the presence of multiple aneurysms and/or dissection and aortic rupture (Figure 5D) and the overall AAD incidence (Figure 5E) were reduced in challenged Nlrp3−/− and Casp1−/− mice when compared with challenged WT mice. In addition, we observed better preserved elastic fiber architecture, decreased elastic fiber fragmentation, less adventitial remodeling (Figure 5F), and reduced inflammatory cell infiltration (data not shown) in the aortas of challenged Nlrp3−/− and Casp1−/− mice compared with the aortas of challenged WT mice, suggesting the importance of the NLRP3–caspase-1 inflammasome in aortic wall damage and remodeling after aortic stress. Together, these findings suggest that the NLRP3–caspase-1 inflammasome cascade is involved in AAD formation.
Figure 5.
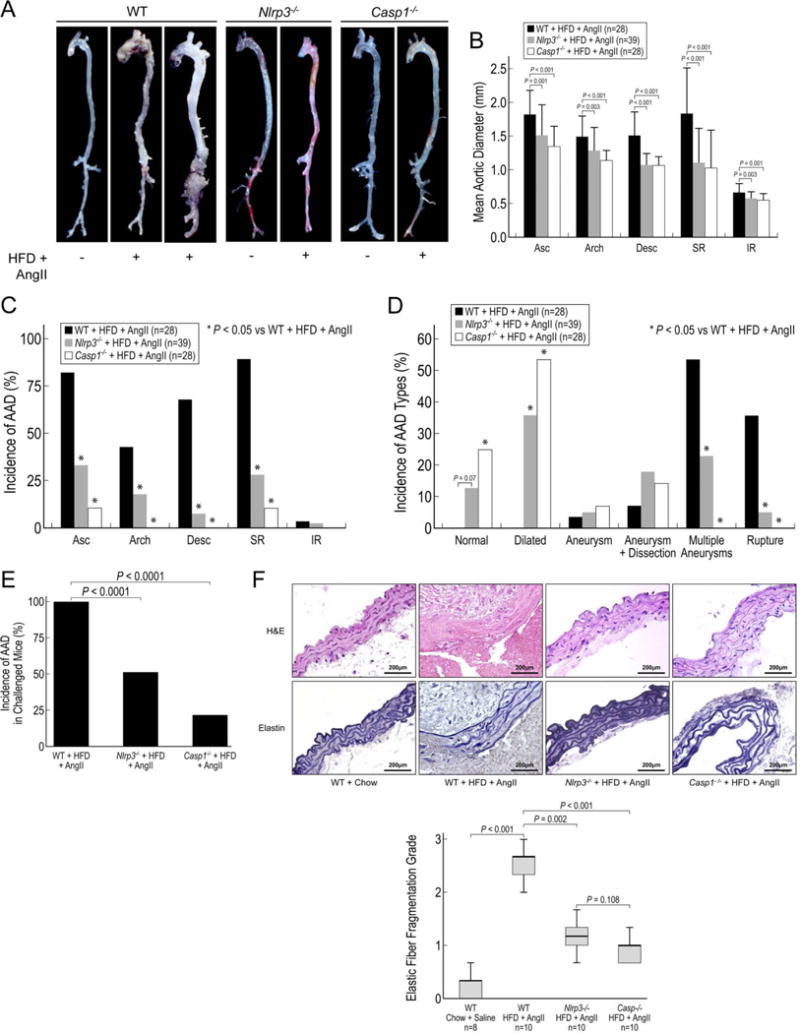
Deficiency in NLRP3–caspase-1 inflammasome reduces aortic aneurysm and dissection (AAD) formation in mice. Wild type (WT), Nlrp3−/−, and Casp1−/− mice were unchallenged or challenged with AngII infusion and a high-fat diet (HFD). A, Representative images of the excised aortas show gross differences among aortas from challenged WT, Nlrp3−/−, and Casp1−/− mice. B, Challenged Nlrp3−/− and Casp1−/− mice have smaller mean aortic diameters in various aortic segments than do challenged WT mice. Asc, ascending; Desc, descending thoracic; SR, suprarenal; and IR, infrarenal. C, The incidence of AAD in different aortic segments is significantly lower in challenged Nlrp3−/− and Casp1−/− mice than in challenged WT mice. D, The severity of types of aortic lesions is reduced in challenged Nlrp3−/− and Casp1−/− mice when compared with that of challenged WT mice. Lesions were classified according to a modified Daugherty system. E, The overall incidence of AAD is reduced in challenged Nlrp3−/− and Casp1−/− mice when compared with that in challenged WT mice. F, Verhoeff–van Gieson elastin staining and hematoxylin and eosin (H&E) staining show the preservation of elastic lamellar architecture and the partial preservation of medial thickness in the aortas of challenged Nlrp3−/− and Casp1−/− mice when compared with aortas of challenged WT mice.
Pharmacologic Blockade of the NLRP3–caspase-1 Inflammasome Cascade Attenuates AAD Formation in Mice
Finally, we tested whether AAD formation in mice can be reduced by the pharmacologic antagonism of the NLRP3–caspase-1 inflammasome cascade. Glyburide is a commonly used anti-diabetic medication that has been shown to stop potassium efflux,15 thereby inhibiting NLRP3–caspase-1 inflammasome complex formation. Glucose levels were measured weekly in challenged WT mice and challenged WT mice treated with glyburide; we found no differences in glucose levels between groups (data not shown). In challenged WT mice, we found that glyburide treatment significantly reduced aortic destruction (Figure 6A), aortic enlargement (Figure 6B), and AAD development in different aortic segments (Figure 6C), as well as the overall incidence (Figure 6D) and the incidence of different severity types (Figure 6E) of aortic disease. Additionally, in the aortas of challenged WT mice treated with glyburide, we observed better preserved elastic fiber architecture and less adventitial remodeling than in the aortas of untreated challenged WT mice (Figure 6F). These findings suggest that glyburide may downregulate the expression and activation of the NLRP3–caspase-1 inflammasome and may have a therapeutic effect on the formation and progression of aortic disease.
Figure 6.
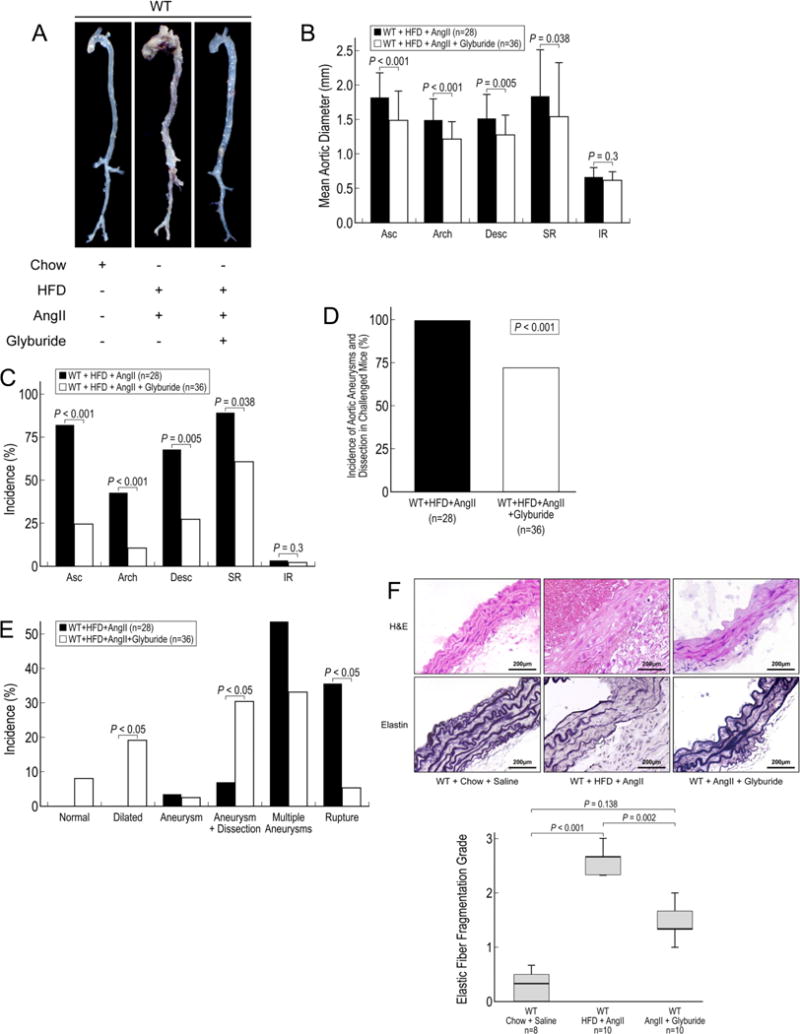
Inflammasome inhibitor glyburide reduces aortic aneurysm and dissection (AAD) development in mice. Wild type (WT) mice that were challenged with AngII and a high-fat diet (HFD) were administered water or glyburide (5 mg/kg/d) by oral gavage for 7 weeks. A, Excised aortas show that glyburide attenuates AAD formation in the thoracic and abdominal aorta. B, Aortic diameters in challenged WT mice are reduced with glyburide treatment. Asc, ascending; Desc, descending thoracic; SR, suprarenal; and IR, infrarenal. C, AAD formation in different aortic segments in challenged WT mice is reduced with glyburide treatment. D, The overall incidence of AAD in challenged WT mice is reduced with glyburide treatment. E, The severity of the types of lesions, classified according to a modified Daugherty system, is shown. F, Hematoxylin and eosin (H&E) staining and Verhoeff–van Gieson elastin staining in the aortas of challenged WT mice show preservation of elastic lamellar architecture in mice treated with glyburide.
Discussion
Recent evidence has suggested that SMC contractile dysfunction2,3,5,6 plays a critical role in AAD development. Mutations in contractile proteins have been identified as the cause of SMC contractile dysfunction in certain genetic aortic disorders. However, the mechanisms for SMC contractile dysfunction in sporadic aortic disease are largely unknown. In this study, we provide evidence suggesting that degradation of SMC contractile proteins, mediated by the NLRP3–caspase-1 inflammasome, may contribute to SMC contractile dysfunction and aortic biomechanical failure during sporadic AAD development.
Altered aortic biomechanics have been described during AAD development. Aortic dysfunction could be secondary to aortic destruction. Interestingly, recent studies have suggested that SMC contractile dysfunction can cause AAD development.2,3,5,6 Studies in genetic TAAD have identified mutations in the genes for smooth muscle alpha-actin (ACTA2),3,5,6 myosin heavy chain 11 (MYH11),2 myosin light chain kinase (MYLK), and cGMP-dependent protein kinase 1 (PRKG1). It has been suggested that dysfunctional mechanical sensing and regulation by intramural cells leads to compromised structural integrity of the aortic wall.16 Although genomic copy number variants that disrupt the cell contractile function have been identified in sporadic TAAD,6 the mechanisms for contractile dysfunction in sporadic AAD are generally unknown. In this study, we found significant contractile protein degradation in aortic tissues from patients with sporadic thoracic aortic aneurysms and dissections. Moreover, in a sporadic AAD model, contractile protein degradation and aortic dysfunction were also observed, even in aortas without dissection and aneurysm. These findings suggest that contractile protein degradation and aortic contractile dysfunction may be an early event, leading to compromised aortic structure and progressive aortic degeneration, and ultimately to AAD development.
What causes aortic contractile protein degradation and dysfunction in sporadic AAD? Our study showed significant activation of the NLRP3–caspase-1 inflammasome cascade in aortic tissues from patients with sporadic thoracic AAD, specifically in aortic medial SMCs of diseased tissues. Interestingly, this cascade was directly involved in SMC contractile protein degradation and SMC contractile dysfunction. We found that caspase-1 bound to tropomyosin and myosin heavy chain in cultured SMCs and in aortic tissues from AAD patients, and caspase-1 directly cleaved these contractile proteins. Knocking down the NLRP3 and caspase-1 in cultured SMCs prevented contractile protein degradation and contractile dysfunction. In an AAD mouse model, we found significant contractile protein degradation and aortic dysfunction in aortas from challenged WT mice. Importantly, NLRP3 and caspase-1 deficiency partially preserved aortic contractile protein integrity and aortic biomechanical functions. Together, our findings suggest that the NLRP3–caspase-1 inflammasome cascade degrades contractile proteins through the actions of caspase-1, leading to SMC and aortic contractile dysfunction during AAD development. Contractile protein degradation has been implicated in cardiac dysfunction during ischemia-reperfusion injury.17 The NLRP3–caspase-1 inflammasome cascade may also contribute to contractile protein degradation and contractile dysfunction in this condition.
We further examined the role of the NLRP3–caspase-1 inflammasome cascade in AAD formation. In our AAD model, mice were fed a HFD and challenged with a high dose of AngII to generate sporadic thoracic and abdominal AAD. AngII has been used extensively in mice to induce abdominal18–20 and thoracic14,21,22 aortic aneurysms, although Ang II-induced AAD development could be independent of increased blood pressure.23,24 Moreover, mice fed a HFD show significantly increased susceptibility to AngII-induced AAA.14,18 In this study, using an approach of combined HFD and high-dose AngII, we successfully generated AAD in the thoracic aorta of mice that resulted in disease development in the ascending, arch, and descending thoracic aorta. In this sporadic AAD model, we showed that NLRP3 and caspase-1 deficiency reduced not only the overall AAD incidence, but also the disease severity and rupture rate. Our findings are consistent with those of recent studies showing that the NLRP3–caspase-1 inflammasome was activated in AAA tissues13,25 and that NLRP3 and caspase-1 deficiency in ApoE−/− mice reduced AAA formation. These studies examined the role of the inflammasome in macrophage activation and aortic inflammation in AAA development.26,27 Here, we proposed a novel role of the inflammasome in SMC contractile protein degradation, aortic contractile dysfunction, and biomechanical failure. We also showed that NLRP3 and caspase-1 deficiency reduced aortic enlargement and AAD development in both the abdominal and thoracic aortic regions and also explored the mechanism of this protection. Together, these findings suggest that the NLRP3–caspase-1 inflammasome cascade contributes to sporadic AAD development. It will be important to examine whether this cascade is activated and plays a role in disease progression in genetically triggered TAA and TAD.
Finally, we assessed whether AAD formation can be reduced by pharmacologic antagonism of the NLRP3–caspase-1 inflammasome cascade. In this study, we used glyburide to treat AAD because it has been shown to inhibit the inflammasome and limit ischemia-reperfusion–induced myocardial injury in mice.28 We found that glyburide treatment in challenged WT mice reduced NLRP3 levels, ASC complex formation, caspase-1 levels and its cleavage/activation, and aortic destruction. Ultimately, glyburide treatment significantly reduced aortic destruction, aortic enlargement, and AAD development in different aortic segments, as well as the overall incidence and severity of aortic disease. Our findings establish that targeting the NLRP3–caspase-1 inflammasome may be a potential therapeutic approach for patients with AAD. However, glyburide is an antidiabetic drug with potential effects, including hypoglycemia; therefore, further studies with more specific NLRP3 inhibitors are warranted.
Diabetes has been consistently shown to be negatively associated with both thoracic29 and abdominal30,31 aortic aneurysm; however, the underlying mechanisms are unclear. Although the abnormal metabolic profile in patients with diabetes (i.e., hyperglycemia and hyperinsulinemia) may have a “protective effect” on the aortic wall, increasing evidence suggests that the protection may come from the medications used to treat diabetes.32–34 In a recent study, the use of antidiabetic medication was analyzed in 4468 diabetes patients with aortic diseases and 4468 matched controls (diabetes patients without aortic diseases). The study shows that the use of metformin or sulfonylurea was associated with a lower risk of developing aortic diseases. The effects of metformin and sulfonylurea on aortic diseases were dose dependent.32 Our study supports the notion that antidiabetic medication may have protective effects against AAD development. Nevertheless, further studies are needed to understand the molecular mechanisms underlying the negative association between diabetes and aortic aneurysm and to define the effects of anti-diabetic medications, insulin signaling/hyperinsulinemia, and hyperglycemia on the structure and function of the aortic wall.
Moreover, it is important to consider the dynamic nature of aortic disease formation in relation to the blockade of the NLRP3–caspase-1 inflammasome as a preventive therapy for aortic disease. Thus, studies are also needed to determine whether the blockade of the NLRP3–caspase-1 inflammasome cascade plays a role in the treatment of acute or chronic aortic disease and whether this blockade inhibits adverse remodeling or promotes protective remodeling.
In conclusion, our findings suggest NLRP3–caspase-1 inflammasome may disrupt aortic wall homeostasis, which promoted SMC contractile protein degradation and dysfunction. We also showed that glyburide reduced the formation of aortic disease in mice, probably in part, by inhibiting NLRP3–caspase-1 inflammasome activation. These findings have important clinical implications and suggest that the blockade of the NLRP3–caspase-1 inflammasome may have therapeutic potential for preventing or treating aortic disease.
Supplementary Material
Table 1.
Patient Characteristics
| Characteristics | Ascending Thoracic Aortic Disease | Descending Thoracic Aortic Disease | |||
|---|---|---|---|---|---|
| Control (n=8) |
aTAA (n=10) |
Control (n=8) |
dTAA (n=10) |
dTAD (n=10) |
|
| Age (y) | 66.6±7.9 | 65.6±6.9 | 61.6±8.2 | 66.7±6.7 | 61.9±4.3 |
| Male | 4 (50%) | 4 (40%) | 3 (38%) | 3 (30%) | 7 (70%) |
| Hypertension | 5 (63 %) | 7 (70%) | 5 (63%) | 10 (100%) | 10 (100%) |
| COPD | 3 (38 %) | 2 (20%) | 1 (13%) | 3 (30%) | 3 (30%) |
| Diabetes mellitus | 0 | 1 (10%) | 1 (13%) | 1 (10%) | 0 |
| History of smoking | 6 (75%) | 5 (50%) | 2 (25%) | 10 (100%) | 6 (60%) |
| Use of anti-lipid medication | 2 (25%) | 5 (50%) | 2 (25%) | 4 (40%) | 4 (40%) |
| Use of COX inhibitor | 4 (50%) | 3 (30%) | 1 (13%) | 3 (30%) | 5 (50%) |
| Aortic diameter (cm) | NA | 5.9±2.3 | NA | 5.6±0.5 | 6.1±1.6 |
Data are expressed as a number (percent) or as the mean ± standard deviation. aTAA, ascending thoracic aortic tissue from patients with ascending thoracic aortic aneurysm; COPD, chronic obstructive pulmonary disease; COX, cyclooxygenase; dTAA, descending thoracic aortic tissue from patients with descending thoracic aortic aneurysm; NA, not available; and dTAD, descending thoracic aortic tissue from patients with descending thoracic aortic dissection.
Highlights.
Significant SMC contractile protein degradation is seen in human sporadic thoracic AAD.
The inflammasome NLRP3- caspase-1 cascade directly degrades contractile proteins, leading to contractile dysfunction in SMCs.
Nlrp3 or caspase-1 deficiency in mice prevents challenge–induced contractile protein degradation, biomechanical dysfunction, and AAD formation in both the thoracic and abdominal aorta.
Treatment with the inflammasome inhibitor, glyburide, reduces challenge–induced AAD formation.
Acknowledgments
We gratefully acknowledge Michael Hughes, BS, for assistance in laboratory work; Scott A. Weldon, MA, CMI, for creating illustrations; and Scientific Publications at the Texas Heart Institute for editorial support.
Funding Sources
This study was supported by NIH grants R01 HL085341, R56/R01 HL127111 and R01HL131980 and St. Luke’s Episcopal Hospital Roderick Duncan MacDonald Research Fund Award 13RDM006. The Thoracic Aortic Disease Tissue Bank at Baylor College of Medicine was supported in part through the Tissue Banking Core of the Specialized Center of Clinically Oriented Research in Thoracic Aortic Aneurysms and Dissections (NIH P50 HL083794). Darrell Wu was supported by a training grant (NIH T32 HL007676) through the Department of Molecular Physiology and Biophysics at Baylor College of Medicine.
Nonstandard Abbreviations and Acronyms
- AAA
abdominal aortic aneurysm
- AAD
aortic aneurysms and dissections
- aTAA
ascending thoracic aortic aneurysm
- BSA
bovine serum albumin
- dTAA
descending thoracic aortic aneurysm
- dTAD
descending thoracic aortic dissection
- ECM
extracellular matrix
- FBS
fetal bovine serum
- HFD
high-fat diet
- IR
infrarenal
- OCT
optimal cutting temperature
- PA
palmitic acid
- PBS
phosphate-buffered saline
- SMC
smooth muscle cell
- SR
suprarenal
- WT
wild type
Footnotes
Journal Subject Codes: aneurysm, aortic dissection, vascular disease, basic science research
Disclosures
None.
References
- 1.Minino AM, Murphy SL, Xu J, Kochanek KD. Deaths: final data for 2008. Natl Vital Stat Rep. 2011;59:1–126. [PubMed] [Google Scholar]
- 2.Zhu L, Vranckx R, Khau Van Kien P, Lalande A, Boisset N, Mathieu F, Wegman M, Glancy L, Gasc JM, Brunotte F, Bruneval P, Wolf JE, Michel JB, Jeunemaitre X. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet. 2006;38:343–349. doi: 10.1038/ng1721. [DOI] [PubMed] [Google Scholar]
- 3.Guo DC, Pannu H, Tran-Fadulu V, et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39:1488–1493. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- 4.Milewicz DM, Guo DC, Tran-Fadulu V, Lafont AL, Papke CL, Inamoto S, Kwartler CS, Pannu H. Genetic basis of thoracic aortic aneurysms and dissections: focus on smooth muscle cell contractile dysfunction. Annu Rev Genomics Hum Genet. 2008;9:283–302. doi: 10.1146/annurev.genom.8.080706.092303. [DOI] [PubMed] [Google Scholar]
- 5.Milewicz DM, Ostergaard JR, Ala-Kokko LM, et al. De novo ACTA2 mutation causes a novel syndrome of multisystemic smooth muscle dysfunction. Am J Med Genet A. 2010;152A:2437–2443. doi: 10.1002/ajmg.a.33657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prakash SK, LeMaire SA, Guo DC, Russell L, Regalado ES, Golabbakhsh H, Johnson RJ, Safi HJ, Estrera AL, Coselli JS, Bray MS, Leal SM, Milewicz DM, Belmont JW. Rare copy number variants disrupt genes regulating vascular smooth muscle cell adhesion and contractility in sporadic thoracic aortic aneurysms and dissections. Am J Hum Genet. 2010;87:743–756. doi: 10.1016/j.ajhg.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coady MA, Rizzo JA, Goldstein LJ, Elefteriades JA. Natural history, pathogenesis, and etiology of thoracic aortic aneurysms and dissections. Cardiol Clin. 1999;17:615–635. vii. doi: 10.1016/s0733-8651(05)70105-3. [DOI] [PubMed] [Google Scholar]
- 8.Mariathasan S, Monack DM. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol. 2007;7:31–40. doi: 10.1038/nri1997. [DOI] [PubMed] [Google Scholar]
- 9.Zhong Z, Zhai Y, Liang S, Mori Y, Han R, Sutterwala FS, Qiao L. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat Commun. 2013;4:1611. doi: 10.1038/ncomms2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 12.Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010 doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roberts RL, Van Rij AM, Phillips LV, Young S, McCormick SP, Merriman TR, Jones GT. Interaction of the inflammasome genes CARD8 and NLRP3 in abdominal aortic aneurysms. Atherosclerosis. 2011;218:123–126. doi: 10.1016/j.atherosclerosis.2011.04.043. [DOI] [PubMed] [Google Scholar]
- 14.Branchetti E, Poggio P, Sainger R, Shang E, Grau JB, Jackson BM, Lai EK, Parmacek MS, Gorman RC, Gorman JH, Bavaria JE, Ferrari G. Oxidative stress modulates vascular smooth muscle cell phenotype via CTGF in thoracic aortic aneurysm. Cardiovasc Res. 2013;100:316–324. doi: 10.1093/cvr/cvt205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lamkanfi M, Mueller JL, Vitari AC, Misaghi S, Fedorova A, Deshayes K, Lee WP, Hoffman HM, Dixit VM. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol. 2009;187:61–70. doi: 10.1083/jcb.200903124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Humphrey JD, Schwartz MA, Tellides G, Milewicz DM. Role of mechanotransduction in vascular biology: focus on thoracic aortic aneurysms and dissections. Circ Res. 2015;116:1448–1461. doi: 10.1161/CIRCRESAHA.114.304936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sawicki G, Leon H, Sawicka J, Sariahmetoglu M, Schulze CJ, Scott PG, Szczesna-Cordary D, Schulz R. Degradation of myosin light chain in isolated rat hearts subjected to ischemia-reperfusion injury: a new intracellular target for matrix metalloproteinase-2. Circulation. 2005;112:544–552. doi: 10.1161/CIRCULATIONAHA.104.531616. [DOI] [PubMed] [Google Scholar]
- 18.Police SB, Thatcher SE, Charnigo R, Daugherty A, Cassis LA. Obesity promotes inflammation in periaortic adipose tissue and angiotensin II-induced abdominal aortic aneurysm formation. Arterioscler Thromb Vasc Biol. 2009;29:1458–1464. doi: 10.1161/ATVBAHA.109.192658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salmon M, Johnston WF, Woo A, Pope NH, Su G, Upchurch GR, Jr, Owens GK, Ailawadi G. KLF4 regulates abdominal aortic aneurysm morphology and deletion attenuates aneurysm formation. Circulation. 2013;128:S163–174. doi: 10.1161/CIRCULATIONAHA.112.000238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang S, Zhang C, Zhang M, Liang B, Zhu H, Lee J, Viollet B, Xia L, Zhang Y, Zou MH. Activation of AMP-activated protein kinase alpha2 by nicotine instigates formation of abdominal aortic aneurysms in mice in vivo. Nat Med. 2012;18:902–910. doi: 10.1038/nm.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis FM, Rateri DL, Balakrishnan A, Howatt DA, Strickland DK, Muratoglu SC, Haggerty CM, Fornwalt BK, Cassis LA, Daugherty A. Smooth muscle cell deletion of low-density lipoprotein receptor-related protein 1 augments angiotensin II-induced superior mesenteric arterial and ascending aortic aneurysms. Arterioscler Thromb Vasc Biol. 2015;35:155–162. doi: 10.1161/ATVBAHA.114.304683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tieu BC, Lee C, Sun H, Lejeune W, Recinos A, 3rd, Ju X, Spratt H, Guo DC, Milewicz D, Tilton RG, Brasier AR. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J Clin Invest. 2009;119:3637–3651. doi: 10.1172/JCI38308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cassis LA, Gupte M, Thayer S, Zhang X, Charnigo R, Howatt DA, Rateri DL, Daugherty A. ANG II infusion promotes abdominal aortic aneurysms independent of increased blood pressure in hypercholesterolemic mice. Am J Physiol Heart Circ Physiol. 2009;296:H1660–1665. doi: 10.1152/ajpheart.00028.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rateri DL, Davis FM, Balakrishnan A, Howatt DA, Moorleghen JJ, O’Connor WN, Charnigo R, Cassis LA, Daugherty A. Angiotensin II induces region-specific medial disruption during evolution of ascending aortic aneurysms. Am J Pathol. 2014;184:2586–2595. doi: 10.1016/j.ajpath.2014.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dihlmann S, Erhart P, Mehrabi A, Nickkholgh A, Lasitschka F, Bockler D, Hakimi M. Increased expression and activation of absent in melanoma 2 inflammasome components in lymphocytic infiltrates of abdominal aortic aneurysms. Mol Med. 2014;20:230–237. doi: 10.2119/molmed.2013.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun W, Pang Y, Liu Z, Sun L, Liu B, Xu M, Dong Y, Feng J, Jiang C, Kong W, Wang X. Macrophage inflammasome mediates hyperhomocysteinemia-aggravated abdominal aortic aneurysm. J Mol Cell Cardiol. 2015;81C:96–106. doi: 10.1016/j.yjmcc.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 27.Usui F, Shirasuna K, Kimura H, Tatsumi K, Kawashima A, Karasawa T, Yoshimura K, Aoki H, Tsutsui H, Noda T, Sagara J, Taniguchi S, Takahashi M. Inflammasome activation by mitochondrial oxidative stress in macrophages leads to the development of angiotensin II-induced aortic aneurysm. Arterioscler Thromb Vasc Biol. 2015;35:127–136. doi: 10.1161/ATVBAHA.114.303763. [DOI] [PubMed] [Google Scholar]
- 28.Marchetti C, Chojnacki J, Toldo S, Mezzaroma E, Tranchida N, Rose SW, Federici M, Van Tassell BW, Zhang S, Abbate A. A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury after ischemia-reperfusion in the mouse. J Cardiovasc Pharmacol. 2014;63:316–322. doi: 10.1097/FJC.0000000000000053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prakash SK, Pedroza C, Khalil YA, Milewicz DM. Diabetes and reduced risk for thoracic aortic aneurysms and dissections: a nationwide case-control study. J Am Heart Assoc. 2012;1 doi: 10.1161/JAHA.111.000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kang SS, Littooy FN, Gupta SR, Johnson GR, Fisher SG, Cote WL, Steffen GF, Mansour MA, Labropoulos N, Maggio JC. Higher prevalence of abdominal aortic aneurysms in patients with carotid stenosis but without diabetes. Surgery. 1999;126:687–691. discussion 691–682. [PubMed] [Google Scholar]
- 31.Shantikumar S, Ajjan R, Porter KE, Scott DJ. Diabetes and the abdominal aortic aneurysm. Eur J Vasc Endovasc Surg. 2010;39:200–207. doi: 10.1016/j.ejvs.2009.10.014. [DOI] [PubMed] [Google Scholar]
- 32.Hsu CY, Su YW, Chen YT, Tsai SH, Chang CC, Li SY, Huang PH, Chen JW, Lin SJ. Association between use of oral-antidiabetic drugs and the risk of aortic aneurysm: a nested case-control analysis. Cardiovasc Diabetol. 2016;15:125. doi: 10.1186/s12933-016-0447-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fujimura N, Xiong J, Kettler EB, Xuan H, Glover KJ, Mell MW, Xu B, Dalman RL. Metformin treatment status and abdominal aortic aneurysm disease progression. J Vasc Surg. 2016;64:46–54 e48. doi: 10.1016/j.jvs.2016.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vasamsetti SB, Karnewar S, Kanugula AK, Thatipalli AR, Kumar JM, Kotamraju S. Metformin inhibits monocyte-to-macrophage differentiation via AMPK-mediated inhibition of STAT3 activation: potential role in atherosclerosis. Diabetes. 2015;64:2028–2041. doi: 10.2337/db14-1225. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.