

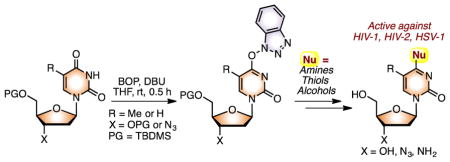
Abstract
Reactions of O-t-butyldimethylsilyl-protected thymidine, 2′-deoxyuridine, and 3′-azidothymidine (AZT) with (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP) leads to activation of the C4 amide carbonyl by formation of putative O4-(benzotriazolyl) derivatives. Subsequent substitution with alkyl and aryl amines, thiols, and alcohols leads to facile functionalization at this position. Reactions with amines and thiols were conducted either as a two-step, one-pot transformation, or as a one-step conversion. Reactions with alcohols were conducted as two-step, one-pot transformations. In the course of these investigations, the formation of 1-(4-pyrimidinyl)-1H-benzotriazole-3-oxide derivatives from the pyrimidine nucleosides was identified. However, these too underwent conversion to the desired products. Products obtained from AZT were converted to the 3′-amino derivatives by catalytic reduction. All products were assayed for their abilities to inhibit cancer cell proliferation and for antiviral activities. Many were seen to be active against HIV-1 and HIV-2, and one was active against herpes simplex virus-1 (HSV-1).
Graphical Abstract
Introduction
Nucleosides are central to the control and treatment of viral diseases, and with the emergence of new viral diseases and development of drug resistance, easy access to novel structural motifs is critical. Within this context, the classical method for modifying the C4 position of pyrimidine nucleosides is via introduction of a leaving group at this position, followed by a displacement with nucleophiles. Typically, electrophilic nucleoside derivatives used for this purpose contain a C4 thione, methylthio, chloro, 1,2,4-triazol-1-yl, 1,2,3,4-tetrazol-1-yl, arylsulfonyl, or N-methylpyrrolidinyl functionality (Figure 1), all generated from the amido group in the nucleobase.
Fig. 1.
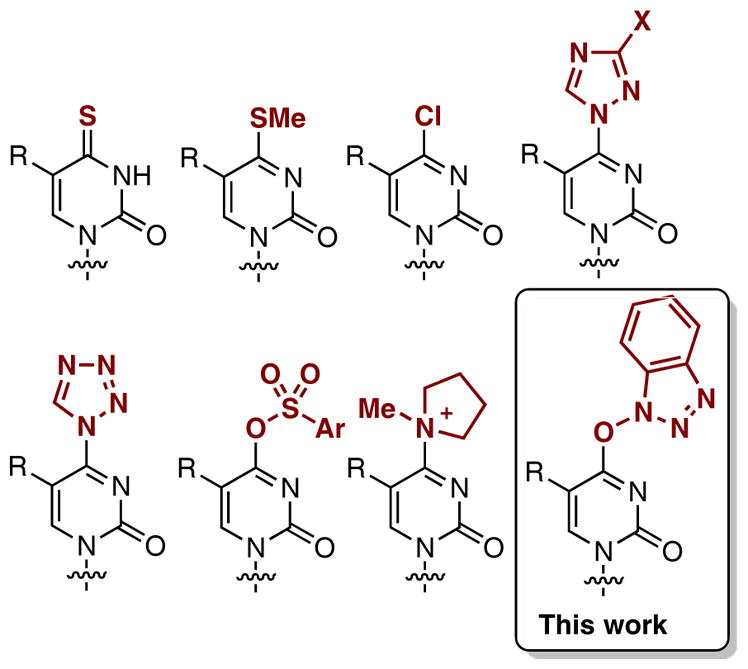
Partial structures of various electrophilic pyrimidine nucleosides developed for C4 functionalization.
Thiones, which can be synthesized by reaction of the amide linkage with P2S51,2 or Lawesson’s reagent,3 have been used for direct displacement reactions.1–3 However, such reactions usually require harsh conditions. Alternatively, the thiones can be S-methylated and subsequently reacted with nucleophiles.4–6 Thiones can also be reacted with dimethyldioxirane and nucleophile, usually an amine or alcohol, leading to modification at the C4.7,8 The C4 chloro derivatives can be prepared by one of two methods, either chlorination with SOCl2/DMF,9–12 or via the Appel reaction with PPh3/CCl4.13–15 However, such chloro compounds seem labile16 and have been used immediately upon preparation. For instance, the C4 chloro derivatives were converted to the C4 ethoxy compounds, which were then used in displacement reactions with nucleophiles.10,12 C4 triazoles17–21 and tetrazoles22–26 are common, stable reactive pyrimidine derivatives that have seen wide applications for nucleoside and nucleic acid modifications. A range of aryl sulfonates (Ar = p-toluenesulfonyl, 2-mesitylenesulfonyl, 2,4,6-triisopropylbenzenesulfonyl),27–32 have also been explored as reactive reagents, not only for direct substitution reactions, but also for cross-coupling chemistry.16,33 Among these, the 2,4,6-triisopropylbenzenesulfonyl derivatives have been shown to be reasonably stable but the 2-mesitylenesulfonyl analogues are not quite as stable.27 By contrast, a p-toluenesulfonyl pyrimidine nucleoside was used directly after preparation.32 Pyrimidine C4 tosylates have also been converted to N-methylpiperidinyl and N-methylpyrrolidinyl salts, as reactive nucleoside analogues.34
We35–41 and others42 have evaluated the use of (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP) for the activation of amide linkages of purine nucleosides, and this approach has now seen general applicability for nucleoside modification.43–47 In our studies, we were the first to report O6-(benzotriazol-1-yl)purine nucleosides as stable, isolable, but reactive nucleoside derivatives.35–37,39–41 In this work, we have evaluated the utility of BOP for activation of amide linkages in pyrimidine nucleosides (Figure 1), as a facile approach to modification at the C4 position.
Results and Discussion
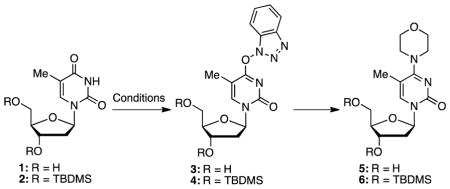
Our studies commenced with the evaluation of conditions for the activation of unprotected thymidine (1) using 2 equiv. each of BOP and a base. Although the reaction with DBU (entry 1 in Table 1) as base proceeded to completion in THF at room temperature, chromatographic purification of the O4-(benzotriazol-1-yl) product 3 was difficult due to its instability towards silica gel chromatography. Next, a one-pot reaction was considered (entry 2) with 4 equiv. of morpholine as a representative nucleophile. This reaction also proceeded but purification of product 5 was difficult due to its polarity and co-elution of impurities.
Table 1.
Activation of the amide linkage in thymidine with BOP and base.a
| ||||
|---|---|---|---|---|
| Entry | Substrate | Conditions | Conversionb | Product: yieldc |
| 1 | 1 | THF, BOP, DBU, 3 h | 100% | 3: decompd |
| 2e | 1 | THF, BOP, DBU, 3 h, then morpholine, 12 h | 100% | 5: ca. 58%f |
| 3 | 2 | THF, BOP, DBU, 0.5 h | 100% | 4: decompd |
|
| ||||
| 4g | 2 | THF, BOP, DBU, 0.5 h, then morpholine, 2 h | 100% | 6: 76% |
|
| ||||
| 5h | 2 | THF, BOP, Cs2CO3, 24 h | ca. 50% | 4: inci |
| 6 | 2 | DME, BOP, DBU, 2 h | ca. 50% | 4: inci |
|
| ||||
| 7h | 2 | MeCN, BOP, Cs2CO3, 0.5 h, then morpholine, 2 h | 100% | 6: 94% |
| 8g | 2 | MeCN, BOP, DBU, 0.5 h, then morpholine, 2 h | 100% | 6: 73% |
| 9j | 2 | THF, BOP, DBU, morpholine, 20 min | 100% | 6: 90% |
|
| ||||
| 10h,j | 2 | MeCN, BOP, Cs2CO3, morpholine, 24 h | ca. 10% | 6: inci |
Reactions were conducted at room temperature with 0.424 M solutions of 1 or 2 in the anhydrous reaction solvent (except when noted otherwise), BOP (2 equiv.), and base (2 equiv.).
As assessed by TLC.
Where reported, yield is of isolated and purified product.
Product was not isolated due to decomposition over silica.
Morpholine (4 equiv.) was added to the reaction mixture after complete conversion of 1 to 3.
Product isolation was difficult as it co-elutes with impurities.
Morpholine (4 equiv.) was added to the reaction mixture, after complete conversion of 2 to 4.
Reaction was conducted with a 0.132 M solution of 2.
Product was not isolated due to incomplete reaction.
After stirring a mixture of 2, BOP (2 equiv.), and base (2 equiv.) in the appropriate solvent for 5 min, morpholine (4 equiv.) was added to the reaction mixture.
This made us consider protected derivative 2. Here, the reaction with BOP proceeded rapidly but isolation of intermediate 4 was not readily possible due to degradation upon chromatography (entry 3). Interestingly, the silyl groups did not offer any stabilization against decomposition, which contrasts with the stabilizing influence of the silyl groups on 8-fluoro-2′-deoxyadenosine.48 Next, formation of the O4-(benzotriazol-1-yl) intermediate 4 was first allowed to proceed to completion (0.5 h), followed by addition of morpholine (entry 4). After an additional 2 h reaction time, a good 76% yield of product 6 was obtained. 1,2-Dimethoxyethane (DME) proved inferior (entry 6) and formation of intermediate 4 was incomplete. Then, in a modification of entry 4, MeCN and Cs2CO3 were used (entry 7), resulting in an excellent 94% yield of product 6. By replacing Cs2CO3 with DBU, a 73% yield of 6 was obtained, comparable to that obtained in THF (compare entries 4 and 8). Interestingly, an excellent 90% yield of 6 was obtained in a very short 20 min reaction time when the entire process was conducted as one-step transformation, with all reactive components present (entry 9). However, this reaction appears sensitive to base and solvent because the MeCN and Cs2CO3 combination resulted in very little conversion even after 24 h (entry 10).
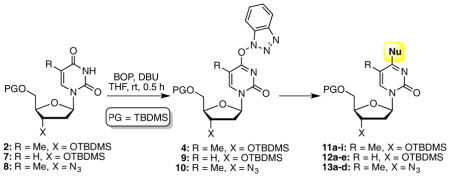
On the basis of the optimizations conducted in Table 1, further generality assessments were conducted. TBDMS-protected thymidine, 2′-deoxyuridine, and 3′-azido-2′-deoxythymidine (AZT), 2, 7, and 8, respectively, were selected. Two approaches were undertaken with amines and thiols. A two-step, one-pot method that involved preformation of the O4-(benzotriazol-1-yl) intermediate, at room temperature (0.5 h reaction). This was followed by addition of the nucleophile and the reaction was conducted until completion. In a second, one-step method, all reactants were added and the reaction was allowed to attain completion. The following conditions were used for the various nucleophiles. Other details are presented under Table 2. (a) For all reactions of alkyl amines, amide-activation was conducted with 2 equiv. each of BOP and DBU, and 4 equiv. of the amine, in THF. (b) With p-toluidine, for the two-step, one pot reaction, formation of the O4-(benzotriazol-1-yl)pyrimidine derivative was accomplished with 2 equiv. each of BOP and DBU in THF, at room temperature. The mixture was evaporated and then the second step was conducted in EtOH (2 mL) with 2 equiv. each of the aryl amine and iPr2NEt. For the one-step method, 2 equiv. of BOP, 4 equiv. of DBU, and 2 equiv. of the aryl amine were used, in MeCN. (c) With EtSH and PhCH2SH, for the two-step one-pot reaction, after amide activation the solvent was evaporated, as with p-toluidine. The second step was conducted with 2 equiv. each of Cs2CO3 and the thiol in DME (2 mL). The one step reaction was conducted with 2 equiv. each of BOP and thiol, and 4 equiv. of DBU, in DME (2 mL). (d) For the two-step, one-pot reaction with MeSNa, as with p-toluidine, amide activation was performed and the solvent was evaporated. The second step was conducted with MeSNa (2 equiv.) in anhydrous DMSO (1 mL). The one-pot reaction was not attempted in this case. The collection of results from these experiments is shown in Table 2.
Table 2.
Results from the two-step, one-pot, and one-step transformations of three pyrimidine nucleosides.a
| ||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Nucleophile | Two step, one potb | One stepc | ||
|
| ||||||
| Time, temp | Product: yieldd | Time, temp | Product: yieldd | |||
| 1 | 2 | MeNH2 | rt, 0.5 h | 11a: 84%e | rt, 45 min | 11a: 90% (97%)e,f |
| 2 | 2 | PhCH2NH2 | rt, 1.5 h | 11b: 72% | rt, 1 h | 11b: 98% |
| 3 | 7 | PhCH2NH2 | rt, 2 h | 12a: 78% | rt, 30 min | 12a: 92% |
| 4 | 2 | Me2NH | rt, 2 h | 11c: 76%e | rt, 1 h | 11c: 93%e |
| 5 | 2 | Et2NH | rt, 2 h | 11d: 78% | rt, 1 h | 11d: 98% |
| 6 | 8 | Et2NH | rt, 1 h | 13a: 57% | rt, 1 h | 13a: 80% |
| 7 | 2 |
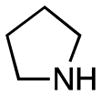
|
rt, 2 h | 11e: 75% | rt, 20 min | 11e: 95% |
| 8 | 7 |
|
rt, 2 h | 12b: 71% | rt, 30 min | 12b: 89% |
| 9 | 8 |
|
rt, 2 h | 13b: 53% | rt, 1 h | 13b: 80% |
| 10 | 2 |
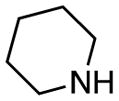
|
rt, 2 h | 11f: 75% | rt, 1 h | 11f: 92% |
| 11 | 7 |
|
rt, 2 h | 12c: 75% | rt, 30 min | 12c: 91% |
| 12 | 8 |
|
rt, 1.5 h | 13c: 63% | rt, 40 min | 13c: 90% |
| 13 | 7 |
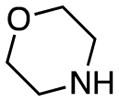
|
rt, 2 h | 12d: 76% | rt, 30 min | 12d: 95% |
| 14 | 8 |
|
rt, 2 h | 13d: 57% | rt, 1 h | 13d: 85% |
| 15 | 2 |
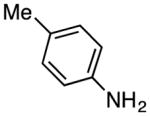
|
50 °C, 16 h | 11g: 62%g | reflux, 12 h | 11g: 67%h |
| 16 | 7 |
|
50 °C, 16 h | 12e: 64%g | reflux, 12 h | 12e: 69%h |
| 17 | 2 | EtSH | rt, 3.5 h | 11h: 68%g | rt, 3 h | 11h: 76%h |
| 18 | 2 | PhCH2SH | rt, 4 h | 11i: 70%g | rt, 3 h | 11i: 82%h |
| 19 | 2 | MeSNa | 50 °C, 2 h | 11j: 49%i | not done | NAj |
Reactions were conducted at 0.424 M nucleoside concentration in the reaction solvent, using 0.212 mmol of 2, 0.219 mmol of 7, or 0.262 mmol of 8.
The substrates were converted to the respective O4-(benzotriazol-1-yl) derivatives using BOP (2 equiv.) and DBU (2 equiv.) in anhydrous THF, and then nucleophile (4 equiv.) was added to the reaction mixture.
The nucleoside, BOP (2 equiv.), DBU (2 equiv.), and nucleophile (4 equiv.) were reacted in the appropriate solvent.
Yields are of isolated and purified products.
The following amine solutions were used: 2 M MeNH2 in THF and 40 wt% Me2NH in H2O.
The yield was based on recovered 2, the reaction was incomplete and ca. 7% of 2 was reisolated.
After formation of the benzotriazolyl derivative, the solvent was evaporated. Then nucleophile (2 equiv.), and base (2 equiv.) were used in the appropriate solvent (2 mL).
DBU (4 equiv.), nucleophile (2 equiv.), and appropriate solvent (2 mL) were used.
After formation of the benzotriazolyl derivative, the solvent was evaporated. Then MeSNa (2 equiv.) was used in anhydrous DMSO (1 mL).
A one-step approach was not attempted in this case.
Next, we queried the applicability of this method for the synthesis of C4 ether derivatives of pyrimidine nucleosides. As we have previously shown, these reactions cannot be conducted as one-step processes, but as a two-step, one-pot reaction.49,50 We had also shown that after formation of the intermediate benzotriazol-1-yl derivative, evaporation of the reaction solvent, addition of the alcohol, and the base allows for conversion to the desired ethers. Thus, such an approach was evaluated in the present cases (Table 3). Although Cs2CO3 was used previously, DBU was also effective and was used here, and good yields were obtained. In reactions with MeOH, allyl alcohol, and benzyl alcohol, 1-alkoxy-1H-benzotriazoles51 were observed to be byproducts (8–20%).
Table 3.
C4 ether derivatives of pyrimidine nucleosides with BOP and alcohols.a
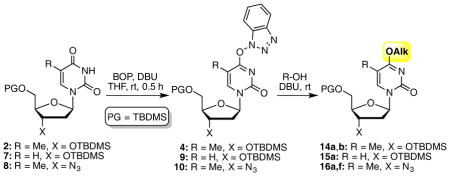
| ||||
|---|---|---|---|---|
| Entry | Substrate | Alcohol | Conditions | Product: yieldb |
| 1 | 2 | MeOH | rt, 0.5 h | 14a: 91%c |
| 2 | 7 | MeOH | rt, 1 h | 15: 85%c |
| 3 | 2 |
|
rt, 1 h | 14b: 88%c |
| 4 | 8 | CH2=CHCH2OH | rt, 1 h | 16a: 56%d |
| 5 | 8 | PhCH2OH | rt, 1 h | 16b: 85%d |
Unless noted otherwise, reactions were conducted at 0.424 M nucleoside concentration in the reaction solvent, using 0.212 mmol of 2, 0.219 mmol of 7, or 0.262 mmol of 8. The substrates were converted to the respective O4-(benzotriazol-1-yl) derivatives using BOP (2 equiv.) and DBU (2 equiv.) in anhydrous THF. After evaporation, alcohol and additional DBU were added.
Yields are of isolated and purified products.
DBU (2 equiv.) and the alcohol (20 equiv.) were used for the second step.
The reaction was conducted at 0.115 M nucleoside concentration in the reaction solvent, and after the formation of O4-(benzotriazol-1-yl) derivative, 2 equiv. of DBU and 2 equiv. of alcohol were added to the reaction mixture.
Reagents such as BOP can contain phosphorus triamides and this is pertinent to some observations that were made. First, an azido group is tolerated under the reaction conditions, despite possible PIII contaminants, and good product yields were obtained with substrate 8 (see Tables 2 and 3). During the synthesis of product 11j, the O4-(benzotriazol-1-yl) intermediate 4 was generated in situ by reaction with 2 equiv. each of BOP and DBU, in THF. Then 2 equiv. of MeSNa was added to the reaction mixture, but the second step did not reach completion, possibly due to the insolubility of MeSNa in THF. In order to circumvent the solubility issue, polar solvents were utilized for the second step. This was done by evaporating THF after the formation of 4 and use of polar solvents such as MeCN, DMF, DMSO, and EtOH. Reactions in MeCN and DMF did not reach completion and the yields of 11j were very low. The reaction in DMSO gave a moderate yield (49%) of 11j along with byproducts, identified as 17 and 18 (Figure 2). N-oxide 17 has the same molecular mass as intermediate 4 but is slower eluting on TLC. Compound 18 has one less oxygen atom in comparison to 4. Increasing the amount of MeSNa from 2 to 4 equiv. did not improve the yield of 11j.
Fig. 2.
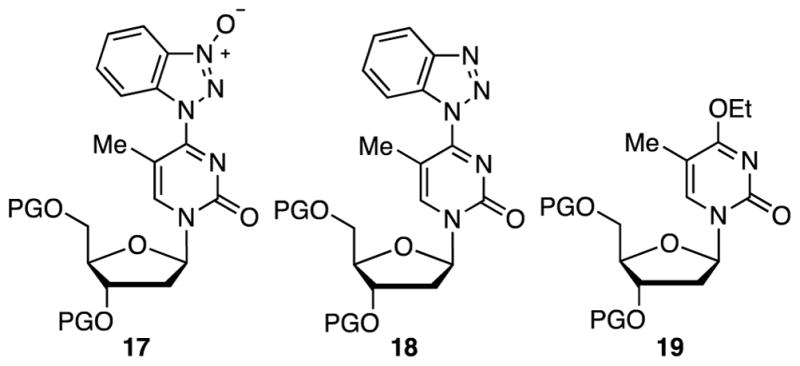
Formation of side products from precursor 2 en route to 11j. PG = TBDMS.
Formation of byproducts 17 and 18 was also observed when reactions were conducted in MeCN and DMF but no attempt was made to isolate them, as they were relatively minor. When EtOH was used as solvent, product 19 was observed. This could result either by displacement of BtO− from 4 or of MeS− from 11j, with EtOH. Compound 11j is known to be susceptible to substitution reactions by suitable nucleophiles.4–6
Compound 17 could originate from a unimolecular isomerization of the O4-(benzotriazol-1-yl) intermediate 4 or en route to 4 (see discussion below). In reactivity studies of phosphorylating reagents in the phosphotriester approach to oligonucleotides, a reagent derived from 1-hydroxy-1H-benzotriazole showed significant reaction at the amide residue of thymidine.52 In that work, O4-(benzotriazol-1-yl)-3′,5′-di-O-acetylthymidine was isolated and it was shown to undergo rearrangement to the 3′,5′-di-O-acetyl equivalent of compound 17, upon heating with 1-methylimidazole in dry pyridine at 50 °C.52
Compound 18 is likely a reduction product, and we have shown that 1-hydroxy-1H-benzotriazoles as well as O6-(benzotriazol-1-yl)purine nucleosides can be reduced with B2(OH)4 and (pinB)2, respectively.53,54 As mentioned earlier, because benzotriazole-based peptide-coupling agents such as BOP could contain PIII contaminants, these could potentially act as reducing agents for a species such as 17 or even intermediate 4. We, therefore, conducted an assessment of BOP obtained from two different sources to evaluate the ratios of these various products (supplier 1: Chem Impex and supplier 2: Sigma-Aldrich).
BOP from supplier 2 out performed that from supplier 1 in a 0.5 h reaction time, in that desired intermediate 4 was formed in ca. 60% from the former. Nearly 20% of the N-oxide 17 and 0–1% of reduction product 18 was formed from both within the short reaction time. At 24 h, a substantial amount of 17 had resulted from both, with an increased amount of the reduction product (more so with BOP from supplier 1). We then evaluated whether DBU had any role in this process, and with the better-performing BOP, the amount of DBU was increased to 4 equiv. After 24 h, none of intermediate 4 was observed, the amount of 17 was lower, and the amount of the reduction product 18 increased. It has been empirically shown that an N-oxide type intermediate from uridine can also react with nucleophiles.52 Thus, one must surmise that DBU can react with 4 and/or 17 (there are numerous examples in the literature where DBU undergoes ring opening to a caprolactam by action as nucleophile). From this, the displaced BtO− could undergo reduction, possibly with HMPT from BOP, forming the benzotriazolide. The benzotriazolide can then function as nucleophile, resulting in compound 18 from any reactive intermediate formed along the reaction path, including a species formed from the reaction of DBU with 4 and/or 17. This would be consistent with a mechanism we have recently proposed for the reduction of O6-(benzotriazol-1-yl)purine nucleosides by (pinB)2.54
Mechanistic Considerations
With the reactions and analysis of plausible modes for formation of byproducts completed, we performed one reaction to confirm that byproducts 17 and 18 are indeed as anticipated. We have previously shown that 1-hydroxy-1H-benzotriazoles can be reduced to 1H-benzotriazoles by reaction with B2(OH)4/Et3N in MeCN.53 Because product 17 is anticipated to be an N-oxide, exposure to just B2(OH)4 in MeCN should result in its reduction. This was indeed the case and a high-yield conversion 17 to 18 was observed (Figure 3).
Fig. 3.

Partial NMR spectra of compounds 17 (in black) and 18 (in blue) in CDCl3. Benzotriazolyl resonances are indicated by dots and TBDMS resonances are not shown.
From a mechanistic standpoint, there are three possible routes to the O4-(benzotriazol-1-yl)pyrimidine derivatives, represented by pathways a, b, and c shown in Scheme 1. Among these, pathway a proceeds through phosphonium ion I. Pathways b and c produce the O4-(benzotriazol-1-yl)pyrimidine derivative II directly, either via direct displacement of HMPA from the benzotriazolyl N1 atom or a SN2′-like process at the N3 atom of that moiety.
Scheme 1.

Some mechanistic considerations.
In our previous reports on purine nucleoside modification, clear formation of a phosphonium ion was evident via 31P{1H} NMR studies, and no N-oxide-type species could be identified.35,40 In contrast, in the present cases, exposure of the pyrimidine nucleoside to BOP and DBU led to concomitant formation of the O4-(benzotriazol-1-yl) intermediate and the N-oxide, but the former was the predominant product as observed by TLC. Although we cannot comment on whether the N-oxide is independently formed in the reaction, it can result from the O4-(benzotriazolyl) intermediate on the basis of the data in Table 4 (increase in the N-oxide at the expense of the O4-(benzotriazolyl) species over a protracted reaction time). Also, we cannot comment on the role of DBU, if any, in this isomerization process. Nevertheless, both species II and III ultimately result in product formation. Phosphonium ion I, if formed, can also lead directly to product.
Table 4.
Evaluating the various products formed with BOP from two sources.a
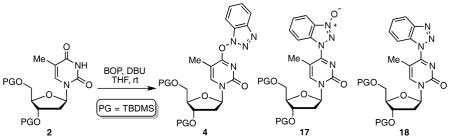
| |||||||
|---|---|---|---|---|---|---|---|
| BOPb | DBU | Time | Yieldc | ||||
|
| |||||||
| 4 | 17 | 18 | Recovered 2 | ||||
| Supplier 1 99.7% pure |
|
2 equiv. | 0.5 h | 33% | 18% | 1% | 14% |
| 2 equiv. | 24 h | 14% | 32% | 10% | – | ||
| Supplier 2 97% pure |
|
2 equiv. | 0.5 h | 59% | 23% | – | – |
| 2 equiv. | 24 h | 22% | 46% | 3% | – | ||
| 4 equiv. | 24 h | – | 8% | 16% | – | ||
Reactions were conducted with 2 equiv. of BOP in THF.
Purities are as stated by the suppliers.
Yields are of isolated and purified products.
In order to gain additional insight in plausible pathways and for comparison to the reactions of purine nucleosides,35,40 several 31P{1H} experiments were undertaken. In one experiment BOP (δ = 43.8 ppm, PF6− δ = −144.5 ppm) was exposed to precursor 2 (Figure 4), with no observable change. However, upon addition of DBU, formation of HMPA was immediately observed (δ = 23.5 ppm), and this increased as BOP diminished.
Fig. 4.

31P{1H} NMR spectra in CD3CN to observe for changes during the course of the reaction at room temperature.
In another experiment, BOP was exposed to DBU and pyrrolidine. Here a trace of HMPA was observed upon contact of all reagents, and after 1 h (Figure 5). Eventually though, BOP diminishes but HMPA formation remains low. This appears to indicate that DBU and pyrrolidine will cause consumption of BOP. But in the presence of the nucleoside, because reaction of the amide linkage is fast (0.5 h), this secondary reaction may be inconsequential. Individually, DBU and pyrrolidine also cause consumption of BOP with formation of HMPA, but those reactions (data not shown) are slower than that with the combination. Importantly though, at least as assessed by 31P{1H} NMR, the mechanism by which BOP reacts with amide groups in the pyrimidine nucleosides studied here, appears to differ from the corresponding reactions of purine nucleosides where discernible phosphorus resonances of nucleoside phosphonium salts can be observed (δ ~ 35 ppm) as the reactions progress.35,40
Fig. 5.

31P{1H} NMR spectra in CD3CN to observe for changes when BOP is in contact with DBU and pyrrolidine at room temperature.
Biological Results
In order to obtain materials for biological assays, compounds were subjected to desilylation (Figure 6). Three different conditions were used for this purpose: n-Bu4NF in THF at room temperature, KF in MeOH at 80 °C, and tris(dimethylamino)sulfonium difluorotrimethylsilicate (TASF) in MeCN from 0 °C to room temperature. Specific conditions used and product yields are summarized in Figure 6. Notably desilylation of 11h with KF/MeOH yielded 23a by substitution of the SEt group.
Fig. 6.

Structures of products prepared, desilylation conditions, reaction times, and yields. Conditions A: n-Bu4NF, THF; rt, B: KF, MeOH, 80 °C; C: TASF, MeCN, 0 °C to rt.
Because C4-modified azido nucleosides were available, we decided to reduce the N-substituted 3′-azido-2′,3′-dideoxy-5-methylcytidine derivatives to the corresponding amino compounds for assessment of biological activities (Scheme 2). 3′-Amino-2′,3′-dideoxy-5-methylcytidine, cytidine, and 5-fluorocytidine have previously been prepared via the C4 triazolyl intermediates obtained from corresponding azido derivatives.21
Scheme 2.
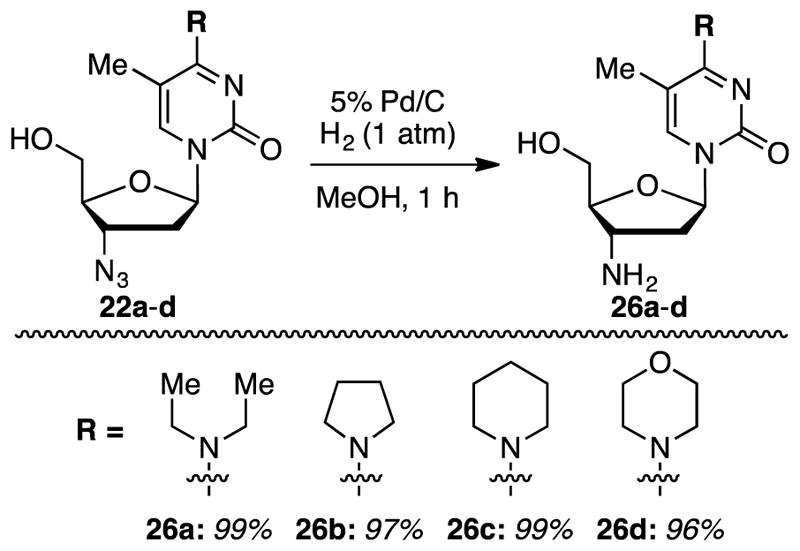
Reduction of the 3′-azido-2′,3′-dideoxy nucleosides to the corresponding amines
Compounds shown in Figure 6 and Scheme 2 were assayed for their abilities to inhibit proliferation of murine leukemia (L1210), human cervix carcinoma (HeLa), and human T-lymphocytic (CEM) cell lines. Modest activity (90 ± 5 to 219 ± 48 μM) was observed for thymidine-derived compounds 5, 20b, d e, f–i, and AZT-derived 25b (see Table 5). Notably, removal of the 5-methyl group abrogated activity in compounds 21a, 21b, 21d, and 21e.
Table 5.
Inhibitory effects of compounds against proliferation of murine leukemia (L1210), human T-lymphocyte (CEM), and human cervix carcinoma (HeLa) cellsa
| Compound | IC50b (μM) | ||
|---|---|---|---|
| L1210 | CEM | HeLa | |
| 5 | 150 ± 56 | >250 | 231 ± 27 |
| 20b | 116 ± 32 | 120 ± 8 | 219 ± 48 |
| 20d | 99 ± 33 | ≥250 | 198 ± 75 |
| 20e | 141 ± 40 | >250 | 120 ± 4 |
| 20f | 130 ± 32 | >250 | >250 |
| 20g | 106 ± 15 | 103 ± 1 | 201 ± 7 |
| 20h | 90 ± 5 | ≥250 | 188 ± 49 |
| 20i | 103 ± 9 | ≥250 | 207 ± 14 |
| 25b | 166 ± 94 | 123 ± 15 | 181 ± 23 |
Only compounds showing some activity for any of the three cell lines are shown. The other synthesized compounds were not inhibitory at the higher concentration evaluated, i.e. 250 μM.
IC50 = 50% Inhibitory concentration or compound concentration required to inhibit tumor cell proliferation by 50%.
All compounds were also tested for their antiviral activities against: (a) cytomegalovirus (CMV), varicella-zoster virus (VZV), herpes simplex virus 1 (HSV-1), herpes simplex virus 2 (HSV-2), vaccinia virus, and vesicular stomatitis virus (VSV) in human embryonic lung (HEL) cells, (b) VSV, coxsackie virus B4, and respiratory syncytial virus in human cervix carcinoma (HeLa) cells, (c) parainfluenza-3 virus, reovirus, sindbis virus, coxsackie B-4 virus, and punta toro virus in green monkey kidney (VERO) cells, (d) feline corona and feline herpes viruses in feline kidney (CRFK) cells, and (e) influenza A H1N1, influenza B H3N2, and influenza B viruses in canine kidney (MDCK) cells. The activity data are summarized in Table 6.
Table 6.
Activities of compounds against HIV-1 and HIV-2a
| Compound | EC50b (μM) | |
|---|---|---|
| HIV-1 | HIV-2 | |
| 20d | 151 ± 85 | 59 ± 46 |
| 22a | 11 ± 7.9 | 13 ± 8.7 |
| 22b | 176 ± 105 | ≥250 |
| 22c | 8.6 ± 2.1 | 23 ± 2.1 |
| 22d | 0.83 ± 0.38 | 1.6 ± 0.57 |
| 25a | 0.44 ± 0.21 | 0.64 ± 0.064 |
| 25b | 0.66 ± 0.087 | 0.74 ± 0.16 |
Only compounds showing some anti-HIV activity are shown. The other synthesized compounds were not inhibitory at the higher concentration evaluated, i.e. 250 μM.
EC50 = 50% effective concentration or compound concentration required to inhibit virus replication by 50%.
Several compounds showed activities against HIV-1 and HIV-2 (Table 6), particularly interesting are azido derivatives 22a (compare with 20d), 22c, 22d, 25a, and 25b. In comparison to diethylamino derivative 20d, presence of a 3′-azide greatly enhances activity in 22a. Piperidinyl derivative 22c was comparable to 22a and both showed higher activity over the pyrrolidinyl analogue 22b. Inclusion of an oxygen atom in the six-membered ring in 22d increased activity by an order of magnitude. Ether derivatives 25a and 25b also displayed significant activity, with the former being higher. In these cases, release of AZT by loss of the ether moiety may be possible.
Among the entire set of compounds tested, none of them showed activity against the other virus tested except for a single C4 ether derivative; compound 23a displayed anti-HSV-1 activity. This compound inhibited HSV-1 Kos strain with an EC50 of 5.4 ± 2.0 μM while the minimum cytotoxic concentration required to cause a microscopically detectable alteration of normal cell morphology was >100 μM. Removal of the 5-methyl unit of 23a, however, caused loss of anti-HSV-1 activity as observed with compound 24. Notably, compound 23a lacked activity against HSV-2 and an acyclovir-resistant (thymidine-kinase deficient) HSV-1 virus.
Conclusions
Herein, we have demonstrated a procedure for the facile introduction of substituents at the C4 position of pyrimidine nucleosides. These include amines, thiols, and alcohols. The approach involves reaction of the amide, but not the urea moiety, with BOP and base, leading to an intermediate O4-(benzotriazol-1-yl)pyrimidine nucleoside derivative, within a short reaction time. With amines and thiols, two-step, one-pot, and one-step processes have been investigated. The latter was marginally to significantly superior to the former in every case studied. With alcohols, only a two-step, on-pot approach is feasible. The method is tolerant of a 3′-azido group in the nucleoside. It appears that prolonged reactions times with BOP and DBU lead to conversion of the O4-(benzotriazol-1-yl)pyrimidine nucleoside to an isomeric benzotriazolyl N-oxide form, although the latter is also expected to be reactive towards displacement by nucleophiles. The N-oxide could undergo reduction with PIII species in BOP, resulting in 4-(benzotriazol-1-yl)pyrimidine nucleosides. That the byproducts from the reactions of the amide groups in pyrimidine nucleosides with BOP are plausibly the benzotriazolyl N-oxide and the 4-(benzotriazol-1-yl) nucleoside derivatives was shown by reduction of the former to the latter by B2(OH)4. Mechanism of the reaction was queried by NMR experiments. All products were desilylated and the azido compounds were reduced to the corresponding amines. The ensuing 29 compounds were assessed for their abilities to inhibit cancer cell proliferation using L1210, HeLa, and CEM cell lines, where some showed modest activity. Antiviral assessments indicated several to possess anti-HIV-1 and HIV-2 activities, and one compound showed anti-HSV-1 activity. In summary, the method described herein offers a generally facile, broadly applicable approach for the C4 functionalization of pyrimidines and pyrimidine nucleosides, which can lead to the discovery of new pharmacologically active agents as well as for other applications.
Supplementary Material
Acknowledgments
This work was supported by National Science Foundation Grant CHE-1265687 to MKL. Infrastructural support at CCNY was provided by National Institutes of Health Grant G12MD007603 from the National Institute on Minority Health and Health Disparities. Dr. Padmanava Pradhan (CCNY) is thanked for assistance with some NMR experiments.
Footnotes
Electronic Supplementary Information (ESI) available: Experimental procedures and copies of NMR spectra of the products.
References
- 1.Fox JJ, Van Praag D, Wempen I, Doerr IL, Cheong L, Knoll JE, Eidinoff ML, Bendich A, Brown GB. J Am Chem Soc. 1959;81:178–187. [Google Scholar]
- 2.Wempen I, Duschinsky R, Kaplan L, Fox JJ. J Am Chem Soc. 1961;83:4755–4766. [Google Scholar]
- 3.Griffon JF, Mathé C, Faraj A, Aubertin AM, De Clercq E, Balzarini J, Sommadossi JP, Gosselin G. Eur J Med Chem. 2001;36:447–460. doi: 10.1016/s0223-5234(01)01238-7. [DOI] [PubMed] [Google Scholar]
- 4.Ueda T, Fox JJ. J Med Chem. 1963;6:697–701. doi: 10.1021/jm00342a015. [DOI] [PubMed] [Google Scholar]
- 5.Wempen I, Miller N, Falco EA, Fox JJ. J Med Chem. 1968;11:144–148. doi: 10.1021/jm00307a034. [DOI] [PubMed] [Google Scholar]
- 6.Perlman ME, Watanabe KA, Schinazi RF, Fox JJ. J Med Chem. 1985;28:741–748. doi: 10.1021/jm00383a009. [DOI] [PubMed] [Google Scholar]
- 7.Saladino R, Mincione E, Crestini C, Mezzeti M. Tetrahedron. 1996;52:6759–6780. [Google Scholar]
- 8.Saladino R, Crestini C, Bernini R, Frachey G, Mincione E. J Chem Soc Perkin Trans 1. 1994:3053–3054. [Google Scholar]
- 9.Žemlička J, Šorm F. Collect Czech Chem Commun. 1965;30:2052–2067. [Google Scholar]
- 10.Robins MJ, Naik SR. Biochemistry. 1971;10:3591–3597. doi: 10.1021/bi00795a017. [DOI] [PubMed] [Google Scholar]
- 11.Lin TS, Mancini WR. J Med Chem. 1983;26:544–548. doi: 10.1021/jm00358a016. [DOI] [PubMed] [Google Scholar]
- 12.Matsuda A, Itoh H, Takenuki K, Sasaki T, Ueda T. Chem Pharm Bull. 1988;36:945–953. doi: 10.1248/cpb.36.945. [DOI] [PubMed] [Google Scholar]
- 13.Appel R. Angew Chem Int Ed Engl. 1975;14:775–843. [Google Scholar]
- 14.De Napoli L, Messere A, Montesarchio D, Piccialli G, Santacroce C. Nucleosides Nucleotides. 1991;10:1719–1728. [Google Scholar]
- 15.Lou C, Dallmann A, Marafini P, Gao R, Brown T. Chem Sci. 2014;5:3836–3844. [Google Scholar]
- 16.Hennecke U, Kuch D, Carell T. Synthesis. 2007:929–935. [Google Scholar]
- 17.Reese CB, Ubasawa A. Nucleic Acids Symp Ser. 1980;7:5–21. [PubMed] [Google Scholar]
- 18.Divakar KJ, Reese CB. J Chem Soc Perkin Trans 1. 1982:1171–1176. [Google Scholar]
- 19.Reese CB, Skone PA. J Chem Soc Perkin Trans 1. 1984:1263–1271. [Google Scholar]
- 20.Sung WL. J Org Chem. 1982;47:3623–3628. [Google Scholar]
- 21.Lin TS, Gao YS, Mancini WR. J Med Chem. 1983;26:1691–1696. doi: 10.1021/jm00366a006. [DOI] [PubMed] [Google Scholar]
- 22.Sung WL, Narang SA. Can J Chem. 1982;60:111–120. [Google Scholar]
- 23.Kamaike K, Takahashi M, Utsugi K, Tomizuka K, Ishido Y. Tetrahedron Lett. 1995;36:91–94. [Google Scholar]
- 24.Ariza X, Vilarrasa J. J Org Chem. 2000;65:2827–2829. doi: 10.1021/jo9918706. [DOI] [PubMed] [Google Scholar]
- 25.Kobori A, Miyata K, Ushioda M, Seio K, Sekine M. J Org Chem. 2002;67:476–485. doi: 10.1021/jo010813l. [DOI] [PubMed] [Google Scholar]
- 26.Mahto SK, Chow C. Bioorganic Med Chem. 2008;16:8795–8800. doi: 10.1016/j.bmc.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bischofberger N. Tetrahedron Lett. 1987;28:2821–2824. [Google Scholar]
- 28.Bischofberger N, Matteucci MD. J Am Chem Soc. 1989;111:3041–3046. [Google Scholar]
- 29.Ding Y, Habib Q, Shaw SZ, Li DY, Abt JW, Hong Z, An H. J Comb Chem. 2003;5:851–859. doi: 10.1021/cc0300199. [DOI] [PubMed] [Google Scholar]
- 30.Verma S, Miller PS. Bioconjugate Chem. 1996;7:600–605. doi: 10.1021/bc960049n. [DOI] [PubMed] [Google Scholar]
- 31.Cismaş C, Gimisis T. Tetrahedron Lett. 2008;49:1336–1339. [Google Scholar]
- 32.Holmes SC, Gait MJ. Eur J Org Chem. 2005;5171–5183 [Google Scholar]
- 33.Kang SB, De Clercq E, Lakshman MK. J Org Chem. 2007;72:5724–5730. doi: 10.1021/jo070843+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsuchiya K, Komatsu H. Nucleic Acids Res Suppl. 2002:135–136. doi: 10.1093/nass/2.1.135. [DOI] [PubMed] [Google Scholar]
- 35.Bae S, Lakshman MK. J Am Chem Soc. 2007;129:782–789. doi: 10.1021/ja064682n. [DOI] [PubMed] [Google Scholar]
- 36.Bae S, Lakshman MK. J Org Chem. 2008;73:1311–1319. doi: 10.1021/jo7021795. [DOI] [PubMed] [Google Scholar]
- 37.Bae S, Lakshman MK. J Org Chem. 2008;73:3707–3713. doi: 10.1021/jo702558n. [DOI] [PubMed] [Google Scholar]
- 38.Bae S, Lakshman MK. Org Lett. 2008;10:2203–2206. doi: 10.1021/ol8006106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bae S, Chaturvedi S, Lakshman MK. Curr Protoc Nucleic Acid Chem. 2009;36:1.22.1–1.2.23. doi: 10.1002/0471142700.nc0122s36. [DOI] [PubMed] [Google Scholar]
- 40.Lakshman MK, Frank J. Org Biomol Chem. 2009;7:2933–2940. doi: 10.1039/b905298d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sakilam SK, Vuram PK, Relangi SS, Gurram V, Zhou H, Kreitman RJ, Martinez Montemayor M, Yang L, Kaliyaperumal M, Shama S, Pottabathini N, Lakshman MK. Molecules. 2015;20:18437–18463. doi: 10.3390/molecules201018437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wan ZK, Wacharasindhu S, Binnun E, Mansour T. Org Lett. 2006;8:2425–2428. doi: 10.1021/ol060815y. [DOI] [PubMed] [Google Scholar]
- 43.Lakshman MK, Singh MK, Parrish D, Balachandran R, Day BW. J Org Chem. 2010;75:2461–2473. doi: 10.1021/jo902342z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ashton TD, Scammells PJ. Aust J Chem. 2008;61:49–58. [Google Scholar]
- 45.Devine SM, May LT, Scammells PJ. Med Chem Comm. 2014;5:192–196. [Google Scholar]
- 46.Krüger S, Meier C. Eur J Org Chem. 2013:1158–1169. [Google Scholar]
- 47.Printz M, Richert C. Chem Eur J. 2009;15:3390–3402. doi: 10.1002/chem.200801587. [DOI] [PubMed] [Google Scholar]
- 48.Ghosh A, Lagisetty P, Zajc B. J Org Chem. 2007;72:8222–8226. doi: 10.1021/jo071121l. [DOI] [PubMed] [Google Scholar]
- 49.Kokatla HP, Lakshman MK. Org Lett. 2010;12:4478–4881. doi: 10.1021/ol101655h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kokatla HP, Lakshman MK. Current Protoc Nucleic Acid Chem. 2012;49:1.26.1–1.26.16. doi: 10.1002/0471142700.nc0126s49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lakshman MK, Singh MK, Kumar M, Chamala RR, Yedulla VR, Wagner D, Leung E, Yang L, Matin A, Ahmad S. Beilstein J Org Chem. 2014;10:1919–1932. doi: 10.3762/bjoc.10.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reese CB, Richards KH. Tetrahedron Lett. 1985;26:2245–2248. [Google Scholar]
- 53.Gurram V, Akula HK, Garlapati R, Pottabathini N, Lakshman MK. Adv Synth Catal. 2015;357:451–462. doi: 10.1002/adsc.201400889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Basava V, Yang L, Pradhan P, Lakshman MK. Org Biomol Chem. 2016;14:7069–7083. doi: 10.1039/c6ob01170e. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.