

Abstract
Background and Purpose
Raloxifene can induce both endothelium‐dependent and ‐independent relaxation in different arteries. However, the underlying mechanisms by which raloxifene triggers endothelium‐independent relaxation are still incompletely understood. The purpose of present study was to examine the roles of NOSs and Ca2+ channels in the relaxant response to raloxifene in the rat isolated, endothelium‐denuded aorta.
Experimental Approach
Changes in isometric tension, cGMP, nitrite, inducible NOS protein expression and distribution in response to raloxifene in endothelium‐denuded aortic rings were studied by organ baths, radioimmunoassay, Griess reaction, western blot and immunohistochemistry respectively.
Key Results
Raloxifene reduced the contraction to CaCl2 in a Ca2+‐free, high K+‐containing solution in intact aortic rings. Raloxifene also acutely relaxed the aorta primarily through an endothelium‐independent mechanism involving NO, mostly from inducible NOS (iNOS) in vascular smooth muscle layers. This effect of raloxifene involved the generation of cGMP and nitrite. Also, it was genomic in nature, as it was inhibited by a classical oestrogen receptor antagonist and inhibitors of RNA and protein synthesis. Raloxifene‐induced stimulation of iNOS gene expression was partly mediated through activation of the NF‐κB pathway. Raloxifene was more potent than 17β‐estradiol or tamoxifen at relaxing endothelium‐denuded aortic rings by stimulation of iNOS.
Conclusions and Implications
Raloxifene‐mediated vasorelaxation in rat aorta is independent of a functional endothelium and is mediated by oestrogen receptors and NF‐κB. This effect is mainly mediated through an enhanced production of NO, cGMP and nitrite, via the induction of iNOS and inhibition of calcium influx through Ca2+ channels in rat aortic smooth muscle.
Abbreviations
- HRT
hormone replacement therapy
- NPLA
Nω‐propyl‐L‐arginine
- PDTC
pyrrolidine dithiocarbamate
- Rf
raloxifene HCl
- SERMs
selective oestrogen receptor modulators
- SNP
sodium nitroprusside
- VSMC
vascular smooth muscle cells
Tables of Links
| TARGETS | |
|---|---|
| Nuclear hormone receptors a | Enzymes b |
| Oestrogen receptors (ERs) | eNOS |
| Oestrogen receptor‐α (ERα) | iNOS |
| nNOS | |
| sGC |
| LIGANDS | |
|---|---|
| 17β‐estradiol | ODQ |
| Acetylcholine | Phenylephrine |
| Aminoguanidine | Raloxifene |
| L‐NAME | Tamoxifen |
| LPS | U46619 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a,b).
Introduction
Throughout the world, cardiovascular disease is the main cause of morbidity and mortality in postmenopausal women. Epidemiological studies show a strong relationship between menopause and the development of cardiovascular disease (Barrett‐Connor and Bush, 1991). Although there is observational evidence to support the therapeutic benefits of hormone replacement therapy (HRT) (Stampfer et al., 1991), the therapeutic role of oestrogens in preventing cardiovascular diseases remains controversial, as several clinical trials have failed to demonstrate the efficacy of HRT (Nabel, 2000; Herrington et al., 2000; Rossouw et al., 2002) and the practice of taking HRT may not be encouraged in postmenopausal women (Rossouw et al., 2002; Lagro‐Janssen et al., 2010). However, HRT has been demonstrated to have more potential benefits on cardiovascular events if it is started around menopause, (Schierbeck et al., 2012; Manson et al., 2013).
In order to search for more effective and selective agents to replace oestrogens, molecules like selective oestrogen receptor (ER) modulators (SERMs), such as raloxifene, have been synthesized because these SERMs possess beneficial effects due to their oestrogenic actions on the skeleton, vascular system and CNS, but are devoid of any harmful effects as they have been shown to have anti‐oestrogenic effects in the breast and uterus. Raloxifene has been approved for the prevention of osteoporosis in postmenopausal women (Genazzani et al., 1999), and raloxifene therapy decreases the risk of cardiovascular events in women who are high risk for cardiovascular diseases (Barrett‐Connor et al., 2002). However, the results of the completed Raloxifene Use for the Heart trial (Barrett‐Connor et al., 2006) showed that raloxifene did not significantly reduce the risk of coronary heart disease in postmenopausal women who have high risk of coronary disease. In fact a new hormone therapy has recently been developed, which entails the use of molecular combinations of various oestrogen receptor modulators in one treatment (Valera et al., 2015). Thus, a better understanding of the mechanisms of raloxifene's actions on vascular tissues is needed in order to clarify the potential benefits of raloxifene therapy for postmenopausal women.
Raloxifene causes endothelium‐independent relaxation in rat cerebral arteries (Tsang et al., 2004), intrarenal arteries (Leung et al., 2005), pulmonary arteries (Chan et al., 2005) and porcine coronary arteries (Leung et al., 2007). The endothelium‐independent relaxation is probably mediated by direct inhibition of voltage‐sensitive Ca2+ channel in vascular smooth muscle cells (VSMC) (Tsang et al., 2004). The role of endothelium in raloxifene‐induced vascular effects is still controversial. On the one hand, raloxifene can rapidly improve endothelial dysfunction caused by oxidative stress (Wong et al., 2008). Raloxifene therapy increases NO concentration and flow‐mediated vasodilatation in healthy postmenopausal women (Saitta et al., 2001). Our group also showed that the chronic oral administration of raloxifene to ageing ovariectomized female rats augmented the bioavailability of endothelial NO in isolated aortic rings and did not affect eNOS expression (Wong et al., 2006). On the other hand, raloxifene was found to enhance NO release in rat aorta by increasing the expression of eNOS mRNA (Rahimian et al., 2002). Therefore, in the present study we have investigated the mechanisms by which raloxifene exerts its NO‐related vascular action. We demonstrated that inducible NO, from iNOS in smooth muscle, and the inhibition of calcium influx through Ca2+ channels are involved in raloxifene‐mediated endothelium‐independent relaxation.
Methods
Vessel preparation
Male Sprague–Dawley rats (250–300 g) were killed by inhalation of pure CO2. The thoracic segment of aorta was dissected out, and the surrounding connective tissues were cleaned off. Each aorta was cut into several ring segments of ~3 mm in length. Rings were then transferred into 10 mL organ baths containing Krebs solution bubbled with a gas mixture of 95% O2 plus 5% CO2. Each ring was mounted horizontally between two L‐shaped stainless steel hooks. One of the hooks was connected to the bottom of the bath, while the other was connected to an FT03 force‐displacement transducer (Grass Instruments Co, USA). Krebs solution contained (in mM): 119 NaCl, 4.7 KCl, 2.5 CaCl2, 1 MgCl2, 25 NaHCO3, 1.2 KH2PO4 and 11 D‐glucose. A basal tension of 1 g was applied to aortic rings. All experiments were performed at 37°C. Twenty minutes after being mounted in the organ baths, the rings were first contracted with 0.3 μM phenylephrine to test the contractility of the vessel and then relaxed by 1 μM acetylcholine, which produced over 85% relaxation, to assess the integrity of the endothelial layer. Thereafter, they were rinsed several times in pre‐warmed Krebs solution until baseline tension was restored and were finally allowed to equilibrate for 60 min. Baseline tension was readjusted to 1 g when necessary during the entire duration of the experiment. In some aortic ring segments, the endothelial layer was mechanically removed by gently rubbing the luminal surface back and forth several times with a stainless steel wire. Functional removal of the endothelium was confirmed by the absence of a relaxant response to 1 μM acetylcholine at the beginning of each experiment. Each experiment was performed on rings obtained from different rats.
These experiments were approved by the Animal Experimentation Ethical Committee of the Chinese University of Hong Kong. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Protocols for measurement of isometric force in vitro
The role of the endothelium was first examined. The effects of raloxifene on concentration‐dependent contractile responses to U46619 (0.3–500 nM) in endothelium‐intact aortic rings were determined. After the first response curve, rings were rinsed. Once baseline tone was re‐established, rings were exposed for 30 min to different concentrations of raloxifene (0.1–10 μM). Thereafter, the second response curve to U46619 was repeated. The effect of raloxifene was also tested on U46619‐contracted endothelium‐denuded rings. The effect of raloxifene on receptor‐independent contraction, induced by smooth muscle membrane depolarization, was also examined in rings precontracted by elevating the extracellular potassium ion concentration.
To determine whether raloxifene has any effects on calcium channels, the effects of 30 min raloxifene (0.1–3 μM) pretreatments were examined on contractile responses, in a Ca2+‐free 60 mM K+ solution, to cumulative addition of CaCl2 (induces concentration‐dependent contractions in endothelium‐intact aortic rings). The role of NO and prostacyclin was also investigated. The initial data showed that removal of the endothelium did not affect raloxifene‐induced aortic relaxation. However, it was still unclear whether endothelium‐derived vasoactive factors were involved. In this set of experiments, after obtaining the first concentration–response curve to U46619, endothelium‐intact rings were incubated for 30 min with L‐NAME (100 μM, NOS inhibitor) or ODQ (3 μM, soluble guanylate cyclase inhibitor) before repeating the second concentration–response curve. In addition, the effect of indomethacin (3 μM, COX inhibitor) on raloxifene (1 μM)‐induced relaxation in endothelium‐intact rings was tested. Our initial results clearly demonstrated that inhibition of NO‐dependent dilation attenuated raloxifene‐induced relaxation, while removal of the endothelium did not affect raloxifene‐induced relaxation, thus suggesting a role of endothelium‐independent NO. In order to investigate this possibility further, most of the following experiments were performed with endothelium‐denuded rings.
The role of iNOS and nNOS was then examined in aortic relaxant response to raloxfene. In this series of experiments, a single concentration of raloxifene (1 μM) was used to induce relaxation in endothelium‐denuded rings that had been treated with vehicle (DMSO), L‐NAME (100 μM), ODQ (3 μM), AMT‐HCl (100 μM, iNOS inhibitor), aminoguanidine (100 μM, iNOS inhibitor), Nω‐propyl‐L‐arginine (NPLA, 100 μM, nNOS inhibitor), ICI 182780 (1 μM, classical oestrogen receptor antagonist), actinomycin D (10 μM, RNA synthesis inhibitor), cycloheximide (10 μM, protein synthesis inhibitor) or pyrrolidine dithiocarbamate (PDTC 3 μM, NF‐κB inhibitor). Rings were exposed to each of the above‐mentioned inhibitors for 30 min before the addition of U46619. Once a steady vessel tension was obtained, application of 1 μM raloxifene caused time‐dependent relaxant response for 3 h. In some experiments, rings were first exposed to 1 mM L‐arginine for 10 min, then to each of the NOS inhibitors for 30 min before the addition of U46619. The inhibitors tested included L‐NAME, AMT‐HCl and aminoguanidine. In some experiments, the effects of various inhibitors were also examined on the relaxant response to 10 μg·mL−1 bacterial LPS (from Escherichia coli, strain 0111:B4), a stimulator of iNOS, which acts as a positive control for the raloxifene‐induced activation of iNOS. The relaxant responses to sodium nitroprusside (1 μM, an exogenous NO donor) were also studied in U46619‐contracted endothelium‐denuded rings after treatment for 30 min with different inhibitors for a comparison with raloxifene action.
cGMP assay
At the end of the organ bath study mentioned above, all rings were rapidly frozen in liquid nitrogen and then homogenized in 0.5 mL of ice‐cold 0.1 M HCl, using a glass homogenizer. The homogenate was centrifuged at 20 000 × g for 10 min at 4°C. The protein content of supernatant was determined using a protein assay kit (Sigma, St. Louis, MO, USA) using BSA as the standard. The remaining supernatant was lyophilized at 4°C. The tissue content of cGMP was assayed by radioimmunoassay using a [125I]‐cGMP RIA kit (DuPont, Wilmington, DE, USA). The concentration of cGMP is presented as pmol mg−1 protein.
Nitrite assay
At the end of the organ bath study, rings were weighed. The incubation medium was removed and centrifuged at 20 000 × g for 10 min at 4°C. The nitrite content in the supernatants was subsequently detected by Griess reagent system (Promega, USA), which contains sulfanilamide solution and NED solution, according to the manufacturer's instructions, and measured in a spectrophotometer at 548 nm. Standard curves were constructed by mixing Krebs solution with different concentrations of sodium nitrite standards.
Western blot analysis
At the end of the experiments performed in organ baths, rings were collected in centrifuge tubes, quickly frozen in liquid nitrogen, and used for sample preparation of aortic tissue proteins as previously described (Wong et al., 2006). For each sample, 80 μg protein was separated under reducing conditions on a 7.5% SDS‐polyacryamide gel. At the same time, a positive control sample for iNOS (LPS‐treated rat macrophage NR 8383 cell lysate) was run in parallel to all samples. The resolved proteins were electrophoretically transferred to a nitrocellulose immobilon‐P PVDF membrane (Millipore). The membranes were blocked with 5% non‐fat milk (Carnation, Nestle, New Zealand) in PBS 0.5% Tween‐20 and subsequently exposed to a polyclonal rabbit anti‐iNOS antibody 1:500 (BD Transduction Laboratories, USA) overnight at 4°C. The membranes were then probed with a secondary anti‐rabbit antibody linked to horseradish peroxidase (DakoCytomation, Denmark) at a dilution of 1:2500 for 1 h at room temperature. The membranes were developed with an enhanced chemiluminescence detection system (ECL reagents; Amersham Pharmacia, USA) and then exposed on X‐ray films (Fuji, Japan). Densitometry was performed for the iNOS band corresponding to 135 kDa using a documentation programme (Fluorchem, Alpha Innotech Corp., USA).
Immunohistochemistry
After being used in the organ bath experiments, the aortic rings were snap‐frozen in optimum cutting temperature compound (embedding medium for frozen tissue specimens, Miles Inc, USA), and 8 μm sections were prepared from the embedded tissues. The sectioned specimens were fixed in a mixture of acetone and methanol (1:1) for 10 min. Endogenous peroxidase was quenched using 3% H2O2 in PBS for 30 min. Non‐specific adsorption was minimized by incubating the section in 20% fetal bovine serum for 1 h at room temperature. The sections were then incubated overnight in a primary anti‐iNOS antibody (1:10 dilution, BD Transduction Laboratories). Specific labelling was detected with 1:75 dilution of biotin‐conjugated goat anti‐rabbit IgG and 1:400 dilution of peroxidase conjugated streptavidin‐biotin. Specific staining of the antigen was visualized by the amnioethyl carbazole substrate kit (Vector Laboratories, USA), which gave a red colour after 5–15 min incubation at room temperature. The positive immunostaining was examined under a light microscope, and areas of interest were recorded using a Leica microscope fitted with a high‐resolution CCD camera. The images were captured as computer files using the SPOT Advanced software (version 3.5.2 for Windows, Diagnostic Instruments Inc., Sterling Heights, MI, USA).
Chemicals and drugs
The following chemical drugs were used in this study: phenylephrine hydrochloride, acetylcholine hydrochloride, L‐NAME, U46619, indomethacin, ODQ, actinomycin D, cycloheximide, PDTC, (LPS) strain 0111:B4 and sodium nitroprusside (SNP) purchased from Sigma (St. Louis, MO, USA); raloxifene HCl (Rf) as a gift from Eli Lily company; AMT AMT‐HCl, aminoguanidine hemisulfate, ICI 182780 and NPLA purchased from Tocris; L‐arginine (RBI, USA). Phenylephrine, acetylcholine, aminoguanidine hemisulfate, AMT‐HCl, L‐arginine, LPS and SNP were prepared in distilled water and the others in DMSO (Sigma).
Statistical analysis
Results are expressed as mean ± SEM of n experiments on the rings prepared from different rats. To study the effect of raloxifene on U46619‐induced contractile responses, the values of pEC50, the negative log of the constrictor concentration that caused 50% the maximum contraction (Emax %), were determined from different curves. These values were compared in the absence and presence of raloxifene. Cumulative concentration–contraction relationships were analysed with a non‐linear regression curve fitting (Prism). The effects of vasodilators are expressed as a % of the agonist‐induced maximal contractile response. Statistical differences between mean values were determined by ANOVA followed by the Student–Newman–Keuls test for comparison of mean values. P < 0.05 was regarded as significant difference between groups. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Results
Role of the endothelium
After a control curve for U46619‐induced contractile responses had been done, endothelium‐intact rings were incubated for 30 min with different concentrations of raloxifene (0.1–10 μM), and a concentration‐contraction curve was again obtained. A concentration of 1 μM raloxifene caused an approximately parallel shift of the U46619 concentration–response curve to the right (Figure S1A, B). At higher concentrations (>3 μM), raloxifene exerted an insurmountable inhibition (P < 0.05 vs. control for Emax), reducing the magnitude of the maximum contraction. Removal of the endothelium did not significantly modify the contractile responses to U46619 (Figure S1C) and 1 μM raloxifene reduced the U46619 contraction to the same extent as that in the presence of endothelium (Figure S1D). The raloxifene‐induced relaxation of U46619‐induced contractile responses was similar in the presence of 100 μM L‐NAME (a NOS inhibitor) (Figure S1E, F) or 3 μM ODQ (a soluble guanylate cyclase inhibitor) (Figure S1G, H). These results indicate that the endothelium may not have an overall influence on raloxifene‐induced relaxation.
Relaxant effect of raloxifene in endothelium‐intact rings
In another set of experiments, once a steady contraction to 20 nM U46619 was obtained, addition of raloxifene caused a slowly developing relaxation. A steady‐state relaxation was achieved over 3 h in endothelium‐intact aortic rings (Figure 1A). Figure 1B presents the maximal relaxation induced by different concentrations of raloxifene. Raloxifene at concentrations greater than 0.2 μM caused the maximal effect even though the onset of the evoked relaxation was faster as the concentration increased from 0.1 to 3 μM (Figure 1A).
Figure 1.
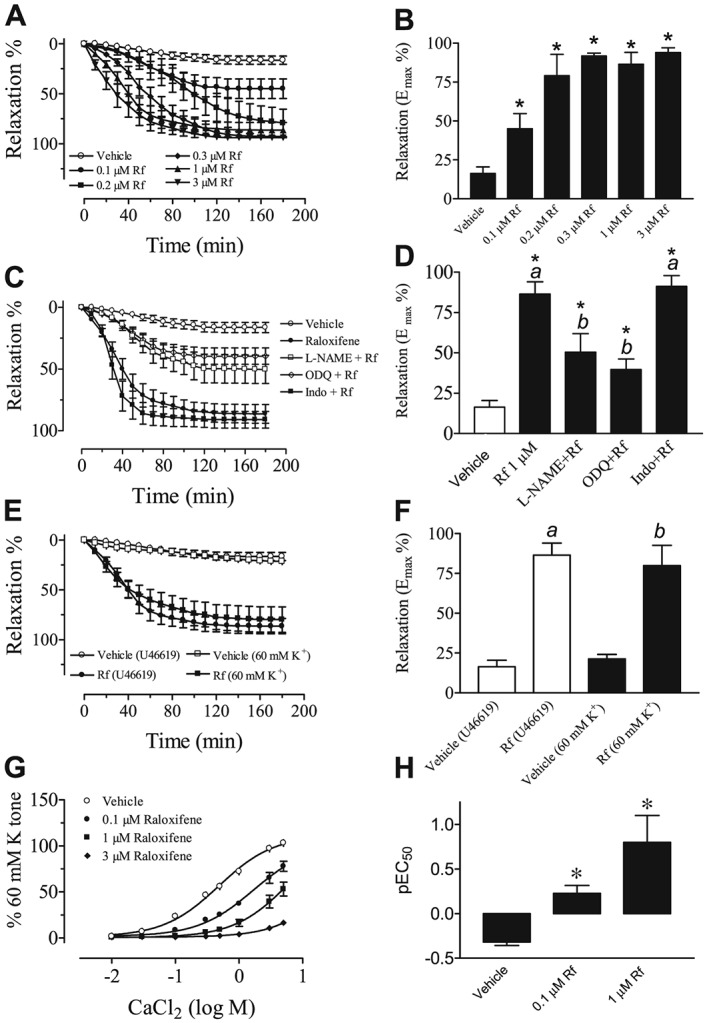
(A) The time course for the relaxant response induced by different concentrations of raloxifene (Rf; 0.1–3 μM) in the U46619‐contracted endothelium‐intact aortic rings. (B) The steady‐state maximum relaxation to raloxifene. Statistical differences are indicated by between vehicle control and raloxifene group (*P < 0.05). Results are mean ± SEM of six to seven rings from different rats. (C) The inhibitory effects of L‐NAME (100 μM) and ODQ (3 μM) on raloxifene (1 μM)‐induced relaxation in endothelium‐intact rat aortic rings. Treatment with indomethacin (Indo, 3 μM) was without effect. (D) Steady‐state maximum relaxation induced by raloxifene after various treatments. Rings were exposed for 30 min to each inhibitor before addition of U46619 to induce a sustained tone. Statistical differences are indicated by a between vehicle control and raloxifene group and b between raloxifene and other treatment groups (*P < 0.05). Results are mean ± SEM of 6–10 rings from different rats. (E) Raloxifene‐induced relaxant effect was similar in endothelium‐intact aortic rings contracted by U46619 and 60 mM K+. (F) Steady‐state maximum relaxation induced by raloxifene after various treatments. Results are mean ± SEM of five to six rings from different rats. (G) Concentration‐dependent inhibition by raloxifene (0.1–3 μM) on CaCl2‐induced contractile responses in Ca2+‐free, 60 mM K+‐containing Krebs solution. (H) pEC50 values for CaCl2‐induced contraction in the absence and presence of raloxifene (0.1–1 μM). *P < 0.05 between control and all treatment curves for CaCl2 responses. Results are mean ± SEM of five to six rings from different rats.
In another series of experiments, a single concentration of raloxifene (1 μM) was used to induce time‐dependent relaxation. Figure 1C shows that treatment of endothelium‐intact rings with L‐NAME or ODQ attenuated raloxifene‐induced relaxation to the same extent. In contrast, 3 μM indomethacin (the COX inhibitor) was without an effect (Figure 1C). The Emax values (the steady‐state maximal relaxation) are summarized in Figure 1D.
Relaxant effects on contractions induced by high K+ and CaCl2
Raloxifene at 1 μM also relaxed rings preconstricted with 60 mM extracellular potassium ions, and this effect was similar to that in U46619‐contracted rings, as shown in Figure 1E (no difference on Emax % for U46619 and high K+ in Figure 1F). In another set of experiments, rings were exposed to a Ca2+‐free 60 mM K+ solution and the cumulative addition of CaCl2 induced concentration‐dependent contractions. Incubation with raloxifene reduced the concentration‐dependent contractile responses to CaCl2, and increasing the raloxifene concentration induced a progressive reduction in the maximal response (Figure 1G). Raloxifene at 3 μM almost abolished CaCl2‐induced contractile responses (pEC50 in Figure 1H).
Effects of inhibitors of NO pathways on relaxant effect of raloxifene in endothelium‐denuded rings
Raloxifene appeared to induce primarily endothelium‐independent relaxation since the maximal relaxant responses were unaffected by removal of the endothelium (Figure S2). However, inhibitors of NO pathways caused a marked reduction in raloxifene‐induced relaxation in the absence and presence of endothelium, thus indicating that NO derived from sources other than the endothelium is involved in this response. Indeed, the representative traces in Figure 2A show that 30 min incubation with L‐NAME or ODQ suppressed raloxifene (1 μM)‐induced relaxation in endothelium‐denuded aortic rings. While exposure to L‐arginine (0.5–1 mM), the NO precusor, significantly antagonized the inhibitory effect of L‐NAME (Figure 2B). In contrast, L‐arginine at 1 mM did not affect the inhibitory effect of ODQ (Figure 2C). The bar graph in Figure 2D summarizes the Emax values for raloxifene‐induced relaxation following various pharmacological interventions.
Figure 2.
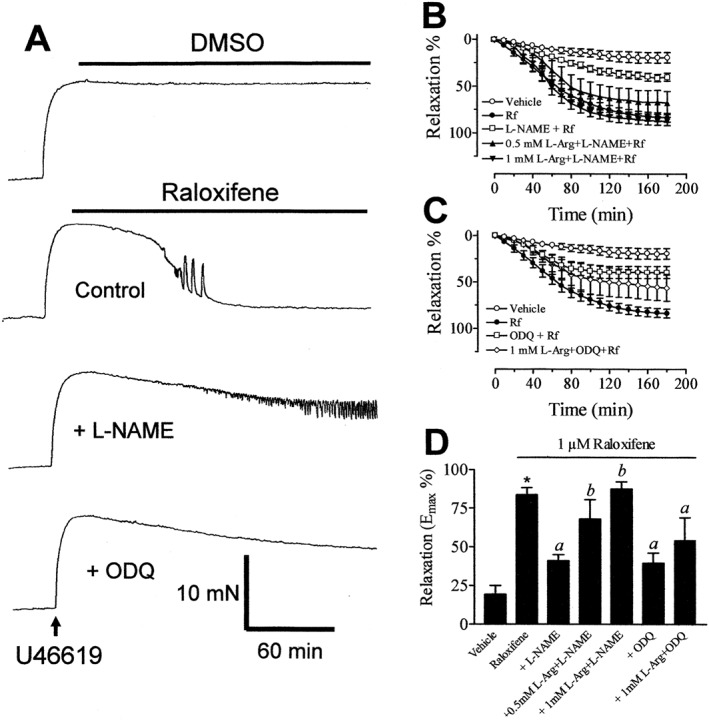
(A) Recordings showing the time course of raloxifene‐induced relaxation in endothelium‐denuded aortic rings in control and in the presence of 100 μM L‐NAME and 3 μM ODQ. (B) Inhibitory effect of L‐NAME on raloxifene (1 μM)‐induced aortic relaxation and L‐arginine (L‐Arg, 0.5–1 mM) antagonized the effect of L‐NAME. (C) The inhibitory effect of ODQ on raloxifene (1 μM)‐induced aortic relaxation and lack of effect of 1 mM L‐arginine. (D) The maximal relaxant effect of raloxifene following different pharmacological manipulations. All experiments were performed on endothelium‐denuded rings. Statistical differences are indicated by *(P < 0.05) between vehicle control and raloxifene group, a (P < 0.05) between raloxifene and other treatment groups and b (P < 0.05) between L‐NAME and treatment groups. Results are mean ± SEM of 6–11 rings from different rats.
Effects of iNOS inhibitors on raloxifene‐induced relaxation
Traces in Figure 3A and graphs in Figure 3B, C show that treatment of endothelium‐denuded rings with 100 μM aminoguanidine or 100 μM AMT‐HCl (both selective inhibitors of iNOS) attenuated raloxifene‐induced relaxation to a similar extent. Their effects were similar to those of L‐NAME and ODQ. The inhibitory effects of both iNOS inhibitors were partially or fully reversed in rings treated with 1 mM L‐arginine (Figure 3B, C). On the contrary, treatment with Nω‐propyl‐L‐arginine (100 μM, a selective inhibitor of nNOS) had no effect on the relaxant responses to raloxifene (Figure 3A, E). The bar graphs in Figure 3D, F summarize the Emax values for raloxifene‐induced relaxation following various pharmacological interventions.
Figure 3.
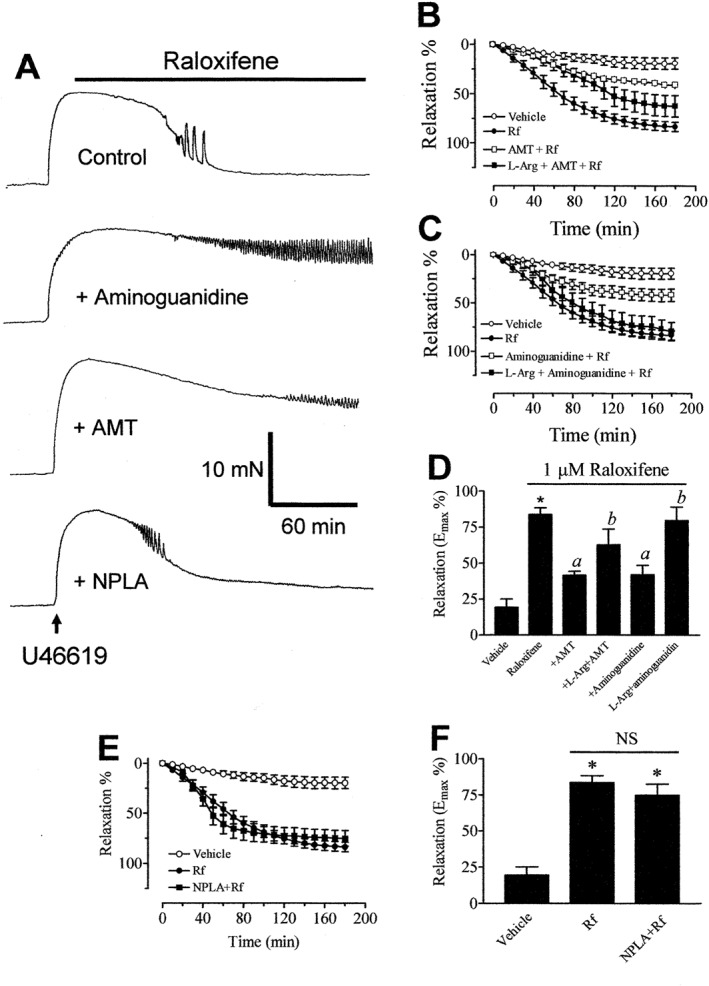
(A) Recordings showing the time course of raloxifene‐induced relaxation in endothelium‐denuded aortic rings in control and in the presence of 100 μM AMT‐HCl or 100 μM aminoguanidine. Inhibitory effect of AMT‐HCl (B) or aminoguanidine (C) on raloxifene (1 μM)‐induced aortic relaxation. (D) The maximal relaxant effect of raloxifene in the absence and presence of iNOS inhibitors. (E) Treatment with NPLA (100 μM) was without effect on raloxifene‐induced relaxation. (F) The maximal relaxant effect of raloxifene in the absence and presence of NPLA. Statistical differences are indicated by * (P < 0.05) between vehicle control and raloxifene group, a (P < 0.05) between raloxifene and other treatment groups and b (P < 0.05) between AMT‐HCl/aminoguanidine and treatment groups. Results are mean ± SEM of 6–11 rings from different rats.
Genomic nature of raloxifene‐induced effect
In endothelium‐denuded aortic rings, the raloxifene‐induced relaxant effects were attenuated after treatment with 10 μM actinomycin D (RNA synthesis inhibitor) (Figure 4A, B, D) or 10 μM cycloheximide (protein synthesis inhibitor) (Figure 4A, C, D). Both inhibitors were equi‐effective.
Figure 4.
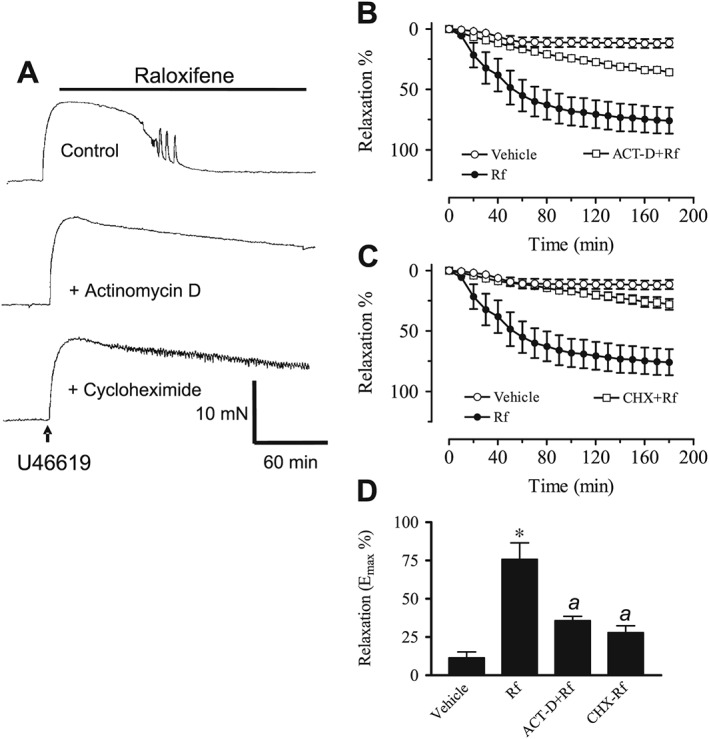
(A) Recordings showing the time course of raloxifene‐induced relaxation in endothelium‐denuded aortic rings in control and in the presence of 10 μM actinomycin D (ACT‐D) or 10 μM cycloheximide (CHX). (B) Inhibitory effect of actinomycin D or cycloheximide on raloxifene (1 μM)‐induced aortic relaxation. (C) The maximal relaxant effect of raloxifene in the absence and presence of actinomycin D/cycloheximide. Statistical differences are indicated by * (P < 0.05) between vehicle control and raloxifene group and a (P < 0.05) between raloxifene and other treatment groups. Results are mean ± SEM of 6–11 rings from different rats.
Involvement of oestrogen receptors and NF‐κB in raloxifene‐induced relaxation
Traces in Figure 5A show that treatment of endothelium‐denuded rings with 1 μM ICI 182780 (a pure oestrogen receptor antagonist) significantly reduced raloxifene‐induced relaxation when compared with the control trace. The time course and Emax values for the relaxant effect of raloxifene are presented in Figure 5B, C. The degree of inhibition was similar to that in rings treated with iNOS inhibitors (see Figure 3). Incubation with 3 μM PDTC (an inhibitor of NF‐κB) also decreased raloxifene‐induced relaxation (Figure 5D–F).
Figure 5.
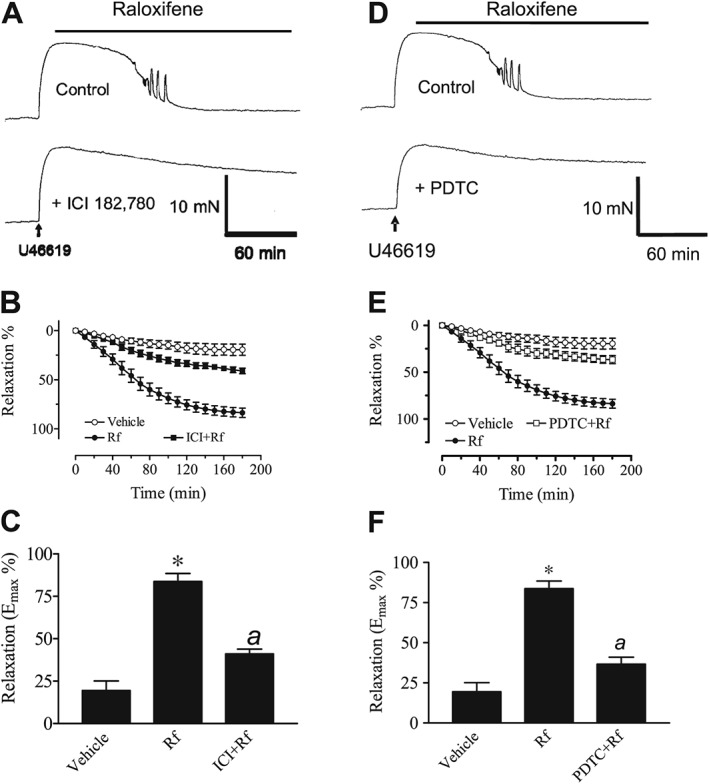
(A) Recordings showing time‐dependent relaxant responses to 1 μM raloxifene in endothelium‐denuded aortic rings in control and in the presence of 1 μM ICI 182780. (B) Inhibitory effect of ICI 182780 on raloxifene‐induced aortic relaxation. (C) The maximal relaxant effect of raloxifene in the absence and presence of ICI 182780. Results are mean ± SEM of six to seven rings from different rats. (D) Recordings showing the time course of raloxifene‐induced relaxation in endothelium‐denuded aortic rings in control and in the presence of 3 μM PDTC. (E) Inhibitory effects of PDTC on relaxant responses to raloxifene in endothelium‐denuded aortic rings. (F) The maximal relaxant effect of raloxifene in the absence and presence of PDTC. Results are mean ± SEM of six rings from different rats. Statistical differences are indicated by * (P < 0.05) between vehicle control and raloxifene group and a (P < 0.05) between raloxifene and other treatment groups.
Relaxant effect of LPS in endothelium‐denuded rings
In various control experiments, LPS was used to induce inducible NO‐mediated aortic relaxation in endothelium‐denuded rings. Traces in Figure S3 show that treatment with ICI 182780 did not attenuate LPS‐induced relaxation. In contrast, this concentration markedly reduced the raloxifene‐induced relaxation, as shown in Figure 5.
The LPS‐mediated relaxation was attenuated by treatment with each of the following inhibitors: PDTC (Figure S4), L‐NAME (Figure S5A), AMT‐HCl (Figure S5A), actinomycin D (Figure S5B) or cycloheximide (Figure S5B). The Emax values are summarized in Figure S5C. The above data suggest that LPS‐induced iNOS expression for endothelium‐independent relaxation acts as a control to show that iNOS is involved in endothelium‐denuded aortic relaxation.
Comparison of the vascular effects of raloxifene and nitroprusside
To compare the relaxant effects induced by raloxifene and SNP, another set of experiments was conducted with the NO donor in endothelium‐denuded aortic rings. Figure S6 summarizes the effects of various inhibitors. Raloxifene‐induced relaxation was inhibited by ODQ, L‐NAME, AMT‐HCl, actinomycin D and cycloheximide (Figure S6A), while SNP‐induced relaxation was abolished by ODQ only (Figure S6B). The other four inhibitors were without effect, thus confirming that the cGMP‐dependent mechanism accounts entirely for the NO donor‐mediated aortic response. These experiments also served to demonstrate the specificity of each inhibitor used in this study.
Effects on tissue content of cGMP
The results of the experiments described above strongly suggest that NO is involved in the raloxifene‐mediated relaxant effect in aortic rings. In most instances, NO‐mediated vasorelaxation is caused by activation of cGMP‐dependent signalling pathways leading to a reduction in the intracellular Ca2+ concentrations in VSMC. The reduced relaxation in aortic rings treated with ODQ also indicates that this pathway is involved in raloxifene‐induced vasorelaxant responses, since ODQ is a highly selective and potent inhibitor of guanylate cyclase, which catalyses the generation of cGMP from GTP. In order to verify this, we measured the total tissue content of cGMP in response to raloxifene, at the concentration that relaxed the endothelium‐denuded rings, after different pharmacological treatments. Figure 6A shows that raloxifene (0.1–1 μM) significantly increased the total tissue content of cGMP in endothelium‐denuded rings, and this increase was inhibited by treatment with the inhibitors that were shown to inhibit raloxifene‐induced aortic relaxation. These inhibitors included L‐NAME, ODQ, AMT‐HCl, aminoguanidine, actinomycin D and cycloheximide. Treatment with L‐arginine (1 mM) partially reversed the inhibitory effect of L‐NAME on cGMP production. In control experiments, LPS (10 μg·mL−1) also stimulated an increase in the cGMP level.
Figure 6.
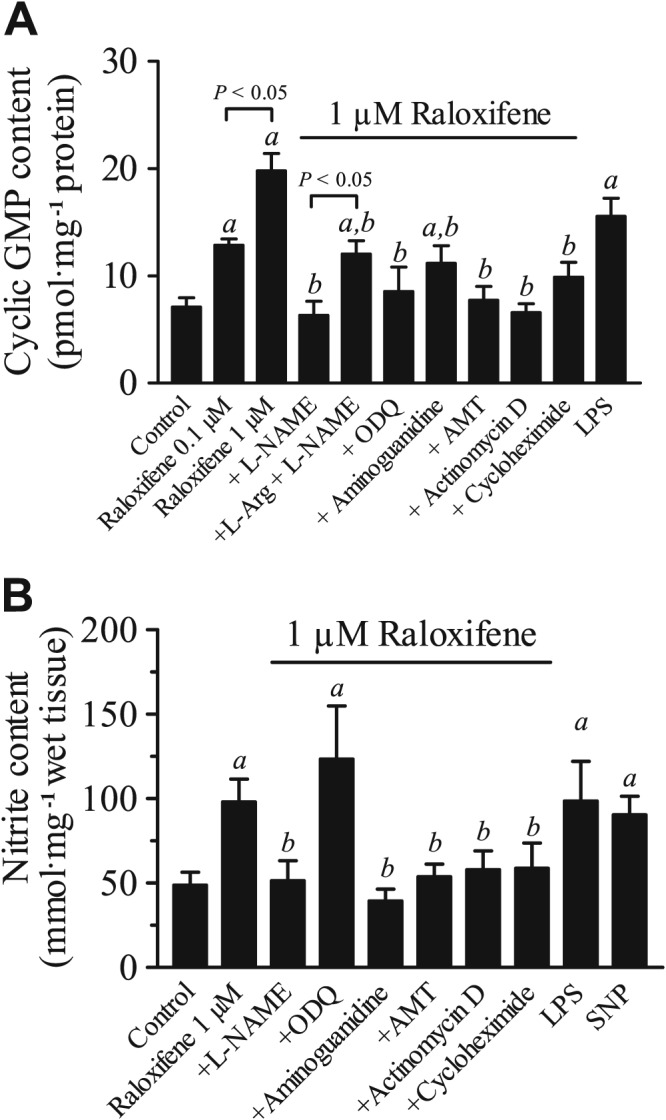
(A) Raloxifene‐stimulated increases in cyclic GMP (cGMP) levels in endothelium‐denuded rat aortic rings and effects of various inhibitors (L‐NAME, ODQ, aminoguanidine, AMT, actinomycin D and cyclohexinide). The effect of LPS (10 μg·mL−1) served as a positive control. Statistical differences (P < 0.05) are indicated by a between control and treatment groups and b between raloxifene (1 μM) and other treatment groups. Results are mean ± SEM of six rings from different rats. (B) Basal and raloxifene‐stimulated release of nitrite and the effects of pharmacological inhibitors on raloxifene‐induced increase in nitrite production. All experiments were performed on endothelium‐denuded rat aortic rings. The effect of LPS (10 μg·mL−1) or SNP (100 nM) served as a positive control. Statistical differences (P < 0.05) are indicated by a between control and treatment groups and b between raloxifene (1 μM) and other treatment groups. Results are mean ± SEM of six to eight rings from different rats.
Effects on nitrite formation in aortic rings
The involvement of NO in raloxifene‐induced relaxation was further supported by the measurement of nitrite, a metabolite of NO degradation. Figure 6B shows that raloxifene (1 μM) induced a significant increase in total nitrite content in the incubation medium bathing the endothelium‐denuded rings, and this increase was inhibited by treatment with L‐NAME, AMT‐HCl, aminoguanidine, actinomycin D or cycloheximide. In positive control experiments, exposure to LPS or SNP (100 nM) increased the nitrite concentration in the incubation medium.
Effects on iNOS protein expression levels
The involvement of iNOS in raloxifene‐induced relaxation was also reflected in the western blotting analysis. Figure 7A shows that a 3 h treatment with 1 μM raloxifene up‐regulated the iNOS protein by increasing its expression in endothelium‐denuded rings, and this change was prevented by pre‐exposure to ICI 182780, PDTC, actinomycin D or cycloheximide. LPS treatment also increased the iNOS protein level in endothelium‐denuded rings. LPS‐treated rat macrophage NR 8383 cells, which expressed iNOS, served as a positive control.
Figure 7.
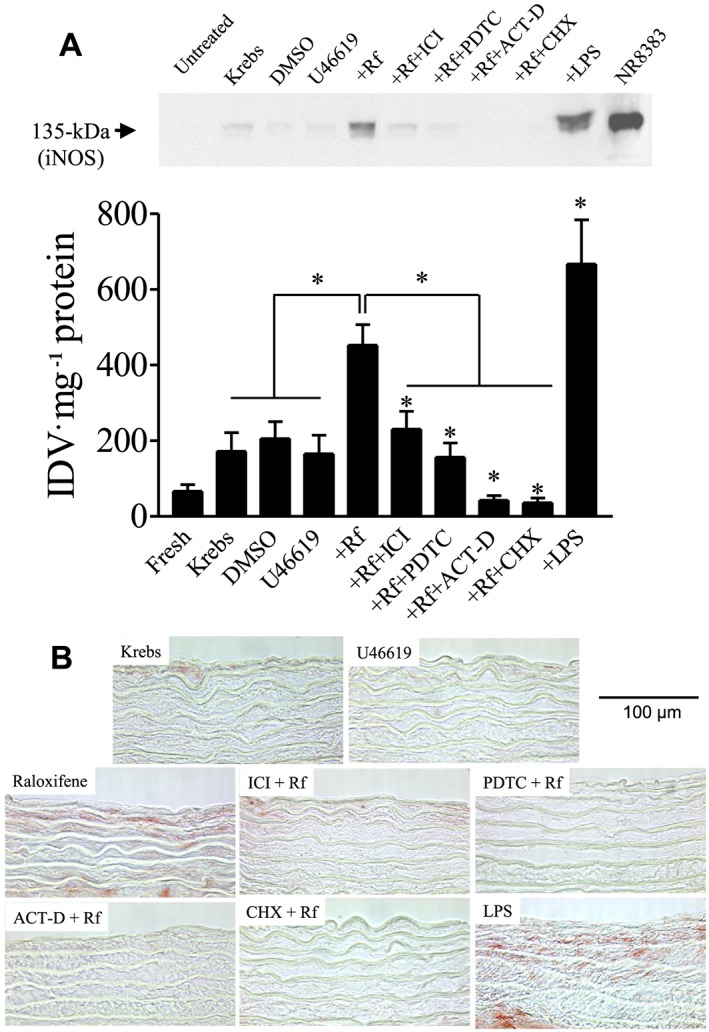
(A) Effects of inhibitors on the increase in iNOS protein expression induced by 1 μM raloxifene after pre‐contracting with 20 nM U46619 in endothelium‐denuded rings. Representative western blot analysis (top panel). Densitometry analysis of results of western blots (bottom panel). Statistical differences are indicated between control and treatment groups, and between raloxifene (1 μM) and other treatment groups (*P < 0.05). Data are expressed as the value of integrated density value (IDV) mg‐1 protein. The results are means ± SEM of 6 to 12 experiments. The position of iNOS with 135 kDa was verified by molecular markers and LPS‐treated rat macrophage NR 8383 cell lysate. (B) Effects of inhibitors on the increase in iNOS protein expression induced by 1 μM raloxifene after pre‐contracting with 20 nM U46619 in endothelium‐denuded rings in vascular smooth muscle. Representative immunohistochemistry sections of endothelium‐denuded rings. Area stained with red colour (like raloxifene and LPS group) represents the location of iNOS induction. Original magnification: ×400. Scale bar: 100 μm. Figure is representative of at least three experiments performed on different experimental days.
Effects on iNOS protein distribution
Immunohistochemical staining of rat aortic rings also showed that raloxifene increased the iNOS protein expression in smooth muscle layers of the endothelium‐denuded rings, and this change was prevented by treatment with ICI 182780, PDTC, actinomycin D or cycloheximide (Figure 7B). In control rings, LPS treatment also increased the level of iNOS protein expression in the smooth muscle layers.
Comparison of the relaxant effects of raloxifene, 17β‐estradiol and tamoxifen
To test the specificity of raloxifene‐induced relaxation, endothelium‐independent relaxations induced by 17β‐estradiol and tamoxifen (oestrogen receptor agonist/antagonist) were tested for comparison. In endothelium‐denuded aortic rings, 1 μM 17β‐estradiol or tamoxifen did not induce any relaxation. However, both of them at 10 μM induced a comparable relaxation response to that of 1 μM raloxifene (Figure S7). Furthermore, 17β‐estradiol (10 μM) and tamoxifen (10 μM)‐induced relaxation were fully inhibited by pretreatment with AMT‐HCl or cycloheximide (Figure S8), suggesting that all of them share a similar genomic mechanism for the inducing iNOS expression and mediating endothelium‐independent relaxation.
The supplemental figures can be found in the online supporting information.
Discussion
VSMC are known to be the primary targets of endothelium‐derived NO during the regulation of vascular tone. The present study provided novel evidence to support the hypothesis that the action of raloxifene in rat isolated aortas is mediated through an induction of iNOS expression and NO production in vascular smooth muscles. This inducible NO subsequently activates a cGMP‐dependent signalling pathway to mediate raloxifene‐induced aortic vasorelaxation.
Relaxation in endothelium‐intact aortic rings
In endothelium‐intact rings, raloxifene‐induced inhibition of U46619‐induced contractile responses was inhibited in the presence of L‐NAME or ODQ, indicating that this effect of raloxifene involves activation of the NOS/guanylate cyclase pathway. However, in the present study it was shown that raloxifene can induce relaxation in both endothelium‐intact and endothelium‐denuded aortas, with the former relaxation being larger and faster during the 3 h treatment with raloxifene. It appears that the area of the gap between the curves of ‘+endo, Rf’ and ‘‐endo, Rf’ in Figure S2 is the relaxation mediated by raloxifene‐induced activation of eNOS. These results indicate that the major source of NO responsible for raloxifene‐induced aortic relaxation is derived from non‐endothelial cells, such as VSMC that are known to express iNOS. The relaxant effect of raloxifene appears to be independent of the types of constrictors used because raloxifene caused almost identical effects in rings precontracted by 60 mM K+.
The role of the endothelium in relaxant responses to raloxifene is controversial. This study showed that the endothelium only has minor role in raloxifene‐induced vasodilatation in rat aorta, as removal of the endothelium only slightly reduced raloxifene‐induced relaxation. The endothelium can also release prostacyclin, which regulates vascular tone in response to some vasoactive agents (Vanhoutte and Mombouli, 1996). In endothelium‐intact rings, treatment with indomethacin (the COX inhibitor used to block prostacyclin biosynthesis) did not affect raloxifene‐induced relaxation. This suggests that the relaxing prostanoids are not involved.
Relaxant effects in endothelium‐denuded aortic rings
In endothelium‐intact or endothelium‐denuded rings, treatment with L‐NAME (non‐specific inhibitor of NOS), or ODQ (inhibitor of guanylate cyclase) attenuated raloxifene‐induced relaxation to the same degree. These data confirmed that the endothelium is not involved in raloxifene‐induced vasodilatation in rat aorta and indicate that VSMC may respond to raloxifene by increasing the production and/or release of NO, which then triggers the cGMP‐dependent pathway for vascular relaxation.
Treatment of endothelium‐denuded rings with aminoguanidine or AMT‐HCl (both are specific iNOS inhibitors, Sung et al., 2002; Md et al., 2003) was equally effective in attenuating raloxifene‐induced relaxation, suggesting that the activation of iNOS is involved in the raloxifene‐induced relaxation. In contrast, treatment with NPLA (a specific nNOS inhibitor, Ferrer et al., 2004) had no effect, indicating neuronal‐derived NO is not involved in this response.
A common molecular target of NO is membrane‐bound guanylate cyclase, an enzyme that catalyses the chemical conversion of GTP into cGMP, the first signalling molecule in the cGMP‐dependent intracellular pathway. Also cGMP‐dependent protein kinase phosphorylates the ion channels to regulate intracellular Ca2+ concentrations (Schroder et al., 2003). Therefore, the tissue levels of cGMP should be increased if NO, regardless of its source, mediates raloxifene‐induced vasorelaxation. Indeed, raloxifene at a concentration that relaxes isolated aortas increased the aortic cGMP content in endothelium‐denuded rings, and this increase was inhibited by L‐NAME, ODQ or iNOS inhibitors (aminoguanidine and AMT‐HCl). These data with iNOS inhibitors further support the notion that activation of iNOS in VSMC elevates cellular guanylate cGMP that then evokes aortic ring relaxation. Nitrite is a stable end product of NO metabolism. The accumulation of nitrite in bathing solution serves as an indicator of NO production. This set of results is consistent with the observed increases in cGMP.
Genomic action
The genomic effects of oestrogens are mediated through activation of nuclear oestrogen receptors. The following findings support the genomic nature of raloxifene‐induced relaxation in endothelium‐denuded aortic rings. Firstly, raloxifene‐induced relaxation was inhibited by ICI 182780 (a selective oestrogen receptor antagonist), actinomycin D (RNA synthesis inhibitor), cycloheximide (protein synthesis inhibitor) and PDTC (an inhibitor of NF‐κB activity). Secondly, all of these inhibitors reduced raloxifene‐stimulated increase in iNOS protein expression in endothelium‐denuded rings. Thirdly, the iNOS inhibitors, actinomycin D, cycloheximide and PDTC but not ICI 182780 nearly abolished LPS‐induced relaxation and iNOS protein expression. LPS acted as a positive control for the stimulation of iNOS leading to vasorelaxation. We compared the effects of the inhibitors on the relaxant responses to raloxifene and sodium nitroprusside, an exogenous NO donor. These results clearly show that the inhibitors acted on specific targets in vascular smooth muscle and that the aortic action of raloxifene is genomic in nature and an NO donor causes non‐genomic relaxation. Although no efforts was made to examine which cell type might have been affected by raloxifene, the immunohistochemical study using an iNOS‐specific antibody showed that iNOS immunoreactivity was clearly localized in vascular smooth muscle layers.
Discrepancies from previous studies
The findings from the present study differ from those of previous studies. In Pinna et al., 2006, raloxifene causes concentration‐dependent relaxation of aorta from oestrogen‐replaced ovariectomized female rats. Parallel to the current study, they also showed that the inhibition of COX has no effect on raloxifene‐induced relaxation, whereas L‐NAME inhibits it. However, this relaxation was inhibited by removal of the endothelium but not by ICI 182780, which is opposite to the results in the current study. Furthermore, in another study (Bolego et al., 2005) in female rat aorta, it was found that the relaxant responses to 17β‐estradiol or the ERα agonist 4,4′,4″‐(4‐propyl‐[1H]pyrazole‐1,3,5‐triyl) tris‐phenol were endothelium‐dependent. Moreover, in male rat aorta, three different SERM agonists, raloxifene, desmethylarzoxifene (DMA) and NO‐DMA, showed similar potency at inducing relaxation, and this effect of DMA was dose‐ and endothelium‐dependent (VandeVrede et al., 2013). Also, raloxifene at 1 μM has been shown to inhibit iNOS expression and NO levels induced by cytokines in rat aortic smooth cells, effects reversed by specific ERα inhibitors (Pinna et al., 2006). Although the concentration of raloxifene and the tissue and species of the cells are same as those used in the current study, the effects of raloxifene on iNOS expression and activity were the opposite of those in the present study. However, in ovariectomized female rat aorta, iNOS expression was found to be increased (Cetinkaya Demir et al., 2013). Also in macrophages, raloxifene has been shown to have an inhibitory, rather than a stimulating effect, on the expression of iNOS via NF‐κB (Lee et al., 2008). Moreover,17β‐estradiol has been shown to inhibit the reduction in contraction evoked by iNOS‐inducers such as TNF‐α or IL‐1β in rat aorta (Kauser et al., 1998). However, the findings of the present study suggest that iNOS is activated by SERMs or 17β‐estradiol. All of these discrepancies may be due to differences in the cell types, species and/or health as well as pathological conditions. Furthermore, the hormonal status of the animals needs to be considered, as it can influence the responsiveness of isolated blood vessels (Shaw et al., 2001). Because oestrogen can influences ER expression in vascular and nonvascular tissues, ER‐dependent responses may be tightly related to the hormonal status of animals (Ihionkhan et al., 2002).
Conclusions
In summary, several novel findings were obtained in this study. They include: (1) raloxifene relaxes the rat aorta primarily through an endothelium‐independent mechanism; (2) raloxifene stimulates an increase in cGMP, NO metabolite (nitrite) and iNOS protein levels in VSMC in vitro through the oestrogen receptor; (3) NF‐κB may be involved in the raloxifene‐mediated activation of iNOS; (4) raloxifene inhibits calcium influx through Ca2+ channels; and (5) raloxifene is more potent than 17β‐estradiol or tamoxifen at relaxing endothelium‐denuded aortic rings by stimulation of iNOS.
Possible clinical implications
Several studies have suggested that activation of iNOS may be beneficial under certain conditions. In contrast to eNOS and nNOS, enhanced iNOS expression leads to an increased production of NO, which causes vasorelaxation of blood vessels (Weigert et al., 1995) and prevents leukocyte adhesion (Hickey et al., 1997) and platelet adhesion (Yan et al., 1996). Thus, the production of NO from VSMC via the iNOS pathway may provide a compensatory mechanism for the reduction in NO generation from dysfunctional endothelial cells under pathological conditions. For example, an increase in iNOS expression or activity of VSMC may substitute for the reduction in endothelium‐derived NO production in the aged organism (Tschudi et al., 1996) and in atherosclerotic lesions (Buttery et al., 1996). However, in general, endothelial dysfunction is associated with pathophysiological conditions often characterized by increased generation of ROS. Under such conditions, increased activity of iNOS may lead to harmful peroxynitrite formation, and hence, iNOS activation is connected with inflammatory conditions. The concentration of the stimuli and the amount of NO production tend to determine whether the induced NO is beneficial (Shears et al., 1997; Yan et al., 1996; Yan and Hansson, 1998) or detrimental (Thiemermann, 1997). Raloxifene‐stimulated iNOS expression may be beneficial in blood vessels with impaired endothelial function if the amount of induced NO does not stimulate vascular inflammatory responses.
Author contributions
C.M.W., C.L.A., S.Y.T. and X.Y. designed the experiments; C.M.W. and C.W.L. performed the experiments; C.M.W. and C.W.L. analysed the data; and C.M.W., A.C.C. and C.Z. wrote the paper.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 A, Concentration‐dependent inhibition of U46619‐induced contractions in endothelium‐intact rat aortic rings by raloxifene (Rf) (n = 6–8). B, pEC50 values for U46619‐induced contraction in the absence and presence of raloxifene (0.1–10 μM, n = 6–8). C, Similar inhibitory effects of 1 μM raloxifene on U46619‐contracted endothelium‐intact (+Endo) and –denuded aortic rings (−Endo) (n = 7–9). D, pEC50 values obtained in (C). Concentration‐response curves for U46619‐induced contractile responses in rat endothelium‐intact aortic rings. Raloxifene reduced the U46619 contraction in the absence and presence of 100 μM L‐NAME (E) or 3 μM ODQ (G). Results are mean ± SEM of 6–9 rings from different rats. F & H, pEC50 values obtained in E & G. Contractions were normalized as percentage of 60 mM K+‐induced tension. Statistical differences are indicated by between different treatment groups (* P < 0.05, ** P < 0.01, *** P < 0.001). NS indicates no significant difference.
Figure S2 The steady‐state maximum relaxation to raloxifene (Rf, 1 μM) in the U46619‐contracted endothelium‐intact (+endo) and endothelium‐denuded (−endo) aortic rings. Statistical differences (P < 0.001) are indicated by a between vehicle control and raloxifene group in endothelium‐intact rings, and b between vehicle control and raloxifene group in endothelium‐denuded rings. Results are mean ± SEM of 6–7 rings from different rats.
Figure S3 A, Recordings showing time‐dependent relaxant responses to 10 μg·mL−1 LPS in endothelium‐denuded aortic rings in control and in the presence of 1 μM ICI 182780. B, No inhibitory effects of ICI 182780 on LPS‐induced aortic relaxation. C, The maximal relaxant effect of LPS in the absence and presence of ICI 182780. Statistical differences are indicated by ** (P < 0.01) or *** (P < 0.001) between vehicle control and treatment groups. Results are mean ± SEM of 6 rings from different rats. NS indicates no significant difference.
Figure S4 A, Recordings showing time‐dependent relaxant responses to 10 μg·mL−1 LPS in endothelium‐denuded aortic rings in control and in the presence of 3 μM PDTC. B, Inhibitory effects of PDTC on LPS‐induced aortic relaxation. C, The maximal relaxant effect of LPS in the absence and presence of PDTC. Statistical differences are indicated by *** (P < 0.001) between vehicle control and LPS group, and a (P < 0.05) between LPS and treatment group. Results are mean ± SEM of 6 rings from different rats.
Figure S5 A, Inhibitory effects of 100 μM L‐NAME or 100 μM AMT‐HCl on LPS (10 μg·mL−1)‐induced relaxation. B, Inhibitory effect of 10 μM actinomycin D or 10 μM cycloheximide on LPS‐induced relaxation. C, The maximal relaxant effect of LPS in the absence and presence of L‐NAME, AMT‐HCl, actinomycin D and cycloheximide. Statistical differences are indicated by *** (P < 0.001) between vehicle control and LPS group, and a (P < 0.05) between LPS and other treatment groups. These experiments were performed on endothelium‐denuded aortic rings. Results are mean ± SEM of 5–6 rings from different rats.
Figure S6 Comparison of relaxations induced by 1 μM raloxifene (A) and 100 nM SNP (B) in endothelium‐denuded aortic rings following different pharmacological treatments. Statistical differences are indicated by * (P < 0.001) between control and treatment groups. Results are mean ± SEM of 6–11 rings from different rats.
Figure S7 A, The time course for the relaxant response induced by raloxifene (1 μM), 17β‐estradiol (E2, 1 and 10 μM) and tamoxifen (Tam, 1 and 10 μM) in the U46619‐contracted endothelium‐denuded aortic rings. B, The steady‐state maximum relaxation to raloxifene, 17β‐estradiol and tamoxifen. Statistical differences are indicated by between vehicle control and other treatment groups (*** P < 0.001). Results are mean ± SEM of 5–6 rings from different rats. NS indicates no significant difference.
Figure S8 A, Inhibitory effects of 100 μM AMT‐HCl or 10 μM cycloheximide on 17β‐estradiol (10 μM)‐induced relaxation in the U46619‐contracted endothelium‐denuded aortic rings. B, The maximal relaxant effect of 17β‐estradiol in the absence and presence of AMT‐HCl and cycloheximide. C, Inhibitory effect of 100 μM AMT‐HCl or 10 μM cycloheximide on tamoxifen (10 μM)‐induced relaxation. D, The maximal relaxant effect of tamoxifen in the absence and presence of AMT‐HCl and cycloheximide. Statistical differences are indicated by *** (P < 0.001) between vehicle control and 17β‐estradiol or tamoxifen group, and a (P < 0.05) between 17β‐estradiol or tamoxifen and other treatment groups. Results are mean ± SEM of 5–6 rings from different rats.
Data S1. Supporting info item
Acknowledgements
C.M.W. was the recipient of a CUHK Postgraduate Studentship. This work is supported by National Natural Science Foundation of China (General Program 21577115 to C.M.W. and 21477101 to A.C.C.); the Research Grant Council of Hong Kong (GRF 463612 and 14104314 to A.C.C.); Faculty Research Grants from the Hong Kong Baptist University (FRG2/15‐16/067 to A.C.C.); Hong Kong Health and Medical Research Fund (HMRF/03144376 to A.C.C.); and HKASO research grant 2015‐16 to A.C.C. We thank Aster Lai‐Fong Chan for excellent technical assistance in immunohistochemistry study.
Wong, C. M. , Au, C. L. , Tsang, S. Y. , Lau, C. W. , Yao, X. , Cai, Z. , and Chung, A. C.‐K. (2017) Role of inducible nitric oxide synthase in endothelium‐independent relaxation to raloxifene in rat aorta. British Journal of Pharmacology, 174: 718–733. doi: 10.1111/bph.13733.
Contributor Information
Chi Ming Wong, Email: henrywong1999@gmail.com.
Arthur Chi‐Kong Chung, Email: chungack@hkbu.edu.hk.
References
- Alexander SPH, Cidlowski JA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors. Br J Pharmacol 172: 5956–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett‐Connor E, Bush TL (1991). Estrogen and coronary heart disease in women. JAMA 265: 1861–1867. [PubMed] [Google Scholar]
- Barrett‐Connor E, Grady D, Sasheyi A, Anderson PW, Cox DA, Hoszowski K et al. (2002). Raloxifene and cardiovascular events in osteoporotic postmenopausal women: four‐year results from the MORE (Multiple Outcomes of Raloxifene Evaluation) randomized trial. JAMA 287: 847–857. [DOI] [PubMed] [Google Scholar]
- Barrett‐Connor E, Mosca L, Collins P, Geiger MJ, Grady D, Kornitzer M et al. (2006). Effects of raloxifene on cardiovascular events and breast cancer in postmenopausal women. N Engl J Med 355: 125–137. [DOI] [PubMed] [Google Scholar]
- Bolego C, Cignarella A, Sanvito P, Pelosi V, Pellegatta F, Puglisi L et al. (2005). The acute estrogenic dilation of rat aorta is mediated solely by selective estrogen‐α agonists and is abolished by estrogen deprivation. J Pharmacol Exp Ther 313: 1203–1208. [DOI] [PubMed] [Google Scholar]
- Buttery L, Springall DR, Chester AH, Evans TJ, Standfield N, Parums DV et al. (1996). Inducibe nitric oxide synthase is present within human atherosclerotic lesions and promotes the formation and activity of peroxynitrite. Lab Invest 75: 77–85. [PubMed] [Google Scholar]
- Cetinkaya Demir B, Uyar Y, Ozbilqin K, Kose C (2013). Effect of raloxifene and atorvastatin in atherosclerotic process in ovarietomized rats. J Obstet Gynaecol Res 39: 229–239. [DOI] [PubMed] [Google Scholar]
- Chan YC, Leung FP, Yao X, Lau CW, Vanhoutte PM, Huang Y (2005). Raloxifene relaxes rat pulmonary arteries and veins: roles of gender, endothelium, and antagonism of Ca2+ influx. J Pharmacol Exp Ther 312: 1266–1271. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer M, Sanchez M, Martin Mdel C, Marquez‐Rodas I, Alonso MJ, Salaices M et al. (2004). Protein kinase A increases electrical stimulation‐induced neuronal nitric oxide release in rat mesenteric artery. Eur J Pharmacol 487: 167–173. [DOI] [PubMed] [Google Scholar]
- Genazzani AR, Gambacciani M (1999). Hormone replacement therapy: the perspectives for 21st century. Maturitas 32: 11–17. [DOI] [PubMed] [Google Scholar]
- Herrington DM, Reboussin DM, Brosnihan KB, Sharp PC, Shumaker SA, Snyder TE et al. (2000). Effects of estrogen replacement on progression of coronary‐artery atherosclerosis. N Engl J Med 343: 522–529. [DOI] [PubMed] [Google Scholar]
- Hickey MJ, Sharkey KA, Sihota EG, Reinhardt PH, Macmicking JD, Nathan C et al. (1997). Inducible nitric oxide synthase‐deficient mice have enhanced leukocyte‐endothelium interactions in endotoxemia. FASEB J 11: 955–964. [DOI] [PubMed] [Google Scholar]
- Ihionkhan CE, Chambliss KL, Gibson LL, Hahner LD, Mendelsohn ME, Shaul PW (2002). Estrogen causes dynamic alternations in endothelial estrogen receptor expression. Circ Res 91: 814–820. [DOI] [PubMed] [Google Scholar]
- Kauser K, Sonnenberg D, Diel P, Rubanyi GM (1998). Effect of 17β‐oestradiol on cytokine‐induced nitric oxide production in rat isolated aorta. Br J Pharmacol 123: 1089–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagro‐Janssen A, Knufing MW, Schreurs L, van Weel C (2010). Significant fall in hormone replacement therapy prescription in general practice. Fam Pract 27: 424–429. [DOI] [PubMed] [Google Scholar]
- Lee SA, Park SH, Kim BC (2008). Raloxifene, a selective estrogen receptor modulator, inhibits lipopolysaccharide‐induced nitric oxide production by inhibiting the phosphatidylinositol 3‐kinase/Akt/nuclear factor kappa B pathway in RAW264.7 macrophage cells. Mol Cells 26: 48–52. [PubMed] [Google Scholar]
- Leung FP, Yao X, Lau CW, Ko WH, Lu L, Huang Y (2005). Raloxifene relaxes rat intrarenal arteries by inhibiting Ca2+ influx. Am J Physiol Renal Physiol 289: F137–F144. [DOI] [PubMed] [Google Scholar]
- Leung HS, Seto SW, Kwan YW, Leung FP, Au AL, Yung LM et al. (2007). Endothelium‐independent relaxation to raloxifene in porcine coronary artery. Eur J Pharmacol 555: 178–184. [DOI] [PubMed] [Google Scholar]
- Manson JE, Chlebowski RT, Stefanick ML et al. (2013). Menopausal hormone therapy and health outcomes during the intervention and extended poststopping phases of the Women's Health Initiative randomized trials. JAMA 310: 1353–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Md S, Moochhala SM, Siew‐Yang KL (2003). The role of inducible nitric oxide synthase inhibitor on the arteriolar hyporesponsiveness in hemorrhagic‐shocked rats. Life Sci 73: 1825–1834. [DOI] [PubMed] [Google Scholar]
- Nabel EG (2000). Coronary heart disease in women – an ounce of prevention. N Engl J Med 343: 572–574. [DOI] [PubMed] [Google Scholar]
- Pinna C, Bolego C, Sanvito P, Pelosi V, Baetta R, Corsini A et al. (2006). Raloxifene elicits combined rapid vasorelaxation and long‐term anti‐inflammatory actions in rat aorta. J Pharmacol Exp Ther 319: 1444–1451. [DOI] [PubMed] [Google Scholar]
- Rahimian R, Dube GP, Toma W, Dos Santos N, McManus BM, van Breemen C (2002). Raloxifene enhances nitric oxide release in rat aorta via increasing endothelial nitric oxide mRNA expression. Eur J Pharmacol 434: 141–149. [DOI] [PubMed] [Google Scholar]
- Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML et al. (2002). Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women's Health Initiative randomized controlled trial. JAMA 288: 321–233. [DOI] [PubMed] [Google Scholar]
- Saitta A, Altavilla D, Cucinotta D, Morabito N, Frisina N, Corrado F et al. (2001). Randomized, double‐blind, placebo‐controlled study on effects of raloxifene and hormone replacement therapy on plasma NO concentrations, endothelin‐1 levels, and endothelium‐dependent vasodilation in postmenopausal women. Arterioscler Thromb Vasc Biol 21: 1512–1519. [DOI] [PubMed] [Google Scholar]
- Schierbeck L, Rejnmark L, Landbo Tofteng C et al. (2012). Effect of hormone replacement therapy on cardiovascular events in recently postmenopausal women: randomized trial. BMJ 345: e6409. [DOI] [PubMed] [Google Scholar]
- Schroder F, Klein G, Fiedler B, Bastein M, Schnasse N, Hillmer A et al. (2003). Single L‐type Ca(2+) channel regulation by cGMP‐dependent protein kinase type I in adult cardiomyocytes from PKG I transgenic mice. Cardiovasc Res 60: 268–277. [DOI] [PubMed] [Google Scholar]
- Shaw L, Taggart M, Austin C (2001). Effects of the oestrous cycle and gender on acute vasodilatory responses of isolated pressurized rat mesenteric arteries to 17β‐oestradiol. Br J Pharmacol 132: 1055–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shears LL, Kawaharada N, Tzeng E, Billiar TR, Watkins SC, Kovesdi I et al. (1997). Inducible nitric oxide synthase suppresses the development of allograft arteriosclerosis. J Clin Invest 100: 2035–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl. Acids Res. 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stampfer MJ, Colditz GA, Willett WC, Manson JE, Rosner B, Speizer FE et al. (1991). Postmenopausal estrogen therapy and cardiovascular disease. Ten‐year follow‐up from the nurses' health study. N Engl J Med 325: 756–762. [DOI] [PubMed] [Google Scholar]
- Sung JM, Low KS, Khoo HE (2002). Characterization of the mechanism underlying stonustoxin‐mediated relaxant response in the rat aorta in vitro. Biochem Pharmacol 63: 1113–1118. [DOI] [PubMed] [Google Scholar]
- Thiemermann C (1997). Nitric oxide and septic shock. Gen Pharmacol 29: 159–166. [DOI] [PubMed] [Google Scholar]
- Tsang SY, Yao XQ, Essin K, Wong CM, Chan FL, Gollasch M et al. (2004). Raloxifene relaxes rat ceberal arteries in vitro and inhibits L‐type voltage‐sensitive Ca2+ channels. Stroke 35: 1709–1714. [DOI] [PubMed] [Google Scholar]
- Tschudi MR, Barton M, Bersinger NA, Moreau P, Cosentino F, Noll G et al. (1996). Effect of age on kinetics of nitric oxide release in rat aorta and pulmonary artery. J Clin Invest 98: 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valera M, Gourdy P, Tremollieres F, Arnal J (2015). From the Women's Health Initiative to the combination of estrogen and selective estrogen receptor modulators to avoid progestin addition. Maturitas 82: 274–277. [DOI] [PubMed] [Google Scholar]
- VandeVrede L, Adelhamid R, Qin Z, Choi J, Piyankarage S, Luo J et al. (2013). An NO donor approach to neuroprotective and precognitive estrogen therapy overcomes loss of NO synthase function and potentially thrombotic risk. PLoS One 8: e70740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhoutte PM, Mombouli JV (1996). Vascular endothelium: vasoactive mediators. Prog Cardiovasc Dis 39: 229–238. [DOI] [PubMed] [Google Scholar]
- Weigert AL, Higa EM, Niederberger M, McMurtry IF, Raynolds M, Schrier RW (1995). Expression and preferential inhibition of inducible nitric oxide synthase in aortas of endotoxemic rats. J Am Soc Nephrol 5: 2067–2072. [DOI] [PubMed] [Google Scholar]
- Wong CM, Yao X, Au CL, Tsang SY, Fung KP, Laher I et al. (2006). Raloxifene prevents endothelial dysfunction in aging ovariectomized female rats. Vascul Pharmacol 44: 290–298. [DOI] [PubMed] [Google Scholar]
- Wong CM, Yung LM, Leung FP, Tsang SY, Au CL, Chen ZY et al. (2008). Raloxifene protects endothelial cell function against oxidative stress. Br J Pharmacol 155: 326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Hansson GK (1998). Overexpression of inducible nitric oxide synthase by neointimal smooth muscle cells. Circ Res 82: 21–29. [DOI] [PubMed] [Google Scholar]
- Yan Z, Yokota T, Zhang W, Hansson GK (1996). Expression of inducible nitric oxide synthase inhibits platelet adhension and restores blood flow in the injured artery. Circ Res 79: 38–44. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 A, Concentration‐dependent inhibition of U46619‐induced contractions in endothelium‐intact rat aortic rings by raloxifene (Rf) (n = 6–8). B, pEC50 values for U46619‐induced contraction in the absence and presence of raloxifene (0.1–10 μM, n = 6–8). C, Similar inhibitory effects of 1 μM raloxifene on U46619‐contracted endothelium‐intact (+Endo) and –denuded aortic rings (−Endo) (n = 7–9). D, pEC50 values obtained in (C). Concentration‐response curves for U46619‐induced contractile responses in rat endothelium‐intact aortic rings. Raloxifene reduced the U46619 contraction in the absence and presence of 100 μM L‐NAME (E) or 3 μM ODQ (G). Results are mean ± SEM of 6–9 rings from different rats. F & H, pEC50 values obtained in E & G. Contractions were normalized as percentage of 60 mM K+‐induced tension. Statistical differences are indicated by between different treatment groups (* P < 0.05, ** P < 0.01, *** P < 0.001). NS indicates no significant difference.
Figure S2 The steady‐state maximum relaxation to raloxifene (Rf, 1 μM) in the U46619‐contracted endothelium‐intact (+endo) and endothelium‐denuded (−endo) aortic rings. Statistical differences (P < 0.001) are indicated by a between vehicle control and raloxifene group in endothelium‐intact rings, and b between vehicle control and raloxifene group in endothelium‐denuded rings. Results are mean ± SEM of 6–7 rings from different rats.
Figure S3 A, Recordings showing time‐dependent relaxant responses to 10 μg·mL−1 LPS in endothelium‐denuded aortic rings in control and in the presence of 1 μM ICI 182780. B, No inhibitory effects of ICI 182780 on LPS‐induced aortic relaxation. C, The maximal relaxant effect of LPS in the absence and presence of ICI 182780. Statistical differences are indicated by ** (P < 0.01) or *** (P < 0.001) between vehicle control and treatment groups. Results are mean ± SEM of 6 rings from different rats. NS indicates no significant difference.
Figure S4 A, Recordings showing time‐dependent relaxant responses to 10 μg·mL−1 LPS in endothelium‐denuded aortic rings in control and in the presence of 3 μM PDTC. B, Inhibitory effects of PDTC on LPS‐induced aortic relaxation. C, The maximal relaxant effect of LPS in the absence and presence of PDTC. Statistical differences are indicated by *** (P < 0.001) between vehicle control and LPS group, and a (P < 0.05) between LPS and treatment group. Results are mean ± SEM of 6 rings from different rats.
Figure S5 A, Inhibitory effects of 100 μM L‐NAME or 100 μM AMT‐HCl on LPS (10 μg·mL−1)‐induced relaxation. B, Inhibitory effect of 10 μM actinomycin D or 10 μM cycloheximide on LPS‐induced relaxation. C, The maximal relaxant effect of LPS in the absence and presence of L‐NAME, AMT‐HCl, actinomycin D and cycloheximide. Statistical differences are indicated by *** (P < 0.001) between vehicle control and LPS group, and a (P < 0.05) between LPS and other treatment groups. These experiments were performed on endothelium‐denuded aortic rings. Results are mean ± SEM of 5–6 rings from different rats.
Figure S6 Comparison of relaxations induced by 1 μM raloxifene (A) and 100 nM SNP (B) in endothelium‐denuded aortic rings following different pharmacological treatments. Statistical differences are indicated by * (P < 0.001) between control and treatment groups. Results are mean ± SEM of 6–11 rings from different rats.
Figure S7 A, The time course for the relaxant response induced by raloxifene (1 μM), 17β‐estradiol (E2, 1 and 10 μM) and tamoxifen (Tam, 1 and 10 μM) in the U46619‐contracted endothelium‐denuded aortic rings. B, The steady‐state maximum relaxation to raloxifene, 17β‐estradiol and tamoxifen. Statistical differences are indicated by between vehicle control and other treatment groups (*** P < 0.001). Results are mean ± SEM of 5–6 rings from different rats. NS indicates no significant difference.
Figure S8 A, Inhibitory effects of 100 μM AMT‐HCl or 10 μM cycloheximide on 17β‐estradiol (10 μM)‐induced relaxation in the U46619‐contracted endothelium‐denuded aortic rings. B, The maximal relaxant effect of 17β‐estradiol in the absence and presence of AMT‐HCl and cycloheximide. C, Inhibitory effect of 100 μM AMT‐HCl or 10 μM cycloheximide on tamoxifen (10 μM)‐induced relaxation. D, The maximal relaxant effect of tamoxifen in the absence and presence of AMT‐HCl and cycloheximide. Statistical differences are indicated by *** (P < 0.001) between vehicle control and 17β‐estradiol or tamoxifen group, and a (P < 0.05) between 17β‐estradiol or tamoxifen and other treatment groups. Results are mean ± SEM of 5–6 rings from different rats.
Data S1. Supporting info item