

Abstract
Neural pathways, especially those in the hypothalamus, integrate multiple nutritional, hormonal, and neural signals, resulting in the coordinated control of body weight balance and glucose homeostasis. Nuclear receptors (NRs) sense changing levels of nutrients and hormones, and therefore play essential roles in the regulation of energy homeostasis. Understanding the role and the underlying mechanisms of NRs in the context of energy balance control may facilitate the identification of novel targets to treat obesity. Notably, NRs are abundantly expressed in the brain, and emerging evidence indicates that a number of these brain NRs regulate multiple aspects of energy balance, including feeding, energy expenditure and physical activity. In this Review we summarize some of the recent literature regarding effects of brain NRs on body weight regulation and discuss mechanisms underlying these effects.
Introduction
Obesity is a serious global health problem due to its increasing prevalence and comorbidities. Over 30% of Americans are obese (1). Obese individuals are at an increased risk for developing type 2 diabetes, cardiovascular disease, and cancer. These observations highlight the urgent need to better understand the physiology of body weight control, which may facilitate the development of more effective therapies for obesity.
The brain, including the hypothalamus, integrates information regarding the body’s nutritional status and orchestrates a series of coordinated endocrine, autonomic, and behavioral responses that regulate body weight (2–4). Development of Cre-loxP conditional gene manipulation and more recent circuit functional mapping using optogenetic and chemogenetic approaches have facilitated advances in our understanding of how complex brain neural circuits regulate energy homeostasis, particularly within the hypothalamus.
Among the numerous hypothalamic nuclei, the arcuate nucleus of the hypothalamus (ARH) has received the most extensive attention. In particular, ARH neurons that express proopiomelanocortin (POMC) and those that co-release neuropeptide Y, agouti-related peptide (AgRP), and GABA (referred to as AgRP neurons in this Review) have long been believed to be the primary central regulators of energy homeostasis (5, 6). POMC neurons synthesize and secrete an anorexigenic peptide, α-melanocyte-stimulating hormone (α-MSH), which prevents overeating and body weight gain (7, 8), while AgRP neurons are indispensable for normal feeding and survival (9, 10), as activation of AgRP neurons rapidly promotes eating even when mice are satiated (11, 12). Inhibitory AgRP neurons project locally to POMC neurons and to a number of distant targets throughout the brain; the latter include the paraventricular nucleus of the hypothalamus (PVH), the paraventricular nucleus of the thalamus (PVT), the parabrachial nucleus (PBN), the bed nucleus of stria terminalis (BNST), the lateral hypothalamic area (LHA), and the medial amygdala (MeA). Many of these projections (e.g., POMC, PVH, PVT, PBN, BNST, and LHA) have been shown to promote feeding behavior (9, 13–15). In addition, subsets of neurons in the PVH project to and activate AgRP neurons to trigger feeding (16). While α-MSH activates melanocortin-4 receptors (MC4Rs) to prevent body weight gain, AgRP (as an endogenous antagonist) inhibits MC4Rs to promote body weight gain. In particular, MC4Rs expressed in the PVH and MeA specifically inhibit food intake (17). In particular, PVH MC4R neurons suppress food intake by projecting to the PBN (18), while the targets mediating anorexigenic effects of MeA MC4R neurons remain unknown. ARH AgRP neurons also provide inhibitory GABAergic inputs to PBN neurons to promote feeding (9, 19). The PBN receives excitatory inputs from glutamatergic neurons in the nucleus tractus solitarius (NTS), and activation of these PBN neurons inhibits feeding (15). The LHA is another complex hypothalamic nucleus that plays an essential role in feeding control. Activation of glutamatergic neurons in the LHA inhibits feeding (20) through their projections to the lateral habenula (21); in contrast, activation of GABAergic neurons in the LHA promotes feeding (22, 23) through their projections to the PVH (22). While we do not intend to systemically review the complex brain neural circuits that regulate energy homeostasis (please see refs. 24–26 for excellent reviews on this topic), it is clear that numerous brain structures, including the ARH, PVH, LHA, MeA, PBN, NTS, and many more, are heavily involved in the regulation of body weight balance (Figure 1).
Figure 1. The complex brain neural circuits regulating body weight balance.

Red lines represent anorexigenic circuits, and blue lines represent orexigenic circuits.
Nuclear receptors (NRs) represent a large family of transcription factors found in all metazoan species (27). Many NRs have known endogenous ligands, including endocrine steroids (i.e., corticosteroids, progesterone, androgens, and estrogens), fat-soluble vitamins A and D, thyroid hormone, fatty acids, oxysterols, bile acids, and numerous diet-derived xenobiotic lipids (28). Additional members of the NR family are called orphan receptors because their ligands remain unknown. NRs govern the expression of genes involved in a broad range of biological processes, including reproduction, development, immune responses, metabolism, and oncogenesis. Among the total 48 human NRs, only three — farnesoid X receptor (FXR), constitutive androstane receptor, and pregnane X receptor — are absent or minimally expressed in the brain (29). Thus, we will not discuss these three NRs in the current Review. However, it is important to note that FXR was recently identified as a key mediator of the metabolic benefits induced by vertical sleeve gastrectomy bariatric surgeries (30). The vast majority of NRs are found to be expressed at modest to high levels in the brain, including in the hypothalamus (29). In particular, recent gene profiling efforts revealed that many genes (including NRs) expressed by AgRP and/or POMC neurons undergo dramatic changes during the feed-fast transition (31). These provide a neuroanatomic basis for detailed investigation of NRs in the brain, including those involved in regulating energy balance. Notably, several groups have recently focused on the complex functions of brain NRs in the regulation of energy homeostasis. It should be noted that the actions of NRs in peripheral tissues (e.g., adipose tissue, muscle, and liver) also are important for regulation of metabolism (readers are directed to refs. 32 and 33 for excellent reviews on this topic), but this Review will focus on the functions of NRs in the brain in the control of body weight.
Steroidogenic factor-1
One of the most studied hypothalamic NRs is steroidogenic factor-1 (SF1, also known as NR5A1). SF1 has a highly restricted expression pattern in the brain, with abundant expression in the ventromedial hypothalamic nucleus (VMH) (34, 35). A recent, detailed lineage-tracing study by Cheung et al. indicates that rodent brains begin to express SF1 prior to the onset of neurogenesis and that SF1-expressing cells give rise to all neurons in the VMH, although a subdivision of VMH, the ventrolateral VMH, further differentiates into a neuronal cluster devoid of SF1 (36). Embryonic deletion of Sf1 is perinatal lethal and results in malformation of the VMH (37, 38), suggesting that early expression of SF1 is required for the normal development of the VMH structure. Interestingly, global Sf1 knockout mice develop obesity if they survive the perinatal period (39), consistent with the notion that VMH neurons are required to maintain normal body weight balance (40). However, the obese phenotype in global Sf1 knockout mice may partly result from the loss of SF1 in peripheral tissues, including the pituitary gland, the adrenal gland, and the gonads (41). To circumvent these issues, Kim et al. crossed nestin-Cre mice with Sf1fl/fl mice to achieve prenatal deletion of Sf1 selectively in the brain VMH (42). This early deletion of Sf1 in mice disrupted the formation of the VMH, resulting in increased body weight gain (compared with controls) when challenged with a high-fat diet (HFD) (42). Furthermore, the same group crossed Camk2a-Cre with Sf1fl/fl mice. Since Camk2a-Cre mice express Cre recombinase at approximately P15, these crosses resulted in a postnatal deletion of Sf1 selectively in the brain VMH (42). Interestingly, postnatal Sf1 deletion does not have any obvious effects on the formation of the VMH structure; however, the mutant mice still show increased sensitivity to diet-induced obesity (42). This obese phenotype is associated with impaired thermogenic responses to HFD feeding as well as blunted leptin-induced anorexia (42). Collectively, these observations indicate that SF1 is essential for the formation of multiple VMH cell types and for the development of the hypothalamic nucleus. In contrast, in the postnatal VMH, SF1 may also play a physiologically relevant role in regulating the functions of VMH neurons, including those regulating energy balance and glucose homeostasis.
The mechanisms by which SF1 regulates VMH neural functions may involve its transcriptional activity on target genes. While this area needs to be explored in depth, a few putative targets have been identified. For example, SF1 has been demonstrated to regulate expression of several VMH genes that are important for body weight control, including brain-derived neurotropic factor (BDNF) (43, 44) and cannabinoid receptor type 1 (CB1) (44). Additionally, Sf1 is required for leptin-induced phosphorylation of STAT3 (42), suggesting that Sf1 also participates in leptin signaling. Further, glutamate release from SF1 neurons has been implicated in the regulation of body weight, glucose balance (45), locomotor activity, and anxiety in mice (46). While these studies shed some light on the molecular mechanisms underlying SF1 functions in VMH neurons, the full details of SF1 actions remain to be elucidated.
Estrogen receptors
Estrogen receptors (ERs) are perhaps among the most studied NRs in the context of energy balance. In particular, ERα is believed to be the primary ER that mediates estrogenic actions to prevent body weight gain. For example, humans or mice with mutations in the ERα gene (Esr1) are obese (47, 48), and Esr1 knockout mice are unresponsive to the anti-obesity effects of 17β-estradiol (E2) replacement (49). ERα expressed in the brain has been implicated in body weight control, as suggested by early observations that microinjections of E2 into various brain regions reduce food intake and body weight in animals (50, 51). Further supporting this notion, we (Xu and Clegg) crossed the nestin-Cre mice and Esr1fl/fl mice to produce mice with selective deletion of ERα in the brain (52). We found that both male and female mutant mice develop obesity associated with increased food intake, decreased energy expenditure, and decreased physical activity (52). Notably, female mutant mice display elevated E2 in their circulation (52), presumably due to impaired negative feedback regulation by estrogen signaling. Given that these mice develop robust obese phenotypes despite the higher E2 in the circulation, we suggest that, compared with ERα expressed in peripheral tissues, brain ERα plays a predominant role in the regulation of energy balance.
Independent efforts from several groups have further narrowed down the critical ERα-expressing cell populations in the brain that regulate body weight balance. Musatov et al. knocked down ERα specifically in the VMH in mice, which resulted in loss of sensitivity to E2-induced weight loss and the development of obesity (53). Interestingly, the obese syndrome in these mice is likely related to decreased physical activity and impaired thermogenesis, whereas food intake is not directly affected (53). In parallel, we crossed Esr1fl/fl mice and SF1-Cre mice to achieve selective deletion of 50% of ERα in the VMH (52). These female mutant mice show modest body weight gain associated with normal food intake but also show profound decreases in basal metabolism and brown adipose tissue–mediated (BAT-mediated) thermogenesis (52). Martínez de Morentin et al. reported that injections of E2 into the VMH promote BAT-mediated thermogenesis in a feeding-independent manner (54). Notably, Correa et al. recently developed a mouse model with loss of 26% of ERα-positive neurons in the VMH; these mice develop profound obesity associated with decreases in physical activity but normal BAT-mediated thermogenesis and food intake (55). Collectively, these observations in different models highlight an important role for VMH ERα signaling in stimulating basal metabolism, thermogenesis, and physical activity.
We recently reported that selective deletion of ERα from the MeA also leads to obesity in mice (56). Interestingly, these mutant mice display normal food intake and thermogenesis but show robust decreases in physical activity (56), indicating that MeA ERα is important to maintain normal physical activity but does not regulate feeding and thermogenesis. Furthermore, we found that the loss of ERα in the MeA causes obesity not only in female mice, but also in male mice (56). Notably, the MeA expresses abundant aromatase (57), an enzyme that converts testosterone into E2. Thus, we speculate that ERα in the male MeA may be exposed to high levels of E2 that is locally synthesized and therefore exerts physiologically relevant functions in the context of body weight control.
Administration of estrogen into the brain also suppresses food intake (58). This E2-induced anorexia appears to be mediated by multiple ERα populations in the brain, including ARH POMC neurons. E2 can increase excitatory synaptic inputs onto ARH POMC neurons (59). In addition, E2 stimulates POMC neurons by rapidly uncoupling GABAB receptors from the G protein–gated inwardly rectifying K+ channels (60). We reported that a selective ERα agonist, propylpyrazole triol, can rapidly depolarize ERα-positive POMC neurons (61). Furthermore, we showed that female mice lacking ERα only in POMC neurons develop hyperphagia and modest body weight gain (52). Moreover, E2-induced anorexigenic effects are blunted in mice lacking ERα only in POMC neurons (62). Taken together, these observations indicate that ERα in POMC neurons mediates estrogenic actions to suppress food intake (52). Notably, E2-induced anorexia is accompanied by increased neural activity in NTS neurons; these responses are abolished in global ERα knockout mice (49, 63). In addition, direct administration of E2 in the NTS potentiates satiety signals (64). Collectively, these findings support the notion that ERα in the NTS may be another physiologically important site to mediate E2-induced anorexia.
Based on the types of studies described above, an interesting segregation model is beginning to emerge (Figure 2) in which ERα in the VMH enhances energy expenditure by stimulating basal metabolism, BAT-mediated thermogenesis, and physical activity. ERα in the MeA specifically stimulates physical activity, while ERα in the ARH and NTS prevents body weight gain primarily by suppressing energy intake. These segregated ERα populations may function complementarily to mediate the full spectrum of estrogenic effects on energy homeostasis.
Figure 2. Neuroanatomic segregation of brain ERα functions in female body weight balance.
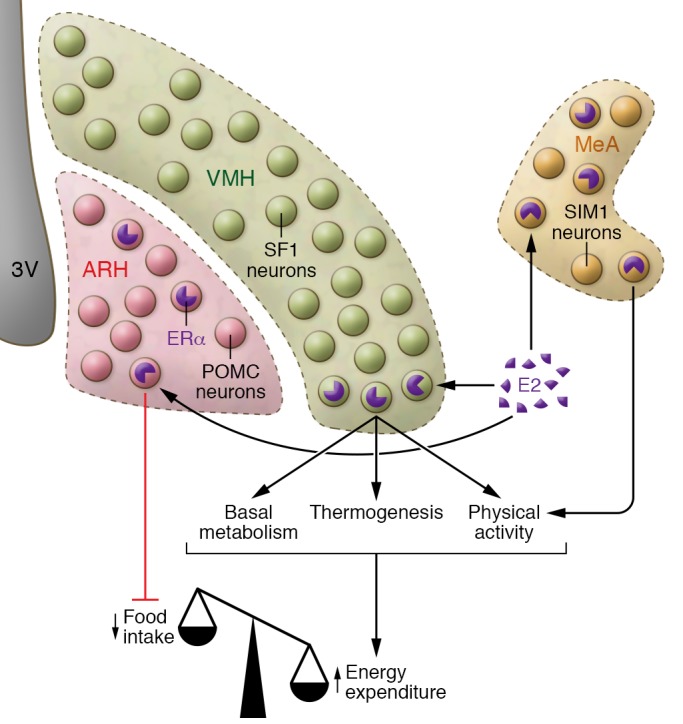
In female brains, ERα expressed by ARH POMC neurons and NTS neurons (not shown in this figure) primarily suppresses food intake; ERα expressed by VMH SF1 neurons enhances basal metabolism, thermogenesis, and physical activity; and ERα expressed by MeA single-minded-1 (SIM1) neurons stimulates physical activity. These distinct ERα populations, as well as other unidentified ERα neurons, function complementarily to mediate the full spectrum of estrogenic effects on female energy homeostasis.
Interestingly, despite the fact that ERα is a classic nuclear receptor, current evidence suggests that transcriptional activity of ERα has only minor effects on body weight balance. This notion primarily arises from observations from a non-classical estrogen receptor knock-in (NERKI) mouse model in which E207A/G208A mutations were introduced in the DNA binding domain of ERα to abolish ERα transcriptional activity (65). Since these NERKI mice display normal body weight, energy expenditure, and physical activity (66), it has been suggested that the direct binding of ERα to DNA and its associated gene transcription are not required to maintain body weight. On the other hand, several rapid intracellular signals coupled to ERα have been implicated in mediating estrogenic actions on body weight balance. For example, E2 stimulates the PI3K/Akt cascade in VMH neurons (66, 67) as well as in ARH POMC neurons (62). Furthermore, E2 rapidly stimulates the firing of VMH neurons (67) and ARC POMC neurons (60, 62); these effects can be blocked by a PI3K inhibitor. Importantly, genetic inhibition of PI3K in VMH or ARH POMC neurons blunts the anti-obesity effects of E2 (62, 67), highlighting an important role of PI3K in mediating the effects of E2 on body weight balance. In parallel, it has been reported that activation of ERα in the VMH leads to inhibition of the AMPK pathway and this AMPK inhibition triggers increased thermogenesis (54). Together, these observations support a model in which ERα-initiated rapid signaling pathways, including PI3K and AMPK, mediate estrogenic actions to prevent body weight gain.
The role of ERβ in body weight control has been less studied. An earlier publication reported normal body weight in global ERβ knockout mice fed regular chow (68). However, a recent study demonstrated that ERβ knockout mice are more sensitive to HFD-induced obesity compared with wild-type mice (69). Consistently, selective ERβ agonists (β-LGNDs) are reported to attenuate HFD-induced body weight gain (70). Thus, while ERβ has a minor effect on body weight balance in animals fed regular diets, it appears to play a more important role in preventing obesity in the face of an obesogenic diet challenge. How much of these ERβ effects are attributed to its direct actions in the brain remains unclear.
Androgen receptor
The role of androgen receptor (AR) in body weight control was suggested by an abnormal body weight profile in male mice with global AR knockout (71, 72). Interestingly, while young mutant mice show decreased body weight, aged mutant mice have increased body weight and adiposity compared with age-matched wild-type males (71, 72). Although selective deletion of AR in the brain (resulting from crosses between synapsin 1-Cre mice and ARfl/y) does not affect body weight in young male mice, such deletion leads to late-onset obesity (73). These observations indicate that brain AR is required to prevent aging-associated obesity, while AR expressed by peripheral tissues (e.g., muscle) may be required to maintain normal body mass during early adulthood (74). Further characterization of the brain-specific AR deletion mouse model revealed increased AgRP expression and impaired insulin sensitivity in the hypothalamus, which presumably contribute to development of obesity in these mice (73). Interestingly, HFD feeding was found to accelerate obesity development, and this phenomenon was associated with a heightened activity of hypothalamic NF-κB and increased expression of protein-tyrosine phosphatase 1B (PTP1B) (73).
Thyroid hormone receptor
Thyroid hormone plays a profound role in the regulation of energy balance, and many of these effects can be attributed to its direct actions on metabolically active tissues in the periphery, such as BAT (reviewed in ref. 75). However, emerging evidence suggests that the CNS also may be a key site of thyroid hormone actions that influence metabolism. For example, glial cells in the ARH produce type 2 deiodinase (D2), an enzyme that catalyzes active thyroid hormone (T3) production, and these D2-producing glial cells are in direct apposition to AgRP neurons (76). Interestingly, food deprivation increases D2 activity and consequently the production of T3 in the ARH, which is associated with increased GDP-regulated UCP2-dependent mitochondrial uncoupling (76). This T3-mediated UCP2 activation leads to mitochondrial proliferation and increased excitability of AgRP neurons, which trigger hungry animals to refeed (76). On the other hand, genetic inhibition of thyroid hormone receptor (TR) via virus-mediated overexpression of a dominant-negative TR in the VMH of rats prevents body weight loss induced by experimental hyperthyroidism; these body weight changes are associated with unchanged food intake but are likely attributed to decreased thermogenesis (77). Interestingly, the same TR inhibition in the VMH does not affect body weight in euthyroid rats (77), suggesting a stronger metabolic function of VMH TR in the hyperthyroid condition. While the overall effects of brain TR actions on energy balance remain to be elucidated, the current data indicate that TR-mediated effects may vary depending on the site of action and/or on various physiologic and pathophysiologic conditions.
Glucocorticoid receptor
It has been long recognized that glucocorticoids play an important role in regulating body weight balance. For example, increased glucocorticoids are associated with obesity in humans, while decreased glucocorticoids are associated with anorexia (78). In rodents, depletion of glucocorticoid via adrenalectomy can prevent obesity induced by genetic mutations (e.g., leptin deficiency) (79, 80). Central administration of glucocorticoids in rats produces sustained body weight gain that is associated with increased food intake and decreased energy expenditure, while peripheral delivery of glucocorticoids decreases food intake and body weight (81). Consistent with the obesogenic effects of central glucocorticoids, brain-specific deletion of the glucocorticoid receptor (GR) in mice (through crosses with nestin-Cre mice and GRfl/fl mice) leads to decreased body weight and food intake as well as increased energy expenditure (82). At least a portion of these central effects of glucocorticoids are mediated by GR expressed by AgRP neurons, since selective deletion of GR from these neurons partially recapitulates the lean phenotypes seen in mice with brain-specific GR deletion (83). Notably, GR in AgRP neurons appears to only affect energy expenditure and has no effect on food intake (83). Thus, other unexplored GR populations in the brain likely mediate the orexigenic effects of glucocorticoids.
Vitamin D receptor
Vitamin D supplementation has been shown to decrease body weight and fat in humans (84–86). Consistently, vitamin D prevents diet-induced obesity in mice (87). These anti-obesity effects of vitamin D may be at least partly mediated by vitamin D receptor (VDR) in the brain because central administration of a small dose of vitamin D produces profound body weight-lowering effects in HFD-fed rats that are associated with decreased food intake (88). Furthermore, vitamin D directly depolarizes ARH POMC neurons, an action that might partly account for the anorexigenic effects of central vitamin D (88). In addition, central administration of vitamin D improves glucose tolerance and markedly increases hepatic insulin sensitivity in mice; these effects are blunted in mice with VDR deleted specifically in the PVH (88). While these studies suggest actions of brain VDR in the prevention of obesity and insulin resistance, more experimentation is needed to reveal the underlying mechanisms.
PPARs
All three PPAR isoforms, PPARα, PPARδ, and PPARγ, have been implicated in the regulation of body weight through functions in the brain. PPARγ agonists, including thiazolidinediones (TZDs), often lead to body weight gain in humans (89). Consistently, chronic peripheral administration of PPARγ antagonists protect mice from diet-induced obesity (90). The obesogenic effects of PPARγ are mediated in part by its actions in the brain. For example, acute and chronic activation of brain PPARγ by either TZDs or hypothalamic overexpression of PPARγ results in increased food intake and body weight gain (91). In contrast, inhibition of endogenous PPARγ activity in the brain with antagonists or by shRNA-mediated knockdown decreases food intake and body weight in HFD-fed rats (91). These body weight–lowering effects of PPARγ inhibition may be attributed to increased leptin actions, because pharmacologic inhibition of brain PPARγ restores leptin sensitivity in HFD-fed rats (91). Consistently, mice with deletion of PPARγ in the brain (crosses between synapsin-1–Cre mice and Ppargfl/fl mice) show reduced body weight associated with decreased food intake and increased energy expenditure (92). These mutant mice are resistant to TZD-induced hyperphagia and weight gain (92). Collectively, these data demonstrate an important role of brain PPARγ in promoting body weight gain. Similarly, hypothalamic PPARα is known to promote obesity, as pharmacologic activation of hypothalamic PPARα increased food intake in a genetic mouse model with increased sensitivity to brain fatty acids (93). On the other hand, brain PPARδ exerts the exact opposite effects to prevent obesity, as demonstrated by the obese phenotypes observed in mutant mice that lack PPARδ only in the brain (crosses between synapsin-1–Cre mice and Ppardfl/fl mice) (94). Interestingly, these mutant mice express higher levels of PPARγ and PPARα in the hypothalamus (94), suggesting that PPARδ may prevent body weight gain by inhibiting the expression of obesogenic PPARγ and PPARα. The detailed molecular interplay among these PPARs in the brain has not been examined fully.
Nerve growth factor 1B
Although the endogenous ligand for nerve growth factor 1B (NGF1B, also known as Nur77) has not been identified, it is thought that NGF1B has constitutive activity (95). NGF1B has been reported to regulate energy metabolism, immunity, cellular stress, memory, insulin sensitivity, and cardiac homeostasis (95). In particular, decreased expression of NGF1B in the hypothalamus has been associated with obesity in mice (96), suggesting that decreased hypothalamic NGF1B levels may contribute to development of obesity. Supporting this notion, global NGF1B knockout mice develop late-onset obesity associated with increased food intake and decreased energy expenditure (96). Furthermore, these mutant mice are insensitive to the body weight–lowering effects of leptin (96). Similarly, knockdown of NGF1B specifically in the hypothalamus blunts the actions of leptin on feeding and body weight (96); the molecular mechanisms involve increased STAT3 activity, a key downstream signal of leptin. NGF1B enhances STAT3 acetylation by recruiting acetylase p300 and disassociating histone deacetylase 1, which results in increased STAT3 transcriptional activity on its target genes, e.g., POMC (96). Therefore, hypothalamic NGF1B may function as a positive modulator in leptin signaling and can contribute to the regulation of body weight balance.
Estrogen-related receptors
The estrogen-related receptors (ERRs) ERRα, ERRβ, and ERRγ are orphan nuclear hormone receptors with no known ligands. While ERRs can bind and regulate gene transcription via the same estrogen response elements as ERs, they do not respond to endogenous estrogens (97). Phytoestrogens have been reported to activate ERRα; ERRβ and ERRγ can be activated by synthetic ligands, including GSK4716 and GSK9089, although the effects of these synthetic ligands on energy balance have not been reported (97). Nevertheless, global Errb knockout mice display decreased body weight, fat mass, and lean mass (98). These phenotypes are likely attributed to marked increases in physical activity and energy expenditure (98). Interestingly, food intake of these mutant mice is increased (98), presumably due to increased energy demands associated with hyperactivity and hypermetabolism. Thus, the overall functions of ERRβ appear to suppress physical activity and energy expenditure in order to promote body weight gain. However, the functions of ERRβ in the brain seem to mediate opposing effects, as selective deletion of Errb in the brain leads to increased body weight (mainly reflected by increases in lean mass but not fat mass) (98). The roles of brain ERRα and ERRγ remain to be elucidated.
Other NRs
Additional NRs have been proposed to regulate energy homeostasis through their actions in the brain. For example, the neuron-derived orphan receptor 1 (NOR1, also known as NR4A3) is known to play important roles in the regulation of cell proliferation, differentiation, metabolism, and apoptosis (99–101). NOR1 may also promote food intake and body weight gain through its actions in the brain, as the injection of a siRNA oligonucleotide against NOR1 into the third cerebral ventricle markedly suppressed food intake and body weight in mice (102). In addition, retinoid X receptor (RXR), the NR activated by 9-cis retinoic acid, may play a similar orexigenic role, since central administration of a pan agonist (LG268) decreases both food intake and body weight gain in obese rats (103). Furthermore, systemic administration of a potent agonist of Rev-erb (SR9011) increases energy expenditure and decreases body weight in mice with diet-induced obesity; these effects are associated with robust alterations in the circadian pattern of core clock gene expression in the hypothalamus, but not in the liver, skeletal muscle, or adipose tissue of mice (104). Although these results suggest potential roles of brain NOR1, RXRs, and Rev-erb in the regulation of energy homeostasis, their functions in the brain need to be further validated through genetic models.
A few other NRs have also been implicated in the regulation of body weight. For example, hepatocyte nuclear factor 4γ (NHF4γ) is thought to bind to linoleic acid as its endogenous ligand (105); global deletion of NHF4γ in mice leads to increased body weight, which is likely attributable to lowered energy expenditure and physical activity (106). In addition, the liver receptor homolog-1 (LRH-1, also known as NR5A2) plays critical roles in the regulation of development, cholesterol transport, bile acid homeostasis, and steroidogenesis (107); heterozygous deletion of LRH-1 causes mild body weight gain in HFD-fed mice (108). Furthermore, chicken ovalbumin upstream promoter transcription factor 2 (COUP-TFII, also known as NR2F2) is essential for the development of multiple tissues and organs (109), and mice with global deficiency in COUP-TFII display reduced body weight (110). While these results indicate that these NRs are required to maintain normal body weight balance, it remains unclear how much of these effects stem from the actions of these NRs in the brain (29).
Conclusions and future directions
As summarized in Table 1, many NRs, including those responding to endogenous ligands or those without identified ligands, have been shown to play important roles in the regulation of body weight balance. Accumulating evidence obtained from genetic mouse models and pharmacologic approaches pinpoints the brain, especially the hypothalamus, as a key site mediator of at least some of their functions. Despite exciting advances in the field, it should be recognized that this is an understudied area and much more needs to be done to fully reveal the physiologic functions of brain NRs in the context of energy homeostasis. For example, in addition to the NRs discussed above, many other NRs are also reported to be expressed in the brain (29). These include mineralocorticoid receptor, testicular receptor 4, nuclear receptor TLX, retinoic acid receptor (RAR), RAR-related orphan receptors, NR-related 1 protein, progesterone receptor, photoreceptor cell–specific nuclear receptor, small heterodimer partner, and liver X receptor. Currently there is no direct evidence of a role for these NRs in the regulation of body weight. Thus, one obvious future direction of this field is to continue to explore the potential functions of these other NRs in the brain.
Table 1. Summary of brain NR effects on energy balance and underlying mechanisms.

For those brain NRs with confirmed effects on energy balance, detailed mechanisms for their actions remain largely unknown. For example, within the same brain structure, neurons with different neurochemical identities may exert opposite effects on energy homeostasis (e.g., POMC neurons vs. AgRP neurons within the ARH). In addition, different brain structures can regulate distinct aspects of energy balance. For example, different ERα populations in the brain regulate feeding, thermogenesis, and physical activity; the overall estrogenic actions on body weight appear to be an additive outcome of these distinct ERα populations. Such similar functional segregations are likely to exist for other brain NRs. Thus, the effects of brain NRs need to be further dissected among relevant brain regions in order to fully appreciate their diverse functions. At the molecular level, the major actions of NRs are to regulate gene transcription, so it is conceivable that many of these brain NRs regulate expression levels of key genes that control energy homeostasis, e.g., SF1 transcribing BDNF and CB1. However, the targeted genes of most brain NRs remain unclear, a topic that warrants further investigation. It is worth noting that some NRs (e.g., ERα and VDR) may also regulate firing activities of neurons via rapid signals that are independent of the classic nuclear receptor transcriptional activities. Thus, it will be necessary to explore further the non-classic mechanisms for certain brain NRs.
Finally, while the majority of brain NRs function to prevent body weight gain and obesity, a number of other brain NRs exert exactly the opposite effects to promote body weight gain (Table 1). It has to be pointed out that there is no clear picture regarding how signals of these brain NRs are integrated to regulate energy homeostasis. Evidence exists that some brain NRs may be coordinated to regulate energy balance. For example, expression levels of orexigenic PPARγ/PPARα appear to be negatively regulated by anorexigenic PPARδ, and the functional balance among these three PPARs contributes to body weight balance. In addition, activation of RXR, VDR, TR, and PPARδ all lead to reductions in body weight. Given that RXR can form heterodimers with VDR, TR, or PPARδ to regulate gene transcription, it also is possible that RXR may function as a common hub to integrate the effects of multiple NRs on energy balance. Other potential hubs for NR functions include nuclear receptor coactivators, e.g., steroid receptor coactivator-1, -2, and -3 (SRC-1, SRC-2, and SRC-3, respectively); these and other coactivators are master integrators of genomic functions in that they have the ability to coordinately activate the diverse DNA-binding transcription factors required for the coordinated gene expression in various physiologic processes (111). Notably, global deletion of SRC-1 in mice causes obesity (112), while mice lacking SRC-2 (112) or SRC-3 (113) are resistant to diet-induced obesity. It is likely that these SRC family coactivators interact with multiple brain NRs to coordinate their effects on energy balance. Thus, we suggest that additional efforts are needed to investigate various mechanisms for the signal integrations among brain NRs that must occur to provide complex homeostatic control of body weight.
Acknowledgments
The authors are supported by grants from the NIH (R01DK093587 and R01DK101379 to YX, R01HD008818 and P01DK059820 to BWO, R01DK100659 and P01DK088761 to JKE, and U24 DK097748 to BWO and JKE) and the USDA Current Research Information System (6250-51000-059-04S to YX).
Footnotes
Conflict of interest: B.W. O’Malley reports receiving a research grant from, and has ownership interest (including patents) in, Coregon, Inc.
Reference information:J Clin Invest. 2017;127(4):1172–1180.https://doi.org/10.1172/JCI88891.
Contributor Information
Yong Xu, Email: yongx@bcm.edu.
Joel K. Elmquist, Email: joel.elmquist@utsouthwestern.edu.
References
- 1.Ogden CL, Carroll MD, Curtin LR, McDowell MA, Tabak CJ, Flegal KM. Prevalence of overweight and obesity in the United States, 1999-2004. JAMA. 2006;295(13):1549–1555. doi: 10.1001/jama.295.13.1549. [DOI] [PubMed] [Google Scholar]
- 2.[No authors listed] Nutrition Classics. The Anatomical Record, Volume 78, 1940: Hypothalamic lesions and adiposity in the rat. Nutr Rev. 1983;41(4):124–127. doi: 10.1111/j.1753-4887.1983.tb07169.x. [DOI] [PubMed] [Google Scholar]
- 3.Elmquist JK, Elias CF, Saper CB. From lesions to leptin: hypothalamic control of food intake and body weight. Neuron. 1999;22(2):221–232. doi: 10.1016/S0896-6273(00)81084-3. [DOI] [PubMed] [Google Scholar]
- 4.Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006;443(7109):289–295. doi: 10.1038/nature05026. [DOI] [PubMed] [Google Scholar]
- 5.Cone RD. The central melanocortin system and energy homeostasis. Trends Endocrinol Metab. 1999;10(6):211–216. doi: 10.1016/S1043-2760(99)00153-8. [DOI] [PubMed] [Google Scholar]
- 6.Huszar D, et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88(1):131–141. doi: 10.1016/S0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 7.Xu AW, et al. Effects of hypothalamic neurodegeneration on energy balance. PLoS Biol. 2005;3(12):e415. doi: 10.1371/journal.pbio.0030415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yaswen L, Diehl N, Brennan MB, Hochgeschwender U. Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat Med. 1999;5(9):1066–1070. doi: 10.1038/12506. [DOI] [PubMed] [Google Scholar]
- 9.Wu Q, Boyle MP, Palmiter RD. Loss of GABAergic signaling by AgRP neurons to the parabrachial nucleus leads to starvation. Cell. 2009;137(7):1225–1234. doi: 10.1016/j.cell.2009.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luquet S, Perez FA, Hnasko TS, Palmiter RD. NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science. 2005;310(5748):683–685. doi: 10.1126/science.1115524. [DOI] [PubMed] [Google Scholar]
- 11.Krashes MJ, et al. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J Clin Invest. 2011;121(4):1424–1428. doi: 10.1172/JCI46229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aponte Y, Atasoy D, Sternson SM. AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nat Neurosci. 2011;14(3):351–355. doi: 10.1038/nn.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Atasoy D, Betley JN, Su HH, Sternson SM. Deconstruction of a neural circuit for hunger. Nature. 2012;488(7410):172–177. doi: 10.1038/nature11270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Betley JN, Cao ZF, Ritola KD, Sternson SM. Parallel, redundant circuit organization for homeostatic control of feeding behavior. Cell. 2013;155(6):1337–1350. doi: 10.1016/j.cell.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu Q, Clark MS, Palmiter RD. Deciphering a neuronal circuit that mediates appetite. Nature. 2012;483(7391):594–597. doi: 10.1038/nature10899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krashes MJ, et al. An excitatory paraventricular nucleus to AgRP neuron circuit that drives hunger. Nature. 2014;507(7491):238–242. doi: 10.1038/nature12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balthasar N, et al. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell. 2005;123(3):493–505. doi: 10.1016/j.cell.2005.08.035. [DOI] [PubMed] [Google Scholar]
- 18.Garfield AS, et al. A neural basis for melanocortin-4 receptor-regulated appetite. Nat Neurosci. 2015;18(6):863–871. doi: 10.1038/nn.4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andrews ZB, et al. UCP2 mediates ghrelin’s action on NPY/AgRP neurons by lowering free radicals. Nature. 2008;454(7206):846–851. doi: 10.1038/nature07181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jennings JH, Rizzi G, Stamatakis AM, Ung RL, Stuber GD. The inhibitory circuit architecture of the lateral hypothalamus orchestrates feeding. Science. 2013;341(6153):1517–1521. doi: 10.1126/science.1241812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stamatakis AM, Van Swieten M, Basiri ML, Blair GA, Kantak P, Stuber GD. Lateral hypothalamic area glutamatergic neurons and their projections to the lateral habenula regulate feeding and reward. J Neurosci. 2016;36(2):302–311. doi: 10.1523/JNEUROSCI.1202-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu Z, et al. GABAergic projections from lateral hypothalamus to paraventricular hypothalamic nucleus promote feeding. J Neurosci. 2015;35(8):3312–3318. doi: 10.1523/JNEUROSCI.3720-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jennings JH, et al. Visualizing hypothalamic network dynamics for appetitive and consummatory behaviors. Cell. 2015;160(3):516–527. doi: 10.1016/j.cell.2014.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Münzberg H, Qualls-Creekmore E, Berthoud HR, Morrison CD, Yu S. Neural control of energy expenditure. Handb Exp Pharmacol. 2016;233:173–194. doi: 10.1007/164_2015_33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grill HJ, Hayes MR. Hindbrain neurons as an essential hub in the neuroanatomically distributed control of energy balance. Cell Metab. 2012;16(3):296–309. doi: 10.1016/j.cmet.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams DL, Schwartz MW. The melanocortin system as a central integrator of direct and indirect controls of food intake. Am J Physiol Regul Integr Comp Physiol. 2005;289(1):R2–R3. doi: 10.1152/ajpregu.00226.2005. [DOI] [PubMed] [Google Scholar]
- 27.Mangelsdorf DJ, et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83(6):835–839. doi: 10.1016/0092-8674(95)90199-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Nuclear receptors and lipid physiology: opening the X-files. Science. 2001;294(5548):1866–1870. doi: 10.1126/science.294.5548.1866. [DOI] [PubMed] [Google Scholar]
- 29.Bookout AL, Jeong Y, Downes M, Yu RT, Evans RM, Mangelsdorf DJ. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell. 2006;126(4):789–799. doi: 10.1016/j.cell.2006.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ryan KK, et al. FXR is a molecular target for the effects of vertical sleeve gastrectomy. Nature. 2014;509(7499):183–188. doi: 10.1038/nature13135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henry FE, Sugino K, Tozer A, Branco T, Sternson SM. Cell type-specific transcriptomics of hypothalamic energy-sensing neuron responses to weight-loss. Elife. 2015;4:09800. doi: 10.7554/eLife.09800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hong SH, Ahmadian M, Yu RT, Atkins AR, Downes M, Evans RM. Nuclear receptors and metabolism: from feast to famine. Diabetologia. 2014;57(5):860–867. doi: 10.1007/s00125-014-3209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McKenna NJ, Evans RM, O’Malley BW. Nuclear Receptor Signaling: a home for nuclear receptor and coregulator signaling research. Nucl Recept Signal. 2014;12:e006. doi: 10.1621/nrs.12006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ikeda Y, Luo X, Abbud R, Nilson JH, Parker KL. The nuclear receptor steroidogenic factor 1 is essential for the formation of the ventromedial hypothalamic nucleus. Mol Endocrinol. 1995;9(4):478–486. doi: 10.1210/mend.9.4.7659091. [DOI] [PubMed] [Google Scholar]
- 35.Stallings NR, Hanley NA, Majdic G, Zhao L, Bakke M, Parker KL. Development of a transgenic green fluorescent protein lineage marker for steroidogenic factor 1. Mol Endocrinol. 2002;16(10):2360–2370. doi: 10.1210/me.2002-0003. [DOI] [PubMed] [Google Scholar]
- 36.Cheung CC, Kurrasch DM, Liang JK, Ingraham HA. Genetic labeling of steroidogenic factor-1 (SF-1) neurons in mice reveals ventromedial nucleus of the hypothalamus (VMH) circuitry beginning at neurogenesis and development of a separate non-SF-1 neuronal cluster in the ventrolateral VMH. J Comp Neurol. 2013;521(6):1268–1288. doi: 10.1002/cne.23226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dellovade TL, et al. Disruption of the gene encoding SF-1 alters the distribution of hypothalamic neuronal phenotypes. J Comp Neurol. 2000;423(4):579–589. doi: 10.1002/1096-9861(20000807)423:4<579::AID-CNE4>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 38.Büdefeld T, Tobet SA, Majdic G. Altered position of cell bodies and fibers in the ventromedial region in SF-1 knockout mice. Exp Neurol. 2011;232(2):176–184. doi: 10.1016/j.expneurol.2011.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Majdic G, et al. Knockout mice lacking steroidogenic factor 1 are a novel genetic model of hypothalamic obesity. Endocrinology. 2002;143(2):607–614. doi: 10.1210/endo.143.2.8652. [DOI] [PubMed] [Google Scholar]
- 40.King BM. The rise, fall, and resurrection of the ventromedial hypothalamus in the regulation of feeding behavior and body weight. Physiol Behav. 2006;87(2):221–244. doi: 10.1016/j.physbeh.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 41.Zhao L, et al. Steroidogenic factor 1 (SF1) is essential for pituitary gonadotrope function. Development. 2001;128(2):147–154. doi: 10.1242/dev.128.2.147. [DOI] [PubMed] [Google Scholar]
- 42.Kim KW, et al. Steroidogenic factor 1 directs programs regulating diet-induced thermogenesis and leptin action in the ventral medial hypothalamic nucleus. Proc Natl Acad Sci U S A. 2011;108(26):10673–10678. doi: 10.1073/pnas.1102364108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tran PV, Akana SF, Malkovska I, Dallman MF, Parada LF, Ingraham HA. Diminished hypothalamic bdnf expression and impaired VMH function are associated with reduced SF-1 gene dosage. J Comp Neurol. 2006;498(5):637–648. doi: 10.1002/cne.21070. [DOI] [PubMed] [Google Scholar]
- 44.Lee J, Yang DJ, Lee S, Hammer GD, Kim KW, Elmquist JK. Nutritional conditions regulate transcriptional activity of SF-1 by controlling sumoylation and ubiquitination. Sci Rep. 2016;6:19143. doi: 10.1038/srep19143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tong Q, et al. Synaptic glutamate release by ventromedial hypothalamic neurons is part of the neurocircuitry that prevents hypoglycemia. Cell Metab. 2007;5(5):383–393. doi: 10.1016/j.cmet.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheung CC, et al. Sex-dependent changes in metabolism and behavior, as well as reduced anxiety after eliminating ventromedial hypothalamus excitatory output. Mol Metab. 2015;4(11):857–866. doi: 10.1016/j.molmet.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Okura T, Koda M, Ando F, Niino N, Ohta S, Shimokata H. Association of polymorphisms in the estrogen receptor alpha gene with body fat distribution. Int J Obes Relat Metab Disord. 2003;27(9):1020–1027. doi: 10.1038/sj.ijo.0802378. [DOI] [PubMed] [Google Scholar]
- 48.Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS. Increased adipose tissue in male and female estrogen receptor-α knockout mice. Proc Natl Acad Sci U S A. 2000;97(23):12729–12734. doi: 10.1073/pnas.97.23.12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Geary N, Asarian L, Korach KS, Pfaff DW, Ogawa S. Deficits in E2-dependent control of feeding, weight gain, and cholecystokinin satiation in ER-α null mice. Endocrinology. 2001;142(11):4751–4757. doi: 10.1210/endo.142.11.8504. [DOI] [PubMed] [Google Scholar]
- 50.Butera PC, Beikirch RJ. Central implants of diluted estradiol: independent effects on ingestive and reproductive behaviors of ovariectomized rats. Brain Res. 1989;491(2):266–273. doi: 10.1016/0006-8993(89)90062-0. [DOI] [PubMed] [Google Scholar]
- 51.Palmer K, Gray JM. Central vs. peripheral effects of estrogen on food intake and lipoprotein lipase activity in ovariectomized rats. Physiol Behav. 1986;37(1):187–189. doi: 10.1016/0031-9384(86)90404-X. [DOI] [PubMed] [Google Scholar]
- 52.Xu Y, et al. Distinct hypothalamic neurons mediate estrogenic effects on energy homeostasis and reproduction. Cell Metab. 2011;14(4):453–465. doi: 10.1016/j.cmet.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Musatov S, et al. Silencing of estrogen receptor α in the ventromedial nucleus of hypothalamus leads to metabolic syndrome. Proc Natl Acad Sci U S A. 2007;104(7):2501–2506. doi: 10.1073/pnas.0610787104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martínez de Morentin PB, et al. Estradiol regulates brown adipose tissue thermogenesis via hypothalamic AMPK. Cell Metab. 2014;20(1):41–53. doi: 10.1016/j.cmet.2014.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Correa SM, et al. An estrogen-responsive module in the ventromedial hypothalamus selectively drives sex-specific activity in females. Cell Rep. 2015;10(1):62–74. doi: 10.1016/j.celrep.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu P, et al. Estrogen receptor-α in medial amygdala neurons regulates body weight. J Clin Invest. 2015;125(7):2861–2876. doi: 10.1172/JCI80941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu MV, et al. Estrogen masculinizes neural pathways and sex-specific behaviors. Cell. 2009;139(1):61–72. doi: 10.1016/j.cell.2009.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Santollo J, Wiley MD, Eckel LA. Acute activation of ERα decreases food intake, meal size, and body weight in ovariectomized rats. Am J Physiol Regul Integr Comp Physiol. 2007;293(6):R2194–R2201. doi: 10.1152/ajpregu.00385.2007. [DOI] [PubMed] [Google Scholar]
- 59.Gao Q, et al. Anorectic estrogen mimics leptin’s effect on the rewiring of melanocortin cells and Stat3 signaling in obese animals. Nat Med. 2007;13(1):89–94. doi: 10.1038/nm1525. [DOI] [PubMed] [Google Scholar]
- 60.Malyala A, Zhang C, Bryant DN, Kelly MJ, Rønnekleiv OK. PI3K signaling effects in hypothalamic neurons mediated by estrogen. J Comp Neurol. 2008;506(6):895–911. doi: 10.1002/cne.21584. [DOI] [PubMed] [Google Scholar]
- 61.Saito K, Cao X, He Y, Xu Y. Progress in the molecular understanding of central regulation of body weight by estrogens. Obesity (Silver Spring) 2015;23(5):919–926. doi: 10.1002/oby.21099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhu L, et al. The ERα-PI3K cascade in proopiomelanocortin progenitor neurons regulates feeding and glucose balance in female mice. Endocrinology. 2015;156(12):4474–4491. doi: 10.1210/en.2015-1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Asarian L, Geary N. Estradiol enhances cholecystokinin-dependent lipid-induced satiation and activates estrogen receptor-alpha-expressing cells in the nucleus tractus solitarius of ovariectomized rats. Endocrinology. 2007;148(12):5656–5666. doi: 10.1210/en.2007-0341. [DOI] [PubMed] [Google Scholar]
- 64.Thammacharoen S, Lutz TA, Geary N, Asarian L. Hindbrain administration of estradiol inhibits feeding and activates estrogen receptor-alpha-expressing cells in the nucleus tractus solitarius of ovariectomized rats. Endocrinology. 2008;149(4):1609–1617. doi: 10.1210/en.2007-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jakacka M, Ito M, Martinson F, Ishikawa T, Lee EJ, Jameson JL. An estrogen receptor (ER)alpha deoxyribonucleic acid-binding domain knock-in mutation provides evidence for nonclassical ER pathway signaling in vivo. Mol Endocrinol. 2002;16(10):2188–2201. doi: 10.1210/me.2001-0174. [DOI] [PubMed] [Google Scholar]
- 66.Park CJ, et al. Genetic rescue of nonclassical ERα signaling normalizes energy balance in obese Erα-null mutant mice. J Clin Invest. 2011;121(2):604–612. doi: 10.1172/JCI41702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Saito K, et al. PI3K in the ventromedial hypothalamic nucleus mediates estrogenic actions on energy expenditure in female mice. Sci Rep. 2016;6:23459. doi: 10.1038/srep23459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ohlsson C, et al. Obesity and disturbed lipoprotein profile in estrogen receptor-α-deficient male mice. Biochem Biophys Res Commun. 2000;278(3):640–645. doi: 10.1006/bbrc.2000.3827. [DOI] [PubMed] [Google Scholar]
- 69.Foryst-Ludwig A, et al. Metabolic actions of estrogen receptor beta (ERβ) are mediated by a negative cross-talk with PPARgamma. PLoS Genet. 2008;4(6):e1000108. doi: 10.1371/journal.pgen.1000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yepuru M, et al. Estrogen receptor-{beta}-selective ligands alleviate high-fat diet- and ovariectomy-induced obesity in mice. J Biol Chem. 2010;285(41):31292–31303. doi: 10.1074/jbc.M110.147850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lin HY, Xu Q, Yeh S, Wang RS, Sparks JD, Chang C. Insulin and leptin resistance with hyperleptinemia in mice lacking androgen receptor. Diabetes. 2005;54(6):1717–1725. doi: 10.2337/diabetes.54.6.1717. [DOI] [PubMed] [Google Scholar]
- 72.Sato T, Matsumoto T, Yamada T, Watanabe T, Kawano H, Kato S. Late onset of obesity in male androgen receptor-deficient (AR KO) mice. Biochem Biophys Res Commun. 2003;300(1):167–171. doi: 10.1016/S0006-291X(02)02774-2. [DOI] [PubMed] [Google Scholar]
- 73.Yu IC, et al. Neuronal androgen receptor regulates insulin sensitivity via suppression of hypothalamic NF-κB-mediated PTP1B expression. Diabetes. 2013;62(2):411–423. doi: 10.2337/db12-0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ophoff J, et al. Androgen signaling in myocytes contributes to the maintenance of muscle mass and fiber type regulation but not to muscle strength or fatigue. Endocrinology. 2009;150(8):3558–3566. doi: 10.1210/en.2008-1509. [DOI] [PubMed] [Google Scholar]
- 75.McAninch EA, Bianco AC. Thyroid hormone signaling in energy homeostasis and energy metabolism. Ann N Y Acad Sci. 2014;1311:77–87. doi: 10.1111/nyas.12374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Coppola A, et al. A central thermogenic-like mechanism in feeding regulation: an interplay between arcuate nucleus T3 and UCP2. Cell Metab. 2007;5(1):21–33. doi: 10.1016/j.cmet.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.López M, et al. Hypothalamic AMPK and fatty acid metabolism mediate thyroid regulation of energy balance. Nat Med. 2010;16(9):1001–1008. doi: 10.1038/nm.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.King BM. Glucocorticoids and hypothalamic obesity. Neurosci Biobehav Rev. 1988;12(1):29–37. doi: 10.1016/S0149-7634(88)80071-X. [DOI] [PubMed] [Google Scholar]
- 79.Tokuyama K, Himms-Hagen J. Enhanced acute response to corticosterone in genetically obese (ob/ob) mice. Am J Physiol. 1989;257(2 pt 1):E133–E138. doi: 10.1152/ajpendo.1989.257.2.E133. [DOI] [PubMed] [Google Scholar]
- 80.Freedman MR, Horwitz BA, Stern JS. Effect of adrenalectomy and glucocorticoid replacement on development of obesity. Am J Physiol. 1986;250(4 pt 2):R595–R607. doi: 10.1152/ajpregu.1986.250.4.R595. [DOI] [PubMed] [Google Scholar]
- 81.Zakrzewska KE, et al. Induction of obesity and hyperleptinemia by central glucocorticoid infusion in the rat. Diabetes. 1999;48(2):365–370. doi: 10.2337/diabetes.48.2.365. [DOI] [PubMed] [Google Scholar]
- 82.Kellendonk C, et al. Inactivation of the GR in the nervous system affects energy accumulation. Endocrinology. 2002;143(6):2333–2340. doi: 10.1210/endo.143.6.8853. [DOI] [PubMed] [Google Scholar]
- 83.Shibata M, et al. AgRP neuron-specific deletion of glucocorticoid receptor leads to increased energy expenditure and decreased body weight in female mice on a high-fat diet. Endocrinology. 2016;157(4):1457–1466. doi: 10.1210/en.2015-1430. [DOI] [PubMed] [Google Scholar]
- 84.Ortega RM, et al. Vitamin D status modification by two slightly hypocaloric diets in young overweight/obese women. Int J Vitam Nutr Res. 2009;79(2):71–78. doi: 10.1024/0300-9831.79.2.71. [DOI] [PubMed] [Google Scholar]
- 85.Zhu W, et al. Calcium plus vitamin D3 supplementation facilitated fat loss in overweight and obese college students with very-low calcium consumption: a randomized controlled trial. Nutr J. 2013;12:8. doi: 10.1186/1475-2891-12-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zemel MB, et al. Dairy-rich diets augment fat loss on an energy-restricted diet: a multicenter trial. Nutrients. 2009;1(1):83–100. doi: 10.3390/nu1010083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sergeev IN, Song Q. High vitamin D and calcium intakes reduce diet-induced obesity in mice by increasing adipose tissue apoptosis. Mol Nutr Food Res. 2014;58(6):1342–1348. doi: 10.1002/mnfr.201300503. [DOI] [PubMed] [Google Scholar]
- 88.Sisley SR, et al. Hypothalamic vitamin D improves glucose homeostasis and reduces weight. Diabetes. 2016;65(9):2732–2741. doi: 10.2337/db16-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lehrke M, Lazar MA. The many faces of PPARγ. Cell. 2005;123(6):993–999. doi: 10.1016/j.cell.2005.11.026. [DOI] [PubMed] [Google Scholar]
- 90.Nakano R, et al. Antagonism of peroxisome proliferator-activated receptor gamma prevents high-fat diet-induced obesity in vivo. Biochem Pharmacol. 2006;72(1):42–52. doi: 10.1016/j.bcp.2006.03.023. [DOI] [PubMed] [Google Scholar]
- 91.Ryan KK, Li B, Grayson BE, Matter EK, Woods SC, Seeley RJ. A role for central nervous system PPAR-γ in the regulation of energy balance. Nat Med. 2011;17(5):623–626. doi: 10.1038/nm.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lu M, et al. Brain PPAR-γ promotes obesity and is required for the insulin-sensitizing effect of thiazolidinediones. Nat Med. 2011;17(5):618–622. doi: 10.1038/nm.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chakravarthy MV, et al. Brain fatty acid synthase activates PPARalpha to maintain energy homeostasis. J Clin Invest. 2007;117(9):2539–2552. doi: 10.1172/JCI31183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kocalis HE, et al. Neuron-specific deletion of peroxisome proliferator-activated receptor delta (PPARΔ) in mice leads to increased susceptibility to diet-induced obesity. PLoS One. 2012;7(8):e42981. doi: 10.1371/journal.pone.0042981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ranhotra HS. The NR4A orphan nuclear receptors: mediators in metabolism and diseases. J Recept Signal Transduct Res. 2015;35(2):184–188. doi: 10.3109/10799893.2014.948555. [DOI] [PubMed] [Google Scholar]
- 96.Chen Y, et al. Enhancement of hypothalamic STAT3 acetylation by nuclear receptor Nur77 dictates leptin sensitivity. Diabetes. 2015;64(6):2069–2081. doi: 10.2337/db14-1206. [DOI] [PubMed] [Google Scholar]
- 97.Ariazi EA, Jordan VC. Estrogen-related receptors as emerging targets in cancer and metabolic disorders. Curr Top Med Chem. 2006;6(3):203–215. doi: 10.2174/1568026610606030203. [DOI] [PubMed] [Google Scholar]
- 98.Byerly MS, et al. Estrogen-related receptor β deletion modulates whole-body energy balance via estrogen-related receptor γ and attenuates neuropeptide Y gene expression. Eur J Neurosci. 2013;37(7):1033–1047. doi: 10.1111/ejn.12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pearen MA, Ryall JG, Maxwell MA, Ohkura N, Lynch GS, Muscat GE. The orphan nuclear receptor, NOR-1, is a target of β-adrenergic signaling in skeletal muscle. Endocrinology. 2006;147(11):5217–5227. doi: 10.1210/en.2006-0447. [DOI] [PubMed] [Google Scholar]
- 100.Pearen MA, Myers SA, Raichur S, Ryall JG, Lynch GS, Muscat GE. The orphan nuclear receptor, NOR-1, a target of β-adrenergic signaling, regulates gene expression that controls oxidative metabolism in skeletal muscle. Endocrinology. 2008;149(6):2853–2865. doi: 10.1210/en.2007-1202. [DOI] [PubMed] [Google Scholar]
- 101.Nomiyama T, et al. The NR4A orphan nuclear receptor NOR1 is induced by platelet-derived growth factor and mediates vascular smooth muscle cell proliferation. J Biol Chem. 2006;281(44):33467–33476. doi: 10.1074/jbc.M603436200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nonogaki K, Kaji T, Ohba Y, Sumii M, Wakameda M, Tamari T. Serotonin 5-HT2C receptor-independent expression of hypothalamic NOR1, a novel modulator of food intake and energy balance, in mice. Biochem Biophys Res Commun. 2009;386(2):311–315. doi: 10.1016/j.bbrc.2009.06.023. [DOI] [PubMed] [Google Scholar]
- 103.Ogilvie KM, Saladin R, Nagy TR, Urcan MS, Heyman RA, Leibowitz MD. Activation of the retinoid X receptor suppresses appetite in the rat. Endocrinology. 2004;145(2):565–573. doi: 10.1210/en.2003-0907. [DOI] [PubMed] [Google Scholar]
- 104.Solt LA, et al. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature. 2012;485(7396):62–68. doi: 10.1038/nature11030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yuan X, et al. Identification of an endogenous ligand bound to a native orphan nuclear receptor. PLoS One. 2009;4(5):e5609. doi: 10.1371/journal.pone.0005609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gerdin AK, et al. Phenotypic screening of hepatocyte nuclear factor (HNF) 4-γ receptor knockout mice. Biochem Biophys Res Commun. 2006;349(2):825–832. doi: 10.1016/j.bbrc.2006.08.103. [DOI] [PubMed] [Google Scholar]
- 107.Fayard E, Auwerx J, Schoonjans K. LRH-1: an orphan nuclear receptor involved in development, metabolism and steroidogenesis. Trends Cell Biol. 2004;14(5):250–260. doi: 10.1016/j.tcb.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 108.Hattori T, Iizuka K, Horikawa Y, Takeda J. LRH-1 heterozygous knockout mice are prone to mild obesity. Endocr J. 2014;61(5):471–480. doi: 10.1507/endocrj.EJ14-0017. [DOI] [PubMed] [Google Scholar]
- 109.Lin FJ, Qin J, Tang K, Tsai SY, Tsai MJ. Coup d’Etat: an orphan takes control. Endocr Rev. 2011;32(3):404–421. doi: 10.1210/er.2010-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li L, et al. The nuclear orphan receptor COUP-TFII plays an essential role in adipogenesis, glucose homeostasis, and energy metabolism. Cell Metab. 2009;9(1):77–87. doi: 10.1016/j.cmet.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.York B, O’Malley BW. Steroid receptor coactivator (SRC) family: masters of systems biology. J Biol Chem. 2010;285(50):38743–38750. doi: 10.1074/jbc.R110.193367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Picard F, et al. SRC-1 and TIF2 control energy balance between white and brown adipose tissues. Cell. 2002;111(7):931–941. doi: 10.1016/S0092-8674(02)01169-8. [DOI] [PubMed] [Google Scholar]
- 113.Coste A, et al. The genetic ablation of SRC-3 protects against obesity and improves insulin sensitivity by reducing the acetylation of PGC-1{alpha} Proc Natl Acad Sci U S A. 2008;105(44):17187–17192. doi: 10.1073/pnas.0808207105. [DOI] [PMC free article] [PubMed] [Google Scholar]