

Abstract
Trifluoroselenomethionine (TFSeM), a novel non-natural amino acid, was synthesized in seven steps from N-(tert-butoxycarbonyl)-L-aspartic acid tert-butyl ester. TFSeM shows enhanced methioninase-induced cytotoxicity toward human colon cancer derived HCT-116 cells compared to selenomethionine (SeM). Mechanistic explanations for this enhanced activity are computationally and experimentally examined. Comparison of TFSeM and SeM by selenium EXAFS and DFT calculations showed them to be spectroscopically and structurally very similar. None-the-less, when two different variants of the protein GB1 were expressed in an E. coli methionine auxotroph cell line using TFSeM and methionine (Met) in a 9:1 molar ratio, it was found that, surprisingly, 85% of the proteins were composed of SeM, even though no SeM had been added, implying loss of the trifluoromethyl group from TFSeM. The recycling of TFSeM to SeM is enzymatically catalyzed by E. coli extracts. However, TFSeM is not a substrate of E. coli methionine adenosyltransferase.
Keywords: trifluoroselenomethionine, selenomethionine, methionine γ-lyase, Fluorinated amino acids, Se-adenosyltrifluoromethylselenomethionine
TOC image
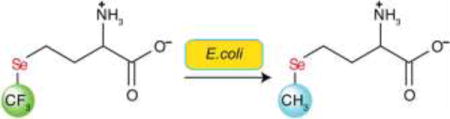
Introduction
Trifluoromethionine (3; TFM; Figure 1) and selenomethionine (5; SeM) are of interest as prodrugs, affording cytotoxic trifluoromethanethiol (4) and methaneselenol (6), respectively, when cleaved by methioninase (MGL; L-methionine γ-lyase; EC 4.4.1.11), analogous to cleavage of methionine (1; Met) to methanethiol (2).[1] In the presence of methioninase, 3 is effective against anaerobic pathogens,[1a] while 5 inhibits cell growth and induces apoptosis in colon cancer derived HCT-116 cells.[1c, 1d] The Km values for 3 and 5 toward methioninase are significantly smaller than that for 1, consistent with higher acidity of 4 and 6 compared to 2.[1a, 1b, 1f] Very different mechanisms have been proposed for the cytotoxicity of 4 and 6: 4 is unstable under physiological conditions, breaking down to the amine cross-linker S=CF2 (9; thiocarbonyl fluoride),[1f] while 6 generates superoxide, inducing oxidative stress as it oxidizes glutathione and other cellular thiols.[2] In other work, 3, incorporated into recombinant proteins by E. coli, provided a 19F NMR spectroscopic probe. It also showed diminished activity at the sulfur atom of 3, compared to 1, toward oxidation and reaction with electrophilic reagents.[3]
Figure 1.
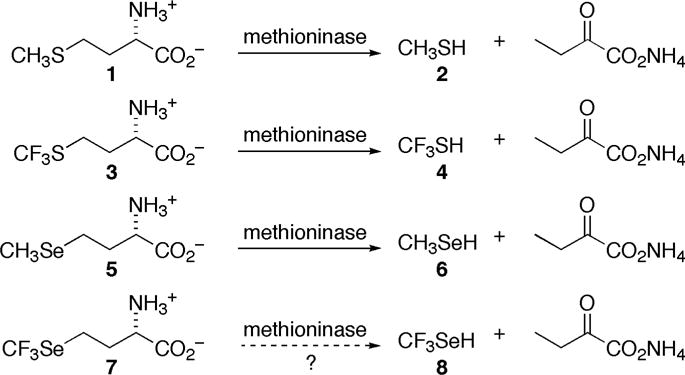
Comparison of methioninase cleavage of methionine (1), trifluoromethionine (3), selenomethionine (5) and trifluoroselenomethionine (7) (as 7b).
Would trifluoroselenomethionine (7, TFSeM; Se-trifluoromethyl-L-homoselenocysteine), combining characteristics of both 3 and 5, show similar biochemistry, affording trifluoromethaneselenol (8)? If so, how would the cytotoxicity of 8 compare to that of 4 and 6? Would 7 be incorporated into recombinant proteins in E. coli, paralleling the case of 3, but with enhanced resistance to oxidation compared to 5? We describe here the first synthesis of 7 (Figure 2), its reactivity toward methioninase, comparative computational studies of one-electron reduction of oxygen by 2, 6, and 8, as well as comparative computational and experimental studies of loss of fluoride ion from 4 and 8 to give thiocarbonyl fluoride, S=CF2 (9) and selenocarbonyl fluoride, Se=CF2 (10), respectively. We also report physical, spectroscopic and calculated structural properties of 7 relative to those of 1, 3 and 5, as well the outcome of efforts to incorporate 7 into recombinant proteins in E. coli.
Figure 2.
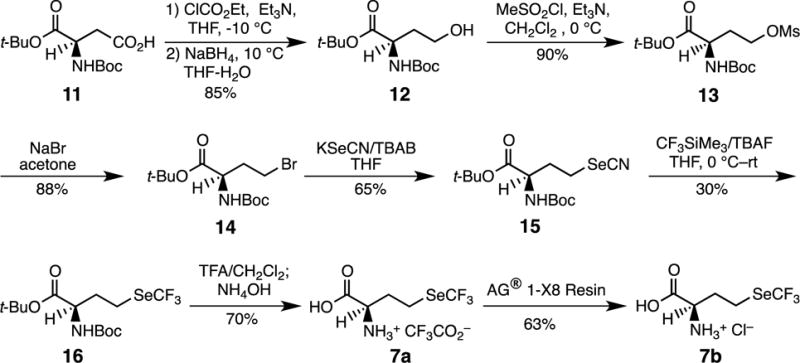
Synthesis of (S)-trifluoroselenomethionine (7).
Experimental Section
Materials and methods
L-Tryptophan, 2-mercaptoethanol, S-adenosyl-L-homocysteine (SAH), adenine, 5′-methylthioadenosine (MTA), and phenylmethanesulfonyl fluoride (PMSF) were purchased from Sigma Corp (St. Louis, MO). Tris(hydroxymethyl)aminomethane (Tris-HCl) and magnesium chloride hexahydrate were purchased from Avantor Performance Materials (Center Valley, PA). Selenomethionine (>98.0%) was purchased from TCI (Tokyo, Japan), ATP (disodium salt) from CalBioChem (San Diego, CA), potassium chloride from EMD Chemicals (Gibbstown, NJ), dithiothreitol (DTT) from Gold Biotechnology (St. Louis, MO), and ammonium acetate (ACS grade) from Affymetrix (Cleveland, OH). All other chemicals were of the highest grade available. S-adenosyl-L-methionine (SAM), and Se-adenosyl-L-selenomethionine (SeSAM) were synthesized and purified as described previously, as was methionine adenosyltransferase (MAT).[4] HPLC methods 1 and 2 were previously described and used without any modifications.[4]
Synthetic procedure
(S)-tert-Butyl 2-(tert-butoxycarbonylamino)-4-hydroxybutanoate (12)
Ethyl chloroformate (1.4 mL, 14.6 mmol) was added at −10 °C to a stirred solution of N-(tert-butoxycarbonyl)-L-aspartic acid 1-tert-butyl ester (11) (4 g, 13.8 mmol) and Et3N (2.0 mL, 14.2 mmol, 1.03 eq) in THF (60 mL) and the mixture was stirred at −10 °C for 30 min. The precipitated mass was filtered and the filtrate was added over a period of 30 min to a solution of NaBH4 (1.6 g, 42.5 mmol, 3.1 eq) in H2O-THF (3 mL:12 mL) at 10–15 °C. The reaction was stirred for 4 h and acidified with HCl, and the THF was removed in vacuo. The aqueous solution was extracted twice with EtOAc (25 mL each time), washed with 10% NaOH (25 mL), H2O (25 mL) and brine (25 mL). The organic solution was dried (Na2SO4) and concentrated to give a clear oil. The crude oil product was purified by flash chromatography on silica (elution with EtOAc and toluene (1:4)) to give the known[5] title compound as a clear, viscous oil (5.93 g, 85%); 1H-NMR (400 MHz, CDCl3) δ=1.45 (s, 9 H), 1.47 (s, 9 H), 2.00–2.22 (m, 2 H), 3.54–3.58 (m, 2 H), 4.20 (m, 1 H), 5.41 ppm (s, 1 H); 13C-NMR (CDCl3, 100 MHz) δ=172.5, 152.9, 82.2, 79.9, 53.2, 36.5, 29.7, 28.4 ppm.
(S)-tert-Butyl 4-bromo-2-(tert-butoxycarbonylamino)butanoate (14)
To a solution containing 12 (1.39 g, 5.04 mmol) in anhydrous CH2Cl2 (60 mL) at 0 °C was added Et3N (1.05 mL, 7.56 mmol, 1.5 eq) followed by CH3SO2Cl (0.39, mL, 2.3 mmol, 2.2 eq). The reaction mixture was stirred at 0 °C under argon atmosphere for 1 h, diluted with brine (100 mL) and the organic layer was separated. The aqueous layer was then back-extracted twice with diethyl ether (50 mL each time) and the combined organic layer was dried (Na2SO4) and concentrated in vacuo to give the known[6] mesylate 13 as a white solid. Sodium bromide (2.07 g, 20.16 mmol, 4 eq) was added to a solution containing the crude mesylate 13 in dry acetone (100 mL). The solution was heated to reflux for 24 h. The cooled reaction mixture was filtered and then concentrated in vacuo. The residue was purified by flash chromatography using EtOAc and hexane (1:4) to give the known[5, 7] title compound as a colorless solid (1.5 g, 88%); 1H-NMR (CDCl3, 400 MHz) δ=1.45 (s, 9 H), 1.46 (s, 9 H), 2.21–2.41 (m, 2 H), 3.43–3.58 (m, 2 H), 4.43 (m, 1 H), 5.13 (s, 1 H) ppm.
(S)-tert-Butyl 4-selenocyanato-2-(tert-butoxycarbonylamino)butanoate (15)
To a solution of (S)-tert-butyl 4-bromo-2-(tert-butoxycarbonylamino)butanoate (1.0 g, 2.96 mmol) in THF (45 mL), KSeCN (0.64 g, 4.5 mmol) and tetra-n-butylammonium bromide (0.096 g, 0.29 mmol) were added, and the mixture was refluxed for 3 h. After completion of the reaction, the solvent was evaporated under reduced pressure. The residue was purified by flash chromatography (1:4 EtOAc:hexane) giving the title compound as a colorless crystalline solid, m.p. 78–80 °C (0.65 g, 65 %); 1H-NMR (CDCl3, 400 MHz) δ=1.44 (s. 9 H), 1.46 (s, 9 H), 2.13–2.42 (m, 2 H), 2.96–3.14 (m, 2 H), 4.31 (m, 1 H), 5.23 ppm (m, 1 H); 13C-NMR (CDCl3, 100 MHz) δ=170.4, 155.7, 102.3, 83.0, 80.3, 53.1, 35.0, 28.2, 27.9, 26.0 ppm; 77Se-NMR (CDCl3, 95.3 MHz) δ=216 ppm; IR: νmax 3387, 2980, 2516, 2159, 1707, 1688, 1503, 1190 cm−1; DART-HR-TOF-MS (M + H+) C14H25O4N2Se: found for 80Se 365.0979, calcd 365.0980. The structure crystallizes in the non-centrosymmetric P212121 space group. Having a heavy Se atom in the light-atoms molecule allowed us to use Mo radiation to determine the absolute configuration by utilizing the anomalous dispersion effects in the x-ray diffraction measurements on the crystal. Flack parameter (refined to essentially zero with low e.s.d −0.010(12)) confirms the correct assignment of the absolute configuration.
(S)-tert-Butyl 4-trifluoromethylselanyl-2-(tert-butoxycarbonylamino)butanoate (16)
To an ice-cooled solution of selenocyanate 15 (500 mg, 1.37 mmol) and CF3SiMe3 (0.44 mL, 2.7 mmol) in anhydrous THF (5 mL) under argon, tetra-n-butylammonium fluoride (TBAF) (0.3 mL, 0.3 mmol) was added dropwise with a syringe pump (1 mL/h). The reaction was monitored by 19F-NMR (CDCl3, CFCl3). After stirring for 1 h at room temperature, the crude mixture was purified by flash chromatography (petroleum ether/ether) giving the title compound as a clear oil (0.32 g, 35%); 1H-NMR (CDCl3, 400 MHz) δ=1.44 (s. 9 H), 1.47 (s, 9 H), 1.99–2.24 (m, 2 H), 2.82–2.94 (m, 2 H), 4.24–4.26 (m, 1 H), 5.11 ppm (bd, J = 6.8, 1 H); 13C-NMR (CDCl3, 100 MHz) δ=170.7, 155.3, 122.6 (q, J=328), 82.5, 79.9, 53.6, 34.3, 28.1, 27.8, 21.1 ppm; 19F-NMR (CFCl3, 376.5 MHz) δ=–35.1 ppm; 77Se-NMR (CDCl3, 95.3 MHz) δ=400 ppm; DART-HR-TOF-MS (M + H+) C14H25O4NF3Se: for 80Se found 408.0900, calcd 408.0901; for 76Se, found 404.0931, calcd 404.0928.
(S)-1-Carboxy-3-((trifluoromethyl)selanyl)propan-1-aminium 2,2,2-trifluoroacetate (7a)
(S)-tert-Butyl 4-trifluoromethylselanyl-2-(tert-butoxycarbonylamino)butanoate (16; 0.2 g, 0.5 mmol) was dissolved in a 10% solution of freshly distilled TFA in dry CH2Cl2 (45 mL) and the mixture was stirred for 3 h. When TLC indicated complete deprotection, the mixture was extracted twice with water (15 mL each time) and the collected aqueous layers were evaporated under reduced pressure to give the title compound as a colorless solid (110 mg; 70%), m.p. 224–226 °C (decomp.); 1H-NMR (D2O, 400 MHz) δ=2.32–2.46 (m, 2 H), 3.08–3.20 (m, 2 H), 3.85 ppm (t, J=6.8, 1 H); 13C-NMR (D2O, 100 MHz) δ=173.5, 162.7 (q, J=38), 122.5 (q, overlap with CF3 of acetate), 115.5 (q, overlap with -SeCF3), 54.3, 31.8, 20.5 ppm; 19F-NMR (D2O, 376.5 MHz) δ=−35.1, −76.0 ppm; 77Se-NMR (D2O, 95.3 MHz) δ=400 ppm; DART-HR-TOF-MS (M + H+) C5H9O2NF3Se for 80Se: found 251.9748, calcd 251.9751; for 76Se: found 247.9748, calcd 247.9778. By absolute qNMR the purity of 7a was determined to be 98% (see SI).
(S)-1-Carboxy-3-((trifluoromethyl)selanyl)propan-1-aminium chloride (7b)
To 100 mg (0.28 mmol) of 7a and AG® 1-X8 Resin (500 mg, chloride form, 50–100 mesh) in a 25 mL round bottomed flask was added 10 mL of water and stirred for 24 h. The heterogeneous mixture was filtered through cotton and aqueous solution was evaporated under reduced pressure to give the title compound as a colorless solid (52 mg; 63%), m.p. 228–231 °C (decomp.); 1H-NMR (D2O, 400 MHz) δ=2.24–2.31 (m, 2 H), 2.96–3.08 (m, 2 H), 3.74 ppm (t, J=6.8, 1 H); 13C-NMR (D2O, 100 MHz) δ=173.5, 122.7(q, J=328), 54.3, 31.8, 20.5 ppm; 19F-NMR (D2O, 376.5 MHz) δ=–34.56 ppm; 77Se-NMR (D2O, 95.3 MHz) δ=400 ppm; DART-HR-TOF-MS (M + H+) C5H9O2NF3Se for 80Se: found 251.9745, calcd 251.9751; for 76Se: found 247.9750, calcd 247.9778. By absolute qNMR the purity of 7b was determined to be 98% (see SI).
3-Selenocyanatopropanenitrile (17)
To a solution of 3-chloropropionitrile (2.0 g, 22.0 mol) in acetone (50 mL) was added potassium selenocyanate (3.2 g, 0.22 mol). The reaction mixture was refluxed overnight, filtered, and evaporated in vacuo. The crude product was purified by flash chromatography (n-pentane/EtOAc) giving the title compound 17 as a yellow oil (2.3 g, 65%); 1H NMR (CDCl3, 400 MHz) δ=3.19–3.24 (m, 2 H), 3.00–3.04 ppm (m, 2 H); 13C NMR (CDCl3, 101 MHz) δ=117.1, 99.9, 22.8, 20.0 ppm; DART-HR-TOF-MS (M+H+) C4H5N2Se: for 80Se found 160.9605, calcd 160.9618; for 76Se, found 156.9630, calcd 156.9645.
3-(Trifluoromethylselanyl)propionitrile (18)
To an ice-cooled solution of selenocyanate 17 (800 mg, 5.0 mmol) and CF3SiMe3 (1.42 g, 10.0 mmol) in anhydrous THF (10 mL) under argon, tetra-n-butylammonium fluoride (TBAF) (2.0 mL, 2.0 mmol) was added dropwise with a syringe pump (1 mL/h). The reaction was monitored by 19F-NMR (CDCl3, CFCl3). After stirring for 1 h at room temperature, the crude mixture was purified by flash chromatography (n-pentane/EtOAc) giving the title compound as a yellow oil (300 mg, 30%); 1H NMR (CDCl3, 400 MHz) δ=3.14–3.17 (m, 2 H), 2.85–2.89 ppm (m, 2 H); 13C NMR (CDCl3, 101 MHz) δ=120.5 (q, J=330, CF3), 117.7, 19.6, 19.5 ppm; 19F NMR (CDCl3, 376 MHz) δ=–34.89 ppm. DART-HR-TOF-MS (M+H+) C4H5F3NSe: found for 80Se 203.9538, calcd 203.9539; for 76Se, found 199.9564, calcd 199.9566.
Generation and decomposition of trifluoromethaneselenol (8); trapping of 3,3-difluoro-2-selenabicyclo[2.2.1]hept-5-ene
Under argon, NaOMe (20 mg, 0.37 mmol) in methanol (2 mL) was added dropwise to 3-(trifluoromethylselanyl)propanenitrile (18) (75 mg, 0.37 mmol), and the mixture was stirred at room temperature for 30 min. Methanol was then removed in vacuo, CD3CN was added under argon, and the solution was directly examined by 19F NMR (376 MHz). The 19F NMR spectrum showed a singlet at δ=–82.71, which can be compared to a signal at δ=–84.48 for a standard solution of KF in CD3CN. In a second experiment, a mixture of cyclopentadiene (2.0 eq) and 18 (75 mg, 0.37 mmol) in a 10 mL flask was frozen using liquid nitrogen and the flask was filled with argon. A solution of NaOMe (0.37 mmol) in methanol (2 mL) was added and the reaction was warmed to room temperature. Analysis of the product by GC-MS showed a molecular ion at m/z 196 and fragment ions at m/z 115 and 95, corresponding to the mass spectrum reported for the F2C=Se (10) cyclopentadiene Diels-Alder reaction product 3,3-difluoro-2-selenabicyclo[2.2.1]hept-5-ene (19) (Figure 4).[8]
Figure 4.
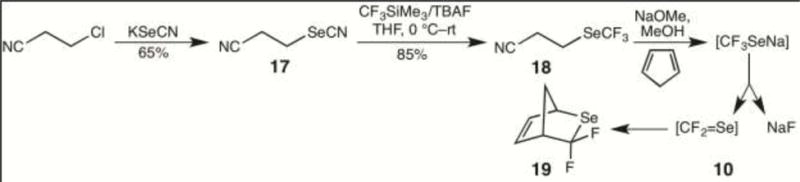
Formation and decomposition of the sodium salt of trifluoromethaneselenol (8).
Cell cultures
HCT116 colorectal carcinoma cells were obtained from American Type Culture Collection and maintained in DMEM (GIBCO Invitrogen, Carlsbad, CA). Stock cells were passaged twice weekly at 80% confluency [0.25% trypsin (GIBCO), 1 mmol/L EDTA, in Ca–Mg–free Hanks’ balanced salt (Sigma Chemical Co., St. Louis, MO); viability was determined by trypan blue exclusion using a hemocytometer] and incubated in a humidified chamber at 36.5 °C, 5% CO2. HCT116 cells with passages 22∼40 and all media were supplemented with 10% fetal bovine serum (FBS) (Sigma Chemical Co., St. Louis, MO).
Methaneselenol and trifluoromethaneselenol generation
Purified recombinant Trichomonas vaginalis methioninase produced in E. coli was purchased from Wako Pure Chemical Industries (Japan). For cell treatments, the enzyme substrate 5 or 7 (1.25 to 5 μmol/L) was added to the culture medium and was immediately followed by the addition of methioninase (40 U/L) for 16 h. The method of methaneselenol generation has been previously described.[1c, 1d]
Statistical analysis
Results are given as means ±SEM. Data were analyzed using mixed model analyses of variance (ANOVA), blocking on group and allowing heterogeneous variances (JMP, version 9.0, SAS Institute, Inc., Cary, NC). If the overall ANOVA was statistically significant, then Tukey’s test for multiple comparisons was used to analyze treatment means. Differences with a p value < 0.05 were considered statistically significant.
X-ray absorption spectroscopy
X-ray absorption measurements of the synthesized samples were carried out at the Stanford Synchrotron Radiation Lightsource (SSRL Stanford, CA, USA) on beamline 7–3 using a Si(220) double-crystal monochromator and Rh-coated silicon mirrors for focusing and harmonic rejection, with the storage ring SPEAR containing 200 mA at 3.0 GeV. During data acquisition samples were maintained at a temperature of approximately 10 K using an Oxford Instruments CF1204 helium cryostat. The incident and transmitted X-ray intensities were measured using nitrogen-filled gas ionization chambers. The X-ray absorption was measured as the Se Kα fluorescence excitation spectrum using an array of 30 Ge detectors (Canberra Industries),[9] with 3 absorption units of As filters. The spectrum of a hexagonal Se foil was recorded simultaneously with each scan. Energy calibration was by reference to the lowest-energy inflection point in the Se K-edge absorption spectrum from the Se foil, assumed to be 12,658 eV.
XAS data analysis
The EXAFS oscillations χ(k) were quantitatively analyzed by curve-fitting using the EXAFSPAK suite of computer programs,[10] using ab initio theoretical phase and amplitude functions calculated with the program FEFF, version 8.25.[11] The threshold energy (k = 0 Å−1) of the EXAFS oscillations was assumed to be 12675.0 eV. Only background-subtracted EXAFS data was analyzed, with no Fourier filtering or smoothing applied.
Density functional structure calculations
Density functional calculations were carried out with the Gaussian09, revision C.01, suite of software.[12] Density functional theory (DFT) provides good estimates of energetic trends, particularly between related molecular structures, and one can generally anticipate bond-length accuracies of better than 0.05 Å. Spin restricted calculations were optimized without geometry or symmetry constraints using the B3LYP hybrid functional method. The 6–311+G(2df,2p) basis set was employed for geometry optimizations, as well as harmonic frequency calculations, which were used to confirm that structures were at a stationary point on the potential energy surface. Structures were considered optimized when the change in energy between subsequent optimization steps fell below 0.2 J/mol. Aqueous solvation calculations were performed using the polarizable continuum method IEFPCM,[13] with the molecular volume defined using united atom radii. Partial atomic charges for each of the isostructural models were calculated with the grid-based CHelpG charge derivation method[14] at the same level of theory as the structure calculations. In order to obtain more accurate geometries for non-bonded complexes involving selenium and the π-electron systems of aromatic rings, we employed Mφller-Plesset perturbation theory[15] at the MP2/6–311+G(2df,2p)-level for geometry optimizations.
Expression and purification of GB1 variants
The gene of the immunoglobulin-binding B1 domain of streptococcal protein G (GB1) was codon optimized for expression in E. coli and the gene synthesized by DNA2.0 (Menlo Park, CA). The synthetic gene of N-terminal hexahistidine-tag tagged GB1 was cloned into the bacterial expression vector pJexpress414 (DNA2.0). The hexahistidine-tag included the tobacco etch virus (TEV) protease cleavage sequence ENLYFQG to allow for removal of the hexahistidine-tag. Following cleavage with TEV protease the resulting GB1 has Gly at position 1 instead of Met, which allowed us to insert a sole Met at specific locations. In addition, the resulting protein also contained a substitution of Gln for Thr at position 2 since the majority of studies on GB1 utilize this variant protein to improve sample homogeneity.[16] This vector was subsequently used as a template to introduce Met at either position Val39Met or position Leu5Met using PCR-based mutagenesis.
For protein expression, the expression plasmid of the appropriate variant was transformed into E. coli methionine auxotroph RF11 cells, in which the gene metA was deleted (Addgene).[17] To enrich the protein with TFSeM, cells were grown in LB media (10 g/L tryptone, 10 g/L NaCl, 5 g/L yeast extract), supplemented with 100 μg/mL ampicillin, with good aeration at 37 °C. At the mid exponential phase of cell growth (when the OD at 600 nm reached 0.6–0.7), the cells were harvested by centrifugation (5000 ×g), and resuspended in an equal volume of a defined media adapted from Studier’s MDAG media.[18] The media contained 25 mM Na2HPO4, 25 mM KH2PO4, 50 mM NH4Cl, 5 mM Na2SO4, 2 mM MgSO4, 0.2× metals, 0.5% glucose (27.8 mM), 0.25 mM aspartate (18.8 mM) and 200 μg/ml each of 17 canonical amino acids (not Cys, Met or Tyr). A 1× concentration of trace metals solutions contained 50 μM FeCl3, 20 μM CaCl2, 10 μM MnCl2, 10 μM ZnSO4, 2 μM CoCl2, 2 μM CuCl2, 2 μM NiCl2, 2 μM Na2MoO4, and 2 μM H3BO3. The media also contained vitamins as described by Studier.[18a]
The resuspended cell solution was divided into three equal aliquots (10 mL each) and supplemented with either 200 μM Met, 200 μM SeM + 22 μM Met, or 200 μM TFSeM (as 7b) + 22 μM Met. Protein expression was induced with 0.5 mM isopropyl-1-thio-β-D-galactopyranoside (IPTG) at 18 °C. The cell paste, following an 18 h expression period, was resuspended in 400 μL B-PER™ Bacterial Protein Extraction Reagent (Thermo Scientific Pierce) supplemented with 1 mM PMSF and 0.5 mM benzamidine. The resuspended cell paste was then lysed using a syringe needle. The cell debris was removed following a 15 min incubation at 25 °C by centrifugation at 15000 ×g for 30 min. The supernatant was mixed with 400 μL 50 mM phosphate buffer (pH 7.5), 200 mM NaCl, and 10 mM imidazole (IMAC buffer). The mixture was incubated with 150 μL IMAC beads (Gold Biotechnology) for 1 h while shaking. The target protein was eluted using IMAC buffer with 500 mM imidiazole. Following elution, the volume was concentrated to 100 μL using a spin concentrator (Amicon ultra-0.5 mL). The buffer was exchanged into 25 mM phosphate buffer (pH 7.5), 50 mM NaCl, and 1 mM EDTA by dialysis and the hexahistidine-tag was cleaved off with TEV protease for 18 h at 4 °C.[19]
A 200 mL scale expression and purification of GB1 Val39Met was carried out as specified above. The growth medium was supplemented with either 22 μM Met, 200 μM TFSeM (as 7b) + 22 μM Met, or 200 μM TFSeM (as 7b).
Mass spectrometry
Mass spectra of intact proteins were obtained using a QTOF Ultima (Waters) mass spectrometer, operating under positive electrospray ionization (+ESI) mode, connected to an LC-20AD (Shimadzu) liquid chromatography unit. Protein samples were separated from small molecules by reverse phase chromatography on a Waters Xbridge BEH C4 column (300 Å, 3.5 μm, 2.1 mm × 50 mm), using an acetonitrile gradient from 30–71.4%, with 0.1% formic acid. Each analysis was 25 min under constant flow rate of 0.2 mL/min at RT. Data were acquired from m/z 350 to 2500, at a rate of 1 sec/scan. Alternatively, spectra were acquired by Xevo G2-S QTOF on a Waters ACQUITY UPLC Protein BEH C4 reverse-phase column (300 Å, 1.7 μm, 2.1 mm × 150 mm). An acetonitrile gradient from 5%–95% was used with 0.1% formic acid, over a run time of 5 min and constant flow rate of 0.5 mL/min at RT. Spectrum were acquired from m/z 350 to 2000, at a rate of 1 sec/scan. The spectra were deconvoluted using maximum entropy in MassLynx.
Analysis and sequencing of peptides was carried out using a Q Exactive Orbitrap interfaced with Ultimate 3000 LC system. Data acquisition by Q Exactive Orbitrap was as follows: 10 μl of trypsin-digested protein was loaded on an Ace UltraCore super C18 reverse-phase column (300 Å, 2.5 μm, 75 mm × 2.1 mm) via an autosampler. An acetonitrile gradient from 5%–95% was used with 0.1% formic acid, over a run time of 45 min and constant flow rate of 0.2 mL/min at RT. MS data were acquired using a data-dependent top10 method dynamically choosing the most abundant precursor ions from the survey scan for HCD fragmentation using a stepped normalized collision energy of 28, 30 35 eV. Survey scans were acquired at a resolution of 70,000 at m/z 200 on the Q Exactive. Theoretical patterns of isotopic patterns of peptides were calculated using UCSF MS-ISOTOPE9 or enviPat Web 2.1.
Time-dependent activity assays of methionine adenosyltransferase with SeM and TFSeM as substrates
The time-dependent formation of SeSAM, Se-adenosyl-trifluoromethylselenomethionine (TFSeSAM), Se-adenosylhomoselenocysteine (SeAH), and methylselenoadenosine (MSeA) was determined from the same reaction mixtures, which contained the following in a volume of 400 μL when analyzed by HPLC: 0.168 mg/mL MAT, 10 mM SeM or TFSeM (as 7b), 100 mM Tris-HCl, pH 8.0, 15 mM ATP, 40 mM MgCl2, 50 mM KCl, 8% v/v 2-mercaptoethanol, and 1 mM tryptophan, used as an internal standard (IS). Reaction mixtures analyzed by LC-MS were identical except that the total reaction volume was 250 μL and the final concentration of SeM or TFSeM (as 7b) was 1 mM. All components except MAT were incubated at 25 °C for 3 min before initiating the reaction with MAT. Aliquots of the reaction mixture (50 μL for HPLC, 20 μL for LC-MS) were withdrawn at various times from 0–18 h and added to 10 μL of 2 M H2SO4 (HPLC) or 10 μL of 1 M H2SO4 (LC-MS) to quench the reaction. Precipitated protein was removed by centrifugation at 18,000 ×g for 15 min, and a 20 μL aliquot of the resulting supernatant was subjected to analysis by HPLC using method 2 or by ESI+ LC-MS in scan mode (m/z 150 – 600) using the method described below. Standards were generated for each analyte and analyzed under identical conditions to determine the identity of the products generated in assays. HPLC elution times are as follows: ATP, 3.2 min; SAM, 11.0 min; SeSAM 12.3 min; adenine, 18.4 min; S-adenosylhomocysteine, (SAH), 21.8 min; tryptophan (IS) 25.1 min; methylthioadenosine (MTA), 29.4 min. The LC-MS buffer system consisted of solvent A (40 mM ammonium acetate and 5% v/v methanol titrated to pH 6.2 with acetic acid) and solvent B (100 % acetonitrile). The column was equilibrated in 98% solvent A, 2% solvent B at a flow rate of 0.5 mL min−1. After sample injection (2 μL), a gradient was applied from 2% solvent B to 45% solvent B over 5 min and then 45% to 5% over 3 min. The observed ions (m/z) and retention times (min), respectively, were 447.1 and 1.1 (SeSAM), 198.0 and 1.3 (SeM), 433.1 and 1.5 (SeAH), 188.0 and 3.0 (tryptophan), 252.0 and 3.1 (TFSeM), and 346.1 and 4.5 (MSeA). Data were analyzed using the Agilent Technologies MassHunter qualitative and quantitative analysis software.
Time-dependent assays of crude lysate, boiled crude lysate, and lysate filtrate with SeM and TFSeM. E. coli B834(DE3)pLysS cells (B834 is a methionine auxotroph[18a]) were grown overnight in M9 minimal media containing 1 μg/mL chloramphenicol and 80 mg/L methionine. The cells were isolated by centrifugation at 10,000 ×g at 4 ˚C for 15 min and then frozen in liquid nitrogen. The frozen cell paste was thawed in lysis buffer containing 50 mM Tris-HCl, pH 8.0, 200 mM KCl, and 10 mM 2-mercaptoethanol. Lysozyme and PMSF were added to final concentrations of 1 mg/mL and 2 mg/mL, respectively, and the cells were lysed by sonication using a Fisher sonic dismembrator (Fisher Scientific). The lysate was transferred to sterile centrifuge tubes and subjected to centrifugation at 50,000 ×g for 1 h at 4 ˚C. The supernatant was kept on ice until use that day. An aliquot of the supernatant was boiled for 10 min and then subjected to centrifugation at 18,000 ×g at 4 ˚C for 30 min. An additional aliquot of the supernatant was transferred to a spin concentrator containing a 3 kDa molecular weight cutoff membrane and subjected to the same centrifugation as the boiled lysate to produce a protein-free filtrate. Aliquots (200 μL) of crude lysate, boiled crude lysate, or filtrate were added to reaction mixtures at 25 ˚C containing 1 mM SeM or 1 mM TFSeM (as 7b) and 100 mM Tris-HCl, pH 8.0. Aliquots (20 μL) were withdrawn at various times from 0–18 h, quenched in 10 μL of 1 M H2SO4, and analyzed by LC-MS as described above.
Results and Discussion
Synthesis of Trifluoroselenomethionine
A short and convenient synthesis of trifluoroselenomethionine (7) was developed from the known, chiral starting material (S)-tert-butyl 2-(tert-butoxycarbonylamino)-4-hydroxybutanoate (12, Figure 2) which, by successive mesylation (13) and treatment with NaBr (14) followed by KSeCN gave the crystalline (S)-tert-butyl 4-selenocyanato-2-(tert-butoxycarbonylamino)butanoate (15; ~80% overall yield), which could be characterized by x-ray crystallography (Figure 3). The highly lipophilic CF3Se group was conveniently introduced giving 16 through reaction of commercially available trifluoromethyl trimethylsilane (CF3SiMe3) with selenocyanate 15 with slow (syringe pump) addition of catalytic (0.2 eq) TBAF (tetrabutylammonium fluoride) at 0 °C.[20] The selenocyanate route is preferred since all of the selenium group is consumed. The synthesis was completed by removal of the protecting groups in 16 with trifluoroacetic acid giving trifluoroacetate salt 7a as a colorless solid, showing 19F-NMR δ −35.12 and −76.07 ppm and 77Se-NMR δ 400 ppm. The 19F NMR signal for alkyl-substituted CF3Se groups generally appears at −35 ppm (relative to CFCl3) compared to −41 ppm for CF3S; trifluoroacetic acid appears at −76.55.[20] Treatment of 7a with the chloride form of AG® 1-X8 Resin gave 7b, the hydrochloride salt of 7, as a colorless solid, showing 19F-NMR δ −35.35 ppm and 77Se-NMR δ 400. Salt 7b was used in all biological studies.
Figure 3.
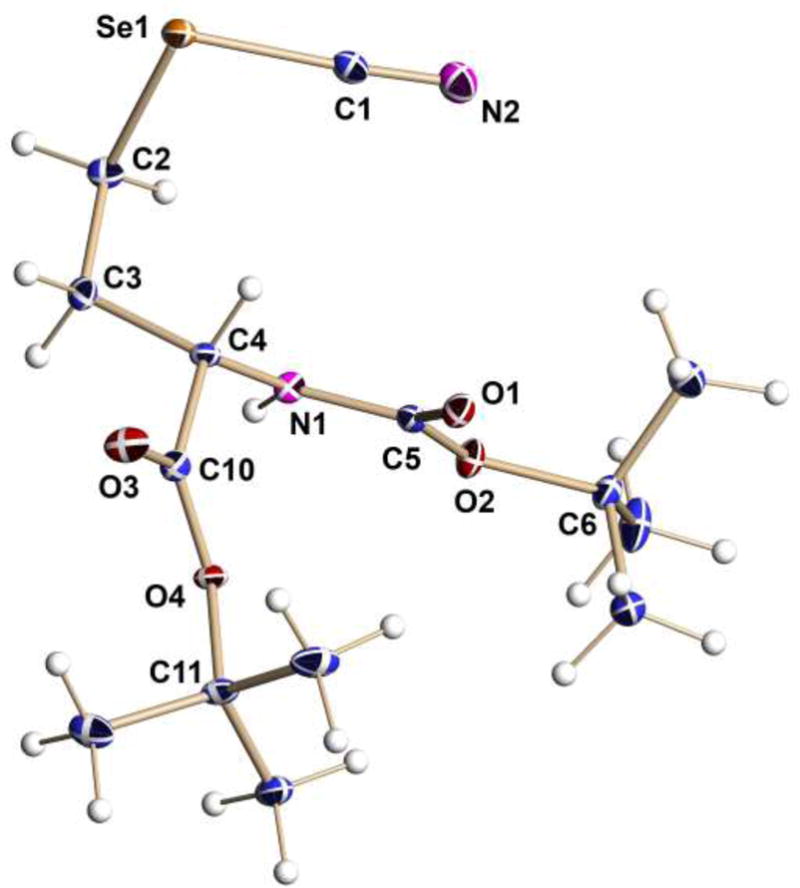
Molecular structure of key intermediate 15 drawn with thermal ellipsoids at the 50% probability level.
Generation and Decomposition of Trifluoromethaneselenol (8)
In order to evaluate the consequences of enzymatic generation of 8 in the presence of human colon cancer derived HCT-116 cells, we sought to generate 8. While 8 has been previously characterized,[21] it is said to be unstable in water[21a] and to decompose to HF and F2C=Se upon heating.[21b] We sought to generate 8, or its sodium salt, by base-catalyzed decomposition of 3-(trifluoromethylselanyl)propionitrile (18), which in turn we proposed to prepare by treatment of 3-selenocyanatopropanenitrile (17) with CF3SiMe3 (Figure 4). In the event, 18 was smoothly prepared from 17, which in turn was easily prepared from 3-chloropropionitrile. When 18 was treated with sodium methoxide and the reaction monitored by 19F NMR, the only 19F signal seen corresponded to fluoride ion, as confirmed by NMR analysis of a standard. When 18 was treated with sodium methoxide in the presence of excess cyclopentadiene at low temperature and then slowly warmed, the known[8] cyclopentadiene adduct of F2C=Se, 3,3-difluoro-2-selenabicyclo[2.2.1]hept-5-ene (19), was detected by GC-MS. Hence decomposition of 8 in solution follows the path seen upon heating.[21b]
Spectroscopic and Computational Comparison of Structures of Selenomethionine (5) and Trifluoroselenomethionine (7)
A combined spectroscopic and computational approach was used to determine the physical and electronic structural changes that occur on replacing the methyl group of 5 with the trifluoromethyl group in 7. In particular, extended X-ray absorption fine structure (EXAFS) measurements of 5 and 7 (as 7b) were conducted on beamline 7–3 of the Stanford Synchrotron Radiation Lightsource (SSRL). The data obtained was analyzed using density functional theory (DFT) calculations and other methods. Analysis of the Se K near-edge spectrum for both 5 and 7, shown in Figure 5, yielded strikingly similar near edge spectra, with no discernible shift in the near edge between the two. The first derivatives for both near edge spectra (inset in Figure 5) are also very similar, such that it would not be possible to distinguish the two chemical species from one another using the Se K near edge spectrum alone.
Figure 5.
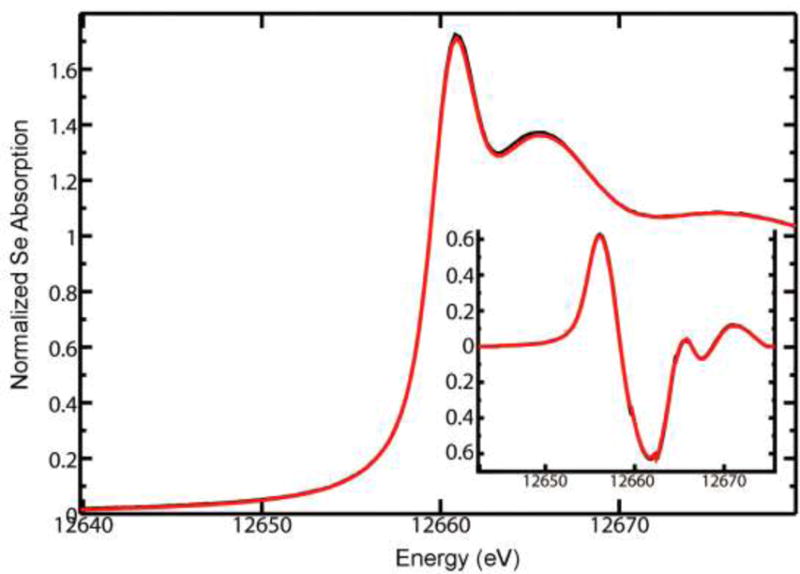
Overlay of Se K near edge spectra for 7 (red curve) and 5 (black curve) for reference. The associated derivative spectra are shown in the inset plot.
Like the near-edge spectra, the EXAFS spectra and associated Fourier transforms (Figure 6), for 5 and 7 were very similar. The theoretical resolution of bond lengths that can be obtained from the EXAFS data (calculated as π/(2Δk), where Δk is the extent of the fitted EXAFS data) for 5 is 0.13 Å (k-range 1–12.5 Å−1) and 0.12 Å (k-range 1–14 Å−1) for 7. Given the bond length resolution of the EXAFS data, both of the Se–C bond lengths in each of the analogs are sufficiently similar that they must be modeled as a single shell of two Se–C scattering interactions, resulting in an average fit distance (R) for the two interactions. The EXAFS curve fitting analysis, using single scattering paths for the two Se–C backscattering interactions yielded very similar bond length results between 5 and 7, with 2 Se–C at 1.957(2) Å for 5 and 2 Se–C at 1.955(4) Å for 7 (Table 1).
Figure 6.
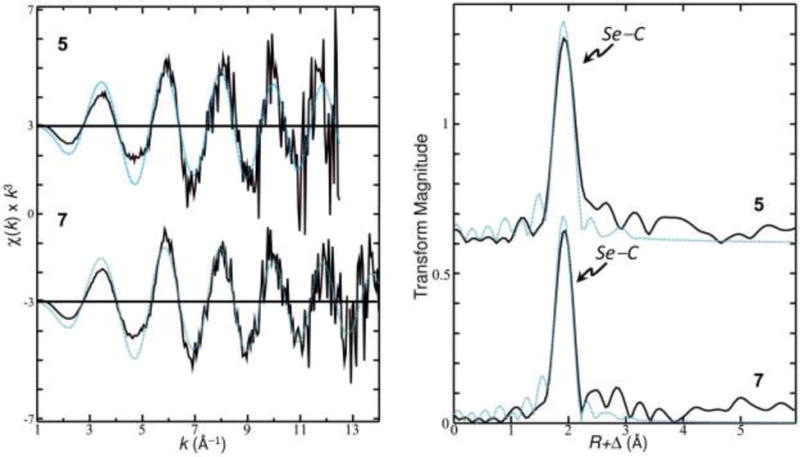
Se K k3-weighted EXAFS for 7 and 5 (left); associated EXAFS Fourier transforms (right). Experimental data shown as a solid line, the results of EXAFS curve fitting are shown as a dashed line.
Table 1.
EXAFS curve-fitting parameters for 5 and 7.a
| Cmpd | Bond | N | R | σ | ΔE0 | F |
|---|---|---|---|---|---|---|
| 5 | Se–C | 2 | 1.957(5) | 0.0021(4) | −6(1) | 0.6337 |
| 7 | Se–C | 2 | 1.955(4) | 0.0024(2) | −6.3(9) | 0.5540 |
Coordination numbers N, interatomic distances R (Å), Debye-Waller factors (the mean-square deviations in interatomic distance) σ2 (Å2), and threshold energy shifts ΔE0 (eV). The values in parentheses are the estimated standard deviations in the last digit obtained from the diagonal elements of the covariance matrix. The fit-error function F is defined by where χ(k) are the EXAFS oscillations and k is the photo-electron wave number given by .
A search of the Cambridge Structural Database (CSD)[22] for Se–C bond lengths from 5 gives an average distance of 1.944 ± 0.006 Å for Se–CH3 and 1.954 ± 0.008 Å for Se–CH2, slightly below the average obtained from EXAFS data fitting (averages from the CSD search were taken from three small molecule crystal structures, all of which contained Met within a peptide chain: MAJTAT C–Se–C = 97.23°, CH3–Se = 1.950, CH2–Se = 1.947; DEYFOD C–Se–C = 98.68°, CH3–Se = 1.939, CH2–Se = 1.963; AVIKOG C–Se–C = 97.94°, CH3–Se = 1.942, CH2–Se = 1.952). The average interatomic distance for Se–C obtained by EXAFS is still within the experimental error obtained from the CSD search for the Se–CH2 bond and only 0.002 Å longer than the maximum value for Se–CH3 found in the CSD search.
DFT Structure Calculations
Gas-phase geometry optimizations were carried out for each of the isostructural charge neutral amino acid models: methionine (1) and selenomethionine (5), as well as each of their CF3-bearing derivatives, 3 and 7, respectively. The calculated Se–CH3 bond length, 1.96(2) Å, presented the largest deviation, ~0.03(8) Å from the EXAFS value of 1.92(8) Å. Other calculated heteroatom carbon bond lengths, summarized in Table 2, were within 0.02 Å of the EXAFS values.
Table 2.
Calculated bond lengths for methionine derivatives.
| 1 | 3 | 5 | 7 | |
|---|---|---|---|---|
| S/Se–CζX3 | 1.817 | 1.811 | 1.962 | 1.966 |
| expt | 1.805 | –– | 1.928 | – |
| S/Se–CδH2 | 1.827 | 1.835 | 1.975 | 1.981 |
| expt | 1.832 | –– | 1.958 | – |
The DFT calculations indicate that when the terminal methyl group in the methionine model is changed to CF3 the sulfur bond length shortens, with concomitant elongation of the S–CH2 bond. Similarly, for the Se-containing analogs the Se–CH2 bond also elongated in the CF3 bearing derivative, relative to 5, while the Se–CH3 and Se–CF3 bond lengths remained largely unchanged. Molecular volume calculations, using the united atom topological model, were performed on each of the optimized structures (Figure 7). Each of the investigated modifications to the methionine side chain results in an increase in side chain volume.
Figure 7.
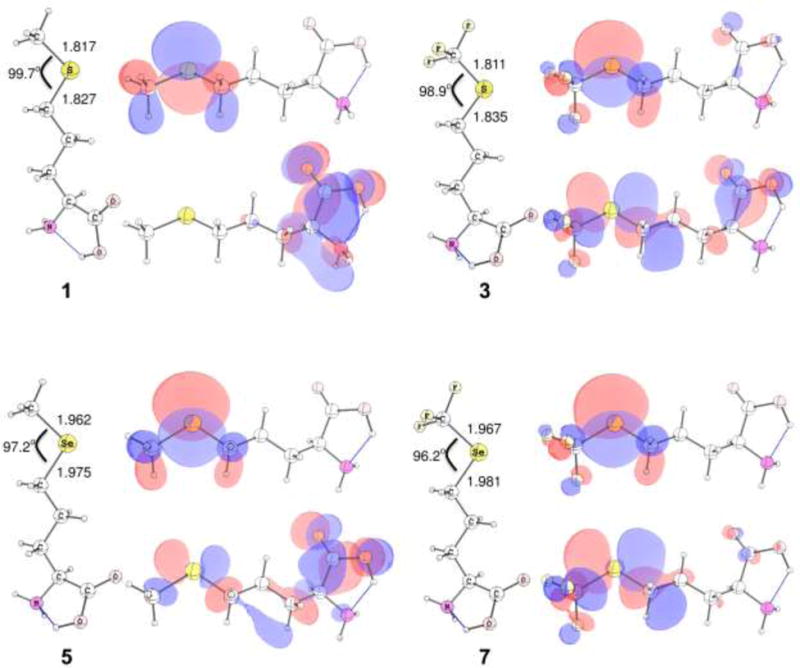
Geometry-optimized density functional structures for 1, 3, 5, and 7 calculated at the B3LYP/6-311+G(2df,2p) level. Additional corresponding HOMO (top) and LUMO (bottom) for each analog, rendered using a 0.03 e–/au3 contour surface.
The molecular volume of amino acid 5 is 104% larger than 1 (111% increase considering only the hetero atom and terminal methyl group moieties). Modification of the methyl group of 1 to CF3 represents a 106% increase in amino acid volume (123% increase for the sulfur and terminal groups), while modification of the methyl group of 5 to CF3 represents a 107% increase in molecular volume (120% increase for Se–CX3). Modification of 1 to 7 represents the most significant increase, at 111% relative to the molecular volume of methionine, and a 133% increase for Se–CF3, relative to S–CH3.
Relative to the sulfur heteroatom in 1, the Se-atom in 5 has low-lying d-orbitals, which are more energetically-accessible than those in the sulfur-containing analog. The result is that the LUMO of 5 is at a lower energy than in the sulfur-containing analog and a greater fraction of the orbital density resides on the Se-atom in the LUMO (Figure 7, LUMO density at the sulfur atom not apparent at the contour value used). Modification of the terminal methyl group in 3 introduces an electron withdrawing moiety to the heteroatom, with the result that in the sulfur-containing methionine analog the LUMO contains some S–C σ* character. These trends are doubly enhanced in the CF3-bearing selenomethionine analog, where the decreased electron density on the Se-atom results in a significant change to the character of the LUMO, such that a significant fraction of the overall density residues on the heteroatom, despite there being no discernable change in the core 1s excitation energy in the Se K near-edge spectrum (Figure 5). The observed C–X–C bond angle is also closer to 90° (see Figure 7).
The HOMO-LUMO gaps for 5, 3, and 7, relative to the calculated HOMO-LUMO gap for 1, are −26.5, +100.9, and +32.5 kJ mol−1. Relative to the methyl-bearing methionine residue, the presence of the electron withdrawing CF3 moiety serves to widen the HOMO/LUMO gap, while the change from sulfur-containing 1 to selenium-containing 5 results in a smaller HOMO-LUMO gap. The HOMO-LUMO gap for 7 supports the trend of the CF3 modification observed in the sulfur-containing models, where the HOMO-LUMO gap is larger than in the methyl-bearing analogs.
Comparing the HOMO-LUMO gap for 7 with 5 gives a relative change of +59.0 kJ mol−1, again mirroring the 1 vs. 3 trends. These data are also summarized in Table S2 in the Supplementary Material. The character of the HOMO of the various analogs changes relatively little between 3, 5 and 7. Comparing the LUMO of the two selenomethionine analogs, both possess some character on the Se-atom; the LUMO of 5 is spread across the amino acid, with some contribution from Se–C σ* as well as the Cα–C(O) π* orbital; however, in the CF3 analog the Se–C σ* contributes significantly to the character of the LUMO, with relatively little π* contribution on the amino acid backbone region. Interestingly, although energetically different, the HOMO and LUMO for 3 and 7 are qualitatively similar, although the HOMO/LUMO gap in 3 is calculated to be ~70 kJ mol−1 greater (see Table S2 in Supporting Material), and unlike 7, the LUMO of 3 has comparatively more π* character on the backbone atoms, similar to 5 (Figure 7).
Leroux previously determined that the past assertion that the CF3 moiety is at least as large as isopropyl[23] is incorrect, and in fact the volume for CF3 is considerably smaller than the volume of isopropyl.[24] We also performed model calculations comparing isopropyl and CF3, and our findings support Leroux’s previous conclusion that the CF3 substituents are indeed larger than CH3, but certainly of smaller volume than models incorporating isopropyl (data not shown). Furthermore, we have compared the molecular cavities for the side chain fragments of the series S–CH3, S–CF3, Se–CH3 and Se–CF3. The cavities of the molecular fragments calculated during the solvation procedure included an additional margin for a solvent probe molecule (based on water). Relative to S–CH3 (47.39 Å3) the volume of the S–CF3 moiety is 123% larger (58.43 Å3), the Se–CH3 moiety is 111% larger (52.60 Å3), and the Se–CF3 is 133% larger (63.22 Å3).
Methioninase Studies
Figure 8 shows that the cell growth rate was inhibited by 16.7%, 26.1%, and 44.6% in HCT116 cells treated by incubating Trichomonas vaginalis methioninase (40 U/L) with 1.25, 2.5 and 5 μmol/L 5, and by 49.4%, 60.0% and 76.3% in the cells treated with 1.25, 2.5 and 5 μ mol/L 7b, respectively. That is, 7b is substantially more effective in reducing the concentration of cells than 5 at all concentrations tested. There are three possible explanations for the enhanced cytotoxicity of 7b compared to 5, if it is assumed that 8 is released from 7b, and 6 from 5, respectively, by the methioninase: 1) CF3SeH 8 is more effective in generating reactive oxygen species than methaneselenol (6) (Figure 9); 2) CF3SeH (8) is assumed to break down under physiological conditions, as it does in our model studies, to Se=CF2 (10), which should act as a potent cross-linker of primary amine groups, as proposed for the cytotoxicity of 4 from 3, through formation of S=CF2 (9) (Figure 10); 3) some combination of the first and second postulated processes occurs.
Figure 8.
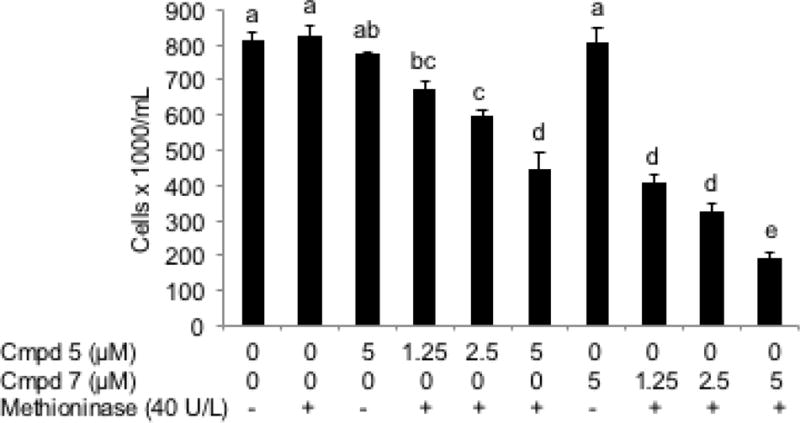
The comparative effect of methaneselenol (6) and trifluoromethaneselenol (8), generated by incubating 40 U/L methioninase with 0, 1.25, 2.5 or 5 μM SeM (5) or TFSeM (7) (as 7b), respectively, on the cell growth of HCT116 human colon cancer cells for 16 h. Values are means ± SEM, n = 4. The letters represent whether measured values are significantly different (p < 0.05, Tukey’s contrasts) between different conditions. If two bars share at least one letter, than the difference between them is not statistically significant (p>0.05), but if they do not have a letter in common, than the measured difference between them is statistically significant (p<0.05). The vertical scale represents the viable cell number of HCT116 human colon cancer cells per mL after 16 h treatment. Each treatment was done with one flask, individually, containing 5 mL media with equal amount of cells prior to treatment.
Figure 9.
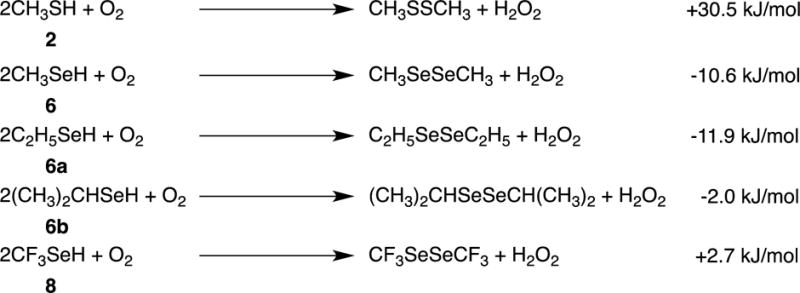
Calculated free energies for reactions of 2, 6, 6a, 6b and 8, with O2.
Figure 10.
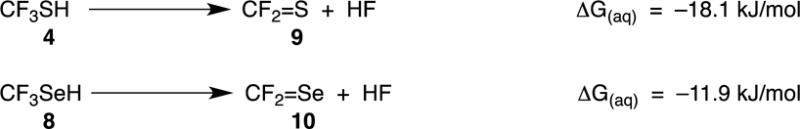
Calculated free energies for conversion of perfluorinated thiol 4 and selenol 8 to the corresponding thio- and selenocarbonyl compounds.
Figure 11 illustrates the proposed redox cycling of selenolate, the anionic form of selenols such as 6 and 8. Figure 9 compares the ease of reduction of dioxygen by methanethiol (2) with selenols 6, 6a, 6b and 8, as calculated by DFT methods. These calculations show that the selenols are superior reducing agents to thiols and that 6 and 6a are comparable (within the error-limits of the method), and slightly superior to 6b. These latter results are in agreement with the findings of the reported relative cytotoxicity 6>6a>6b>2.[2] The fact that the relative observed cytotoxicity of 8 is not correctly predicted by the DFT calculations of Figure 9 suggests that additional, unique properties of 8 need to be considered, e.g., loss of fluoride ion to give selenocarbonyl fluoride 10 (as calculated in Figure 10, and previously reported for 4[1a]), as we have confirmed experimentally for 8. A third possibility is that the cytotoxicity associated with 8 is due to its enhanced acidity (as calculated in Figure 10) or to redox properties of its corresponding anion.[25] The reported pKa values of 2, 4, 6, and 8, are 10.09 ± 0.10, 2.77 ± 0.10, 6.50 ± 0.70, and -1.29 ± 0.70, respectively.
Figure 11.
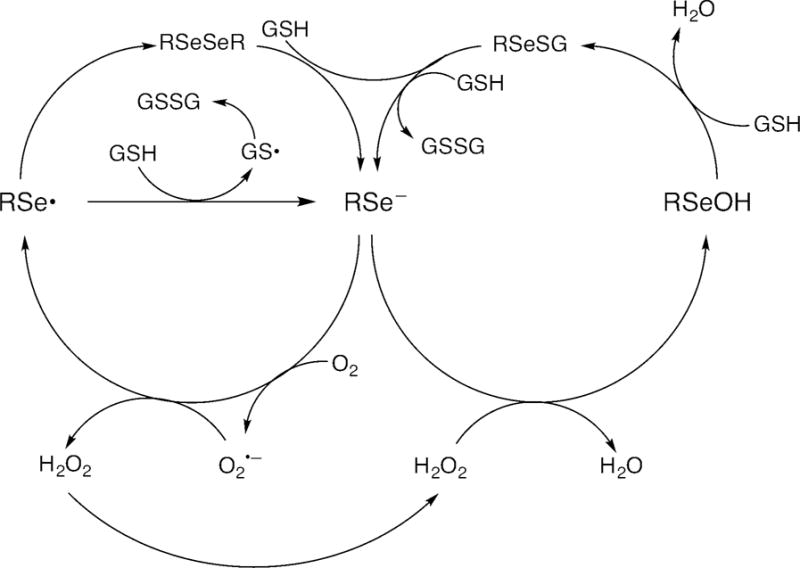
Proposed redox cycling of RSe–, where R = CH3 or CF3.
Incorporation of TFSeM into proteins in E. coli
Incorporation of fluorinated amino acids into proteins provides a structural probe as well as potentially introduces useful functional changes.[26] The methionyltRNA synthetase (MetRS) is among the more promiscuous aminoacyl-tRNAsynthetases,[27] though it still exercises selectivity based on the length and geometry of the side chain.[4] Given that difluoromethionine and trifluoromethionine (3) have been successfully incorporated into proteins,[3, 28] it was of interest to determine if TFSeM (7) could be similarly incorporated. The standard procedure to substitute a given amino acid with its fluorinated analog, involves expressing the target recombinant protein in an E. coli auxotroph that is unable to synthesize the conventional amino acid to be substituted. The fluorinated amino acid is added to a defined media, taken up by the cell, and subsequently incorporated into the protein.[29] The successful substitution relies on both the ability of the respective aminoacyltRNA synthetase to utilize the fluorinated analog and the capacity of the nascent chain to fold into a stable three-dimensional structure when the analog is inserted. The incorporation ratio using this procedure tends to be low due to challenges in loading the fluorinated amino acid to the respective aminoacyltRNA synthetase.[30] Previous publications reported 10–15% successful incorporation of 3 into green fluorescent protein[28a] and 31–70% incorporation into different positions in bacteriophage lambda lysozyme.[28c] These studies suggest that the incorporation of fluorinated amino acids would be most efficient for solvent-exposed positions where the larger volume and the change in hydrophobicity may be better tolerated.
To assess the incorporation of 7, we chose as a target the compact, 56 amino acid protein immunoglobulin-binding B1 domain of streptococcal protein G (GB1). GB1 possesses a remarkably stable fold and is often employed in studies of protein folding and thermal stability.[31] We have constructed variants of GB1 which, following purification, have a single Met present in a unique location in the protein: a Val39Met variant in which the Met is located in a loop and a Leu5Met variant in which the Met is located in the hydrophobic core of the protein (Figure 12A). Similar substitutions have been shown to be stable and well-folded.[31–32] Comparing the expression level of the two different variants allows for a test of whether incorporation of 7 was position dependent.
Figure 12.
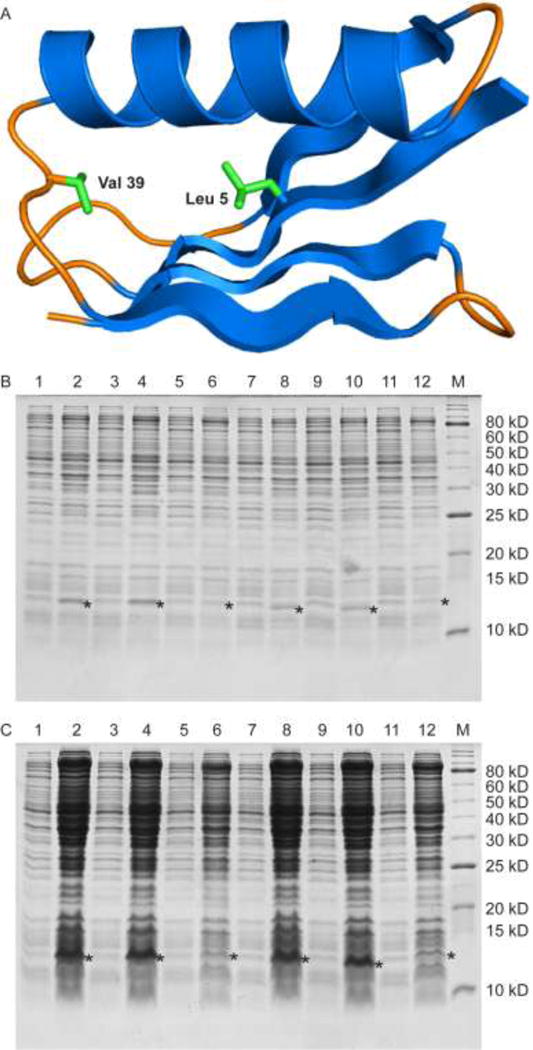
Expression levels of GB1 Val39Met and GB1 Leu5Met. (A) Ribbon diagram of GB1 (PDB ID: 2QMT).[37] In green are the side chains of the residues mutated to Met in the two variants. Val 39 is located on an internal loop between the α-helix and a β-strand while Leu 5 is located on a β-strand and is packed against the hydrophobic core of the protein. (B) Expression levels of GB1 Val39Met and GB1 Leu5Met assessed by a reducing 16% Tris-glycine gel. Lanes 1, 3, 5 are samples of GB1 Val39Met taken before induction, and lanes 2, 4, 6 are samples of GB1 Val39Met taken after induction with 200 μM Met, 200 μM SeM + 22 μM Met, and 200 μM TFSeM (as 7b) + 22 μM Met, respectively. Lanes 7, 9, 11 are samples of GB1 Leu5Met taken before induction, and lanes 8, 10, 12 are samples of GB1 Leu5Met taken after induction with 200 μM Met, 200 μM SeM + 22 μM Met, and 200 μM TFSeM + 22 μM Met, respectively. Induced and uninduced samples were adjusted to have the same optical density to aid the comparison of expression levels. M denotes molecular weight markers. The star denotes the location of expressed GB1 variants. The molecular weight of GB1 Val39Met with the hexahistidine tag is 8823 Da and of GB1 Leu5Met is 8809 Da. Both run with an apparent molecular weight of 12 kDa due to dimerization. (C) Same as in B but the samples’ optical density was not adjusted to the same value.
We followed the expression procedure reported for incorporation of 3 and other methionine analogs[4, 28a, 28c, 33] with the following modifications. First, we utilized a C43(DE3)-based E. coli amino acid auxotrophic host strain named RF11 in which metA was deleted.[17] Second, Met was included in the growth media since a previous study had shown that including 10% Met in the growth media reduced the toxicity of SeM while allowing for a high SeM incorporation ratio.[18a] In order to reduce the toxicity of 7, in the form of its hydrochloride salt 7b, the Met auxotroph RF11 was grown in LB media and switched into a defined media at the mid-logarithmic phase of growth. The defined media was supplemented with either Met, SeM and Met at a 9:1 molar ratio, or TFSeM (as 7b) and Met at a 9:1 molar ratio. The expression levels were compared using SDS–PAGE analysis (Figure 12B and 12C). The expression level of GB1 grown in the presence of TFSeM (as 7b) was lower than that detected in the presence of Met alone or SeM and Met, indicating that TFSeM was more cytotoxic than SeM, in agreement with previous reports about fluorinated amino acids toxicity.
To determine the incorporation rate of TFSeM (as 7b), the two proteins were purified by IMAC chromatography as detailed in the experimental section. The mass spectra of the purified GB1 Leu5Met and GB1 Val39Met grown with 9:1 molar ratio of TFSeM (as 7b):Met contained no clear evidence for TFSeM insertion (Figure 13B and 13D). Instead, the molecular mass of the protein suggested that SeM was incorporated. When cells were grown in the presence of a 9:1 molar ratio of SeM:Met, GB1 Leu5Met and GB1 Val39Met contained primarily SeM reflecting its abundance in the defined media compared to that of Met (Fig. 13A and 13C).
Figure 13.
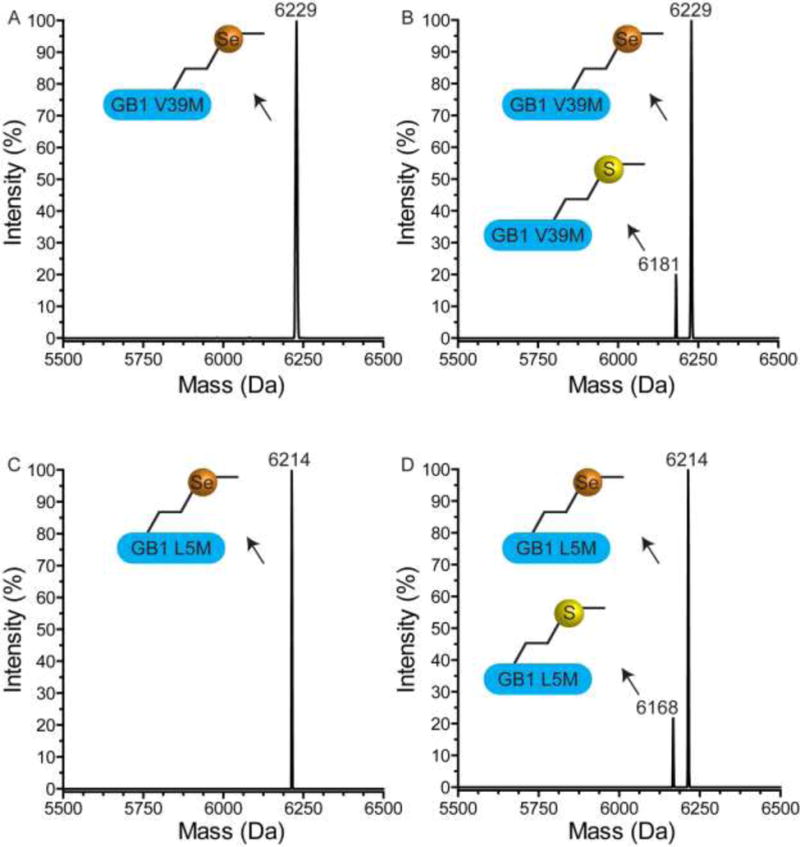
Evaluation of TFSeM (as 7b) incorporation into GB1 by ESI-MS. While TFSeM incorporation was not observed, SeM’s presence was detected even though it was not included in the defined growth media. (A) Deconvoluted mass spectrum showing GB1 Val39Met grown with a 9:1 ratio of SeM:Met in RF11. The expected and observed molecular weight for GB1 Val39Met with Met is 6181 Da and with SeM is 6228 Da (observed 6229 Da). (B) Mass spectrum showing GB1 Val39Met grown with a 9:1 ratio of TFSeM:Met in RF11. The expected molecular weight GB1 Val39Met with TFSeM is 6282 Da (not observed). (C) Deconvoluted mass spectrum showing GB1 Leu5Met grown with a 9:1 ratio of SeM:Met in RF11. The expected and observed molecular weight for GB1 Leu5Met with Met is 6167 Da and with SeM is 6214 Da. (D) Deconvoluted mass spectrum showing GB1 Leu5Met grown with a 9:1 ratio of TFSeM:Met in RF11. The expected molecular weight GB1 Leu5Met with TFSeM is 6268 Da (not observed).
Since neither SeM nor any other source of selenium was included in the growth medium of the Met auxotroph, if SeM is indeed incorporated into GB1 then the observed SeM inserted in GB1 must have originated from TFSeM. To confirm the identity of the protein species detected by ESI-MS the preparation of GB1 Val39Met in RF11 was scaled up. The resulting samples were analyzed by both intact protein ESI-MS and sequencing of trypsin-derived peptides by tandem MS. GB1 Val39Met was grown with solely 22 μM Met (Figure 14A), 22 μM Met and 200 μM TFMet (as 7b) (Figure 14B), and solely 200 μM TFMet (as 7b) (Figure 14C). The predominant protein form observed in this experiment was GB1 Val39Met (calculated molecular mass is 6179 Da and observed is 6180 Da). Also present were lower molecular weight forms that are likely to arise from a loss of methyl (6166 Da) and water (6161 Da) from the protein. These modifications may be attributed to the stress of the Met auxotroph growing with low Met concentrations. The protein forms at 6220/1 Da are most likely due to acetylation in vivo or interactions with acetonitrile used for LC. Only when GB1 Val39Met was grown with 22 μM Met and 200 μM TFMet was it possible to detect the 6227 Da peak suspected as containing SeM. The sample shown in Figure 14B was trypsin digested and sequenced by tandem MS. As Figure 14D shows, the peptide QYANDNG(SeM39)GDEWTYDDATK was identified based on its molecular weight and the distinct isotope pattern of selenium. It was not possible to fully sequence the peptide due to its low abundance in the sample. Interestingly, even though this sample also contains a species with 6280 Da, matching the calculated MW of GB1 Val39TFSeM, the corresponding peptide QYANDNG(TFSeM39)GDEWTYDDATK was not detected. Hence, we could not confirm experimentally that TFSeM was incorporated into GB1 under these specific growth conditions.
Figure 14.
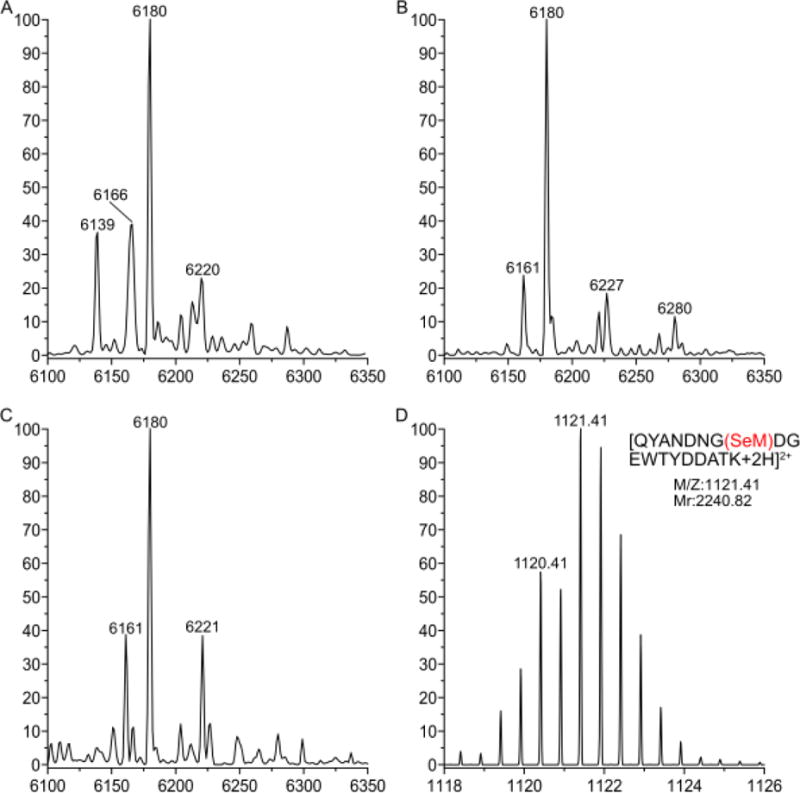
Analysis of the molecular forms detected for GB1 Val39Met grown in the Met auxotroph RF11. Molecular species: GB1 Val39Met calculated mass is 6181 Da and detected is 6180 Da; GB1 Val39SeM calculated mass is 6228 Da and detected is 6227 Da. Other putative assignments are: GB1 Val39CF3Sem calculated molecular mass is 6282 Da, GB1 Val39Met with a loss of a methyl group is 6166 Da, GB1 Val39Met with a loss of water is 6161 Da; and GB1 Val39Met with acetylation or an acetonitrile adduct is 6221 Da. (A) Deconvoluted mass spectrum showing GB1 Val39Met grown with solely 22 μM Met. (B) Deconvoluted mass spectrum showing GB1 Val39Met grown with 22 μM Met and 200 μM TFSeM. (C) Deconvoluted mass spectrum showing GB1 Val39Met grown with solely 200 μM TFSeM. (D) ESI-MS spectrum of the peptide QYANDNG(SeM)DGEWTYDDATK derived from trypsin digestion of the sample shown in panel B. The observed isotope pattern matches the calculated one (the predicted pattern is not shown).
Since as mentioned neither SeM nor any other source of selenium was included in the growth medium, and proteins with SeM were not observed when GB1 Val39Met was grown without TFSeM, we reasoned that the fraction of TFSeM recycled to SeM in vivo. The incorporation of SeM into GB1 Val39Met and Leu5Met during its expression in E. coli cultured in the presence of TFSeM might be envisaged to take place via intermediate formation of TFSeSAM. In such a pathway, the trifluoromethyl group could be removed by one or more of the S-adenosylmethionine (SAM)-dependent methyltransferases (MTases) that normally transfer the methyl moiety of SAM to a target acceptor (Figure 15).[34] The resulting molecule, Se-adenosylhomoselenocysteine (SeAH), would then be acted upon by S-adenosylhomocysteine/methylthioadenosine nucleosidase to yield adenine and Se-ribosylhomoselenocysteine, the latter of which would be converted to homoselenocysteine and (4S)-4,5-dihydroxypentan-2,3-dione by ribosylhomocysteine lyase. Homoselenocysteine would then be methylated by either the cobalamin-dependent or the cobalamin-independent methionine synthase to afford selenomethionine. Methylation of homoselenocysteine could also take place by lesser appreciated mechanisms, such as homocysteine S-methyltransferase, the product of the mmuM gene in E. coli, which uses L-S-methylmethionine or the S(+) isomer of SAM as the methyl donor.[35]
Figure 15.
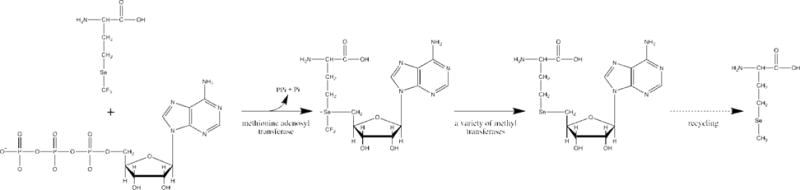
Proposed metabolic path for recycling TFSeM into SeM.
To determine whether TFSeSAM is a key intermediate in the formation of SeM from TFSeM, the ability of E. coli methionine adenosyltransferase (MAT) to convert TFSeM into TFSeSAM was assessed. MAT catalyzes the condensation of L-methionine and ATP to SAM, and also yields pyrophosphate and inorganic phosphate as byproducts. Shown in 16A is an HPLC elution profile of the products of a MAT reaction incubated for 30 or 60 min at 25 °C under turnover conditions in the presence of TFSeM (as 7b). A trace containing authentic standards of related adenine-containing compounds (dashed line) and tryptophan, used as an internal standard, indicates that SAM and SeSAM elute between 11 and 13 min. In both the 30 and 60 min time points, no peaks are present in the region corresponding to SAM or SeSAM in the reaction containing TFSeM, and observable peaks throughout the chromatogram are present before initiation of the reaction (0 min) and do not increase with time (blue, yellow, and red lines). An HPLC elution profile of a control reaction, conducted under identical conditions, but containing SeM rather than TFSeM, shows robust and time-dependent formation of SeSAM (Figure 16B, red, yellow, green, and blue traces), indicating that the MAT preparation is active. The unknown peak eluting just after SAH is also present in the TFSeM reaction, and is most likely a contaminant in the MAT preparation. The observation of SeSAH suggests that the MAT preparation is also contaminated with one or more methyltransferases.
Figure 16.
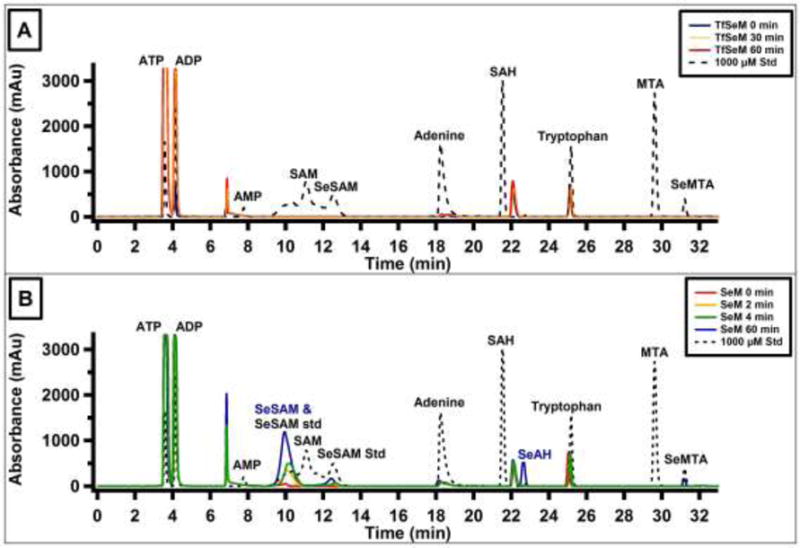
HPLC elution profiles of MAT time-dependent reactions with (A) TFSeM (as 7b) or (B) SeM as substrates. Authentic standard compounds are shown in the black dotted traces. Elution times in minutes are as follows: 3.7 (ATP); 4.2 (ADP); 7.8 (AMP); 11.5 (SAM); 9.8 and 12.3 (SeSAM); 18.2 (adenine); 21.8 (SAH); 25.2 (tryptophan IS); 29.7 (MTA); 31.3 (SeMTA). TFSeM resulted in no apparent formation of TFSeSAM or SeSAM over the course of 60 min, whereas SeM was converted to SeSAM by MAT as evidenced by the time-dependent formation of SeSAM (elution times of 9.8 and 12.3 min). The two elution times for SeSAM match that of the authentic standard; under Method 1 conditions SeSAM elutes as one sharp, defined peak, indicating the double peak is a result of the chromatographic conditions.
Similar reactions were also analyzed by LC-MS to assess whether the concentration of TFSeM (as 7b) changes as a function of time. Shown in Figure S1A is a total ion chromatogram of a MAT reaction at various extents of incubation in the presence of SeM. Peaks at 1.14, 1.39, 1.54, and 4.46 min undergo time-dependent changes. The peak at 1.39 min (m/z 198.1) corresponds to SeM, and decreases as a function of time, while peaks at 1.14 (m/z 447.1), 1.54 (m/z 433.1) and 4.46 (m/z 346.1) correspond to SeSAM, SeAH, and methylselenoadenosine (MSeA), respectively, and increase as a function of time. The formation of SeSAH and MSeA is not unexpected, given the methyltransferase contamination in the MAT preparation and the propensity of SeSAM to form MSeA and homoserine lactone nonenzymatically.[36] By contrast, the total ion chromatogram of a MAT reaction at various extents of incubation in the presence of TFSeM shows that the concentration of TFSeM does not change significantly during 18 h of incubation (Figure S1B).
Given that E. coli MAT does not appear to use TFSeM as a substrate, experiments were conducted to assess whether other proteins in E. coli can act on it. In these experiments, 1 mM TFSeM (as 7b) was incubated for various extents of time with crude lysate from E. coli methionine auxotroph strain B834(DE3)pLysS with a metE deletion, and the reactions were analyzed by LC-MS. As shown in Figure 17A and 17B, both TFSeM and SeM are consumed as a function of time. When similar reactions were conducted with TFSeM using crude lysate that had been boiled for 10 min or the filtrate from crude lysate that had been subjected to centrifugation in a spin concentrator containing a membrane with a 3 kDa molecular weight cutoff, the concentration of both SeM and TFSeM stayed constant throughout the 18 h incubation (Figure S2A–D). This behavior strongly suggests that TFSeM is acted upon by an unidentified protein in E. coli. Together, these results indicate that although TFSeM is not converted into TFSeSAM, it is a target of another protein in E. coli that most likely converts it into homoselenocysteine or a precursor of this metabolite.
Figure 17.
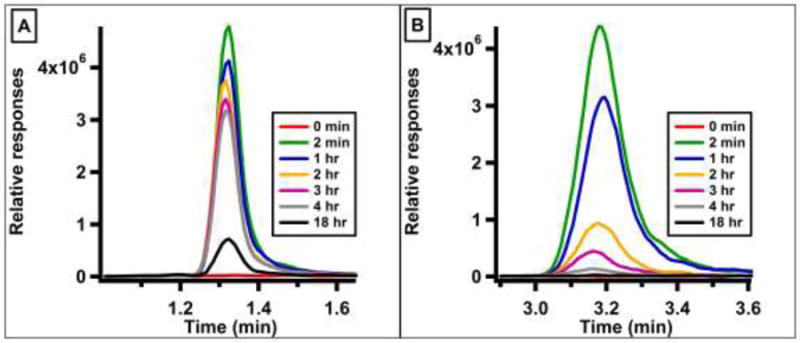
Extracted ion chromatograms for (A) SeM (m/z 198.1) and (B) TFSeM (m/z 252.0) in time-dependent reactions with E. coli B834(DE3)pLysS crude lysate. Both SeM and TFSeM are consumed as a function of time.
Conclusion
Trifluoroselenomethionine (7), a novel non-natural amino acid, was synthesized in seven steps from N-(tert-butoxycarbonyl)-L-aspartic acid tert-butyl ester. Trifluoroselenomethionine shows enhanced methioninase-induced cytotoxicity toward human colon cancer derived HCT-116 cells compared to selenomethionine (5). Mechanistic explanations for this enhanced activity were computationally and experimentally examined. Comparison of 5 and 7 by selenium EXAFS and DFT calculations showed them to be spectroscopically and structurally very similar. However, when two different mutants of the protein GB1 were expressed in the E. coli methionine auxotroph cell line RF11 using 7 (as 7b) and methionine in a 9:1 ratio, it was found that, surprisingly, over 80% of the proteins were composed of 5, even though no 5 had been added, implying loss of the trifluoromethyl group from 7. A yet unidentified enzyme catalyzes the process. It is of interest to determine which enzyme is responsible for the loss of trifluoromethyl from 7, as this chemical group is present in many drugs and thus is of inherent medicinal interest.
Supplementary Material
Acknowledgments
We thank Alexander Filatov for determining the X-ray structure of compound 15 and Dr. Jesse McAtee for CI/MS accurate mass measurement.
Funding Sources
Work at the University of Albany was supported by grants from the National Science Foundation (CHE-0744578; CHE-1265679; CHE-1337594 [MRI]; CHE-1429329 [MRI]) and the University at Albany (E.B.). Work at Grand Forks Human Nutrition Research Center was supported by USDA-ARS, CRIS project (5450-51000-045-00D) (H.Z.). Work at the University of Saskatchewan was supported by the Natural Sciences and Engineering Research Council of Canada, the Canadian Institutes for Health Research (CIHR) and the Saskatchewan Health Research Foundation (SHRF). G.N.G. is a Canada Research Chair. Computational aspects of this research were enabled by support provided by WestGrid (www.westgrid.ca) and Compute Canada/Calcul Canada (www.computecanada.ca). Synchrotron experiments were carried out at the Stanford Synchrotron Radiation Lightsource (SSRL), which is funded by the U.S. Department of Energy (DOE), Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the DOE, Office of Biological and Environmental Sciences, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program. Work at the University of Delaware was supported by the National Science Foundation CAREER grant MCB-1054447 (S.R.) and by the Delaware COBRE program, with a grant from the National Institute of General Medical Sciences – NIGMS (1 P30 GM110758-01) from the National Institutes of Health. Work at the Pennsylvania State University was supported by the National Health Institute grant GM-103268.
Abbreviations
- EXAFS
extended X-ray absorption fine structure
- DFT
density function theory
- GB1
immunoglobulin-binding B1 domain of streptococcal protein G
- Met
methionine
- MGL
L-methionine γ-lyase
- TFM
trifluoromethionine
- SeM
selenomethionine
- TFSeM
trifluoroselenomethionine
- TFSeSAM
Se-adenosyltrifluoromethylselenomethionine
- XAS
X-ray absorption spectroscopy
Footnotes
Supporting Information. X-ray Crystal Data and Structure Refinement, supporting information for DFT calculation, MAT activity assays and information about synthesis and compounds purity. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interest.
References
- 1.a) Moya IA, Westrop GD, Coombs GH, Honek JF. Biochemical J. 2011;438:513–521. doi: 10.1042/BJ20101986. [DOI] [PubMed] [Google Scholar]; b) Esaki N, Tanaka H, Uemura S, Suzuki T, Soda K. Biochemistry. 1979;18:407–410. doi: 10.1021/bi00570a003. [DOI] [PubMed] [Google Scholar]; c) Zeng H, Briske-Anderson M, Wu M, Moyer MP. Nutr Cancer. 2012;64:128–135. doi: 10.1080/01635581.2012.630555. [DOI] [PubMed] [Google Scholar]; d) Zeng H, Cheng WH, Johnson LK. J Nutr Biochemistry. 2013;24:776–780. doi: 10.1016/j.jnutbio.2012.04.008. [DOI] [PubMed] [Google Scholar]; e) Miki K, Xu M, Gupta A, Ba Y, Tan Y, Al-Refaie W, Bouvet M, Makuuchi M, Moossa AR, Hoffman RM. Cancer Res. 2001;61:6805–6810. [PubMed] [Google Scholar]; f) Coombs GH, Mottram JC. Antimicrob Agents Chemother. 2001;45:1743–1745. doi: 10.1128/AAC.45.6.1743-1745.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spallholz JE, Palace VP, Reid TW. Biochem Pharmacol. 2004;67:547–554. doi: 10.1016/j.bcp.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 3.Duewel H, Daub E, Robinson V, Honek JF. Biochemistry. 1997;36:3404–3416. doi: 10.1021/bi9617973. [DOI] [PubMed] [Google Scholar]
- 4.van Hest JCM, Kiick KL, Tirrell DA. J Am Chem Soc. 2000;122:1282–1288. [Google Scholar]
- 5.Salituro GM, Townsend CA. J Am Chem Soc. 1990;112:760–770. [Google Scholar]
- 6.Flohr A, Aemissegger A, Hilvert D. J Med Chem. 1999;42:2633–2640. doi: 10.1021/jm991008q. [DOI] [PubMed] [Google Scholar]
- 7.Vaněk V, Buděšínský M, Kabeleová P, Šanda M, Kožíšek M, Hančlová I, Mládková J, Brynda J, Rosenberg I, Koutmos M, Garrow TA, Jiráček J. J Med Chem. 2009;52:3652–3665. doi: 10.1021/jm8015798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grobe J, Levan D, Welzel J. J Organomet Chem. 1988;340:153–160. [Google Scholar]
- 9.Cramer SP, Tench O, Yocum M, George GN. Nucl Instrum Meth. 1988;A266:586–591. [Google Scholar]
- 10.http://ssrl.slac.stanford.edu/exafspak.html
- 11.Rehr JJ, Mustre de Leon J, Zabinsky SI, Albers RC. J Am Chem Soc. 1991;113:5135–5140. [Google Scholar]
- 12.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro FÉß, Bearpark MJ, Heyd J, Brothers EN, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas ÉÄÉn, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09. Gaussian, Inc.; Wallingford, CT, USA: 2009. [Google Scholar]
- 13.a) Mennucci B, Tomasi J. J Chem Phys. 1997;106:5151–5158. [Google Scholar]; b) Mennucci B, Cancès E, Tomasi J. J Phys Chem B. 1997;101:10506–10517. [Google Scholar]; c) Tomasi J, Mennucci B, Cancès E. J Mol Struct (Theochem) 1999;464:211–226. [Google Scholar]; d) Barone V, Cossi M, Tomasi J. J Chem Phys. 1997;107:3210–3221. [Google Scholar]; e) Scalmani G, Frisch MJ. J Chem Phys. 2010;132:114110. doi: 10.1063/1.3359469. [DOI] [PubMed] [Google Scholar]
- 14.Breneman CM, Wiberg KB. J Comput Chem. 1990;11:361–373. [Google Scholar]
- 15.a) Mφller C, Plesset MS. Phys Rev. 1934;46:618–622. [Google Scholar]; b) Head-Gordon M, Pople JA, Frisch MJ. Chem Phys Lett. 1988;153:503–506. [Google Scholar]
- 16.Smith CK, Withka JM, Regan L. Biochemistry. 1994;33:5510–5517. doi: 10.1021/bi00184a020. [DOI] [PubMed] [Google Scholar]
- 17.Lin MT, Sperling LJ, Frericks Schmidt HL, Tang M, Samoilova RI, Kumasaka T, Iwasaki T, Dikanov SA, Rienstra CM, Gennis RB. Methods. 2011;55:370–378. doi: 10.1016/j.ymeth.2011.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Studier FW. Prot Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]; b) Li F, Lutz PB, Pepelyayeva Y, Arnér ESJ, Bayse CA, Rozovsky S. Proc Natl Acad Sci U S A. 2014;111:6976–6981. doi: 10.1073/pnas.1319022111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a) Kapust RB, Tözser J, Fox JD, Anderson DE, Cherry S, Copeland TD, Waugh DS. Protein Engineering. 2001;14:993–1000. doi: 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]; b) Blommel PG, Fox BG. Protein Expression and Purification. 2007;55:53–68. doi: 10.1016/j.pep.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Billard T, Langlois BR. Tetrahedron Lett. 1996;37:6865–6868. [Google Scholar]
- 21.a) Dale JW, Emeleus HJ, Haszeldine RN. J Chem Soc. 1958:2939–2945. [Google Scholar]; b) Gómez Castaño JA, Romano RM, Beckers H, Willner H, Della Védova CO. Inorg Chem. 2010;49:9972–9977. doi: 10.1021/ic101254k. [DOI] [PubMed] [Google Scholar]
- 22.Bruno IJ, Cole JC, Edgington PR, Kessler M, Macrae CF, McCabe P, Pearson J, Taylor R. Acta Cryst. 2002;B58:389–397. doi: 10.1107/s0108768102003324. [DOI] [PubMed] [Google Scholar]
- 23.Smart BE. In: Chemistry of Organofluorine Compounds. Hudlicky M, editor. American Chemical Society; Washington DC: 1995. [Google Scholar]
- 24.Leroux F. ChemBioChem. 2004;5:644–649. doi: 10.1002/cbic.200300906. [DOI] [PubMed] [Google Scholar]
- 25.McCarty MF, Whitaker JW. Alt Med Rev. 2010;15:264–649. [PubMed] [Google Scholar]
- 26.Salwiczek M, Nyakatura EK, Gerling UIM, Ye SJ, Koksch B. Chem Soc Rev. 2012;41:2135–2171. doi: 10.1039/c1cs15241f. [DOI] [PubMed] [Google Scholar]
- 27.van Hest JCM, Tirrell DA. Chem Commun. 2001:1897–1904. doi: 10.1039/b105185g. [DOI] [PubMed] [Google Scholar]
- 28.a) Budisa N, Pipitone O, Siwanowicz I, Rubini M, Pal PP, Holak TA, Gelmi ML. Chem Biodivers. 2004;1:1465–1475. doi: 10.1002/cbdv.200490107. [DOI] [PubMed] [Google Scholar]; b) Vaughan MD, Cleve P, Robinson V, Duewel HS, Honek JF. J Am Chem Soc. 1999;121:8475–8478. [Google Scholar]; c) Duewel HS, Daub E, Robinson V, Honek JF. Biochemistry. 2001;40:13167–13176. doi: 10.1021/bi011381b. [DOI] [PubMed] [Google Scholar]
- 29.Merkel L, Schauer M, Antranikian G, Budisa N. Chembiochem. 2010;11:1505–1507. doi: 10.1002/cbic.201000295. [DOI] [PubMed] [Google Scholar]
- 30.Yoder NC, Kumar K. Chem Soc Rev. 2002;31:335–341. doi: 10.1039/b201097f. [DOI] [PubMed] [Google Scholar]
- 31.Gronenborn AM, Frank MK, Clore GM. FEBS Letters. 1996;398:312–316. doi: 10.1016/s0014-5793(96)01262-8. [DOI] [PubMed] [Google Scholar]
- 32.Chiu HP, Kokona B, Fairman R, Cheng RP. J Am Chem Soc. 2009;131:13192–13193. doi: 10.1021/ja903631h. [DOI] [PubMed] [Google Scholar]
- 33.Budisa N, Steipe B, Demange P, Eckerskorn C, Kellermann J, Huber R. Eur J Biochem. 1995;230:788–796. doi: 10.1111/j.1432-1033.1995.tb20622.x. [DOI] [PubMed] [Google Scholar]
- 34.a) Martin JL, McMillan FM. Curr Opin Struct Biol. 2002;12:783–793. doi: 10.1016/s0959-440x(02)00391-3. [DOI] [PubMed] [Google Scholar]; b) Schubert HL, Blumenthal RM, Cheng XD. Trends Biochem Sci. 2003;28:329–335. doi: 10.1016/S0968-0004(03)00090-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.a) Neuhierl B, Thanbichler M, Lottspeich F, Böck A. J Biol Chem. 1999;274:5407–5414. doi: 10.1074/jbc.274.9.5407. [DOI] [PubMed] [Google Scholar]; b) Sekowska A, Kung HF, Danchin A. J Mol Microbiol Biotechnol. 2000;2:145–177. [PubMed] [Google Scholar]
- 36.Iwig DF, Booker SJ. Biochemistry. 2004;43:13496–13509. doi: 10.1021/bi048693+. [DOI] [PubMed] [Google Scholar]
- 37.Schmidt HLF, Sperling LJ, Gao YG, Wylie BJ, Boettcher JM, Wilson SR, Rienstra CA. J Phys Chem B. 2007;111:14362–14369. doi: 10.1021/jp075531p. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.