

Abstract
Key points
Endothelial inwardly rectifying K+ (Kir2.1) channels regulate flow‐induced vasodilatation via nitric oxide (NO) in mouse mesenteric resistance arteries.
Deficiency of Kir2.1 channels results in elevated blood pressure and increased vascular resistance.
Flow‐induced vasodilatation in human resistance arteries is also regulated by inwardly rectifying K+ channels.
This study presents the first direct evidence that Kir channels play a critical role in physiological endothelial responses to flow.
Abstract
Inwardly rectifying K+ (Kir) channels are known to be sensitive to flow, but their role in flow‐induced endothelial responses is not known. The goal of this study is to establish the role of Kir channels in flow‐induced vasodilatation and to provide first insights into the mechanisms responsible for Kir signalling in this process. First, we establish that primary endothelial cells isolated from murine mesenteric arteries express functional Kir2.1 channels sensitive to shear stress. Then, using the Kir2.1+/− heterozygous mouse model, we establish that downregulation of Kir2.1 results in significant decrease in shear‐activated Kir currents and inhibition of endothelium‐dependent flow‐induced vasodilatation (FIV) assayed in pressurized mesenteric arteries pre‐constricted with endothelin‐1. Deficiency in Kir2.1 also results in the loss of flow‐induced phosphorylation of eNOS and Akt, as well as inhibition of NO generation. All the effects are fully rescued by endothelial cell (EC)‐specific overexpression of Kir2.1. A component of FIV that is Kir independent is abrogated by blocking Ca2+‐sensitive K+ channels. Kir2.1 has no effect on endothelium‐independent and K+‐induced vasodilatation in denuded arteries. Kir2.1+/− mice also show increased mean blood pressure measured by carotid artery cannulation and increased microvascular resistance measured using a tail‐cuff. Importantly, blocking Kir channels also inhibits flow‐induced vasodilatation in human subcutaneous adipose microvessels. Endothelial Kir channels contribute to FIV of mouse mesenteric arteries via an NO‐dependent mechanism, whereas Ca2+‐sensitive K+ channels mediate FIV via an NO‐independent pathway. Kir2 channels also regulate vascular resistance and blood pressure. Finally, Kir channels also contribute to FIV in human subcutaneous microvessels.
Keywords: endothelial cells, flow‐induced vasodilation, K+ channels
Key points
Endothelial inwardly rectifying K+ (Kir2.1) channels regulate flow‐induced vasodilatation via nitric oxide (NO) in mouse mesenteric resistance arteries.
Deficiency of Kir2.1 channels results in elevated blood pressure and increased vascular resistance.
Flow‐induced vasodilatation in human resistance arteries is also regulated by inwardly rectifying K+ channels.
This study presents the first direct evidence that Kir channels play a critical role in physiological endothelial responses to flow.
Abbreviations
- ACh
acetylcholine
- CO
cardiac output
- COX
cyclooxygenase
- DAR4M
diaminorhodamine‐4M
- EC
endothelial cell
- EDHF
endothelial derived hyperpolarizing factor
- eNOS
endothelial nitric oxide synthase
- FIV
flow‐induced vasodilatation
- KCa
Ca2+‐activated K+ channel
- Kir
inwardly rectifying K+ channel
- l‐NAME
N ω‐nitro‐l‐arginine methyl ester hydrochloride
- MAP
mean arterial pressure
- NO
nitric oxide
- PVR
peripheral vascular resistance
- SK
small conductance Ca2+‐activated K+ channels
- SNP
sodium nitroprusside
- WT
wild type
Introduction
Inwardly rectifying K+ channels (Kir) are a major type of K+ channel that are ubiquitously expressed in a variety of cell types (Kubo et al. 2005; Hibino et al. 2010). Endothelial Kir channels drew significant attention after it was discovered that these channels are sensitive to fluid shear stress, a major factor regulating endothelial function (Olesen et al. 1988; Davies et al. 2005). However, little is known about the physiological role of Kir channels in flow‐induced endothelial responses.
A hallmark of endothelial response to flow is flow‐induced vasodilatation (FIV), which is one of the essential physiological mechanisms for the control of blood flow to the microcirculation. One of the major mechanisms contributing to FIV is the generation of endothelial‐derived hyperpolarizing factor (EDHF), which may be generated by endothelial K+ channels (Luksha et al. 2009). Vascular endothelial cells (ECs) express two major classes of K+ channel; Kir channels characterized by inward rectification and sensitivity to fluid shear stress (Olesen et al. 1988; Nilius & Droogmans, 2001; Fang et al. 2005) and Ca2+‐activated K+ (KCa) channels that are sensitive to the level of intracellular Ca2+ (Wulff & Köhler, 2013). Earlier studies established that genetic deficiency of KCa channels significantly impairs endothelium‐dependent vasodilatation (Taylor et al. 2003; Si et al. 2006). Moreover, it was further shown that among different subtypes of KCa, small‐conductance KCa (SK) channels mainly contribute to FIV (Brahler et al. 2009). However, a lack of an appropriate genetic model for Kir channels constituted until recently a major constraint for determining the role of Kir channels in this process. A recent study by Sonkusare et al. (2016) demonstrated that endothelial‐specific loss of Kir2.1 channels results in the loss of Kir currents in mesenteric ECs and a significant reduction in vasodilatation by muscarinic receptor activation. Our study focuses on the role of Kir channels in flow‐induced vasodilatation.
Structurally, Kir channels are divided into seven subfamilies (Kir1–7) identified by distinct biophysical properties (Kubo et al. 2005). Most types of Kir channels are blocked by Ba2+ (Kubo et al. 2005) and in the absence of more specific blockers, Ba2+ sensitivity has been used in multiple studies to identify Kir channels in different cell types. Specifically, strongly rectifying, Ba2+‐sensitive Kir currents have been found in bovine, porcine and human aortic ECs (Romanenko et al. 2002; Fang et al. 2005), and in rat microvascular cerebral and mesenteric ECs (Crane et al. 2003; Millar et al. 2008) and hamster cremaster arterial ECs (Jackson, 2005) but not in rabbit or mouse aortic endothelium (Rusko et al. 1992; Ledoux et al. 2008). Biophysical characterization of these currents indicated that they are underlain by Kir2 channels, a subfamily of Kir, characterized by strong inward rectification. Further, in human aortic ECs Kir current is underlain by Kir2.1 and Kir2.2 (Fang et al. 2005) and Kir2.1 channels were shown to be expressed in microvascular ECs (Eschke et al. 2002; Yang et al. 2003; Jackson, 2005).
In this study, we took advantage of a heterozygous Kir2.1+/− mouse model and human blood vessel model to test the hypothesis that the functional expression of Kir2.1 channels in the vascular endothelium is critical for FIV in the microcirculation. We also further sought to (1) determine if Kir channels and nitric oxide generation during flow are linked in mesenteric resistance arteries and (2) explore the relationship between Kir and SK channels in FIV of murine and human resistance arteries. These data provide novel insights on the role of Kir channels in human and murine vascular reactivity.
Methods
Ethical approval
The University of Illinois Animal Care Committee (ACC) approved ethical issues for all animal experiments (ACC#13‐209). All mice were housed under pathogen‐free conditions at the University of Illinois Animal Care Vivarium, which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC International) and adheres to the standards of the Animal Welfare Act, the Public Health Service Policy, and the NIH Guide for the Care and Use of Laboratory Animals. We treated all mice humanely in accordance with institutional guidelines, and performed all animal experiments based on the Guide for the Care and Use of Laboratory Animals. We took all steps to minimize the animals’ pain and suffering. All animals were purchased from Jackson Laboratory. All experimental animals were killed before the tissue extraction using 30% CO2 followed by cervical dislocation. All experimental mice were anaesthetized with the isoflurane inhalation (3.5% isoflurane in 96.5% oxygen) during the experiments (echocardiogram, 15 min; tail‐cuff blood pressure measurement, 20 min; carotid artery blood pressure measurement, 30 min). All mice were located on a heating pad to maintain their temperature at 37°C when they were anaesthetized. We conformed to the principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology (Grundy, 2015). The study protocol and procedures were approved by the University of Illinois at Chicago Institutional Review Board (IRB 2007‐0867, 2010‐1010). IRB approved the human experimental procedures based on the Declaration of Helsinki and good clinical practice.
Reagents and antibodies
BaCl2, apamin, N ω‐nitro‐l‐arginine methyl ester hydrochloride (l‐NAME), acetylcholine, sodium nitroprusside (SNP), endothelin‐1 (ET‐1), ouabain, papaverine hydrochloride, and mouse monoclonal anti‐αSMA antibodies were obtained from Sigma‐Aldrich (St Louis, MO, US). Rabbit monoclonal antibodies for mouse inwardly rectifying K+ channel 2.1 (Kir2.1) were obtained from Abcam (Cambridge, MA, US). Rabbit polyclonal anti‐mouse GAPDH, rabbit polyclonal anti‐Akt1, rabbit polyclonal anti‐pAkt1, rabbit polyclonal anti‐peNOS antibodies were purchased from Cell Signalling Technology (Danvers, MA, US) and rabbit polyclonal anti‐eNOS antibodies were purchased from Santa Cruz Biotechnology (Paso Robles, CA, US). Rabbit polyclonal anti‐mouse vWF antibodies were purchased from Dako (Canada), and rat polyclonal anti‐mouse CD31 antibodies were purchased from Beckton Dickinson (Franklin Lakes, NJ, US). Prime Time qPCR predesigned primers for mouse Kir2.1, 2.2, 2.3, CD31, αSMA and GAPDH were purchased from IDT (Coralville, IA, US). pcDNA3‐Kir2.1‐HA was a gift from Dr Carol Vandenberg (University of California, Santa Barbara). Adenoviral vector expressing Kir2.1‐HA with VE‐Cadherin EC‐specific promoter was generated by Vector BioLabs (Malvern, PA, US). Cells or vessels were transfected with VE‐Cad Kir2.1 vector with 100 MOI for 48 h.
Isolation of mesenteric arteries
Mice were killed using CO2 followed by cervical dislocation. The abdominal skin and muscle membrane were opened sufficiently widely to retain a view of the whole of the mesentery. All mesenteric tissues and part of the pancreatic tissues were taken out by cutting the edge of mesenteric fat tissues to the intestine. Mesenteric tissues were transferred to Hepes buffer for the careful dissection of isolated arteries. Connective tissue and fat surrounding each blood vessels was carefully removed and the tissues were rinsed to remove any remaining blood cells.
Endothelial cell isolation and immunostaining
Endothelial cell isolation
Six mesenteric arteriole beds were taken from WT and Kir2.1+/− mice, respectively. Arteriole beds were washed in Ca2+‐ and Mg2+‐ free PBS and transferred to 1 ml of collagenase/dispase digestion solution (100 mg ml−1) (Roche, Burgess Hill, UK). Using sharp tip scissors, arteriolar branches were broken into small pieces. The tubes were incubated in 37°C shaking incubator for 1 h. Digested tissues and cells were spun down in 300 g for 10 min. Supernatants were removed carefully and 1 ml of 0.025% trypsin‐EDTA was added to the tubes. The tubes were incubated in 37°C shaking incubator for 15 min. The solution was gently mixed with 1000p pipette, and sieved through 40‐μm cell‐sieve. The cell‐sieves were washed with 10 ml of DMEM, 10% FBS to neutralize the trypsin effect. Cells were spun down in 300 g for 10 min and then isolated using AutoMACs system and/or MiniMACs system from Miltenyi Biotech (Bisley, UK) with magnetic beads conjugated polyclonal anti‐mouse PECAM 1 IgG (Miltenyi Biotech). As recommended by the company protocol (Miltenyi Biotec) to increase the purity of the EC population, we used a two‐step separation method, in which the cell suspension mixed with the magnetic CD31 beads is run sequentially through two magnetic columns. Purity of the isolated ECs was tested with flow cytometry by staining with monoclonal anti‐mouse CD31‐PE IgG and with immunofluorescence staining with anti‐mouse vWF and PECAM 1 IgGs. Flow cytometry was performed using CyAn ADP (Beckman Coulter). After the isolation, cells were maintained in EGM‐2 endothelial cell growth media (Lonza, Cambridge, UK). Cells for experiments were used between passage numbers 5 and10.
Immunostaining
Briefly, cells were fixed with 4% paraformaldehyde for 15 min, permeablized with 0.5% Triton‐X100 in PBS for 5 min and then incubated with the primary antibodies (rat anti‐mouse PECAM 1 IgG, mouse anti‐αSMA IgG and rabbit anti‐mouse vWF IgG) at 4°C overnight followed by incubation with secondary antibodies (Alexafluor 488 anti‐rat IgG, Alexafluor 555 anti‐mouse IgG and Alexafluor 568 anti‐rabbit IgG, Invitrogen) for 2 h at room temperature. Fluorescence images were taken through a Zeiss Axiovert 200M microscope with an AxioCam MRm camera.
PCR and Western blot analysis
qPCR
To measure the mRNA expression of PECAM1 (CD31), smooth muscle α‐actin (αSMA), Kir2.1, 2.2 and 2.3 in isolated mouse endothelial cells, quantitative PCR was performed. Briefly, endothelial cell RNA from WT and Kir2.1+/− mouse arteries was extracted using the RNeasy mini kit (Qiagen) according to the manufacturer's instructions, including an on‐column DNase treatment step. The quantity and quality of extracted RNA were measured using Nanodrop (Thermo Fisher). cDNAs were generated with random primers using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Quantitative PCR was performed with Universal SYBR green Supermix (Biorad). Each sample was triplicated for analysis and at least three different sets of cells were tested. After initial denaturation at 95°C, 40 cycles of 95°C for 30 s and annealing temperature of 60°C for 30 s were performed, using the Viia 7 real‐time PCR system (Applied Biosystems). Acquired fluorescence data were analysed using 2−ΔΔCt methods, normalized by GAPDH.
Western blot
Cells were rinsed with ice‐cold PBS (pH 7.4) and homogenized in ice‐cold RIPA buffer (50 mm Tris–HCl, 150 mm NaCl, 1% NP‐40, 0.5% Na‐Doc (sodium deoxycholate), 0.1% SDS, 1 mm EDTA, at pH 7.6) containing 2 mm phenylmethylsulfonyl fluoride with protease inhibitor tablets (Roche). Samples were boiled in SDS sample buffer. Samples were resolved by SDS–PAGE (10% acrylamide gel) and transferred onto a PVDF membrane. The transferred blots were blocked with 4% non‐fat milk and then incubated for overnight with primary antibody. After washing, the blots were incubated with peroxidase‐conjugated secondary antibodies for 2 h and developed using the ECL detection system (Thermo Scientific).
Exposure to shear stress
ECs were exposed to laminar flow using a cone and plate apparatus system described previously (Dai et al. 2004; Wu et al. 2015). Briefly, a flow device consisting of a computerized stepper motor UMD‐17 (Arcus Technology) and a 1 deg tapered stainless steel cone. The flow devices were placed in a 37°C incubator with 5% CO2. Aliquots of 1 × 106 ECs from WT and Kir2.1+/− mice were seeded in a gelatin (0.2%) coated well of a 6‐well plate and cultured at 37°C for 72 h. Shear stress (20 dyn cm−2) was applied for 30 min. Cells were lysed immediately after shear exposure on ice using RIPA buffer with protease and phosphatase inhibitors.
Electrophysiology
Inwardly rectifying currents were recorded from WT and Kir2.1+/− freshly isolated and low‐passage cultured endothelial cells using the whole‐cell patch clamp configuration. Pipettes were pulled (SG10 glass, 1.20 mm ID, 1.60 mm; Richland Glass, Richland, NJ, USA) to give a final resistance of 2–4 MΩ. Currents were recorded using an EPC10 amplifier (HEKA Electronik, Lambrecht, Germany) and accompanying acquisition and analysis software (Pulse & PulseFit, HEKA Electronik). Pipette and whole‐cell capacitances were automatically compensated. Currents were low pass filtered at 2 kHz and recordings were digitized at 20 kHz. Traces were accepted for offline analysis if whole‐cell membrane resistance was at least 500 MΩ and access resistance was between 5 and 10 MΩ throughout the experiment.
Solutions and voltage protocol for low‐passage EC recordings
The following solutions for whole‐cell electrophysiology of primary cultured ECs were used (Millar et al. 2008): the external solution contained (in mm): 140 KCl, 5 NaCl, 1.0 MgCl2, 1.0 CaCl2, 10 Hepes, 5 glucose, 20 mannitol, at pH 7.4 (pH adjusted with NaOH), and was perfused at a rate of 1–2 ml min−1 using a flow‐regulated gravity delivery system. The pipette solution contained (mm): 135 KCl, 1.0 MgCl2, 5.0 glucose, 10 Hepes, 5.0 EGTA, at pH 7.2 (pH adjusted with KOH). The external and bath solutions were such that E K was slightly positive (<1 mV) in order to maximize inward K+ currents. Currents were elicited with 1 s linear voltage ramps from −140 to +20 mV at an interpulse interval of 5 s from a holding potential of −60 mV.
Preparing freshly isolated ECs for electrophysiological recordings
Mesenteric adipose tissues from WT or Kir2.1+/− mice were dissected and arteries cleaned of fat before storing in ice‐cold Hepes. Seven to ten 1st order arteries were cut into two or three pieces and digested as previously reported (Sonkusare et al. 2016). Briefly, arteries were enzymatically digested in dissociation solution (in mm: 55 NaCl2, 5.6 KCl, 2 MgCl2, 80 sodium glutamate, 10 Hepes, and 10 glucose, at pH 7.3) containing 0.5 mg ml−1 each of neutral protease and elastase (Worthington) for 1 h at 37⁰ C. For the final 2 min, 0.5 mg ml−1 Type I collagenase (Worthington) was added before mechanical dissociation to liberate 1st order mesenteric artery endothelial cells.
Electrophysiological recordings of freshly isolated ECs under shear stress
Shear stress was applied to the freshly isolated cells using the minimally invasive flow (MIF) device designed to allow electrophysiological recordings to be made under well‐defined shear stress (Levitan et al. 2000). Briefly, a large aliquot of cell suspension was allowed to adhere to the patch chamber before assembly. Upon whole‐cell access, several currents were recorded in a static bath until a stable baseline was achieved. Application of shear stress was implemented by gravity perfusion to induce shear‐activated inward K+ currents. Shear stress (τ; 0.74 dyn cm−2) was calculated using the equation τ = 6μ h 2 w where μ is the fluid viscosity (0.009 g cm−1 s−1), is the flow rate (0.3 ml s−1 applied by gravity perfusion), h is the height (0.1 cm) of the MIF chamber, and w is the width (2.2 cm) of the MIF chamber. Figure 2 C is a picture of the MIF device with a pipette electrode entering one of three 2 mm slits which provide access to endothelial cells for whole‐cell patch clamp experiments. For these experiments, as described previously (Sonkusare et al. 2016), freshly isolated endothelial cells were held at −30 mV and a voltage ramp of −140 to +40 mV was administered after correction of the junction potential. The bath contained (in mm): 10 Hepes, 80 NaCl, 60 KCl, 1 MgCl2, 2 CaCl2, 10 glucose, at pH 7.4. The pipette solution contained (in mm): 10 Hepes, 30 KCl, 10 NaCl, 110 potassium aspartate, 1 MgCl2, at pH 7.2.
Figure 2. Reduced endothelial Kir2.1 expression in Kir2.1+/− mouse mesenteric arteries.
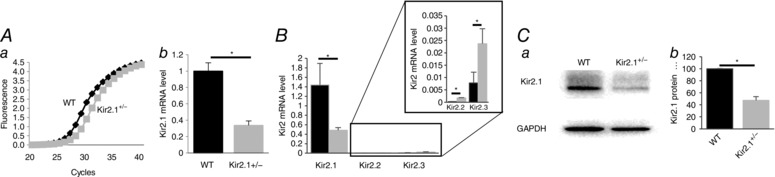
A, Kir2.1 mRNA expression in endothelial cells isolated from WT and Kir2.1+/− mesenteric arteries: a, amplification plots of qPCR; b, relative mRNA expression normalized to GAPDH (n = 3, * P < 0.05). B, Kir2.1, 2.2 and 2.3 mRNA expression levels in endothelial cells from WT and Kir2.1+/− mice (n = 3 per group, * P < 0.05). C, Kir2.1 protein expression in the same cells: a, representative Western blot of Kir2.1 (53 kD); b, densitometry analysis of Kir2.1 expression normalized to GAPDH (n = 3, * P < 0.05).
Vasodilatation measurement in mouse mesenteric arteries
Vasodilatation of mouse mesenteric arteries was measured as described in our previous studies (Miura et al. 2001 a; Phillips et al. 2007; Liu et al. 2011; Goslawski et al. 2013). Briefly, resistance arteries (between 75 and 150 μm) from mesenteric arteriole beds were isolated and cannulated in an organ chamber with glass micropipettes filled with Krebs solution (pH 7.40). The tips of the cannulating pipettes were always arranged with smaller pipettes upstream and larger pipettes downstream. The average tip size of cannulation arteries was 20.1 ± 0.4 μm at the upstream end and 23.6 ± 0.4 μm at the downstream end. Both ends of the vessel were secured, and the vessel was maintained at an intraluminal pressure of 60 cmH2O by elevating two Krebs‐containing reservoirs 60 cm above the organ chamber. The reservoirs and the vessel lumen generate an open circuit (see schematic presentation of the circuit in Fig. 4, inset). Vessels are equilibrated in this way for 30 min. After the 30‐min equilibration period, vessels were constricted (up to 50% of baseline diameter) with endothelin (ET‐1) (120–160 pm final concentration), and flow‐induced vasodilatation (FIV) was determined by exposing the vessels to incremental pressure gradients of Δ10, Δ20, Δ40, Δ60 and Δ100 cmH2O. The rationale for using ET‐1 rather than intraluminal pressure is the design of our preparation which generates intraluminal flow by gravity (the reservoirs are moved in equal and opposite directions), we found that pre‐constriction with ET‐1 provides an alternative and more convenient approach, as described in our previous studies (Phillips et al. 2007). Several criteria were used to select functional vessels, as described in previous studies (Miura et al. 2001 b; Grizelj et al. 2015): (i) all vessels were pre‐constricted with a low dose of ET‐1 (120 pm) and only the vessels that developed 40–60% constriction from the baseline were accepted for an experiment; (ii) at the end of each experiment all vessels were tested with papaverine to induce at least 90% dilatation; (iii) vessels with leaks in the wall of the vessel were discarded; (iv) all vessels had a clear segment without branches for measurement. As expected, no difference was observed between WT and Kir2.1+/− arteries (pre‐constricted by 49 ± 0.6% vs. 49 ± 0.3%, respectively, n = 9), because ET‐1‐induced constriction is mediated by the G‐protein coupled receptor and the downstream signalling is unrelated to Kir channels. Intraluminal pressure was maintained at 60 cmH2O by changing the distance between the reservoirs in equal and opposite directions (e.g. to induce intraluminal flow of ∆10 cmH2O, one reservoir is elevated to 65 cmH2O while the other is lowered simultaneously to 55 cmH2O), as described previously (Liu et al. 2003; Goslawski et al. 2013; Grizelj et al. 2015). FIV was determined in the presence and absence of external perfusion of BaCl2 (30 and/or 300 μm), l‐NAME (100 μm), apamin (20 nm), and a combination of BaCl2 and l‐NAME, BaCl2 and apamin, and apamin and l‐NAME. All inhibitors and blockers were incubated with the arteries for 30 min before the application of flow. Papaverine (100 μm) was added during exposure at the maximal flow (Δ100 cmH2O) to test maximal endothelium‐independent dilatation. Resistance arteries were monitored continuously, and internal diameters were measured at the maximal diameter after each pressure gradient applied. EC function was tested by performing a dose–response relationship for ACh after the completion of the flow protocol. Preparations were visualized with video cameras and monitors (model VIA‐100, Boeckeler).
Figure 4. Role of Kir2.1 in flow‐induced vasodilatation in mouse mesenteric arteries.
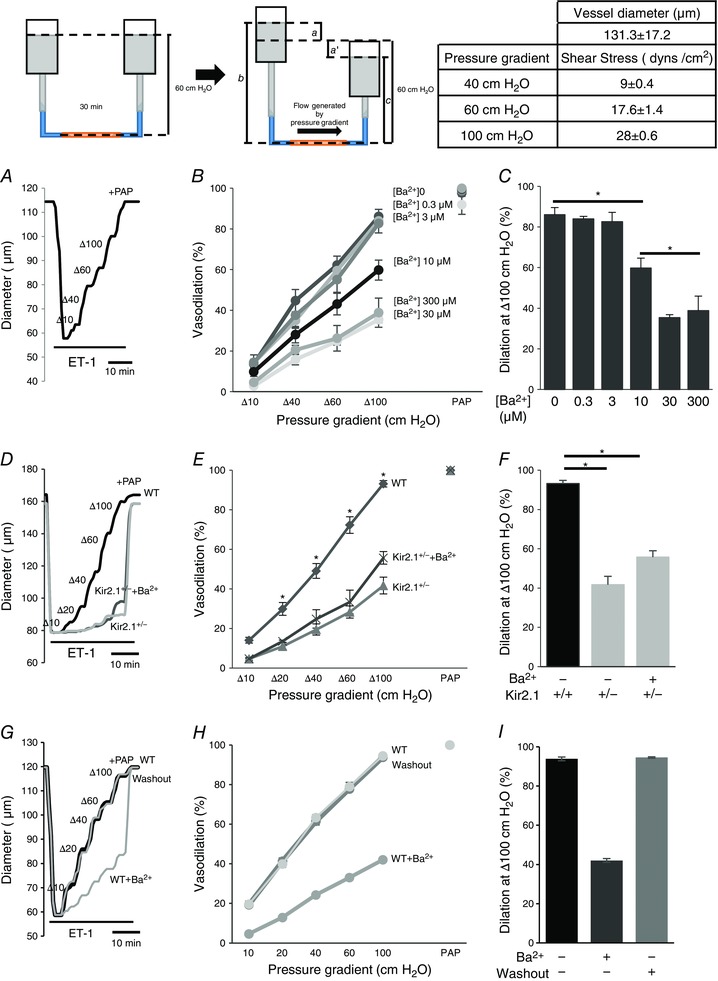
Insets, left: schematic images of flow generation by pressure gradients in FIV. The cannulated mesenteric artery was incubated at 60 cmH2O intraluminal pressure for 30 min. ET‐1 (120 pm) was applied externally to pre‐constrict the artery before the FIV measurement. To generate the flow, one side of the reservoir is moved up a cm and the other side of the reservoir is simultaneously moved down a′ cm from 60 cm position. The pressure gradient between the two reservoirs is defined as [a + a′] cmH2O and is responsible for generating flow inside the cannulated artery. Intraluminal pressure is constant and defined as (b + c)/2 cmH2O, where b = 60 + a cm and c = 60 − a′ cm. If a and a′ are equal in magnitude and opposite in direction, (b + c) is always 120 cm, and intraluminal pressure is thusly maintained at 60 cmH2O. During the experiment, a increased by 5, 10, 20, 30, or 50 cm, and a′ decreased in the same manner to generate the desired pressure gradients Δ10, Δ20, Δ40, Δ60, and Δ100 cmH2O and ultimately intraluminal flow (inset, right). Table, right: shear stress values for pressure gradients (Δ40, Δ60, and Δ100 cmH2O) and the mean ± SEM size of arteries for experiments. A, representative FIV trace in the absence of Ba2+ ([Ba2+]0). Vessel diameters are normalized to diameters stabilized at 60 cmH2O and the intraluminal pressure is maintained while intraluminal flow is applied. Arteries are pre‐constricted with ET‐1 and Ba2+ is perfused extravascularly. B, dose–response curve for Ba2+ performed during flow‐induced dilatations of pre‐constricted mesenteric arteries harvested from WT mice (n = 5, * P < 0.05). D, representative FIV trace of pre‐constricted mesenteric arteries harvested from WT or Kir2.1+/− mice with and without 300 μm Ba2+. E, average flow‐induced dilatations of mesenteric arteries harvested from WT or Kir2.1+/− mice with and without 300 μm Ba2 (n = 6 per group, * P < 0.05). G, representative FIV trace of pre‐constricted mesenteric arteries harvested from WT before, during and after the washout 300 μm Ba2+. H, average flow‐induced dilatations of mesenteric arteries described above (n = 4 per group). C, F and I: average dilatations at Δ100 cmH2O pressure gradient for the same experimental conditions as described in B, E, and H, respectively. ET‐1, endothelin‐1; PAP, papaverine (added at the end of experiments to test the vessels’ maximal dilatation).
Endothelium denuded arteries
Mechanical endothelium denudation was performed by perfusing the artery with 3 ml of air, a common method, previously described (Phillips et al. 2007). Denuded arteries were cannulated in an organ chamber and the dose–response relationships for acetylcholine and the nitric oxide (NO) donor sodium nitroprusside (SNP) (109 to 104 M) were measured. Papaverine (100 μm) was applied with the highest doses of SNP or acetylcholine to confirm the potential of endothelium‐independent vessel relaxation.
EC‐specific Kir2.1‐HA adenoviral transfection
EC‐specific Kir2.1‐HA adenoviral constructs were applied intraluminally for 48 h at 37°C. Then, following pressurization of vessels, FIV was measured with and without BaCl2 (30 μm).
Dominant negative (dn) Kir2.1 lentiviral transfection
dnKir2.1 lentiviral constructs were applied intraluminally overnight at 37°C. Then, following pressurization of vessels, FIV was measured with and without BaCl2 (300 μm).
NO measurements
Resistance arteries (between 75 and 150 μm) from mesenteric circulation were isolated and incubated with NO specific fluorescence dye (diaminorhodamine‐4M, DAR4M; Enzo Scientific, Exeter, UK) in Hepes at room temperature for 2 h in the dark. The arteries were cannulated in an organ chamber and incubated for 30 min with or without a pressure gradient of 60 cmH2O in the dark. Arteries were removed from the chamber and mounted on glass slides for image acquisition by fluorescence microscopy (Nikon eclipse 80i). The NO fluorescent product was excited by a 650 nm wavelength light with an emission spectrum of 670 nm. Acquired images were analysed for fluorescence intensity while correcting for background auto fluorescence using NIH image software (NIH Image J).
Blood pressure measurement and vascular resistance measurement
Blood pressure from WT and Kir2.1+/− mice was measured from carotid arteries. Briefly, mice were anaesthetized with 3.5% isoflurane in 96.5% oxygen and carotid arteries were exteriorized for cannulation with polyethylene tubing with a continuous blood pressure monitor, as described previously (Parasuraman & Raveendran, 2012). In separate experiments blood flow rates and blood pressure were measured by volume–pressure recording with occlusion tail‐cuff, as described in (Sharma et al. 2011). The tail‐cuff measurements were also done in anaesthetized mice. Peripheral vascular resistance is defined as the ratio of mean blood pressure (mmHg) to blood flow rate (μl min−1) (Rindler et al. 2011).
Echocardiogram
Wild‐type mice and Kir2.1+/− mice aged 3–5 months were used for the experiments. Transthoracic echocardiography was conducted using a 40 MHz transducer (MS550D) on VisualSonics’ Vevo 2100 ultrasound machine (VisualSonics, Toronto, Canada). Mice were sedated in an induction chamber using 3.5% isoflurane and then placed in the supine position on a heated stage. The heated stage maintained body temperature at 37°C, measured the electrocardiogram via embedded electrodes and recorded the respiration waveform via impedance pneumography. The anaesthetic plane of the mice was maintained using 1.5% isoflurane in 98.5% oxygen delivered via a nosecone. After the mice were depilated, B‐mode and M‐mode images from the parasternal short axis view were obtained using the integrated rail system. All cardiac parameters were calculated using VisualSonics’ Vevo 2100 analysis software (v. 1.6) with a cardiac measurements package (Pistner et al. 2010).
Vasodilatation measurement of human subcutaneous arteries
All participants were between 26 and 48 years old. Volunteers were excluded if they had diabetes mellitus, cancer, heart disease, a history of smoking, kidney or liver disease, gallbladder disease, autoimmune or other inflammatory disease. After written informed consent was obtained, biopsy samples of subcutaneous adipose were collected from during bariatric surgery at the University of Illinois Hospital and Health Sciences Center. Subcutaneous adipose tissue was obtained from the lower abdominal wall. Biopsies were placed in cold (4°C) Hepes buffer solution. Arteries were cleaned of fat and connective tissue and prepared for continuous measurement of internal luminal diameter as previously described (Grizelj et al. 2015). The internal luminal diameter of each arteriole was initially measured after 30 min of stabilization at 60 cmH2O and following administration of endothelin‐1 (ET‐1; 100–200 pm) to constrict microvessels to 30–50% of their internal luminal diameter. The diameter of human arteries was 112 ± 20 μm. Pipettes were prepared exactly the same way as for mouse arteries (average tip size 19.1 ± 0.2 μm at the upstream end and 22 ± 0.3 μm at the downstream end). Flow was produced by simultaneously changing the heights of the reservoirs in equal and opposite directions to generate a pressure gradient of ∆10–∆100 cmH2O with and without with or without external perfusion of BaCl2 (300 μm), apamin (20 nm), or both drugs. At the end of each experiment, maximal dilatation was determined in the presence of papaverine (10−4 M). Shear stress was calculated from the flow rate in the vessel lumen and the diameter of the vessels using the equation (Schmid‐Schönbein & Granger, 2003): τ = 4(μ)/(πr 3) where μ is viscosity, is volumetric flow rate, and r is internal radius of the vessel. The volumetric flow rate was measured as the volume of the flow‐through at different pressures (Δ40, Δ60 and Δ100 cmH2O). Vessel diameter was measured in all experiments. The shear stress range was 9–28 dyn cm−2 for the pressure gradients of Δ40‐100 cmH2O.
Statistics
Data are presented as means ± SEM unless otherwise stated. The calculations for percentage vasodilatation were performed as described previously (Goslawski et al. 2013). A two‐factor ANOVA with or without repeated measures was used where appropriate to compare FIV‐generated curves. A Bonferroni post hoc analysis revealed significant differences. Linear regression analysis was used to compare the slopes of Ba2+‐sensitive, current–voltage relationships. When appropriate, Kruskal‐Wallis rank tests were performed on group data which had uneven distributions. For all other comparisons, a Student's t test was performed. Statistical significance was accepted when P < 0.05.
Results
Functional expression of Kir2.1 in murine mesenteric ECs
The expression profile of Kir2 channels in freshly isolated mesenteric ECs was characterized by real‐time PCR for three major subtypes of Kir2 channels, Kir2.1, 2.2 and 2.3. The purity of the isolation was first verified by flow cytometry to identify cells positive for an endothelial marker PECAM1 (CD31) (Fig. 1 A) and then by the real‐time PCR (Fig. 1 B). It was also further confirmed by immunofluorescence for PECAM1, an additional endothelial marker, VWF, and a smooth muscle marker, α‐smooth muscle actin (αSMA) (Fig. 1 C). As expected, the endothelial population (CD31+) showed strong expression of CD31 and no detectable expression of αSMA, suggesting high purity EC isolation. In contrast, the residual population of cells (CD31−) showed strong expression of αSMA and low expression of CD31, as expected if a small portion of ECs were not picked up by the isolation procedure. Furthermore, and consistent with these observations, purified ECs showed clear CD31‐specific fluorescence and no detectable αSMA fluorescence. The dominant Kir2 channel in these cells is Kir2.1, whereas Kir2.3 is also expressed but expression is several orders of magnitude lower than Kir2.1. Expression of Kir2.2 was not detected (Fig. 1 D and E). The same expression profile of Kir2 channels was maintained in low‐passage endothelial cultures. Also, consistent with previous studies (Smith et al. 2008), strong expression of Kir2.1 was observed in ECs but not in VSMCs isolated from mouse mesenteric artery (Fig. 1 F). Functional expression of Kir2.1 in freshly isolated/low passage mesenteric ECs was tested electrophysiologically. As expected, Kir currents in mesenteric endothelium were blocked by Ba2+ with the IC50 of 4.8 μm, similar to the Ba2+ sensitivity of Kir in aortic endothelium (Fang et al. 2005) and typical for Kir2.1(Liu et al. 2001) (Fig. 1 G and H).
Figure 1. Kir2.1 functional expression in mouse mesenteric arterial endothelial cells.
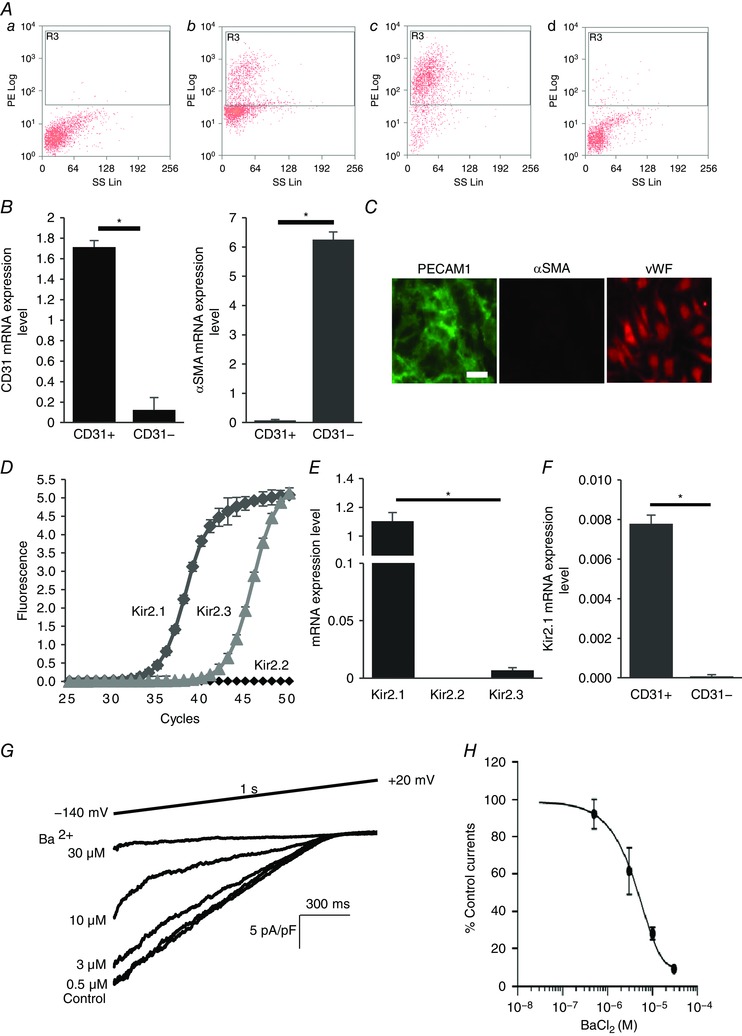
A, mouse mesenteric arterial endothelial cells are isolated by magnetic cell sorter (AutoMACs, Miltenyi Biotech) with polyclonal anti‐mouse platelet endothelial cell adhesion molecule 1 (PECAM1) IgG conjugated to magnetic beads. The purity of the isolated endothelial cells is identified by flow cytometry using PE‐conjugated monoclonal anti‐mouse PECAM1 IgG: a, unstained digested cells; b, PECAM1 positive cells before magnetic cell sorting; c, PECAM1 positive cells (endothelial cells) after magnetic cell sorting (∼93% purity); d, PECAM1 negative cells after magnetic cell sorting. B, real‐time PCR confirmation of isolated pure endothelial cells from the murine mesenteric arterial bed. mRNA levels of endothelial cell marker (PECAM1, CD31) and smooth muscle cell marker smooth muscle α‐actin (αSMA) were normalized by GAPDH (CD31+, purified endothelial cells; CD31−, residual cells; n = 4, * P < 0.05). C, further identification of freshly isolated microvascular endothelial cells with immunofluorescence staining for the smooth muscle cell marker αSMA (negative control, red) and the endothelial cell marker PECAM1 (CD31, green). Another endothelial marker, von Willebrand factor (vWF, red), stained in different sets of the experiment (scale bar = 100 μm). D, amplification plots of qPCR for Kir2.1, 2.2 and 2.3 from freshly isolated cells. E, mRNA expression levels of Kir2.1, 2.2 and 2.3 are compared to Kir2.1 expression levels, after normalizing to GAPDH (n = 3 experiments from 15 mice, * P < 0.05). F, Kir2.1 mRNA expression levels in purified endothelial cells (CD31+) and residual cells (CD31−) (n = 4, * P < 0.05). G, representative whole‐cell patch clamp recording of a dose–response curve for Ba2+ (0.5, 3, 10, and 30 μm) from WT mice‐derived mesenteric arterial endothelial cells. A ramp protocol (−140 to +20 mV) and a high K+ bath (E K = +0.91 mV) were used to optimize Kir currents. The holding potential in all experiments was −60 mV. H, inhibitory dose–response curve elicited by Ba2+ and derived from electrophysiological recording at −100 mV. IC50 was calculated from the curve (n = 12 from 4 different experiments). [Colour figure can be viewed at wileyonlinelibrary.com]
Functional expression of Kir2.1 in mesenteric ECs was verified using a genetic Kir2.1 partial knockout mouse model, the Kir2.1+/− heterozygous mouse. Kir2.1 expression at the mRNA level is reduced by 60–65% in Kir2.1+/− mice in isolated mesenteric ECs (Fig. 2 A). The loss of Kir2.1 expression in Kir2.1+/− mesenteric ECs was accompanied by a slight increase in the expression levels of Kir2.2 and Kir2.3 channels but overall the expression of Kir2.2 and Kir2.3 was still orders of magnitude lower than that of Kir2.1 (Fig. 2 B). Protein expression of Kir2.1 was also reduced by 50% (Fig. 2 C).
The density of Kir currents was measured in freshly isolated ECs both under basal static conditions and in cells exposed to shear stress. The electrophysiological recordings were performed immediately after the isolation on the same day. The effect of shear stress on the currents was measured in real time using a modified parallel‐plate flow chamber, designed in our previous studies to perform electrophysiological experiments under well‐defined flow (Fig. 3, inset; Levitan et al. 2000). Consistent with the Kir2.1 mRNA and protein expression, basal Kir currents recorded under static conditions are significantly reduced in ECs isolated from Kir2.1+/− arteries (34 ± 4 pA pF−1 vs. 17 ± 5 pA pF−1 in WT and Kir2.1+/− cells respectively (Fig. 3 A–C). Shear stress of 0.7 dyn cm−2 (flow rate of 300 μl s−1) was applied after recording a stable baseline of the currents. The same level of shear stress was used in earlier studies (Olesen et al. 1988). Higher levels of shear stress were too detrimental for obtaining stable electrophysiological recordings. We show here that application of this level of shear stress significantly increased Kir current density in ECs isolated from the WT mesenteric arteries (29 ± 7% increase in current density) (Fig. 3 A and B, bottom panels; Fig. 3 C). A significant increase in Kir current density is observed immediately after the initiation of the flow, with a gradual return to baseline after the cessation of the flow within 15–20 s. In contrast to WT, ECs isolated from Kir2.1+/− mice showed no significant increase at the same level of shear stress (Fig. 3 A and B, upper panels; Fig. 3 C). Current traces recorded during the flow protocol at specified time points (a, b and c) are shown as insets to Fig. 3 A. Larger scale representative current traces recorded under static and flow conditions are shown in Fig. 3 B. Mean capacitance of the freshly isolated ECs from WT and Kir2.1+/− mice were 13.4 ± 1.1 pF and 17.4 ± 3.4 pF, respectively. The difference between WT and Kir2.1+/− cells remains significant also in low‐passage cultured ECs. The latter observation allows us to test the role of Kir2.1 in flow‐induced signalling events in cultured cells in vitro, as described below.
Figure 3. Reduction of flow‐stimulated Kir current in endothelial cells of Kir2.1+/− mice.
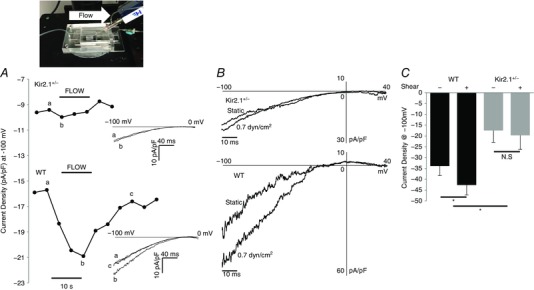
Inset, installed MIF device for electrophysiology recordings as it appears on the microscope head stage. A, time course of flow‐activated Kir current from Kir2.1+/− (upper trace) and WT (lower trace) showing current densities before, during and after flow. The time course is representative of individual data points taken from the ramp protocols (−100 to 0 mV) at −100 mV shown to the right of each time course. Current traces at time points designated by lower case letters (a, b, c) are shown as insets to the panel. B, representative recording of flow stimulated Kir currents in freshly isolated ECs from Kir2.1+/− and WT mouse mesenteric arteries. C, average Kir current density at −100 mV in the freshly isolated ECs from WT and Kir2.1+/− mouse mesenteric arteries (WT, n = 5 cells from 3 mice; Kir2.1+/−, n = 5 cells from 2 mice, * P < 0.05).
Kir2.1 is essential for flow‐induced vasodilatation
FIV was measured in pressurized mesenteric arteries of diameter 100–200 μm isolated from WT and Kir2.1+/− mice (a circuit for generating intraluminal flow is shown schematically in the inset of Fig. 4). Average diameter of the vessels used in this study 131 ± 17 μm. The flow rates generated in the system for Δ40–100 cmH2O were 0.25 ± 0.08 μl s−1, 0.53 ± 0.23 μl s−1, and 0.81 ± 0.34 μl s−1, respectively, generating shear stress levels of 9–28 dyn cm−2 (Fig. 4, inset right). Similar flow rates were reported in previous studies (Liu et al. 2003) and the values of shear stress are within the range reported for mesenteric arteries in vivo (Reneman & Hoeks, 2008). In WT mice, FIV was inhibited by Ba2+ with an IC50 of 7.2 μm, similar to the IC50 of Ba2+‐dependent block of the Kir currents in mesenteric endothelium shown above (Fig. 4 A–C). Downregulation of Kir2.1 expression in Kir2.1+/− mice also results in a significant reduction of FIV (Fig. 4 D–F). To validate further that FIV inhibition in Kir2.1+/− mice can be attributed to the loss of the Kir channels, the vessels were exposed to Ba2+, demonstrating that there is no further inhibition. An endothelial‐independent vasodilatation induced by papaverine was unaltered by downregulation of Kir2.1 or by Ba2+. No difference, however, was observed in the baseline artery diameters after the application of Ba2+ (166 ± 9 μm vs. 166 ± 9 μm, respectively, n = 9). In terms of Ba2+ concentration, no significant difference in FIV was observed for 30 μm or 300 μm Ba2+ exposures. Ba2+ had no effect in Kir2.1+/− arteries, suggesting that Kir2.1 were the only Ba2+‐sensitive channels in these arteries. Flow‐induced vasodilatation was fully reproducible in the same arteries with multiple applications, as well as after the application and washout of Ba2+ (Fig. 4 G–I). To determine whether the effect of Kir2.1 on FIV is endothelium dependent, the vessels were denuded of endothelium and vasodilatation was tested in response to SNP, a known NO donor. No difference was observed in smooth muscle response to SNP between WT and Kir2.1+/− vessels (Fig. 5 A and B). Ba2+ also had no effect on SNP‐induced vasodilatation. As expected, endothelium‐intact vessels respond to ACh with dose‐dependent vasodilatation but denuded vessels have no response (Fig. 5 C). A full response of intact vessels to ACh was observed both before and after the application of the flow. Papaverine controls were performed in all experiments and there was no difference between WT and Kir2.1+/− vessels.
Figure 5. The role of Kir2.1 function in mesenteric artery dilatation is endothelial dependent.
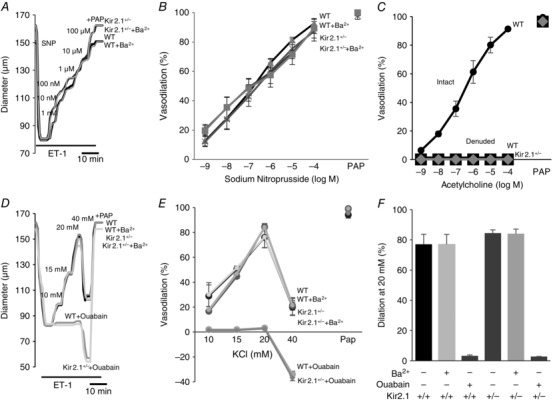
A, representative sodium nitroprusside (SNP)‐induced vasodilatation of pre‐constricted denuded mesenteric arteries. SNP was perfused extravascularly. The endothelium was mechanically denuded by forcing 5 ml of air through the vessel lumen. B, average dilatations as a function of SNP dose (n = 5 per group). C, average dilatations of intact and denuded vessels in response to ACh (n = 4 per group). D, representative K+‐induced vasodilatation of pre‐constricted denuded mesenteric arteries from WT and Kir2.1+/− mice. E, average dilatations of arteries from WT and Kir2.1+/− mice as a function of K+ dose (KCl) (n = 5 arteries per condition). F, average dilatations at 20 mm KCl. ET‐1, endothelin‐1; PAP, papaverine (added at the end of experiments to test the vessels’ maximal dilatation).
We also tested whether Kir2.1 channels might contribute to K+‐induced vasodilatation in denuded mesenteric arteries, as was shown previously in cerebral and coronary arterial beds (Burns et al. 2004; Filosa et al. 2006). As expected, elevating extracellular K+ concentration up to 20 mm resulted in significant vasodilatory effect but this effect was abolished by 10 μm ouabain, an inhibitor of Na+–K+‐ATPase, and not by Ba2+, indicating that it is not mediated by Kir channels (Fig. 5 D–F). Furthermore, no inhibition of K+‐induced vasodilatation was observed in denuded mesenteric arteries isolated from Kir2.1+/− mice. In contrast, K+‐induced vasodilatation of denuded cerebral arteries isolated from the same mice were inhibited by Ba2+, as reported previously (Fig. 6). These observations suggest that smooth muscle Kir do not contribute to K+‐induced vasodilatation of mesenteric arteries. As was shown previously, an increase in the extracellular K+ concentration to 40 mm or above results in vasoconstriction.
Figure 6. Kir2.1 function in cerebral artery dilatation may be endothelial independent.
A, average K+‐induced dilatation of pre‐constricted denuded cerebral arteries from WT mice as a function of K+ doses of 10, 15, 20 and 40 mm (n = 5 arteries per condition). B, average dilatations in response to 20 mm KCl. PAP, papaverine (added at 40 mm KCl to check the vessels’ maximum dilatation).
Finally, to eliminate the possibility that the loss of FIV in Kir2.1+/− mice could be a result of non‐specific changes in this genetic model, we determined whether infecting the WT vessels with the dominant negative Kir2.1 mutant also reduces the FIV. It is well established that the substitution of the GYG region of the pore with AAA in the selectivity filter of Kir2.1 channels results in a specific dominant negative effect on Kir2.1 current (Schram et al. 2002). Our previous studies showed that a lentiviral construct of dnKir2.1 efficiently downregulates Kir2 channel function in mouse macrophages (Thomas et al. 2011). Here we show that infecting endothelial cells with dnKir2.1 lentiviral construct also strongly reduced the Kir current compared to cells infected with an empty virus (Fig. 7). Similar to its effect on the vessels isolated from Kir2.1+/− mice, infecting WT vessels with dnKir2.1 resulted in a significant reduction of FIV. A similar decrease in FIV was observed in vessels exposed to Ba2+. Infecting the vessels with an empty virus had no effect. No difference was observed between control and dnKir2.1‐infected vessels in endothelium‐independent papaverine‐induced vasorelaxation.
Figure 7. Loss of Kir2.1 function with dominant negative Kir2.1 results in the inhibition of FIV in mesenteric arteries.
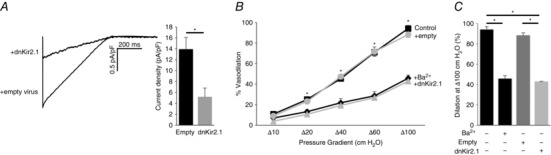
A, representative whole‐cell Kir current traces elicited by linear voltage ramps recorded from human aortic endothelial cells with dnKir2.1 and empty lentiviral constructs. Shown are average Kir current densities in human aortic endothelial cells with dnKir2.1 and empty lentiviral constructs at −90 mV (empty, n = 13 in 3 independent experiments; dnKir2.1, n = 9 in 2 independent experiments; * P < 0.05). B, dnKir2.1 or empty lentiviral constructs were intraluminally applied to intact mesenteric arteries for 24 h. Shown are average flow‐induced dilatation of pre‐constricted lentiviral constructs‐treated and non‐treated mesenteric arteries harvested from WT mice, as a function of pressure gradient. Vessel diameters are normalized to diameters stabilized at 60 cmH2O and the intraluminal pressure was maintained at the same pressure. Ba2+ (300 μm) was perfused extravascularly on to WT mesenteric arteries without the transfection of lentiviral constructs (n = 3 per group, * P < 0.05). C, average dilatations at Δ100 cmH2O.
Rescue of FIV in Kir2.1+/− arteries by endothelial‐specific Kir2.1 expression
Mesenteric arteries freshly isolated from Kir2.1+/− mice were transfected with Kir2.1‐HA using adenoviral vector with EC‐specific VE‐Cad promoter. EC‐specific Kir2.1 expression was confirmed with immunostaining of ECs and SMCs using anti‐HA tag antibody. Endothelial cells (HAECs) showed a clear expression of Kir2.1‐HA whereas VSMCs isolated from mouse mesenteric arteries had no detectable Kir2.1‐HA expression (Fig. 8 A and B). HAECs were used as a well‐established model of ECs. The identity of VSMCs was confirmed with αSMA expression. Functional expression of the channels in endothelial cells was also confirmed electrophysiologically (Fig. 8 C and D). Most importantly, endothelial specific expression of Kir2.1 resulted in full recovery of FIV in Kir2.1+/− arteries (Fig. 8 E–G). Control cells and arteries were transfected with the empty vector. Blocking Kir channels with Ba2+ abrogated the rescue effect of FIV.
Figure 8. Recovery of Kir2.1 function with EC‐specific WT Kir2.1‐HA adenoviral vector in mesenteric arteries and arterial ECs isolated from Kir2.1+/− mice.
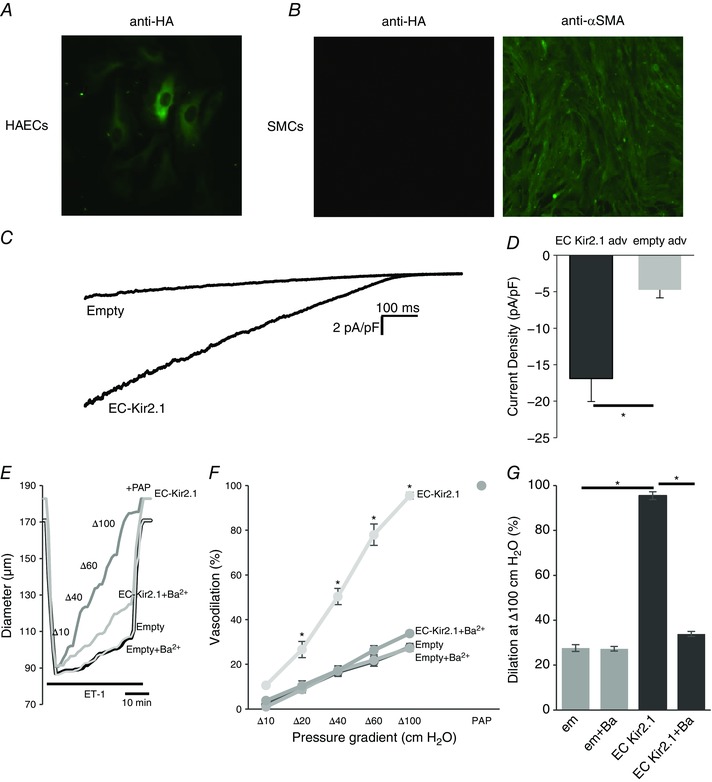
A and B, EC‐specific expression was confirmed using WT Kir2.1‐HA adenoviral vector in human aortic endothelial cells (HAECs; A) and murine mesenteric smooth muscle cells (SMCs; B). C, representative Kir current traces in HAECs with empty or EC‐specific WT Kir2.1‐HA adenoviral vector confirms incorporation of functional Kir channels into EC membranes. D, average Kir current densities recorded from virus‐transfected HAECs (empty adv, n = 6; EC Kir2.1 adv, n = 7 cells in 3 independent experiments, * P < 0.05). E, representative FIV trace of empty or EC‐specific WT Kir2.1‐HA transfected mesenteric arteries harvested from Kir2.1+/− mice. F, average flow‐induced dilatations of empty or EC‐specific WT Kir2.1‐HA transfected mesenteric arteries harvested from Kir2.1+/− mice with and without 30 μm Ba2+ (n = 4 per group, * P < 0.05). G, average dilatations of virus‐transfected arteries at Δ100 cmH2O. [Colour figure can be viewed at wileyonlinelibrary.com]
Complementary roles of Kir2.1 and KCa channels in flow‐induced vasodilatation
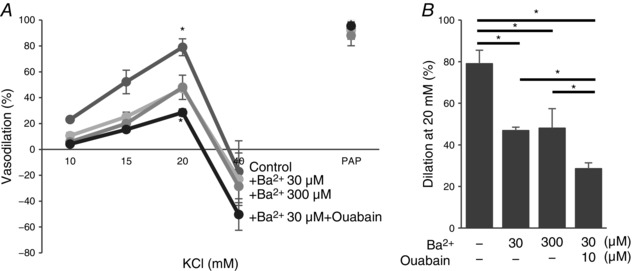
Earlier studies showed that SK channels play a significant role in FIV (Brahler et al. 2009; Milkau et al. 2010; Wulff & Köhler, 2013). We tested, therefore, whether Kir2.1 and SK channels regulate FIV by the same pathway. To address this question, pressurized preconstricted vessels were exposed to flow in the presence of: (i) 20 nm apamin, a blocker of SK channels, (ii) 300 μm Ba2+ or (iii) the combination of the two. Consistent with previous studies, application of apamin resulted in a significant inhibition of FIV, similar to that of Ba2+. However, a combined application of apamin and Ba2+ resulted in a complete abrogation of FIV in the WT vessels, suggesting an additive effect of apamin and Ba2+ on FIV (Fig. 9 A–C). Importantly, the vessels maintained their ability to relax, as evidenced by a full response to papaverine.
Figure 9. Kir2.1 and SK regulate flow‐induced vasodilatation by NO‐dependent and NO‐independent pathways.
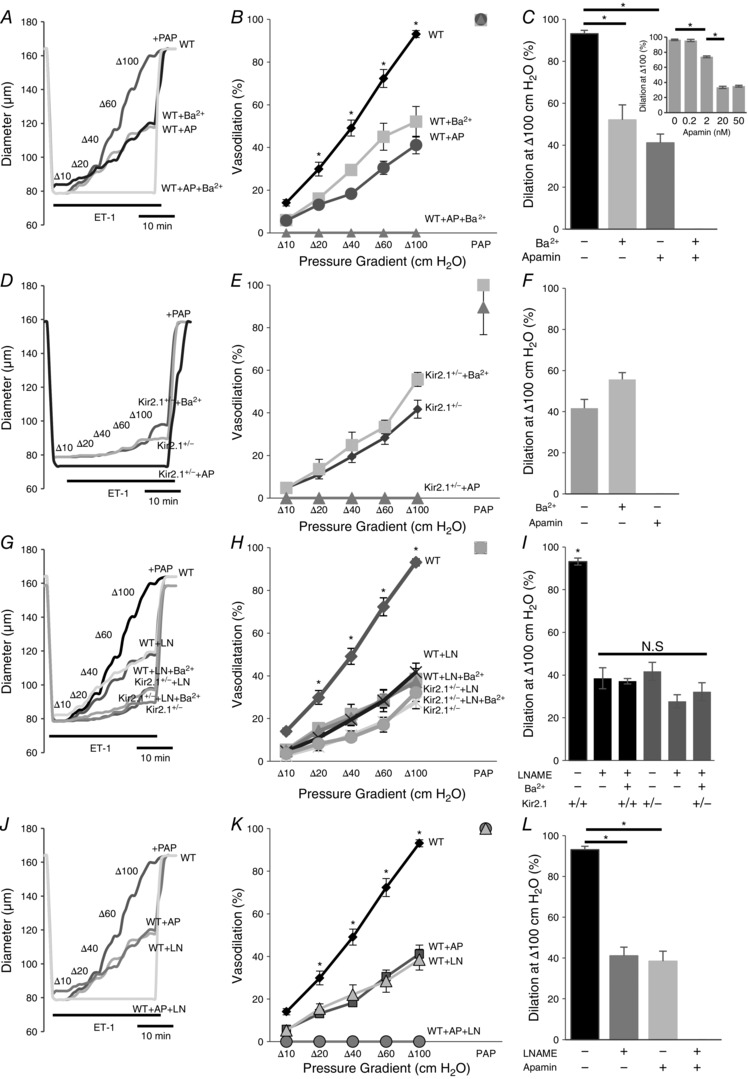
A, representative FIV trace of pre‐constricted (ET‐1) mesenteric arteries harvested from WT mice with and without 20 nm apamin (AP), 300 μm Ba2+, or both. B, average flow‐induced dilatations of arteries harvested from WT mice with the same experimental conditions described in A (n = 4 arteries per condition, * P < 0.05). D, representative FIV trace of mesenteric arteries harvested from Kir2.1+/− mice exposed to the same experimental conditions described in A. E, average flow‐induced dilatations arteries harvested from Kir2.1+/− mice exposed to the same experimental conditions described in A (n = 4 arteries per condition, * P < 0.05). G, representative FIV trace of arteries harvested from WT or Kir2.1+/− mice with and without 100 μm l‐NAME (LN). H, average flow‐induced dilatations of arteries from WT or Kir2.1+/− mice with the same experimental conditions described in G (n = 4 arteries per condition). J, representative FIV trace of arteries harvested from WT mice with and without 20 nm apamin, 100 μm l‐NAME, or both. K, average flow‐induced dilatations of mesenteric arteries harvested from WT mice with the same experimental conditions described in G (n = 5 arteries per condition, * P < 0.05). C, F, I and L: average dilatations at Δ100 cmH2O for the same experimental conditions as described in B, E, H and K, respectively. C, inset: dose–response curve for apamin of FIV in WT mesenteric arteries at average dilatations at Δ100 cmH2O (n = 4, * P < 0.05). Apamin was perfused extravascularly. ET‐1, endothelin‐1; PAP, papaverine (added at Δ100 cmH2O to test the vessels’ maximal dilatation).
In addition, we also tested the effect of apamin in vessels isolated from Kir2.1+/− mice. In this case, apamin completely inhibited the residual vasodilatation observed after the loss of Kir2.1 (Fig. 9 D–F). These observations are fully consistent with the additive effects of apamin and Ba2+ observed in the WT vessels, as described above. Also, as shown above, no difference was observed in endothelial‐independent vasodilatation in response to papaverine.
Next we addressed the question of whether Kir2.1 channels regulate FIV by an NO‐dependent mechanism. As expected, application of N ω‐nitro‐l‐arginine‐methyl‐ester hydrochloride (l‐NAME) to WT arteries results in a significant decrease in FIV. However, we show here that the FIV of vessels isolated from Kir2.1+/− mice was insensitive to l‐NAME. Specifically, as described above, Kir2.1+/− vessels showed significantly lower FIV than WT vessels but there is no further decrease after l‐NAME application (Fig. 9 G–I). A similar effect was observed when WT vessels were exposed to a combination of Ba2+ and l‐NAME. These observations indicate that Kir2.1 and NO regulate FIV through the same pathway. In contrast, application of l‐NAME to vessels after they were exposed to apamin (SK inhibitor) resulted in significant additional inhibition leading to virtual abrogation of any FIV response (Fig. 9 J–L). Inhibition of SK channels and of eNOS had no effect on endothelium‐independent papaverine‐induced vasodilatation. To verify that the dosage used to determine the contribution of SK channels to the FIV response in mesenteric arteries (20 nm) elicits maximal effect, we measured the FIV dose–response relationship for apamin (Fig. 9 C, inset). There was no further decrease of FIV above 20 nm of apamin, which suggested that SK channels involved in FIV were blocked.
Flow‐induced eNOS activation is regulated by Kir2.1 via Akt1 phosphorylation
Since, as described above, we have shown that arteries isolated from Kir2.1+/− mice are not sensitive to the inhibition of eNOS, we tested next whether deficiency in Kir2.1 affects flow‐induced eNOS activation. ECs from WT and Kir2.1+/− mice were exposed to 20 dyn cm−2 shear stress generated by the cone apparatus for 30 min. As expected, in WT ECs, exposure to shear stress resulted in eNOS phosphorylation on S1177, a well‐known eNOS phosphorylation site that leads to eNOS activation (Fisslthaler et al. 2000). In contrast, no shear stress‐induced eNOS phosphorylation on S1177 was observed in cells isolated from Kir2.1+/− arteries (Fig. 10 A and B). Consistent with these observations, blocking Kir channels with Ba2+ also results in the inhibition of flow‐induced eNOS phosphorylation in WT ECs but had no effect in ECs isolated from Kir2.1+/− arteries. Furthermore, blocking Akt with MK2206 results in the inhibition of eNOS phosphorylation in WT cells but has no effect on eNOS in Kir2.1+/− cells. Furthermore, transfecting ECs isolated from Kir2.1+/− arteries with Kir2.1 viral construct resulted in full recovery of flow‐induced eNOS phosphorylation (Fig. 10 C and D). The recovery effect is abrogated, however, by blocking Kir with Ba2+ or by the inhibition of Akt.
Figure 10. Flow‐induced eNOS activation by Akt phosphorylation.
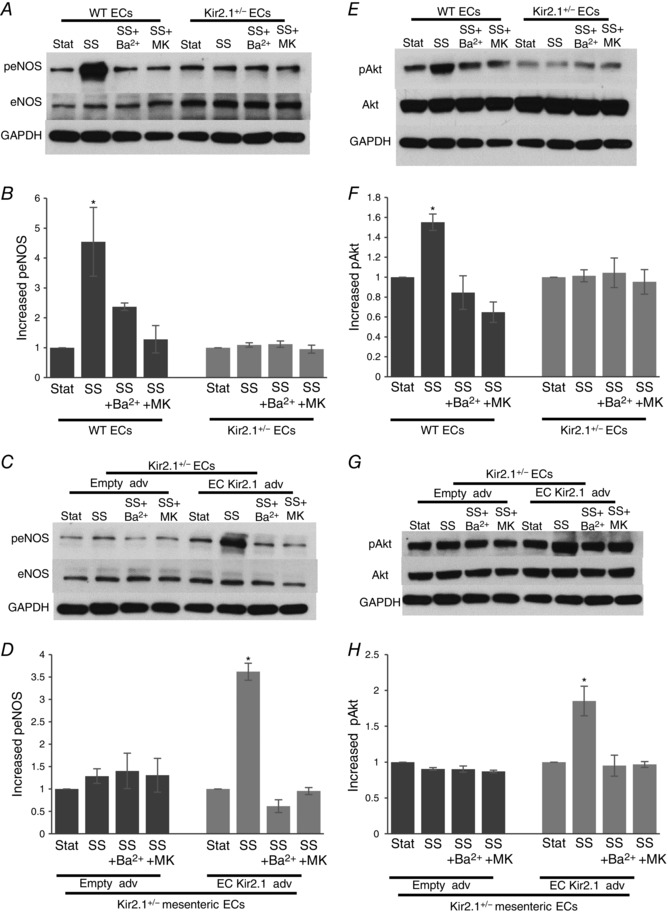
A, representative Western blot for eNOS phosphorylation by shear stress (20 dyn cm−2, 30 min) with or without Ba2+ or Akt inhibitor (MK2206) in murine mesenteric arterial ECs isolated from WT and Kir2.1+/− mice. B, densitometry for group data represented in A (n = 4 per group, * P < 0.05). C, representative Western blot for eNOS phosphorylation by shear stress (20 dyn cm−2, 30 min) with or without Ba2+ or Akt inhibitor (MK2206) in the adenoviral vector (empty or Kir2.1‐HA) transfected murine mesenteric arterial ECs from Kir2.1+/− mice. D, densitometry for the group data represented in C (n = 4 per group, * P < 0.05). E, representative Western blot for Akt phosphorylation in the same experimental samples shown in A. F, densitometry for the group data represented in E (n = 4 per group, * P < 0.05). G, representative Western blot for Akt phosphorylation in the same experimental samples shown in C. H, densitometry for the group data represented in G (n = 4 per group, * P < 0.05). SS, shear stress; MK, MK2206.
Consequently, we tested next the effect of Kir2.1 deficiency on flow‐induced Akt phosphorylation. As was shown in previous studies (Dimmeler et al. 1999), exposure to shear stress resulted in a strong increase in Akt phosphorylation at S473, in WT ECs. No increase in Akt phosphorylation was observed in Kir2.1+/− ECs or in WT cells exposed to Ba2+. The efficiency of MK2206 inhibitor is also verified by testing its effect on Akt phosphorylation (Fig. 10 E and F). Similar to the effects on eNOS phosphorylation, transfecting ECs isolated from Kir2.1+/− arteries with Kir2.1 viral construct resulted in full recovery of the flow‐induced response (Fig. 10 G and H).
Kir 2.1, but not KCa, regulates flow‐induced NO generation in mesenteric arteries
Kir2.1
Isolated intact mesenteric arteries were loaded with a membrane‐permeable NO‐sensitive fluorescent dye, diaminorhodamine‐4M (DAR4M), and then pressurized and exposed to flow generated by a pressure gradient (Δ60 cmH2O) for 30 min. Control vessels were pressurized but not exposed to flow and maintained under the same pressure in static conditions for the same period of time. In WT vessels, there was a significant increase in NO‐specific fluorescence in response to flow (Fig. 11 A; WT) and it was abrogated by the presence of 100 μm l‐NAME, applied for the duration of the experiment (Fig. 11 A; WT±LN). In contrast, there was no increase in flow‐induced NO fluorescence in vessels isolated from Kir2.1+/− mice, indicating that Kir2.1 is essential for this response (Fig. 11 A; Kir2.1+/−). Additionally, WT vessels exposed to Ba2+ had no significant increase in flow‐induced NO fluorescence, which also supports the importance of Kir2.1 in flow‐induced NO generation (Fig. 11 A; WT±Ba2+). Normalized NO‐specific fluorescence is shown in Fig. 11 B. Endothelial denudation of the vessels abrogated flow‐induced increase in NO‐specific fluorescence.
Figure 11. Kir2.1, but not SK, controls flow‐induced nitric oxide production.
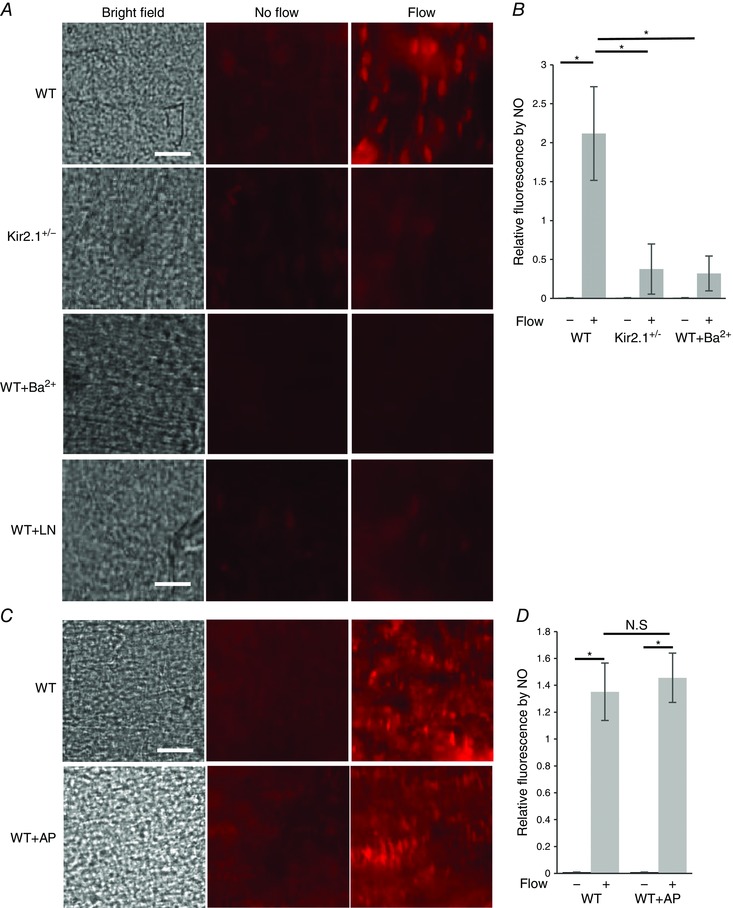
A, images of NO‐specific fluorescence in response to flow in WT and Kir2.1+/− mesenteric arteries: left panels show the inner surface of the arteries in bright field, middle panels show NO‐specific fluorescence stained by DAR4M in arteries not exposed to flow, right panels show NO‐specific fluorescence in arteries exposed to flow generated by a pressure gradient of Δ60 cmH2O for 30 min with or without Ba2+ (30 μm) or l‐NAME (100 μm) (scale = 50 μm). B, quantification of NO‐specific fluorescence in the same arteries normalized to ‘No flow’ control (n = 6 arteries per condition, * P < 0.05). C, images of NO specific fluorescence of WT arteries maintained under static conditions (middle panels) or exposed to 30 min of flow (right panels) with or without application of apamin (20 nm). D, quantification of NO‐specific fluorescence in the same arteries (n = 5 arteries per condition, * P < 0.05). [Colour figure can be viewed at wileyonlinelibrary.com]
KCa
In a parallel series of experiments, flow‐induced NO production was measured in pressurized vessels in the presence and in the absence of 20 nm apamin, the same concentration that had significantly inhibited FIV. In this case, however, there was virtually no reduction in flow‐induced NO production (Fig. 11 C and D).
Kir2.1 contributes to the control of blood pressure
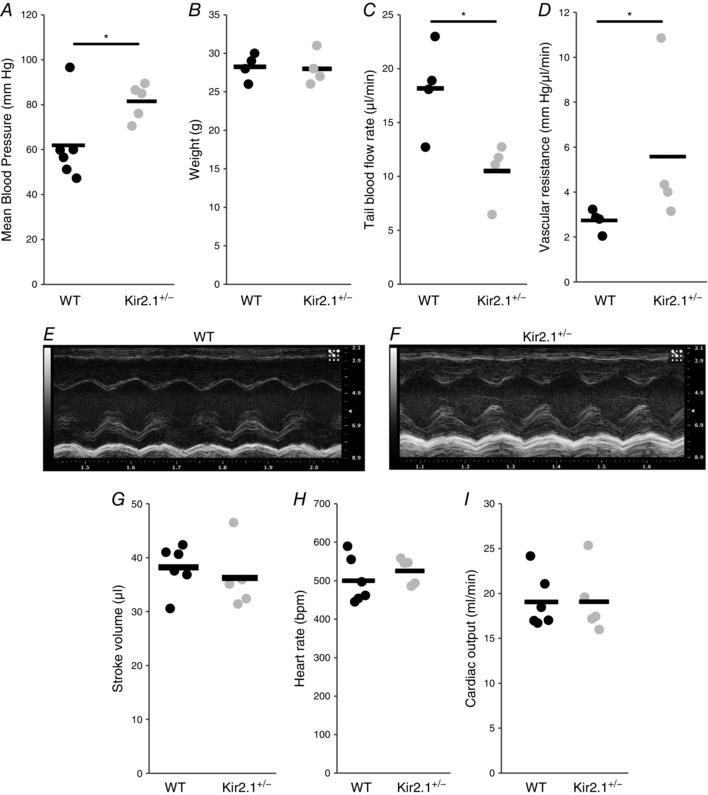
Since, significant loss of FIV is observed in resistance arteries isolated from Kir2.1 deficient mice, we tested whether Kir2.1 deficiency also results in increased blood pressure. Hence, we compared mean arterial pressures (MAP) measured in carotid arteries using carotid cannulation procedure in WT and Kir2.1+/− mice. Our results reveal that Kir2.1 deficiency results in significant elevation of MAP (Fig. 12 A). All experiments were performed on age‐matched male mice and were independent of the body weight (Fig. 8 B).
Figure 12. Deficiency in Kir2.1 increases blood pressure and peripheral vascular resistance, but cardiac function does not contribute to increased blood pressure in Kir2.1 deficient mice.
A, mean arterial blood pressure measured in anaesthetized mice via carotid artery catheterization (each point represents one animal; WT, n = 6; Kir2.1+/−, n = 5; * P < 0.05). B, body weights of WT and Kir2.1+/− animals (n = 4 per group). C, tail blood flow rates are determined by volumetric pressure recording tail‐cuff (n = 4 per group, * P < 0.05). D, peripheral vascular resistance calculated from mean blood pressure divided by blood flow rate, measured by volumetric pressure recording tail‐cuff (n = 4 per group, * P < 0.05). E, representative M‐mode echocardiogram of the left ventricle from WT mouse. F, representative M‐mode echocardiogram of the left ventricle from Kir2.1+/− mouse. G, stroke volumes of WT and Kir2.1+/− mice are measured by M‐mode echocardiogram from the parasternal short axis view (WT, n = 6; Kir2.1+/−, n = 5). H, heart rates are measured during M‐mode echocardiogram (WT, n = 6; Kir2.1+/−, n = 5). I, cardiac outputs of WT and Kir2.1+/− mice are determined from stroke volume multiplied by heart rate (WT, n = 6; Kir2.1+/−, n = 5).
To evaluate further the physiological mechanism of increased blood pressure in Kir2.1+/− mice, we tested whether Kir2.1 deficiency results in an increase in peripheral vascular resistance, a major component of elevated blood pressure (Messerli, 1982). Blood flow was measured after pressurized occlusion of the tail (Fig. 12 C) and vascular resistance was calculated as a ratio of blood flow to mean blood pressure taken from the same animal (Fig. 8 D). Consistent with an increase in the mean blood pressure, blood flow rate is significantly decreased and peripheral vascular resistance is significantly elevated in Kir2.1+/− mice.
Furthermore, we tested the effect of Kir2.1 deficiency on the key parameters of cardiac function, such as stroke volume, heart rate and cardiac output that could also contribute to the elevation of blood pressure. These parameters were measured using an echocardiogram on lightly sedated mice (1.5% isoflurane). Typical left ventricular M‐mode images of the echocardiogram for WT and Kir2.1+/− mice are shown in Fig. 12 E and F. More specifically, the images show the oscillatory movement of the ventricular wall that occur during the cardiac cycle (i.e. systole and diastole), and the stroke volume is calculated based on the difference between the chamber volumes during diastole and systole. As expected, the heart rate of the mice was in the range of ∼500 beats min−1 and no difference was observed between WT and Kir2.1+/− mice (Fig. 12 G). Also, there is no difference between WT and Kir2.1+/− mice in the stroke volume and cardiac output (Fig. 12 H and I).
Contributions of Kir2.1 and KCa to FIV of human arteries
Our previous studies have found that NO contributes to FIV in subcutaneous adipose arteries from patients (Grizelj et al. 2015). In order to determine the contribution of Kir and SK channels in FIV in human arteries, we isolated arteries from subcutaneous biopsies using previously described methodologies (Goslawski et al. 2013; Grizelj et al. 2015). The average diameter of human arteries was 112 ± 20 μm and the shear stress range was 9–28 dyn cm−2 for pressure gradients of Δ40–100 cmH2O, similar to the mouse vessels described above (pipettes were prepared in exactly the same way as for mouse arteries; average tip size 19.1 ± 0.2 μm at the upstream end and 22 ± 0.3 μm at the downstream end). Similar to arteries isolated from WT mice, Fig. 13 A and B shows that FIV in arteries from subcutaneous fat isolated from human patients is significantly reduced in the presence of the Kir inhibitor Ba2+ or the SK inhibitor apamin. The combination of Ba2+ and apamin resulted in an additional inhibition of FIV compared to either inhibitor alone, demonstrating that Kir and SK channels have additive effects.
Figure 13. Kir and SK channels contribute to flow‐induced vasodilatation of human microvessels.
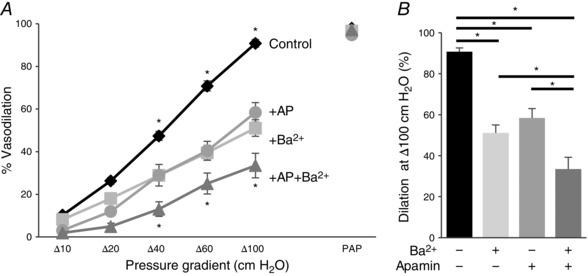
A, average flow‐induced dilatations of pre‐constricted subcutaneous vessels harvested from human lower abdominal adipose tissue with and without 20 nm apamin (AP), 300 μm Ba2+, or both, as a function of pressure gradient. Vessel diameters are normalized to diameters stabilized at 60 cmH2O. Intralumenal pressure is maintained as 60 cmH2O. Apamin and Ba2+ perfused extravascularly and maintained during the experiments. Papaverine (100 μm) is added at Δ100 cmH2O at the end of the experiment to verify the maximal dilatation of arteries (n = 9 vessels per condition, * P < 0.05). B, maximal dilatation at Δ100 cmH2O pressure gradient for the same experimental conditions.
Discussion
This study presents the first specific evidence for the role of Kir channels in FIV of resistance arteries. The main findings of the study are: (i) Kir channels expressed in primary endothelial cells freshly isolated from resistance arteries show clear sensitivity to shear stress; (ii) Kir, and specifically Kir2.1, are essential for NO‐dependent vasodilatation, eNOS and Akt phosporylation and NO production in murine mesenteric arteries in response to flow; (iii) Kir2.1 genetic deficiency in mice results in a significant increase in peripheral resistance and elevation of blood pressure; (iv) Kir and Ca2+‐sensitive K+ channels act in parallel through two distinct NO‐dependent and NO‐independent pathways, respectively, to induce vasodilatation in response to flow; (v) blocking Kir channels results in a strong negative effect on flow‐induced vasodilatation in human microvasculature. These observations demonstrate that Kir channels play a major role in flow‐induced endothelium‐dependent vasodilatation and provide the first insights into the mechanism of this effect.
Expression of Kir2 channels in microvascular endothelial cells
Several studies have reported the presence of Ba2+‐sensitive Kir currents in multiple, but not all subtypes of endothelial cells (Rusko et al. 1992; Romanenko et al. 2002; Crane et al. 2003; Fang et al. 2005; Ledoux et al. 2008; Millar et al. 2008). Here, we present a molecular profile of Kir2 channels in murine mesenteric endothelial cells: at a messenger level, mesenteric endothelial cells express three Kir2 subtypes, Kir2.1, Kir2.2 and Kir2.3, but expression of Kir2.1 is severalfold higher than that of the other two channels. Expression of Kir2.1 is also confirmed on the protein level. Functionally, we show for the first time that primary mesenteric ECs express shear stress‐sensitive Kir currents. These currents significantly reduced in ECs isolated from vessels of Kir2.1+/− heterozygous mice. Taken together, these observations indicate that Kir2.1 is the major functional Kir channel in mesenteric endothelium.
Consideration of the Kir2.1+/− model
Since a full loss of Kir2.1 channels is lethal hours after birth (Zaritsky et al. 2000), we considered using a Kir2.1+/− heterozygous mice that are fully viable. We found that endothelial cells isolated from these mice express significantly decreased levels of Kir2.1, both at the messenger (∼60–65% decrease) and protein (∼50% decrease) levels, as well as in electrophysiological data (∼50% decrease). The reduction of Kir2.1 expression in Kir2.1+/− mice is accompanied by 2‐ to 3‐fold increases in the expression of Kir2.2 and Kir2.3 channels but since their expression level is very low compared to Kir2.1, this increased expression does not compensate for functional Kir currents. The sensitivity of Kir channels to shear stress also seem to be decreased in Kir2.1+/− ECs, even though the basis of this reduced sensitivity to shear is not clear. Importantly, consistent with previous studies (Crane et al. 2003; Smith et al. 2008), mouse mesenteric VSMCs do not express Kir2.1, which allows isolation of the effects of endothelial Kir2.1 on the vessel function in this model. Nevertheless, in a global knockout model, there is always a concern about the tissue specificity of the observed effects. This concern is addressed in our study by EC‐specific viral overexpression of Kir2.1 in excised arteries. Thus, Kir2.1+/− mice in combination with the EC‐specific Kir2.1 overexpression provides a strong model to study the role of Kir2.1 channels in endothelial function.
Role of endothelial Kir2.1 in flow‐induced vasodilatation
This study demonstrates that a decrease in Kir2.1 expression, with the corresponding reduction of shear stress‐sensitive Kir current, results in a dramatic decrease in FIV of mesenteric arteries. Several lines of evidence indicate that this effect should be attributed to the endothelial Kir. First, endothelium‐denuded arteries maintain their full reactivity in response to an NO donor, an assay that is typically used to ascertain that smooth muscle reactivity is not affected (Ignarro & Kadowitz, 1985). In addition, while expression of Kir2 channels in vascular smooth muscle cells is tissue specific, no Kir2.1 was detected in VSMCs of mouse mesenteric arteries in previous studies (Crane et al. 2003; Jackson, 2005; Smith et al. 2008; Dunn & Nelson, 2010). Consistent with these studies, we also show here that mouse mesenteric VSMCs have no detectable Kir2.1 expression. Moreover, to exclude the possibility that other subtypes of Kir2 channels might be expressed in mesenteric VSMCs and contribute to FIV via an NO‐independent mechanism, we tested K+‐induced vasodilatation. The rationale of this approach is that VSMCs Kir2 channels may contribute to NO‐independent K+‐induced vasodilatation due to their sensitivity to extracellular K+, which can be elevated due to K+ efflux from neighboring cells (Quayle et al. 1993; Edwards et al. 1998; Burns et al. 2004; Filosa et al. 2006). Alternatively, K+‐induced vasodilatation may also be mediated by Na+–K+‐ATPase (Burns et al. 2004). To test this idea, denuded vessels were exposed to elevated levels of extracellular K+. As expected, a small elevation in extracellular K+ (<20 mm) resulted in a vasodilatory response whereas higher K+ levels resulted in vasoconstriction. The latter is expected because high extracellular K+ level results in depolarization of smooth muscle cells, which triggers constriction. Our data show, however, that K+‐induced vasodilatation of denuded mesenteric vessels is sensitive to ouabain but not to Ba2+, indicating that it should be attributed to the inhibition of Na+–K+‐ATPase and not to Kir2 channels. In contrast, as expected, we observed Ba2+‐sensitive K+‐induced vasodilatation in denuded cerebral arteries from the same animals. Furthermore, a rescue of the FIV with EC‐specific overexpression of Kir2.1 provides compelling evidence for the critical role of endothelial Kir2.1 in FIV in this vascular bed.
It is important to note that while our study provides direct evidence that Kir channels play a major role in flow‐induced endothelial response, this does not mean that Kir channels are the primary shear stress sensors and this question still needs to be investigated. An earlier study showed shear stress may activate Kir channels in a cell‐attached mode though the channels in the pipette are not exposed to shear directly, suggesting that shear stress can activate these channels indirectly (Jacobs et al. 1995). Possible upstream mechanosensors include but are not limited to integrins, glycocalyx, other ion channels, and G proteins (Ando & Yamamoto, 2009; Sriram et al. 2016). Mechanosensitive ion channels, specifically those permeable to Ca2+, are believed to be a major source of entry for Ca2+ into ECs upon exposure to shear stress (Wiesner et al. 1997); however, any existing link between mechanosensitive Ca2+ channel activation and subsequent Kir channel activation has yet to be investigated. Shear also activates endothelial GPCRs and integrins that converge onto PIP2 to promote IP3 and PIP3 formation, respectively (Chachisvilis et al. 2006; Loufrani et al. 2008; Sriram et al. 2016), but a decrease in PIP2 availability would be expected to deactivate rather than activate Kir channels. Another open question is whether an increase in Kir current is the result of an increase in the open probability of the channels, activation of previously dormant channels or insertion of new channels into the plasma membrane. Clearly, more studies are needed to elucidate the mechanism responsible for shear stress‐induced activation of endothelial Kir channels.
Endothelial Kir2.1 channels control FIV by regulating Akt/eNOS signalling and NO generation
Earlier studies showed that activation of endothelial K+ channels may act as the endothelium‐derived hyperpolarizing factor (EDHF) responsible for the NO‐independent, cyclooxygenase (COX)‐independent mechanism, a residual vasodilatation that is observed when both NO and COX are inhibited (Brandes et al. 2000). This mechanism has been well established for Ca2+‐sensitive K+ channels in earlier studies demonstrating that blocking or genetic loss of small and intermediate‐conductance KCa channels result in almost complete loss of EDHF‐mediated (NO‐independent) vasodilatation of carotid artery and cremaster arteries (Miura et al. 2001 a; Brahler et al. 2009; Wölfle et al. 2009). Interestingly, it was also shown that while intermediate‐conductance KCa channels play the dominant role in acetylcholine‐induced EDHF response (Brahler et al. 2009; Wölfle et al. 2009), small conductance KCa channels are responsible for the shear stress‐mediated EDHF response (Brahler et al. 2009). Our current study demonstrates, however, that this is not the case for endothelial Kir2 channels: in the presence of eNOS inhibitor, neither blocking Kir2 channels with Ba2+, nor the loss of Kir2.1 expression in Kir2.1+/− mice have any effect on the NO‐independent FIV component. Consistent with these observations, we also demonstrate directly that flow‐induced NO generation is significantly inhibited in mesenteric arteries isolated from Kir2.1+/− mice. As expected, since eNOS is expressed selectively in endothelial cells, denudation of the arteries eliminates any flow‐induced NO generation and abrogates the effect of Kir2.1+/−. Taken together, these data indicate that in mesenteric circulation, Kir2 channels regulate FIV by regulating endothelial NO generation.
In terms of further insights into the mechanism, we show that downregulation or blocking of Kir2.1 prevents flow‐induced phosphorylation of eNOS and Akt in mesenteric ECs, with both effects fully rescued by Kir2.1 overexpression in Kir2.1‐deficient ECs. Moreover, consistent with earlier studies (Fleming et al. 1998; Dimmeler et al. 1999), Akt phosphorylation was essential for flow‐induced eNOS phosphorylation. Blocking Akt phosphorylation abrogated the Kir2.1 overexpression rescue effect. Together, these data indicate that Kir2.1 channels regulate flow‐induced activation of eNOS in mesenteric ECs via Akt phosphorylation. It is not clear, however, what is the mechanistic link between Kir channels and Akt. In general, earlier studies implied that K+ channels regulate cellular functions by regulating Ca2+ influx into the cells (Adams et al. 1989; Luckhoff & Busse, 1990; Lin et al. 1993). However, it was also previously shown that shear stress‐induced FIV is Ca2+ independent (Stepp et al. 1999) and that shear stress‐induced eNOS activation is Akt dependent and Ca2+ independent (Dimmeler et al. 1999). Consistent with this study, receptor‐mediated eNOS activation also has a component that is Akt dependent and Ca2+ independent, further supporting the notion that Akt activation is Ca2+ independent (Fleming et al. 1999; Takahashi & Mendelsohn, 2003). Based on these studies, we suggest that Kir channels regulate eNOS through Akt and not through Ca2+. Clearly, further studies are needed to elucidate the mechanism by which Kir channels regulate Akt. This is a novel pathway that is distinct from previously described roles of Kir2.1 as a hyperpolarization booster that is downstream of SK channels (Sonkusare et al. 2016). Our data indicate that another NO‐dependent pathway is the major mechanism, by which Kir2 channels regulate FIV.
Our study also shows that regulation of NO generation and of NO‐dependent vasodilatation is specific for Kir2 channels and that Kir2 and Ca2+‐sensitive K+ channels contribute to FIV via NO‐dependent and NO‐independent pathways, respectively. This conclusion is consistent with earlier observations that NO‐dependent regulation of blood pressure is still intact in mice lacking KCa channels (Kohler & Ruth, 2010). Importantly, blocking both Kir2 and SK channels simultaneously fully abrogates flow‐induced vasorelaxation in murine mesenteric arteries, indicating that these two pathways are the major contributors and are fully responsible for FIV in this vascular bed. While the mechanistic basis for differential effects of Kir2.1 and SK channels on FIV is not clear, one possibility is that there are differential intracellular distributions of these channels. Our earlier studies have shown that Kir2.1 channels partially partition into cholesterol‐rich membrane domains (Tikku et al. 2007). More recently, we also found that Kir2.1 interacts with caveolin‐1 and partially partition into caveolae (Han et al. 2014). Interaction of eNOS of caveolin‐1 is well established (Garcia‐Cardena et al. 1996; Ghosh et al. 1998; Gratton et al. 2000). However, since SK channels were also shown to partition into caveolae (Absi et al. 2007; Goedicke‐Fritz et al. 2015), this alone cannot explain the functional difference between Kir2.1 and SK channels in activating eNOS. Decreased Kir current density together with decreased sensitivity to flow in Kir2.1+/− ECs may underlie the loss of Kir contribution to FIV in Kir2.1+/− arteries. Further studies are needed to understand how different types of K+ channels regulate FIV by distinct mechanisms.
Role of Kir2.1 channels in regulation of blood pressure and vascular resistance
The loss of Kir2.1 channels also results in significant elevation (∼20 mmHg) of mean arterial blood pressure, indicating that Kir2 channels play an important role in control of vascular tone in vivo. A hypertensive shift in Kir2.1 deficient mice is comparable to the elevation in blood pressure that was observed earlier in mice deficient in Ca2+‐sensitive K+ channels (Brahler et al. 2009). The latter is consistent with our observation that Kir2 and SK contribute about equally to FIV. Interestingly, while Kir2 channels play a major role in flow‐induced FIV, we saw no Ba2+ effect on the baseline diameters of the mesenteric arteries. Most likely, it means that other mechanisms have a predominant effect on the resting tone, and should be investigated in the future.
Moreover, since we show here that the loss of Kir2.1 channels results in significant impairment of FIV in resistance arteries and since it is known that resistance arteries are a major source of resistance in the circulation (Mayet & Hughes, 2003), we hypothesized that elevation of blood pressure in Kir2.1+/− mice should be accompanied by increased vascular resistance in vivo. Our data show that this is indeed the case and that Kir2.1+/− mice have significantly decreased flow rate and increased vascular resistance as measured using volumetric pressure recording. Furthermore, since the two key components that contribute to mean arterial pressure (MAP) are cardiac output (CO) and peripheral vascular resistance (PVR) (MAP = CO × PVR) (Mayet & Hughes, 2003), we also determined whether Kir2.1 deficiency in Kir2.1+/− mice may result in an increase in stroke volume and/or heart rate resulting in increased cardiac output. Our results show, however, that this is not the case and that the cardiac physiological parameters are not altered in Kir2.1+/− mice. Taken together, these observations indicate that Kir2.1 deficiency results in increased blood pressure via increased vascular resistance.
Role of Kir channels in flow‐induced vasorelaxation of human resistance arteries
Importantly, the Kir2 dependency of FIV that we observed in mouse mesenteric arteries is confirmed in human resistance arteries isolated from subcutaneous fat of humans. Vasodilatation in response to flow is an important physiological regulator of tissue perfusion and in human subcutaneous adipose we have previously found that FIV in subcutaneous resistance arteries is dependent on NO (Grizelj et al. 2015). However, the upstream mechanisms of NO‐dependent FIV in these vessels are unknown. We found, similar to the mouse mesenteric circulation, that FIV of human arteries is reduced in the presence of barium or apamin, suggesting the role of Kir2 and SK channels in regulating FIV in human resistance arteries. Previous studies have found that KCa channels are involved in FIV in human coronary arteries of patients with coronary disease (Liu et al. 2011). However, this mechanism is likely to involve endothelial‐derived H2O2 stimulation of large conductance potassium channels on the smooth muscle cell. Future studies exploring the relationship between Kir2 channel activation and the regulation of FIV during cardiovascular disease human arteries are warranted.
Additional information
Competing interests
None declared.
Author contributions
The work reported in this paper was carried out at the laboratories of I.L. S.A.P. and the Physiology Core Laboratory, Department of Physiology, College of Medicine in University of Illinois at Chicago. l S. J. A., I. S. F., S. A. P., I. L. contributed to conception or design of the work. S. J. A., I. S. F., J. B., C. X. Z., S. S., R. G., S. A. P., I. L. contributed to acquisition, analysis, or interpretation of data for the work. S. J. A., I. S. F., J. B., C. X. Z., S. S., R. G., S. A. P., I. L. contributed to drafting the work or revising it critically for important intellectual content. All authors approved the final version of the manuscript. All authors agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work were appropriately investigated and resolved. All persons designated as authors qualify for authorship.
Funding
This study is supported by NIH grants HL‐073965 and HL‐083298 to I.L. and T32 DK080674 to I.S.F.
Acknowledgements
We thank Ms Catherine Osborn for her help with electrophysiological recordings and Drs Nattel and Ordog for a generous gift of a lentivirus expressing dnKir2.1.
References
- Absi M, Burnham MP, Weston AH, Harno E, Rogers M & Edwards G (2007). Effects of methyl β‐cyclodextrin on EDHF responses in pig and rat arteries; association between SKCa channels and caveolin‐rich domains. Br J Pharmacol 151, 332–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams DJ, Barakeh J, Laskey R & Van Breemen C (1989). Ion channels and regulation of intracellular calcium in vascular endothelial cells. FASEB J 3, 2389–2400. [DOI] [PubMed] [Google Scholar]
- Ando J & Yamamoto K (2009). Vascular mechanobiology: endothelial cell responses to fluid shear stress. Circ J 73, 1983–1992. [DOI] [PubMed] [Google Scholar]
- Brahler S, Kaistha A, Schmidt VJ, Wolfle SE, Busch C, Kaistha BP, Kacik M, Hasenau AL, Grgic I, Si H, Bond CT, Adelman JP, Wulff H, de Wit C, Hoyer J & Kohler R (2009). Genetic deficit of SK3 and IK1 channels disrupts the endothelium‐derived hyperpolarizing factor vasodilator pathway and causes hypertension. Circulation 119, 2323–2332. [DOI] [PubMed] [Google Scholar]
- Brandes RP, Schmitz‐Winnenthal FH, Feletou M, Godecke A, Huang PL, Vanhoutte PM, Fleming I & Busse R (2000). An endothelium‐derived hyperpolarizing factor distinct from NO and prostacyclin is a major endothelium‐dependent vasodilator in resistance vessels of wild‐type and endothelial NO synthase knockout mice. Proc Natl Acad Sci USA 97, 9747–9752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns WR, Cohen KD & Jackson WF (2004). K+‐induced dilation of hamster cremasteric arterioles involves both the Na+/K+‐ATPase and inward‐rectifier K+ channels. Microcirculation 11, 279–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chachisvilis M, Zhang YL & Frangos JA (2006). G protein‐coupled receptors sense fluid shear stress in endothelial cells. Proc Natl Acad Sci USA 103, 15463–15468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane GJ, Walker SD, Dora KA & Garland CJ (2003). Evidence for a differential cellular distribution of inward rectifier K channels in the rat isolated mesenteric artery. J Vasc Res 40, 159. [DOI] [PubMed] [Google Scholar]
- Dai G, Kaazempur‐Mofrad MR, Natarajan S, Zhang Y, Vaughn S, Blackman BR, Kamm RD, Garcia‐Cardena G & Gimbrone MA Jr (2004). Distinct endothelial phenotypes evoked by arterial waveforms derived from atherosclerosis‐susceptible and ‐resistant regions of human vasculature. Proc Natl Acad Sci USA 101, 14871–14876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies PF, Spaan JA & Krams R (2005). Shear stress biology of the endothelium. Ann Biomed Eng V33, 1714. [DOI] [PubMed] [Google Scholar]
- Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R & Zeiher AM (1999). Activation of nitric oxide synthase in endothelial cells by Akt‐dependent phosphorylation. Nature 399, 601–605. [DOI] [PubMed] [Google Scholar]
- Dunn KM & Nelson MT (2010). Potassium channels and neurovascular coupling. Circ J 74, 608–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards G, Dora KA, Gardener MJ, Garland CJ & Weston AH (1998). K+ is an endothelium‐derived hyperpolarizing factor in rat arteries. Nature 396, 269–272. [DOI] [PubMed] [Google Scholar]
- Eschke D, Richter M, Brylla E, Lewerenz A, Spanel‐Borowski K & Nieber K (2002). Identification of inwardly rectifying potassium channels in bovine retinal and choroidal endothelial cells. Ophthalmic Res 34, 343–348. [DOI] [PubMed] [Google Scholar]
- Fang Y, Schram G, Romanenko VG, Shi C, Conti L, Vandenberg CA, Davies PF, Nattel S & Levitan I (2005). Functional expression of Kir2.x in human aortic endothelial cells: the dominant role of Kir2.2. Am J Physiol Cell Physiol 289, C1134–C1144. [DOI] [PubMed] [Google Scholar]
- Filosa JA, Bonev AD, Straub SV, Meredith AL, Wilkerson MK, Aldrich RW & Nelson MT (2006). Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat Neurosci 9, 1397. [DOI] [PubMed] [Google Scholar]
- Fisslthaler B, Dimmeler S, Hermann C, Busse R & Fleming I (2000). Phosphorylation and activation of the endothelial nitric oxide synthase by fluid shear stress. Acta Physiol Scand 168, 81–88. [DOI] [PubMed] [Google Scholar]
- Fleming I, Bauersachs J, Fisslthaler B & Busse R (1998). Ca2+‐independent activation of the endothelial nitric oxide synthase in response to tyrosine phosphatase inhibitors and fluid shear stress. Circ Res 82, 686–695. [DOI] [PubMed] [Google Scholar]
- Fleming I, Bauersachs J, Schafer A, Scholz D, Aldershvile J & Busse R (1999). Isometric contraction induces the Ca2+‐independent activation of the endothelial nitric oxide synthase. Proc Natl Acad Sci USA 96, 1123–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Cardena G, Oh P, Liu J, Schnitzer JE & Sessa WC (1996). Targeting of nitric oxide synthase to endothelial cell caveolae via palmitoylation: implications for nitric oxide signaling. Proc Natl Acad Sci USA 93, 6448–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Gachhui R, Crooks C, Wu C, Lisanti MP & Stuehr DJ (1998). Interaction between caveolin‐1 and the reductase domain of endothelial nitric‐oxide synthase. Consequences for catalysis. J Biol Chem 273, 22267–22271. [DOI] [PubMed] [Google Scholar]
- Goedicke‐Fritz S, Kaistha A, Kacik M, Markert S, Hofmeister A, Busch C, Banfer S, Jacob R, Grgic I & Hoyer J (2015). Evidence for functional and dynamic microcompartmentation of Cav‐1/TRPV4/KCa in caveolae of endothelial cells. Eur J Cell Biol 94, 391–400. [DOI] [PubMed] [Google Scholar]
- Goslawski M, Piano MR, Bian J‐T, Church EC, Szczurek M & Phillips SA (2013). Binge drinking impairs vascular function in young adults. J Am Coll Cardiol 62, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratton JP, Fontana J, O'Connor DS, Garcia‐Cardena G, McCabe TJ & Sessa WC (2000). Reconstitution of an endothelial nitric‐oxide synthase (eNOS), hsp90, and caveolin‐1 complex in vitro. Evidence that hsp90 facilitates calmodulin stimulated displacement of eNOS from caveolin‐1. J Biol Chem 275, 22268–22272. [DOI] [PubMed] [Google Scholar]
- Grizelj I, Cavka A, Bian JT, Szczurek M, Robinson A, Shinde S, Nguyen V, Braunschweig C, Wang E, Drenjancevic I & Phillips SA (2015). Reduced flow‐and acetylcholine‐induced dilations in visceral compared to subcutaneous adipose arterioles in human morbid obesity. Microcirculation 22, 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H, Rosenhouse‐Dantsker A, Gnanasambandam R, Epshtein Y, Chen Z, Sachs F, Minshall RD & Levitan I (2014). Silencing of Kir2 channels by caveolin‐1: cross‐talk with cholesterol. . J Physiol 592, 4025–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I & Kurachi Y (2010). Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90, 291–366. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ & Kadowitz PJ (1985). The pharmacological and physiological role of cyclic GMP in vascular smooth muscle relaxation. Annu Rev Pharmacol Toxicol 25, 171–191. [DOI] [PubMed] [Google Scholar]
- Jackson WF (2005). Potassium channels in the peripheral microcirculation. Microcirculation 12, 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs ER, Cheliakine C, Gebremedhin D, Birks EK, Davies PF & Harder DR (1995). Shear activated channels in cell‐attached patches of cultured bovine aortic endothelial cells. Pflugers Archiv 431, 129–131. [DOI] [PubMed] [Google Scholar]
- Kohler R & Ruth P (2010). Endothelial dysfunction and blood pressure alterations in K+‐channel transgenic mice. Pflugers Archiv 459, 969–976. [DOI] [PubMed] [Google Scholar]
- Kubo Y, Adelman JP, Clapham DE, Jan LY, Karschin A, Kurachi Y, Lazdunski M, Nichols CG, Seino S & Vandenberg CA (2005). International Union of Pharmacology. LIV. Nomenclature and molecular relationships of inwardly rectifying potassium channels. Pharmacol Rev 57, 509–526. [DOI] [PubMed] [Google Scholar]
- Ledoux J, Bonev AD & Nelson MT (2008). Ca2+‐activated K+ channels in murine endothelial cells: block by intracellular calcium and magnesium. J Gen Physiol 131, 125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitan I, Helmke BP & Davies PF (2000). A chamber to permit invasive manipulation of adherent cells in laminar flow with minimal disturbance of the flow field. Ann Biomed Eng 28, 1184–1193. [DOI] [PubMed] [Google Scholar]
- Lin CS, Boltz RC, Blake JT, Nguyen M, Talento A, Fischer PA, Springer MS, Sigal NH, Slaughter RS & Garcia ML (1993). Voltage‐gated potassium channels regulate calcium‐dependent pathways involved in human T lymphocyte activation. J Exp Med 177, 637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GX, Derst C, Schlichthorl G, Heinen S, Seebohm G, Bruggemann A, Kummer W, Veh RW, Daut J & Preisig‐Muller R (2001). Comparison of cloned Kir2 channels with native inward rectifier K+ channels from guinea‐pig cardiomyocytes. J Physiol 532, 115–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Bubolz AH, Mendoza S, Zhang DX & Gutterman DD (2011). H2O2 is the transferrable factor mediating flow‐induced dilation in human coronary arterioles. Circ Res 108, 566–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhao H, Li H, Kalyanaraman B, Nicolosi AC & Gutterman DD (2003). Mitochondrial sources of H2O2 generation play a key role in flow‐mediated dilation in human coronary resistance arteries. Circ Res 93, 573–580. [DOI] [PubMed] [Google Scholar]
- Loufrani L, Retailleau K, Bocquet A, Dumont O, Danker K, Louis H, Lacolley P & Henrion D (2008). Key role of α1β1‐integrin in the activation of PI3‐kinase‐Akt by flow (shear stress) in resistance arteries. Am J Physiol Heart Circ Physiol 294, H1906–H1913. [DOI] [PubMed] [Google Scholar]
- Luckhoff A & Busse R (1990). Calcium influx into endothelial cells and formation of endothelium‐derived relaxing factor is controlled by the membrane potential. Pflugers Arch 416, 305–311. [DOI] [PubMed] [Google Scholar]
- Luksha L, Agewall S & Kublickiene K (2009). Endothelium‐derived hyperpolarizing factor in vascular physiology and cardiovascular disease. Atherosclerosis 202, 330. [DOI] [PubMed] [Google Scholar]
- Mayet J & Hughes A (2003). Cardiac and vascular pathophysiology in hypertension. Heart 89, 1104–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messerli FH (1982). Cardiovascular effects of obesity and hypertension. Lancet 1, 1165–1168. [DOI] [PubMed] [Google Scholar]
- Milkau M, Kohler R & de Wit C (2010). Crucial importance of the endothelial K+ channel SK3 and connexin40 in arteriolar dilations during skeletal muscle contraction. FASEB J 24, 3572–3579. [DOI] [PubMed] [Google Scholar]
- Millar ID, Wang S, Brown PD, Barrand MA & Hladky SB (2008). Kv1 and Kir2 potassium channels are expressed in rat brain endothelial cells. Pflugers Arch 456, 379–391. [DOI] [PubMed] [Google Scholar]
- Miura H, Wachtel RE, Liu Y, Loberiza FR Jr, Saito T, Miura M & Gutterman DD (2001. a). Flow‐induced dilation of human coronary arterioles: important role of Ca2+‐activated K+ channels. Circulation 103, 1992–1998. [DOI] [PubMed] [Google Scholar]
- Miura H, Wachtel RE, Liu Y, Loberiza FR Jr, Saito T, Miura M & Gutterman DD (2001. b). Flow‐induced dilation of human coronary arterioles: important role of Ca2+‐activated K+ channels. Circulation 103, 1992–1998. [DOI] [PubMed] [Google Scholar]
- Nilius B & Droogmans G (2001). Ion channels and their functional role in vascular endothelium. Physiol Rev 81, 1415–1459. [DOI] [PubMed] [Google Scholar]
- Olesen S‐P, Clapham DE & Davies PF (1988). Hemodynamic shear stress activates a K+ current in vascular endothelial cells. Nature 331, 168–170. [DOI] [PubMed] [Google Scholar]
- Parasuraman S & Raveendran R (2012). Measurement of invasive blood pressure in rats. J Pharmacol Pharmacother 3, 172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips SA, Hatoum OA & Gutterman DD (2007). The mechanism of flow‐induced dilation in human adipose arterioles involves hydrogen peroxide during CAD. Am J Physiol Heart Circ Physiol 292, H93–H100. [DOI] [PubMed] [Google Scholar]
- Pistner A, Belmonte S, Coulthard T & Blaxall B (2010). Murine echocardiography and ultrasound imaging. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quayle JM, McCarron JG, Brayden JE & Nelson MT (1993). Inward rectifier K+ currents in smooth muscle cells from rat resistance‐sized cerebral arteries. Am J Physiol Cell Physiol 265, C1363–C1370. [DOI] [PubMed] [Google Scholar]
- Reneman RS & Hoeks AP (2008). Wall shear stress as measured in vivo: consequences for the design of the arterial system. Med Biol Eng Comput 46, 499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rindler TN, Lasko VM, Nieman ML, Okada M, Lorenz JN & Lingrel JB (2011). Knockout of the Na,K‐ATPase α2‐isoform in cardiac myocytes delays pressure overload‐induced cardiac dysfunction. Am J Physiol Heart Circ Physiol 304, H1147–H1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanenko VG, Rothblat GH & Levitan I (2002). Modulation of endothelial inward rectifier K+ current by optical isomers of cholesterol. Biophys J 83, 3211–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusko J, Tanzi F, van Breemen C & Adams DJ (1992). Calcium‐activated potassium channels in native endothelial cells from rabbit aorta: conductance, Ca2+ sensitivity and block. J Physiol 455, 601–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid‐Schönbein G & Granger DN (2003). Molecular Basis for Microcirculatory Disorders. Springer‐Verlag Paris, Paris, France. [Google Scholar]
- Schram G, Melnyk P, Pourrier M, Wang Z & Nattel S (2002). Kir2.4 and Kir2.1 K+ channel subunits co‐assemble: a potential new contributor to inward rectifier current heterogeneity. J Physiol 544, 337–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma H, Pathan RA, Kumar V, Javed S & Bhandari U (2011). Anti‐apoptotic potential of rosuvastatin pretreatment in murine model of cardiomyopathy. Int J Cardiol 150, 193–200. [DOI] [PubMed] [Google Scholar]
- Si H, Heyken WT, Wolfle SE, Tysiac M, Schubert R, Grgic I, Vilianovich L, Giebing G, Maier T, Gross V, Bader M, de Wit C, Hoyer J & Kohler R (2006). Impaired endothelium‐derived hyperpolarizing factor‐mediated dilations and increased blood pressure in mice deficient of the intermediate‐conductance Ca2+‐activated K+ channel. Circ Res 99, 537–544. [DOI] [PubMed] [Google Scholar]
- Smith PD, Brett SE, Luykenaar KD, Sandow SL, Marrelli SP, Vigmond EJ & Welsh DG (2008). KIR channels function as electrical amplifiers in rat vascular smooth muscle. J Physiol 586, 1147–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonkusare SK, Dalsgaard T, Bonev AD & Nelson MT (2016). Inward rectifier potassium (Kir2.1) channels as end‐stage boosters of endothelium‐dependent vasodilators. J Physiol 594, 3271–3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram K, Laughlin JG, Rangamani P & Tartakovsky DM (2016). Shear‐induced nitric oxide production by endothelial cells. Biophys J 111, 208–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepp DW, Nishikawa Y & Chilian WM (1999). Regulation of shear stress in the canine coronary microcirculation. Circulation 100, 1555–1561. [DOI] [PubMed] [Google Scholar]
- Takahashi S & Mendelsohn ME (2003). Synergistic activation of endothelial nitric‐oxide synthase (eNOS) by HSP90 and Akt: calcium‐independent eNOS activation involves formation of an HSP90‐Akt‐CaM‐bound eNOS complex. J Biol Chem 278, 30821–30827. [DOI] [PubMed] [Google Scholar]
- Taylor MS, Bonev AD, Gross TP, Eckman DM, Brayden JE, Bond CT, Adelman JP & Nelson MT (2003). Altered expression of small‐conductance Ca2+‐activated K+ (SK3) channels modulates arterial tone and blood pressure. Circ Res 93, 124–131. [DOI] [PubMed] [Google Scholar]
- Thomas J, Epshtein Y, Chopra A, Ordog B, Ghassemi M, Christman JW, Nattel S, Cook JL & Levitan I (2011). Anthrax lethal factor activates K+ channels to induce IL‐1β secretion in macrophages. J Immunol 186, 5236–5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikku S, Epshtein Y, Collins H, Travis AJ, Rothblat GH & Levitan I (2007). Relationship between Kir2.1/Kir2.3 activity and their distribution between cholesterol‐rich and cholesterol‐poor membrane domains. Am J Physiol Cell Physiol 293, C440–C450. [DOI] [PubMed] [Google Scholar]
- Wiesner TF, Berk BC & Nerem RM (1997). A mathematical model of the cytosolic‐free calcium response in endothelial cells to fluid shear stress. Proc Natl Acad Sci USA 94, 3726–3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wölfle SE, Schmidt VJ, Hoyer J, Köhler R & Wit CD (2009). Prominent role of KCa3.1 in endothelium‐derived hyperpolarizing factor‐type dilations and conducted responses in the microcirculation in vivo . Cardiovasc Res 82, 476–483. [DOI] [PubMed] [Google Scholar]
- Wu C, Huang RT, Kuo CH, Kumar S, Kim CW, Lin YC, Chen YJ, Birukova A, Birukov KG, Dulin NO, Civelek M, Lusis AJ, Loyer X, Tedgui A, Dai G, Jo H & Fang Y (2015). Mechanosensitive PPAP2B regulates endothelial responses to atherorelevant hemodynamic forces. Circ Res 117, e41–e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff H & Köhler R (2013). Endothelial small‐conductance and intermediate‐conductance KCa channels: an update on their pharmacology and usefulness as cardiovascular targets. J Cardiovasc Pharmacol 61, 102–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, MacCallum DK, Ernst SA & Hughes BA (2003). Expression of the inwardly rectifying K+ channel Kir2.1 in native bovine corneal endothelial cells. Invest Ophthalmol Vis Sci 44, 3511–3519. [DOI] [PubMed] [Google Scholar]
- Zaritsky JJ, Eckman DM, Wellman GC, Nelson MT & Schwarz TL (2000). Targeted disruption of Kir2.1 and Kir2.2 genes reveals the essential role of the inwardly rectifying K+ current in K+‐mediated vasodilation. Circ Res 87, 160–166. [DOI] [PubMed] [Google Scholar]