

Abstract
The centrosome plays a critical role in various cellular processes including cell division and cilia formation, and deregulation of centrosome homeostasis is a hallmark feature of many human diseases. Here, we show that centrosomal protein of 78 kDa (Cep78) localizes to mature centrioles and directly interacts with viral protein R binding protein (VprBP). Although VprBP is a component of two distinct E3 ubiquitin ligases, EDD‐DYRK2‐DDB1Vpr BP and CRL4Vpr BP, Cep78 binds specifically to EDD‐DYRK2‐DDB1Vpr BP and inhibits its activity. A pool of EDD‐DYRK2‐DDB1Vpr BP is active at the centrosome and mediates ubiquitination of CP110, a novel centrosomal substrate. Deregulation of Cep78 or EDD‐DYRK2‐DDB1Vpr BP perturbs CP110 ubiquitination and protein stability, thereby affecting centriole length and cilia assembly. Mechanistically, ubiquitination of CP110 entails its phosphorylation by DYRK2 and binding to VprBP. Cep78 specifically impedes the transfer of ubiquitin from EDD to CP110 without affecting CP110 phosphorylation and binding to VprBP. Thus, we identify Cep78 as a new player that regulates centrosome homeostasis by inhibiting the final step of the enzymatic reaction catalyzed by EDD‐DYRK2‐DDB1Vpr BP.
Keywords: centrosome, Cep78, E3 ligase, inhibitor, ubiquitin
Subject Categories: Cell Cycle; Post-translational Modifications, Proteolysis & Proteomics
Introduction
The centrosome is the major microtubule‐organizing center in most eukaryotic cells and controls a number of cellular processes including cell division, cell shape, motility and polarity, and cilia formation 1. Beginning in G1 phase, a cell possesses one centrosome comprising two centrioles, the mother and daughter centrioles, embedded in the pericentriolar material (PCM) 2. Centrosome duplication commences in S phase, wherein a new centriole or procentriole grows perpendicularly at the proximal end of each existing centriole. Procentrioles elongate in S and G2 phases, while the daughter centriole matures into a mother centriole. At the same time, parental centrioles recruit and accumulate increasing amounts of PCM. Two functional centrosomes, fully capable of nucleating and organizing microtubules, are formed at the G2/M phase. At the onset of mitosis, the two centrosomes separate, migrating to the opposite end of a cell and establishing the mitotic spindle. These events ensure accurate chromosome segregation and allow each of the two incipient daughter cells to receive a diploid set of DNA along with a single centrosome. When a cell exits the cell cycle and enters the G0 phase, the centrosome migrates to the cell cortex where the mother centriole templates the assembly of a cilium, a cellular antenna critical for locomotion and sensation 3. Because centrosome duplication occurs once per cell cycle, the number of centrosomes in a cell is under strict control. Defects in centrosome duplication or function can give rise to human disorders such as cancer, ciliopathies, and microcephaly 4, 5, 6, 7.
The molecular makeup of the centrosome is complex, consisting of hundreds of proteins as revealed by recent bioinformatic, genomic, transcriptomic, and proteomic studies 8, 9, 10. Centrosomal protein of 78 kDa (Cep78) was first discovered as a novel component through proteomic analysis of isolated centrosomes 9. Human full‐length Cep78 is composed of 722 amino acids and possesses a putative coiled‐coil (CC) domain and a leucine‐rich repeat (LRR) domain with six consecutive LRR repeats. Deregulation of Cep78 causes retinal generation and hearing loss 11, 12, 13 and is associated with prostate and colorectal cancer 14, 15. The Cep78 gene is found in ciliated organisms but absent from non‐ciliated organisms, suggesting that the encoded product could be involved in cilia biogenesis and/or function 16. Another recent study shows that Cep78 is involved in regulating centrosome duplication 17. Despite these observations, the biological function of Cep78 remains poorly characterized.
Ubiquitination is common mechanism for regulating protein stability and function 18. The process of ubiquitination is controlled by three main classes of enzymes wherein ubiquitin (Ub) is transferred from an activating enzyme E1 to a conjugating enzyme E2 and finally to lysine residue(s) of the target substrate via a ligating enzyme E3. E3 ligases are responsible for substrate recognition and can be categorized into three types, really interesting new gene (RING), homologous to the E6AP carboxyl terminus (HECT), and RING‐between‐RING (RBR), depending on the presence of functional domains and the mechanism of Ub transfer to the substrate 19. Given that deregulation of some E3 ligases is associated with cancer, it is therefore critical to understand how their activities are controlled at the molecular level. The connection between Cep78 and protein ubiquitination is unknown.
In this study, we identified Cep78 as a new player that controls protein ubiquitination at the centrosome. We found that (i) Cep78 mostly associates with parental centrioles; (ii) Cep78 directly interacts with viral protein R binding protein/DDB1 and Cullin4‐associated factor 1 (VprBP/DCAF1), a subunit of two distinct E3 ligases: the HECT‐type EDD‐DYRK2‐DDB1VprBP consisting of EDD, DYRK2, DDB1 and VprBP, and the RING‐type CRL4VprBP consisting of Roc1, Cullin4A, DDB1, and VprBP 20; (iii) Cep78 specifically interacts with EDD‐DYRK2‐DDB1VprBP; (iv) Cep78 is not a substrate of EDD‐DYRK2‐DDB1VprBP; (v) Cep78 inhibits EDD‐DYRK2‐DDB1VprBP; (vi) a fraction of EDD‐DYRK2‐DDB1VprBP is present at the centrosome and mediates ubiquitination of a novel substrate CP110; (vii) deregulation of Cep78 or EDD‐DYRK2‐DDB1VprBP alters CP110 ubiquitination, thereby disrupting protein stability and cellular processes that are dependent on CP110. Finally, we dissected the molecular mechanism by which Cep78 inhibits ubiquitination of CP110.
Results
Cep78 localizes to parental centrioles and is periodically expressed in the cell cycle
We previously used a combination of biochemistry, cell biology, and proteomics to characterize Cep76, a protein that suppresses centriole amplification 21, 22. During the course of these studies, we identified Cep78 from a proteomic screen for Cep76‐interacting partners. The interaction between Cep78 and Cep76 was subsequently confirmed by immunoprecipitation (IP)/Western blot (WB) (Fig 1A). Furthermore, both recombinant and endogenous Cep78 associated with a known Cep76‐interacting protein CP110 22 (Figs 1A and 5A). By transfecting a plasmid expressing Flag‐tagged Cep78 into normal diploid RPE‐1 cells and performing immunofluorescence (IF) experiments, we found that the staining of recombinant Cep78 overlaps with a distal centriolar marker CP110 but not with a proximal centriolar marker C‐Nap1 (Fig 1B). Likewise, IF experiments performed with an anti‐Cep78 antibody revealed that endogenous Cep78 co‐localizes with a distal centriolar marker centrin but not with C‐Nap1 or another proximal marker glutamylated tubulin (GT335) (Fig 1C). A second anti‐Cep78 antibody also exhibited staining that overlapped with centrin (Fig EV1A). Cep78 is an intrinsic component of centrosomes since its localization was not affected by treatment with nocodazole, a microtubule‐depolymerizing drug (Fig EV1B). Examination of endogenous Cep78 staining pattern at different stages of the cell cycle revealed one bright dot or two dots, one bright and one weak, in G0 cells (Fig 1D). The bright dot is always associated with the mother centriole able to template a cilium (Fig 1C). This staining pattern of Cep78 remains unchanged in G0, G1, and S phases (Fig 1D). In late G2 and M phases, two additional weak dots, presumably associated with maturing procentrioles, could be detected occasionally (Fig 1D). By quantifying the intensity of Cep78 IF in the vicinity of centrosomes, we showed that the Cep78 signal is low in G0 and G1, increases in S and G2, and diminishes in M phase (Fig 1E). Similarly, Cep78 protein levels were low in G0 and G1, increased in G1/S, peaked in S and G2, and decreased in M and the next G1 phase (Fig 1F). Cep78 IF and WB signals were greatly diminished in Cep78‐depleted cells (Fig EV1C and D), indicating that our antibody specifically recognizes endogenous Cep78. Taken together, our data suggest that Cep78 is a distal centriolar protein primarily associated with parental centrioles and that it may function in late G1, S, and G2 phases.
Figure 1. Cep78 interacts with Cep76 and CP110, localizes to the distal region of centrioles, and is cell cycle regulated.
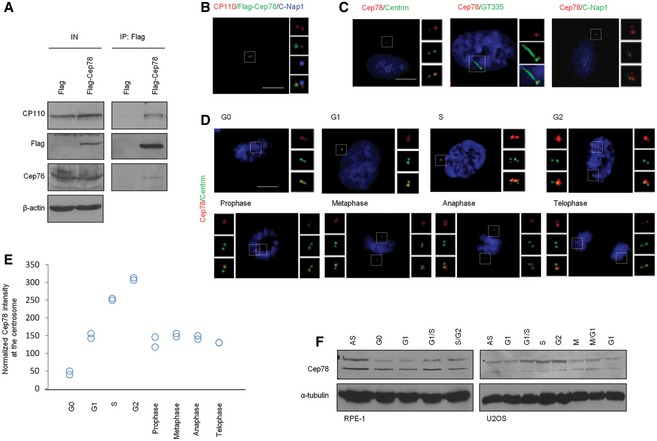
- Flag or Flag‐Cep78 was expressed in HEK293 cells. Lysates were immunoprecipitated with an anti‐Flag antibody and Western blotted with the indicated antibodies. IN, input. β‐actin was used as a loading control.
- RPE‐1 cells expressing Flag‐Cep78 were stained with antibodies against CP110 (red), Flag (green), and C‐Nap1 (blue). Scale bar, 1 μm.
- RPE‐1 cells were stained with DAPI (blue) and antibodies against Cep78 (red) and centrin, glutamylated tubulin (GT335), or C‐Nap1 (green). Scale bar, 1 μm.
- RPE‐1 cells in different phases of the cell cycle were stained with DAPI (blue) and antibodies against Cep78 (red) and centrin (green). Scale bar, 1 μm.
- Cep78 fluorescence intensity at centrosomes was measured and quantitated. At least 75 cells were quantitated in each cell cycle phase and two independent experiments were performed.
- RPE‐1 (left) and U2OS (right) lysates from different cell cycle phases were Western blotted with antibodies against Cep78. α‐tubulin was used as a loading control. AS, asynchronous. *denotes full‐length Cep78.
Source data are available online for this figure.
Figure 5. EDD‐DYRK2‐DDB1Vpr BP‐mediated ubiquitination and degradation of CP110 are regulated by Cep78.
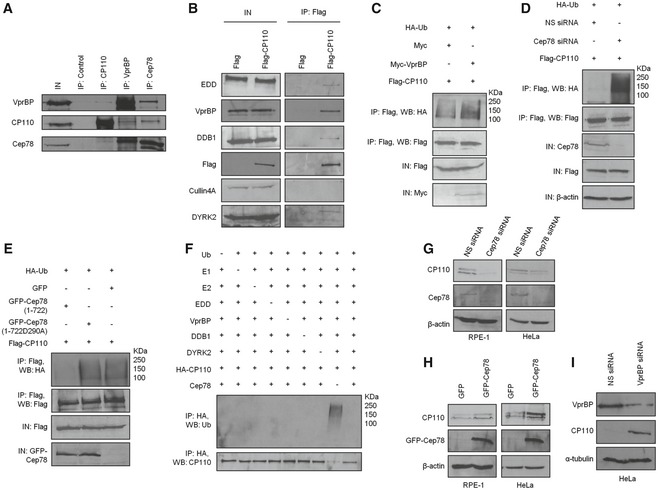
- HEK293 lysates were immunoprecipitated with an irrelevant (control), anti‐CP110, anti‐VprBP or anti‐Cep78 antibody, and Western blotted with the indicated antibodies. IN, input.
- Flag or Flag‐CP110 was expressed in HEK293 cells. Lysates were immunoprecipitated with an anti‐Flag antibody and Western blotted with the indicated antibodies. IN, input.
- Flag‐CP110 was co‐expressed with HA‐Ub and Myc or Myc‐VprBP in HEK293 cells. Lysates were immunoprecipitated with an anti‐Flag antibody in 1% SDS and Western blotted with the indicated antibodies. IN, input.
- HEK293 cells were transfected with NS siRNA or Cep78 siRNA and constructs expressing Flag‐CP110 and HA‐Ub. Lysates were immunoprecipitated with an anti‐Flag antibody in 1% SDS and Western blotted with the indicated antibodies. IN, input. β‐actin was used as a loading control.
- Flag‐CP110 was co‐expressed with HA‐Ub and GFP, GFP‐tagged Cep78 wild type (1–722), or mutant refractory to VprBP binding (1‐722D290A) in HEK293 cells. Lysates were immunoprecipitated with an anti‐Flag antibody in 1% SDS and Western blotted with the indicated antibodies. IN, input.
- In vitro ubiquitination assays were performed with HA‐CP110 as a substrate in the presence of purified Ub, E1, E2, EDD, DYRK2, DDB1, VprBP, and Cep78 in various combinations. Ubiquitinated and non‐ubiquitinated CP110 were detected by immunoblotting with anti‐Ub and anti‐CP110 antibodies, respectively.
- RPE‐1 or HeLa cells were transfected with NS siRNA or Cep78 siRNA. Lysates were Western blotted with the indicated antibodies. β‐actin was used as a loading control.
- RPE‐1 or HeLa cells were transfected with construct expressing GFP or GFP‐Cep78. Lysates were Western blotted with the indicated antibodies. β‐actin was used as a loading control.
- HeLa cells were transfected with NS siRNA or VprBP siRNA. Lysates were Western blotted with the indicated antibodies. α‐tubulin was used as a loading control.
Source data are available online for this figure.
Figure EV1. Characterization of Cep78.
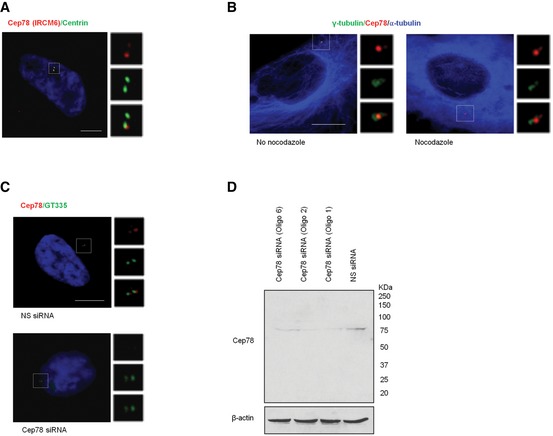
- RPE‐1 cells were stained with DAPI (blue) and antibodies against centrin (green) and Cep78 raised against the C‐terminal region of the protein (IRCM6, red). Scale bar, 1 μm.
- RPE‐1 cells untreated or treated with 10 μM nocodazole for 1 h to induce microtubule depolymerization were stained with antibodies against γ‐tubulin (green), α‐tubulin (blue), and Cep78 (red). Scale bar, 1 μm.
- RPE‐1 cells transfected with NS siRNA or Cep78 siRNA were stained with DAPI (blue) and antibodies against Cep78 (red) and polyglutamylated tubulin (GT335, green). Scale bar, 1 μm.
- RPE‐1 cells were transfected with NS siRNA or Cep78 siRNA (oligo 1, 2 or 6). Lysates were Western blotted with antibody against Cep78. β‐actin was used as a loading control.
Source data are available online for this figure.
Cep78 specifically interacts with the EDD‐DYRK2‐DDB1VprBP E3 ligase at the centrosome
To obtain insights into the biological relevance of Cep78, we performed a proteomic screen for Cep78‐interacting partners. We identified three putative partners—EDD, DDB1, and VprBP (Fig 2A)—components of EDD‐DYRK2‐DDB1VprBP 23. Because the highest number of peptides recovered in our mass spectrometric analysis besides Cep78 corresponded to VprBP, we first tested and confirmed the interaction between endogenous Cep78 and VprBP in multiple cell lines, including HEK293, HeLa, and RPE‐1 (Fig EV2A). To further explore whether Cep78 binds to EDD‐DYRK2‐DDB1VprBP, we transfected a plasmid expressing Flag‐Cep78 into cells, performed anti‐Flag IPs, and showed that Flag‐Cep78 co‐immunoprecipitates with endogenous DYRK2, EDD, DDB1, and VprBP (Fig 2B). In striking contrast, Flag‐Cep78 did not co‐immunoprecipitate with Cullin4A, a component of CRL4VprBP (Fig 2B). Similarly, endogenous Cep78, VprBP, DDB1, and EDD were detected in Flag‐DYRK2 immunoprecipitates (Fig EV2B). The interaction between Cep78 and EDD‐DYRK2‐DDB1VprBP is physiologically relevant since endogenous Cep78 specifically co‐immunoprecipitated with endogenous DYRK2, EDD, DDB1, and VprBP (Fig 2C) and these proteins co‐fractionated in a discrete protein complex at ~670 kDa (Fig 2D). Of note, the interaction between Cep78 and VprBP appeared to be very robust (Fig 2B and C), suggesting that these two proteins may directly interact. Indeed, in vitro binding experiments using purified Cep78 and individual EDD‐DYRK2‐DDB1VprBP subunits revealed that Cep78 directly binds to VprBP only (Fig 2E). Furthermore, since VprBP is thought to directly interact with DDB1, which in turn binds to DYRK2/EDD 23, we determined whether Cep78 interacts with DDB1 through VprBP. The Cep78–VprBP interaction remained intact upon DDB1 depletion (Fig EV2C), whereas the interaction between Cep78 and DDB1, DYRK2 or EDD was substantially reduced in VprBP‐depleted cells (Fig EV2D). Thus, our results suggest that Cep78 specifically binds to EDD‐DYRK2‐DDB1VprBP and that VprBP likely forms a scaffold linking Cep78 to DDB1/DYRK2/EDD (Fig 2F).
Figure 2. Cep78 interacts with EDD‐DYRK2‐DDB1Vpr BP through VprBP .
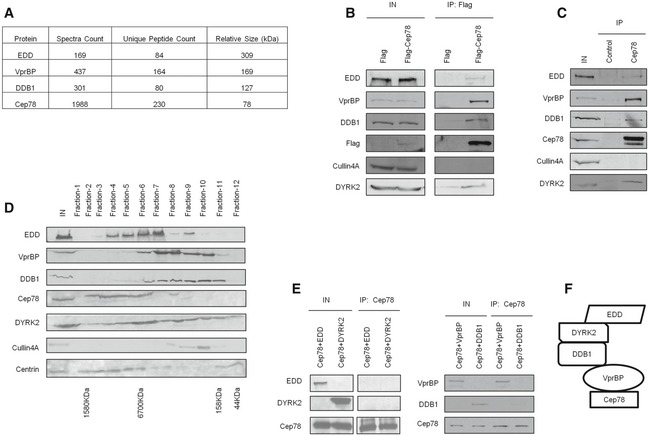
- Flag‐Cep78 protein complexes from HEK293 cells were immunopurified and subjected to mass spectrometric analysis.
- Flag or Flag‐Cep78 was expressed in HEK293 cells. Lysates were immunoprecipitated with an anti‐Flag antibody and Western blotted with the indicated antibodies. IN, input.
- HEK293 lysates were immunoprecipitated with an irrelevant (control) or anti‐Cep78 antibody and Western blotted with the indicated antibodies. IN, input.
- HEK293 cell extracts were chromatographed on a Superpose‐6 column, and the resulting fractions (Fraction‐1 to ‐12) were Western blotted with the indicated antibodies. IN, input.
- Purified Cep78 was mixed with purified EDD, DYRK2, DDB1, or VprBP. Proteins were immunoprecipitated with an anti‐Cep78 antibody and Western blotted with the indicated antibodies. IN, input.
- Proposed architecture of the Cep78‐bounded EDD‐DYRK2‐DDB1VprBP complex.
Source data are available online for this figure.
Figure EV2. Cep78 binds to EDD‐DYRK2‐DDB1Vpr BP through VprBP .
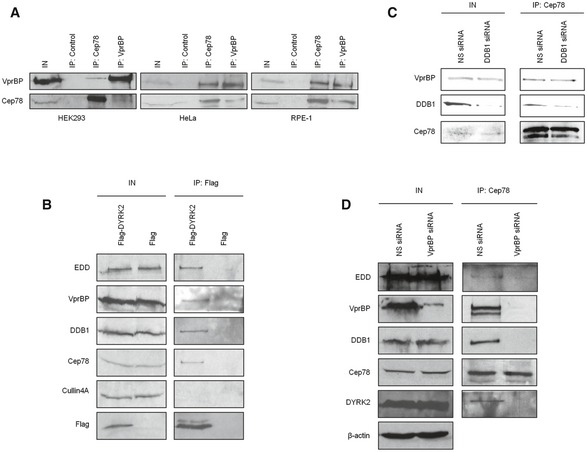
-
AHEK293, HeLa, or RPE‐1 lysates were immunoprecipitated with an anti‐Flag (control), anti‐Cep78 or anti‐VprBP antibody, and Western blotted with the indicated antibodies. IN, input.
-
BFlag or Flag‐DYRK2 was expressed in HEK293 cells. Lysates were immunoprecipitated with an anti‐Flag antibody and Western blotted with the indicated antibodies. IN, input.
-
C, DHEK293 cells were transfected with NS, DDB1, or VprBP siRNA. Lysates were immunoprecipitated with an anti‐Cep78 antibody and Western blotted with the indicated antibodies. IN, input. β‐actin was used as a loading control.
Source data are available online for this figure.
Although EDD‐DYRK2‐DDB1VprBP subunits are present in the nucleus and cytoplasm 24, 25, 26, 27, their localization to the centrosome has not been documented. Since EDD‐DYRK2‐DDB1VprBP binds to Cep78, we speculate that a pool of this E3 ligase is targeted to the centrosome. To biochemically demonstrate the localization of EDD‐DYRK2‐DDB1VprBP, we purified centrosomes through sucrose gradient centrifugation and found that endogenous Cep78, EDD, DYRK2, DDB1, and VprBP co‐sedimented with γ‐tubulin and centrin on a sucrose gradient (Fig 3A). As a negative control, these gradient fractions were devoid of a non‐centrosomal marker giantin (Fig 3A). Furthermore, IF experiments revealed that in addition to their nuclear localization, endogenous DYRK2, EDD, and VprBP co‐localize with centrin in about 5–65% of cells depending on the phase of the cell cycle (Fig 3B and C). Together, these data suggest that Cep78 likely interacts with EDD‐DYRK2‐DDB1VprBP at the centrosome.
Figure 3. EDD‐DYRK2‐DDB1Vpr BP is present at the centrosome and mapping functional domains of Cep78 and VprBP .
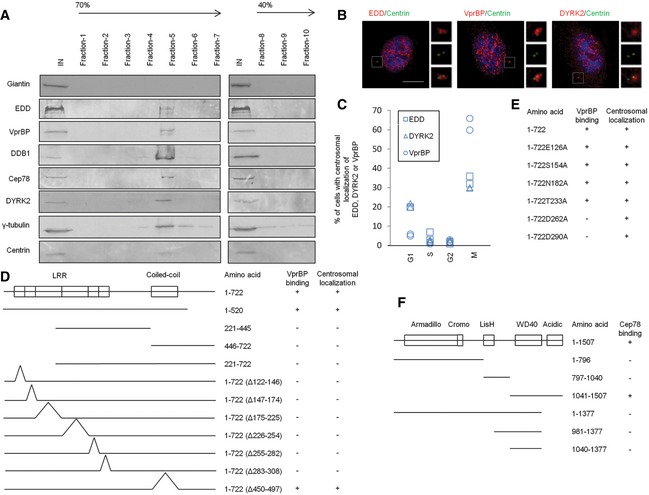
-
ACentrosomes from Jurkat cells were purified through a 40–70% sucrose gradient, and the resulting fractions were Western blotted with the indicated antibodies. IN, input. γ‐tubulin and centrin, positive control; giantin, negative control.
-
BRPE‐1 cells were stained with DAPI (blue) and antibodies against centrin (green) and EDD, DYRK2 or VprBP (red). Scale bar, 1 μm.
-
CThe percentage of cells with centrosomal EDD, DYRK2, or VprBP staining across the cell cycle. At least 100 cells were scored in each cell cycle phase and two independent experiments were performed.
-
D, EThe ability of various Cep78 truncated, deletion, and point mutants to interact with VprBP and localize to centrosomes. 1–722, full‐length; Δ122–146, deletion of the first LRR repeat; Δ147–174, deletion of the second LRR repeat; Δ175–225, deletion of the third LRR repeat; Δ226–254, deletion of the fourth LRR repeat; Δ255–282, deletion of the fifth LRR repeat; Δ283–308, deletion of the sixth LRR repeat; Δ450–497, deletion of the coiled‐coil domain. E126A, S154A, N182A, T233A, D262A, and D290A, point mutations of the first, second, third, fourth, fifth, and sixth LRR repeats, respectively.
-
FThe ability of various VprBP truncates to interact with Cep78. 1–1,507, full‐length.
Source data are available online for this figure.
Mapping domains critical for the function of Cep78 and its interaction with EDD‐DYRK2‐DDB1VprBP
To delineate the functional domain(s) of Cep78 that enable its localization to the centrosome and association with VprBP, a series of Flag‐tagged truncated, deletion, and point mutants of Cep78 were expressed in cells. After IPs with anti‐Flag antibodies, we found that only a fragment (1–520) containing the entire LRR domain can interact with endogenous VprBP (Figs 3D and EV3A) and that deletion of any one repeat within the domain is sufficient to abolish VprBP binding (Figs 3D and EV3B). Deletion of the CC domain, in contrast, did not affect binding to VprBP (Figs 3D and EV3B). Furthermore, disruption of the LRR domain but not the CC domain compromised centrosomal localization (Figs 3D and EV3C), suggesting that the LRR domain or the horseshoe conformation it adopts is critical for centrosomal targeting and VprBP binding. To further dissect the centrosomal localization and VprBP‐binding domains, we mutated single amino acids within the LRR domain that lie on the concave or convex surface of the horseshoe 28 and thus are predicted to induce minimal structural perturbations. We found that a point mutation in the fifth (D262A) or sixth (D290A) LRR repeat is sufficient to abrogate VprBP binding without affecting centrosomal localization (Figs 3E and EV4A and B). Likewise, depletion of VprBP did not impinge on Cep78 localization (Fig EV5A), suggesting that centrosomal targeting of Cep78 and its association with VprBP are separate events.
Figure EV3. Mapping of the centrosomal localization and VprBP‐binding domains of Cep78.
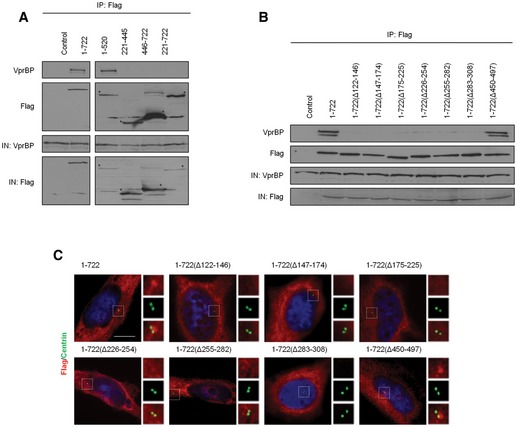
-
A, BFlag (control), Flag‐Cep78 full‐length (1–722), or Flag‐Cep78 truncated/deletion mutants were expressed in HEK293 cells. Lysates were immunoprecipitated with an anti‐Flag antibody and Western blotted with the indicated antibodies. IN, input. *denotes bands corresponding to the expected proteins.
-
CRPE‐1 cells expressing Flag‐Cep78 full‐length or truncated/deletion mutants were stained with DAPI (blue) and antibodies against Flag (red) and centrin (green). Scale bar, 1 μm.
Source data are available online for this figure.
Figure EV4. Mapping of interaction domains of Cep78 and EDD‐DYRK2‐DDB1Vpr BP .

- Flag (control), Flag‐Cep78 full‐length (1–722), or Flag‐Cep78 point mutants were expressed in HEK293 cells. Lysates were immunoprecipitated with an anti‐Flag antibody and Western blotted with the indicated antibodies. IN, input.
- RPE‐1 cells expressing Flag (control), Flag‐Cep78 full‐length, or point mutants were stained with DAPI (blue) and antibodies against Flag (red) and centrin (green). Scale bar, 1 μm.
- Flag (control), Flag‐VprBP full‐length (1–1,507), or Flag‐VprBP truncated mutants were co‐expressed with GFP‐Cep78 in HEK293 cells. Lysates were immunoprecipitated with an anti‐Flag antibody and Western blotted with the indicated antibodies. IN, input.
- Myc, Myc‐VprBP full‐length, or Myc‐VprBP truncated mutants were co‐expressed with Flag‐Cep78 in HEK293 cells. Lysates were immunoprecipitated with an anti‐Flag antibody and Western blotted with the indicated antibodies. IN, input. *denotes bands corresponding to the expected proteins.
Source data are available online for this figure.
Figure EV5. Cep78 and VprBP are independently recruited to the centrosome and Cep78 is not an EDD‐DYRK2‐DDB1Vpr BPsubstrate.
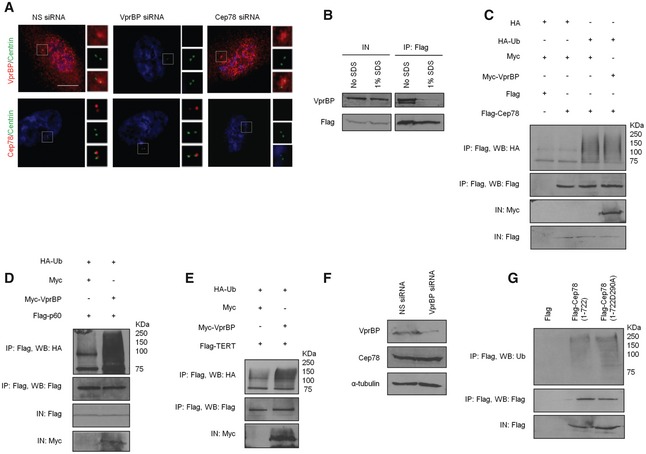
-
ARPE‐1 cells transfected with NS siRNA, Cep78 siRNA, or VprBP siRNA were stained with DAPI (blue) and antibodies against centrin (green) and Cep78 or VprBP (red). Scale bar, 1 μm.
-
BFlag‐Cep78 was expressed in HEK293 cells. Lysates were immunoprecipitated with an anti‐Flag antibody in the presence or absence of 1% SDS and Western blotted with the indicated antibodies. IN, input.
-
CHEK293 cells expressing Flag or Flag‐Cep78, HA or HA‐Ub, and Myc or Myc‐VprBP were synchronized in mitosis with nocodazole. Lysates were immunoprecipitated with an anti‐Flag antibody in 1% SDS and Western blotted with the indicated antibodies. IN, input.
-
D, EHA‐Ub, Flag‐katanin p60 or Flag‐TERT, and Myc or Myc‐VprBP were co‐expressed in HEK293 cells. Lysates were immunoprecipitated with an anti‐Flag antibody in 1% SDS and Western blotted with the indicated antibodies. IN, input.
-
FHEK293 cells transfected with NS siRNA or VprBP siRNA were synchronized in mitosis. Lysates were Western blotted with the indicated antibodies. α‐tubulin was used as a loading control.
-
GHEK293 cells expressing Flag, Flag‐Cep78 wild type (1–722), or mutant (1‐722D290A) were synchronized in mitosis. Lysates were immunoprecipitated with an anti‐Flag antibody in 1% SDS and Western blotted with the indicated antibodies. IN, input.
Source data are available online for this figure.
VprBP possesses an armadillo‐like and a cromo‐like domain in the N‐terminal region, a LisH domain in the middle region, and a WD40 and an acidic domain in the C‐terminal region 20. When VprBP fragments of various sizes were expressed, we observed that the C‐terminal end (1,377–1,507) encompassing the acidic domain is responsible for Cep78 binding (Figs 3F and EV4C and D). The Cep78‐binding domain of VprBP is therefore distinct from the WD40 domain required for DDB1 binding 29, 30.
Cep78 is not a substrate of EDD‐DYRK2‐DDB1VprBP
To explore the functional relationship between Cep78 and EDD‐DYRK2‐DDB1VprBP, we first asked if Cep78 is a substrate of EDD‐DYRK2‐DDB1VprBP by setting up in vivo ubiquitination assays. Flag‐Cep78, HA‐Ub, and Myc‐VprBP expressed in cells were left untreated or treated with MG132 to stabilize ubiquitinated products. Lysates were immunoprecipitated with anti‐Flag in the presence of 1% SDS to prevent interacting proteins such as VprBP from co‐immunoprecipitating with Flag‐Cep78 (Fig EV5B). Under this assay condition, any observable ubiquitinated products with an apparent molecular weight above 78 kDa were likely attributed to Flag‐Cep78 ubiquitination. Although ubiquitination of Flag‐Cep78 could be detected, the levels of ubiquitination did not change upon VprBP expression irrespective of MG132 (Fig 4A). Likewise, expression of VprBP did not significantly increase the levels of Cep78 ubiquitination in mitosis (Fig EV5C) during which Cep78 was down‐regulated (Fig 1E and F). In contrast, two previously known EDD‐DYRK2‐DDB1VprBP substrates, katanin p60 and TERT 23, 31, became highly ubiquitinated in the presence of VprBP (Fig EV5D and E). Consistent with the notion that VprBP does not impinge on Cep78 ubiquitination, depletion or expression of VprBP did not alter the protein levels or centrosomal localization of Cep78 (Figs 4B and EV5A). Depletion of VprBP also did not affect Cep78 protein levels in mitosis (Fig EV5F), and both wild‐type Cep78 and VprBP‐binding mutant of Cep78 (D290A) exhibited similar levels of ubiquitination in mitosis (Fig EV5G). Moreover, depletion or expression of Cep78 had no effect on VprBP protein levels or centrosomal localization (Fig EV5A and Appendix Fig S1A and B). From these results, we conclude that Cep78 is not a substrate of EDD‐DYRK2‐DDB1VprBP and that neither protein affects the localization or stability of the other.
Figure 4. Cep78 is a negative regulator but not a substrate of EDD‐DYRK2‐DDB1Vpr BP .
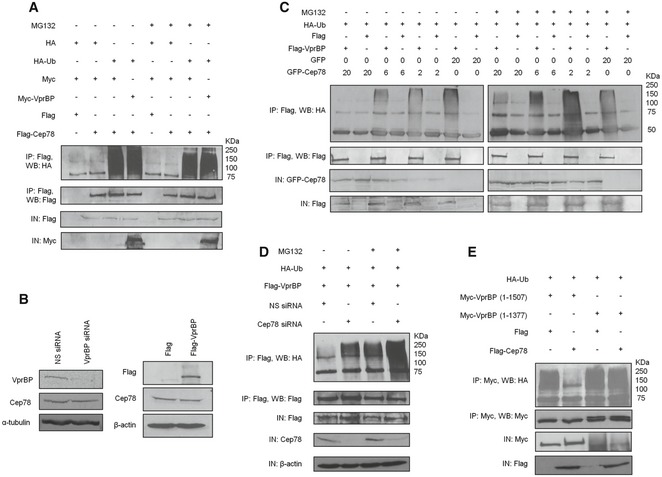
- Flag or Flag‐Cep78 was co‐expressed with HA or HA‐Ub and Myc or Myc‐VprBP in HEK293 cells untreated or treated with MG132. Lysates were immunoprecipitated with an anti‐Flag antibody in 1% SDS and Western blotted with the indicated antibodies. IN, input.
- HEK293 cells were transfected with NS siRNA or VprBP siRNA (left) or construct expressing Flag or Flag‐VprBP (right). Lysates were Western blotted with the indicated antibodies. α‐tubulin (left) or β‐actin (right) was used as a loading control.
- HEK293 cells transfected with HA‐Ub, Flag or Flag‐VprBP, and the indicated amount of GFP or GFP‐Cep78 construct in μg were left untreated or treated with MG132. Lysates were immunoprecipitated with an anti‐Flag antibody without SDS and Western blotted with the indicated antibodies. IN, input.
- HEK293 cells transfected with NS siRNA or Cep78 siRNA and constructs expressing Flag‐VprBP and HA‐Ub were left untreated or treated with MG132. Lysates were immunoprecipitated with anti‐Flag antibody without SDS and Western blotted with the indicated antibodies. IN, input. β‐actin was used as a loading control.
- Flag or Flag‐Cep78 was co‐expressed with HA‐Ub and Myc‐tagged VprBP full‐length (1–1,507) or VprBP refractory to Cep78 binding (1–1,377) in HEK293 cells. Lysates were immunoprecipitated with anti‐Myc antibody without SDS and Western blotted with the indicated antibodies. IN, input.
Source data are available online for this figure.
Cep78 prevents EDD‐DYRK2‐DDB1VprBP from ubiquitinating substrates
Next, we investigated whether Cep78 can modulate the activity of EDD‐DYRK2‐DDB1VprBP. For this purpose, we performed in vivo ubiquitination assays without SDS to immunoprecipitate recombinant VprBP and associated proteins from cells expressing recombinant VprBP and Ub. VprBP co‐immunoprecipitated with ubiquitinated products having apparent molecular weights from 50 to 250 kDa (Fig 4C). These products were present in greater abundance in MG132‐treated cells as opposed to untreated cells (Fig 4C) and likely represent substrates that get ubiquitinated by, and remain bounded to, VprBP throughout IP. Remarkably, co‐expression of Cep78 dramatically reduced the levels of ubiquitinated products in a dose‐dependent manner (Fig 4C). In contrast, the D290A mutant was less efficient in preventing the appearance of ubiquitinated products compared to wild‐type Cep78 (Appendix Fig S1C). Likewise, depletion of Cep78 under the same assay condition led to enhanced accumulation of ubiquitinated products (Fig 4D and Appendix Fig S1D), and these results together suggest that Cep78 suppresses EDD‐DYRK2‐DDB1VprBP. To further confirm a role of Cep78 in suppressing EDD‐DYRK2‐DDB1VprBP, we examined the effects of expressing a VprBP mutant refractory to Cep78 binding (1–1,377) on the levels of ubiquitinated products. Importantly, while the levels of ubiquitinated products associated with wild‐type VprBP were drastically reduced in the presence of Cep78, those associated with 1–1377 exhibited no reduction (Fig 4E). Collectively, our data support the notion that Cep78 inhibits the ubiquitination activity of EDD‐DYRK2‐DDB1VprBP by directly binding to VprBP.
To unambiguously and biochemically proof that Cep78 regulates EDD‐DYRK2‐DDB1VprBP but not CRL4VprBP, we found that depletion of Cep78 leads to enhanced ubiquitination of katanin p60 and TERT, whereas its overexpression diminishes ubiquitination (Appendix Fig S2A–D). On the other hand, the ubiquitination levels of a CRL4VprBP substrate MCM10 32 remained unaffected by Cep78 (Appendix Fig S2E and F). In light of these results, we suggest that Cep78 specifically regulates ubiquitination of EDD‐DYRK2‐DDB1VprBP substrates in a negative manner.
A Cep78‐interacting protein CP110 is a novel EDD‐DYRK2‐DDB1VprBP substrate
Next, we sought to determine whether EDD‐DYRK2‐DDB1VprBP could target certain substrates for ubiquitination at the centrosome and whether this event is regulated by Cep78. To this end, we demonstrated that recombinant or endogenous VprBP interacts with a Cep78‐interacting protein CP110 (Fig 5A and Appendix Fig S3A), and conversely, recombinant or endogenous CP110 associates with VprBP (Fig 5A and B). Furthermore, CP110 co‐immunoprecipitated with VprBP, DDB1, DYRK2, and EDD but not with Cullin4A (Fig 5B), consistent with the idea that it specifically binds to EDD‐DYRK2‐DDB1VprBP. Ectopic expression of VprBP or depletion of Cep78 enhanced CP110 ubiquitination (Fig 5C and D), whereas expression of wild type but not mutant (D290A) Cep78 reduced ubiquitination (Fig 5E), suggesting that CP110 is an EDD‐DYRK2‐DDB1VprBP substrate. Indeed, CP110 ubiquitination could be induced by EDD‐DYRK2‐DDB1VprBP and suppressed by Cep78 in vitro using purified proteins (Fig 5F). Ubiquitination of CP110 likely signals the protein for proteasomal degradation since ectopic expression of EDD, DDB1 or VprBP, or depletion of Cep78, decreased the steady‐state levels of CP110 in HEK293, RPE‐1, and HeLa cells (Fig 5G and Appendix Fig S3A–D). The decrease in CP110 levels caused by Cep78 depletion could be rescued by expression of exogenous Cep78 (Appendix Fig S3D). In sharp contrast, expression of Cep78 or depletion of VprBP increased CP110 levels in these cell lines (Figs 1A and 5H and I). As a control, we found that the levels of another Cep78‐interacting protein Cep76 are not affected by Cep78 or VprBP (Appendix Fig S4A and B). Taken together, our results indicate that EDD‐DYRK2‐DDB1VprBP specifically ubiquitinates CP110, leading to its degradation, and these effects can be counteracted by Cep78.
To validate these results, we were able to distinguish the activity of EDD‐DYRK2‐DDB1VprBP from Cyclin F and Neuralized homologue 4 (Neurl4), two E3 ligases previously known to modulate CP110 ubiquitination and stability 33, 34. Cyclin F ubiquitinates CP110 in G2 phase, whereas Neurl4 may ubiquitinate CP110 throughout the cell cycle. We showed that Cep78 specifically suppresses ubiquitination of CP110 induced by EDD‐DYRK2‐DDB1VprBP, but not by Cyclin F or Neurl4 (Appendix Fig S5A). Furthermore, although CP110 protein levels decreased in the presence of VprBP, they could be further reduced by co‐expression with Cyclin F or Neurl4 (Appendix Fig S5B). Thus, the mechanism by which EDD‐DYRK2‐DDB1VprBP ubiquitinates CP110 appears to be unique since only this enzyme is subjected to regulation by Cep78.
To further interrogate the relationship between Cep78, EDD‐DYRK2‐DDB1VprBP and CP110, we investigated the functional consequences of CP110 degradation in vivo. We employed two sensitive assays to monitor the loss of CP110. First, it has been shown that ablation of CP110 or a CP110 chaperone Cep97 leads to excessive growth or elongation of centrioles in non‐ciliated HeLa cells 35, 36, 37, 38. We thus examined the effects of manipulating the protein levels of Cep78 or EDD‐DYRK2‐DDB1VprBP on the formation of elongated centrioles in this cell line. Remarkably, depletion of Cep78 promoted centriole elongation (Fig 6A and B, and Appendix Fig S6A–D) which could be rescued by expression of exogenous Cep78 (Appendix Fig S6C and D), whereas ectopic expression of wild‐type Cep78 but not the D290A mutant suppressed the elongation phenotype induced by Cep97 loss (Fig 6C and D). Similarly, expression of EDD, DDB1, or VprBP provoked elongation of centrioles (Fig 6E and F). Second, it is known that CP110 can suppress cilia assembly in RPE‐1 cells able to form cilia 35, 39. A loss of CP110 therefore results in ectopic formation of cilia in proliferating cells, while its expression inhibits cilia assembly in quiescent cells. We demonstrated that expression of wild‐type Cep78, in contrast to the D290A mutant, efficiently inhibits cilia formation in quiescent RPE‐1 cells (Fig 6G and H). Conversely, expression of EDD, DDB1, and VprBP promoted cilia formation in proliferating cells (Fig 6I and J). Taken together, these data suggest that Cep78 maintains centrosome homeostasis by counteracting EDD‐DYRK2‐DDB1VprBP‐mediated ubiquitination and degradation of CP110.
Figure 6. Cep78 and EDD‐DYRK2‐DDB1Vpr BP control CP110‐dependent centriole elongation in non‐ciliated cells and cilia assembly in ciliated cells.
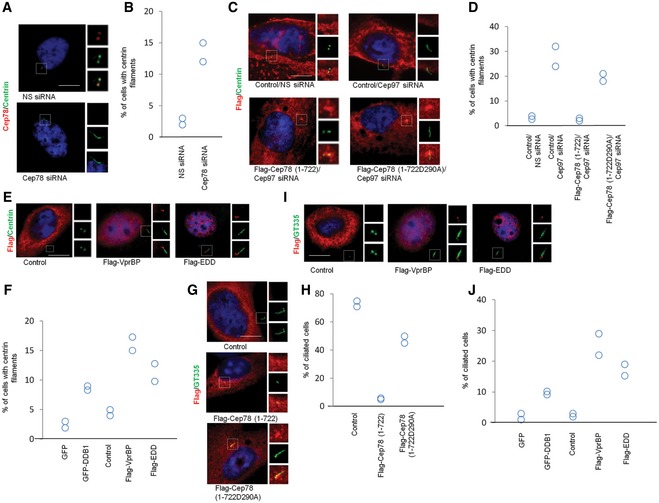
-
A, BHeLa cells transfected with NS siRNA and Cep78 siRNA were stained with DAPI (blue) and antibodies against Cep78 (red) and centrin (green). Scale bar, 1 μm. The percentage of cells with elongated centrioles (centrin filaments) was determined in (B).
-
C, DHeLa cells transfected with NS siRNA or Cep97 siRNA and construct expressing an irrelevant Flag‐tagged protein (control), Flag‐Cep78 (1–722), or Flag‐Cep78 (1‐722D290A) were stained with DAPI (blue) and antibodies against Flag (red) and centrin (green). Scale bar, 1 μm. The percentage of cells with elongated centrioles (centrin filaments) was determined in (D).
-
E, FHeLa cells transfected with construct expressing an irrelevant Flag‐tagged protein (control), Flag‐EDD, or Flag‐VprBP were stained with DAPI (blue) and antibodies against Flag (red) and centrin (green). Scale bar, 1 μm. The percentage of cells with elongated centrioles (centrin filaments) was determined in (F).
-
G, HQuiescent RPE‐1 cells expressing an irrelevant Flag‐tagged protein (control), Flag‐Cep78 (1–722), or Flag‐Cep78 (1‐722D290A) were stained with DAPI (blue) and antibodies against Flag (red) and glutamylated tubulin (GT335, green). Scale bar, 1 μm. The percentage of ciliated cells was determined in (H).
-
I, JProliferating RPE‐1 cells expressing an irrelevant Flag‐tagged protein (control), Flag‐EDD, or Flag‐VprBP were stained with DAPI (blue) and antibodies against Flag (red) and glutamylated tubulin (GT335, green). Scale bar, 1 μm. The percentage of ciliated cells was determined in (J).
Mechanism of EDD‐DYRK2‐DDB1VprBP inhibition by Cep78
To obtain mechanistic details into how Cep78 precludes CP110 ubiquitination, we revisited our current knowledge of substrate ubiquitination carried out by EDD‐DYRK2‐DDB1VprBP 23, 31. First, it is believed that the substrate must be phosphorylated by DYRK2 prior to ubiquitination. Substrate recognition by VprBP is also important since VprBP helps bring the substrate into close proximity with EDD. Once the substrate is physically close to EDD, Ub is transferred from EDD to the substrate. CP110 contains five putative DYRK2 phosphorylation sites (Fig 7A) and was readily phosphorylated by DYRK2 alone or EDD‐DYRK2‐DDB1VprBP in vitro (Fig 7B), suggesting that it is a DYRK2 substrate. When four of these sites were mutated to alanine (S287AS366AS551AS906A), phosphorylation of the mutant protein was significantly diminished (Fig 7C). The mutant protein exhibited impaired binding to VprBP (Fig 7D) and became less susceptible to ubiquitination (Fig 7E), suggesting that CP110 phosphorylation is a prerequisite for VprBP binding and subsequent ubiquitination. Importantly, Cep78 suppressed ubiquitination of CP110 (Fig 5E) without compromising its ability to undergo phosphorylation (Fig 7F) and associate with VprBP (Fig 7G). These results imply that Cep78 specifically prevents the transfer of Ub to CP110.
Figure 7. Mechanism underlying EDD‐DYRK2‐DDB1Vpr BP inhibition by Cep78.
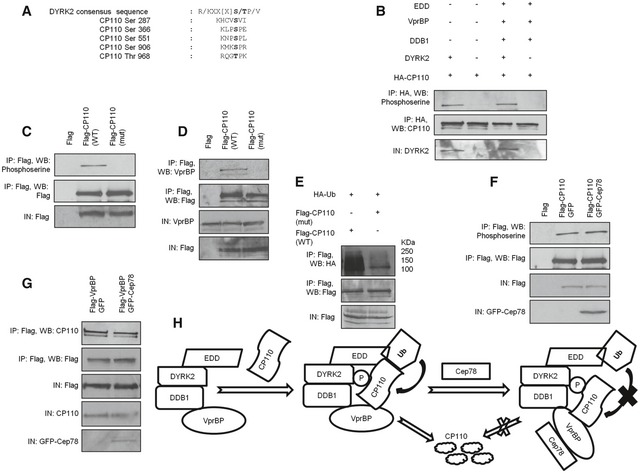
-
APutative DYRK2 phosphorylation sites of CP110.
-
BIn vitro kinase assays were performed using HA‐CP110 as a substrate in the presence of DYRK2 or DYRK2, EDD, DDB1, and VprBP. Phosphorylation of CP110 was detected by immunoblotting with an anti‐phosphoserine antibody. IN, input.
-
C, DFlag, Flag‐CP110 wild type (WT), or S287AS366AS551AS906A mutant (mut) were expressed in HEK293 cells. Lysates were immunoprecipitated with an anti‐Flag antibody and Western blotted with the indicated antibodies. IN, input.
-
EFlag‐CP110 wild type (WT) or S287AS366AS551AS906A mutant (mut) were co‐expressed with HA‐Ub in HEK293 cells. Lysates were immunoprecipitated with an anti‐Flag antibody in 1% SDS and Western blotted with the indicated antibodies. IN, input.
-
FFlag‐CP110 was co‐expressed with GFP or GFP‐Cep78 in HEK293 cells. Lysates were immunoprecipitated with an anti‐Flag antibody and Western blotted with the indicated antibodies. IN, input.
-
GFlag‐VprBP was co‐expressed with GFP or GFP‐Cep78 in HEK293 cells. Lysates were immunoprecipitated with an anti‐Flag antibody and Western blotted with the indicated antibodies. IN, input.
-
HA model illustrating the role of Cep78 in regulating the activity of EDD‐DYRK2‐DDB1VprBP. Upon binding to VprBP, Cep78 induces a conformational change in VprBP. As a result, EDD is no longer in close proximity to CP110 and Ub cannot be transferred to CP110. P, phosphate group.
Source data are available online for this figure.
Discussion
The centrosome is a dynamic organelle which undergoes numerical and structural changes during the cell cycle. To achieve dynamicity, the stability and function of proteins involved in different aspects of centrosome biology must be kept in check at all times. Failure to achieve or maintain centrosome homeostasis can have deleterious consequences and can give rise to human diseases such as cancer and ciliopathies. E3 ligases play an important role in modulating protein stability and function 19, and in this study, we demonstrated that a pool of the E3 ligase EDD‐DYRK2‐DDB1VprBP operates at the centrosome and that a poorly studied protein Cep78 is dedicated to controlling its activity. To our knowledge, Cep78 is the first centrosomal protein shown to possess inhibitory activity against an E3 ligase through direct binding.
Although the precise manner in which EDD‐DYRK2‐DDB1VprBP ubiquitinates its substrates is not fully clear, distinct components of this E3 ligase are thought to participate in substrate phosphorylation, binding to VprBP and ubiquitination 23, 31. Our data show that although Cep78 inhibits CP110 ubiquitination, it does not prevent CP110 from undergoing phosphorylation or from interacting with VprBP. Therefore, it appears that Cep78 explicitly hinders the addition of Ub to CP110, the last step of the enzymatic reaction catalyzed by EDD‐DYRK2‐DDB1VprBP. We postulate that Cep78 binding to VprBP triggers a conformational change in VprBP, thereby precluding CP110 from staying close to EDD (Fig 7H). As a result, the transfer of Ub from EDD to CP110 is hampered.
One interesting finding from our work is that the C‐terminal acidic domain of VprBP binds to Cep78 and plays a crucial role in controlling the ubiquitination activity of EDD‐DYRK2‐DDB1VprBP. In this regard, it is intriguing that Merlin, a protein that inhibits CRL4VprBP, binds to the same domain on VprBP 40. It has been proposed that Merlin acts as a competitive inhibitor to prevent substrates from binding to VprBP 40. This mode of inhibition is undoubtedly different from that of Cep78 which appears to be non‐competitive. Since deregulation of some E3 ligases has been linked to cancer and very few inhibitors of the HECT‐type E3 ligases are currently available 41, 42, our results on Cep78 may provide new insights into targeting this particular type of E3 ligases for therapeutic intervention.
Although we do not yet know how many centrosomal substrates EDD‐DYRK2‐DDB1VprBP can target, this E3 ligase seems to be highly selective since it specifically ubiquitinates and influences the steady‐state levels of CP110 (this work) and katanin p60 23 but not Cep76 (this work), all of which are centrosomal proteins. EDD‐DYRK2‐DDB1VprBP is believed to function during mitosis 23, 31. We postulate that EDD‐DYRK2‐DDB1VprBP is responsible for directing ubiquitination of a subset of substrates in mitosis when Cep78 levels are low, and that regulation of this E3 ligase by Cep78 in other phases of the cell cycle is critical to maintain centrosome homeostasis. In support of this idea, the protein levels of Cep78 correlate with those of CP110 43 and the two cellular processes associated with CP110 function, centriole length, and cilia assembly, go awry when there is insufficient or excessive amount of Cep78 or EDD‐DYRK2‐DDB1VprBP. Future proteomic studies will identify additional EDD‐DYRK2‐DDB1VprBP substrates at the centrosome.
Materials and Methods
Cell culture and plasmids
Human U2OS, hTERT RPE‐1, HeLa, Jurkat, and HEK293 cells were grown in DMEM supplemented with 5% FBS at 37°C in a humidified 5% CO2 atmosphere. To generate Flag‐tagged Cep78 fusion proteins, human Cep78 cDNA fragments encoding residues 1–722 (full‐length), 1–520, 221–445, 446–722, and 221–722 were amplified by PCR using Phusion High‐Fidelity DNA polymerase (New England Biolabs) and cloned into mammalian expression vector pCBF‐Flag. Full‐length cDNA was also subcloned into pEGFP‐C1 vector to generate GFP‐Cep78. The following Cep78 deletions/point mutations (Δ122–146, Δ147–174, Δ175–225, Δ226–254, Δ255–282, Δ283–308, Δ450–497, E126A, S154A, N182A, T233A, D262A, and D290A) and CP110 mutation (S287AS366AS551AS906A) were introduced into full‐length cDNA by employing a two‐step PCR mutagenesis strategy and subcloned into pCBF‐Flag or pEGFP‐C1. To generate Flag‐VprBP fusion proteins, human VprBP cDNA fragments encoding residues 1–1,507 (full‐length), 1–796, 797–1,040, and 1,041–1,507 were cloned into pCBF‐Flag. All constructs were verified by DNA sequencing. The following proteins were also expressed from plasmids in mammalian cells: HA‐Ub (J. Archambault), Flag‐TERT (C. Autexier), GFP‐DDB1, Myc‐VprBP (1–1,507), Myc‐VprBP (1–1,377), Myc‐VprBP (981–1,377), Myc‐VprBP (1,040–1,377) (E. Cohen), HA‐CP110, Flag‐CP110, Flag‐SCAPER, Flag‐Cyclin F and Flag‐Neurl4 (B. Dynlacht), Myc‐MCM10 (S. Saxena), Flag‐katanin p60 (J. Singer), Flag‐EDD (D. Saunders and C. Watts, Addgene plasmid #37188), and Flag‐DYRK2 (A. Rao, Addgene plasmid #20005).
Antibodies
Antibodies used in this study included anti‐CP110, anti‐Cep78, anti‐VprBP, anti‐EDD, anti‐DDB1, anti‐Cullin4A (Bethyl Laboratories), anti‐centrin (Millipore), anti‐GFP (Roche), anti‐C‐Nap1, anti‐HA, anti‐myc, anti‐phosphoserine (Santa Cruz), anti‐α‐tubulin, anti‐Flag, anti‐γ‐tubulin, anti‐β‐actin (Sigma‐Aldrich), anti‐glutamylated tubulin GT335 (Cedarlane), anti‐DYRK2 (Abcam), and anti‐Ub (Dako). To generate rabbit anti‐Cep78 antibodies, a glutathione‐S‐transferase (GST) fusion protein containing residues 590–722 (IRCM6) of Cep78 was expressed in E. coli and purified to homogeneity. Antibodies against Cep78 were purified by affinity chromatography.
Mass spectrometric identification of Cep78 interacting proteins
To identify interacting proteins, Flag‐Cep78 was expressed in HEK293 and immunoprecipitated with anti‐Flag agarose beads (Sigma‐Aldrich) for 2 h at 4°C. Bounded proteins were eluted with Flag peptide for 30 min, and the resultant eluates were precipitated with trichloroacetic acid and fractionated by SDS–PAGE. Six gel slices containing polypeptides were excised after Coomassie staining and subjected to proteolytic digestion mass spectrometric analysis. Analyses were performed at the mass spectrometry core facility from IRCM by micro‐capillary LC/MS/MS.
Immunoprecipitation, immunoblotting, and immunofluorescence microscopy
Immunoprecipitation, immunoblotting, and immunofluorescence were performed as described 44, 45. Briefly, cells were lysed with lysis buffer (50 mM HEPES/pH 7.4, 250 mM NaCl, 5 mM EDTA/pH 8, 0.1% NP‐40, 1 mM DTT, 0.2 mM AEBSF, 2 μg/ml leupeptin, 2 μg aprotinin, 10 mM NaF, 50 mM β‐glycerophosphate, and 10% glycerol) at 4°C for 30 min and extracted proteins were recovered in the supernatant after centrifugation at 16,000 g. For immunoprecipitation, 2 mg of the resulting supernatant was incubated with an appropriate antibody at 4°C for 1 h and collected using protein A‐ or G‐Sepharose beads. The beads were washed with lysis buffer, and bound proteins were analyzed by SDS–PAGE and immunoblotting with primary antibodies and horseradish peroxidase‐conjugated secondary antibodies (VWR). 100 μg of lysate was typically loaded into the input (IN) lane. For indirect immunofluorescence, cells were grown on glass coverslips, fixed with cold methanol, and permeabilized with 1% Triton X‐100/PBS. Slides were blocked with 3% BSA in 0.1% Triton X‐100/PBS prior to incubation with primary antibodies. Secondary antibodies used were Cy3‐, Cy5‐, or Alexa488‐conjugated donkey anti‐mouse, anti‐rat, or anti‐rabbit IgG (Jackson Immunolabs and Molecular Probes). Cells were then stained with DAPI (Sigma), and slides were mounted, observed, and photographed using a Leitz DMRB (Leica) microscope (100×, NA 1.3) equipped with a Retiga EXi cooled camera. To quantify Cep78 signal at the centrosome, the background fluorescence between slides was normalized and the function measurement by region of interest (ROI) from the software Volocity was used.
Cell cycle synchronization and FACS analysis
To obtain U2OS cells synchronized in the G1, G1/S, S, G2, M, M/G1, and next G1 phases, cells were treated with 0.4 mM mimosine for 24 h, 2 mM HU for 24 h, 2 mM HU for 24 h and release for 5 h, 2 mM HU for 24 h and release for 9 h, 40 ng/ml nocodazole for 24 h, 40 ng/ml nocodazole for 24 h and release for 4 h, and 40 ng/ml nocodazole for 24 h and release for 9 h, respectively. To obtain RPE‐1 cells synchronized in the G0, G1, G1/S, S/G2, and M phases, cells were brought to quiescence by serum starvation for 48 h and re‐stimulated for 0, 12, 24, 28, and 34 h. HEK293 cells were synchronized in mitosis with 40 ng/ml nocodazole for 24 h. Cell cycle distribution was confirmed by FACS as described previously 46.
RNA interference
Synthetic siRNA oligonucleotides were purchased from GE Dharmacon. The 21‐nucleotide siRNA sequence for the non‐specific (NS) control was 5′‐AATTCTCCGAACGTGTCACGT‐3′. The 21‐nucleotide siRNA sequences for Cep78 were 5′‐GAGGAGTTGTCCAGAAATA‐3′ (oligo 1), 5′‐GCGATAAGATACAAAGATG‐3′ (oligo 2), 5′‐GGTCGTTCTGGATATAAGA‐3′ (oligo 6), and 5′‐CAAAGAAACTAGGGAAACTAG‐3′ (oligo 7). Oligo 1 was used unless stated otherwise. Oligo 7 targets the 3′UTR of Cep78 mRNA and was used in rescue experiments. The siRNAs for VprBP, DDB1, and Cep97 were described previously 23, 35.
Production of recombinant proteins
Purified DDB1 and DYRK2 were obtained from Cedarlane. Flag‐Cep78, Flag‐EDD, or Flag‐VprBP expressed in HEK293 cells was immunoprecipitated with anti‐Flag beads for 2 h. Beads were washed twice with lysis buffer containing 500 mM NaCl. Proteins were eluted with Flag peptide for 30 min and collected through poly‐prep chromatography columns (Bio‐Rad). A small sample was run on a gel and Coomassie stained to ensure protein purity.
In vitro binding assay
1 μg of purified Cep78 protein was mixed with 1 μg of purified EDD, DYRK2, DDB1, or VprBP at 4°C for 1 h, followed by incubation with an anti‐Cep78 antibody for 1 h and Protein A beads for 2 h. After extensive washing with lysis buffer, bound proteins were analyzed by SDS–PAGE and immunoblotting.
Size exclusion chromatography
2 mg of cell extract was chromatographed (ÄKTA FPLC; GE Healthcare) over a Superose‐6 10/300 GL column (GE Healthcare). 1 ml of fractions was collected, and proteins were precipitated with trichloroacetic acid and analyzed by SDS–PAGE. The column was calibrated with Gel Filtration Standard (Bio‐Rad) containing a mixture of molecular weight markers from 17 to 670 kDa.
Centrosome purification
Centrosomes were isolated as described 47. Briefly, approximately 1 × 109 Jurkat cells treated with 0.2 μM nocodazole and 1 μg/ml cytochalasin D at 37°C for 1 h were collected and lysed. The lysate was treated with 1 mg/ml DNase I before placing onto a 60% sucrose cushion. After centrifugation, the bottom ¼ of the suspension was added to a discontinuous sucrose gradient consisting of 5 ml of 70% sucrose solution at the bottom, 3 ml of 50% sucrose solution in the middle, and 3 ml of 40% sucrose solution at the top. 1 ml of fractions was collected after another round of centrifugation.
In vivo ubiquitination assay
HEK293 cells were typically transfected with various combinations of plasmids including HA‐Ub. 48 h after transfection, cells were left untreated or treated with 10 μM MG132 for 6 h and lysed with lysis buffer. The desired protein was immunoprecipitated without SDS or with 1% SDS to prevent interacting partners from co‐immunoprecipitating with the desired protein. After extensive washing, bound proteins were analyzed by SDS–PAGE and immunoblotting with an anti‐HA antibody.
In vitro ubiquitination assay
HA‐CP110 bound to beads was used as substrate for the assay. Briefly, HA‐CP110 expressed in HEK293 cells was immunoprecipitated with anti‐HA beads, and beads were washed twice with lysis buffer containing 500 mM NaCl. Reactions were performed at 30°C for 1 h in 30 μl of ubiquitination buffer (40 mM Tris–HCl/pH 7.6, 2 mM DTT, 5 mM MgCl2, 0.1 M NaCl, 2 mM ATP) containing various combinations of the following components: 100 μM Ub (Boston Biochem), 20 nM E1/UBE1 (Boston Biochem), 100 nM UbcH5b (Boston Biochem), 50 ng EDD, 50 ng DYRK2 (Abcam), 50 ng DDB1 (Abnova), 50 ng VprBP, and 50 ng Cep78. After the reaction, beads were washed with lysis buffer and bound proteins were analyzed by SDS–PAGE and immunoblotting.
In vitro kinase assay
HA‐CP110 bound to beads was used as substrate for the assay. Reactions were performed at 30°C for 1 h in 30 μl of kinase buffer (25 mM Tris–HCl/pH 7.5, 2 mM DTT, 10 mM MgCl2, 5 mM β‐glycerophosphate, 0.1 mM Na3VO4) containing 50 ng DYRK2 or 50 ng each of EDD, DYRK2, DDB1, and VprBP. After the reaction, beads were washed with lysis buffer and bound proteins were analyzed by SDS–PAGE and immunoblotting.
Author contributions
WYT designed the experiments. DH, YJE, AD, and WYT conducted the experiments. DH and WYT wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We thank all members of the Tsang laboratory for constructive advice; N. Al‐Bader, M. Hudson, H. Ling, R. Rouget, J. Song, and X. Deng for initial efforts on this project; J. Archambault, C. Autexier, E. Cohen, B. Dynlacht, A. Rao, D. Saunders, S. Saxena, J. Singer, and C. Watts for providing plasmids; E. Cohen for critical reading of the manuscript; and Denis Faubert for assistance with mass spectrometric identification of Cep78‐interacting proteins. W.Y.T. was a Canadian Institutes of Health Research New Investigator and a Fonds de recherche Santé Junior 1 Research Scholar. This work was supported by the Canadian Institutes of Health Research (MOP‐115033 to WYT) and the Natural Sciences and Engineering Research Council of Canada.
EMBO Reports (2017) 18: 632–644
References
- 1. Bornens M (2012) The centrosome in cells and organisms. Science 335: 422–426 [DOI] [PubMed] [Google Scholar]
- 2. Fu J, Hagan IM, Glover DM (2015) The centrosome and its duplication cycle. Cold Spring Harb Perspect Biol 7: a015800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Goetz SC, Anderson KV (2010) The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet 11: 331–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barbelanne M, Tsang WY (2014) Molecular and cellular basis of autosomal recessive primary microcephaly. Biomed Res Int 2014: 547986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nigg EA, Raff JW (2009) Centrioles, centrosomes, and cilia in health and disease. Cell 139: 663–678 [DOI] [PubMed] [Google Scholar]
- 6. Gonczy P (2015) Centrosomes and cancer: revisiting a long‐standing relationship. Nat Rev Cancer 15: 639–652 [DOI] [PubMed] [Google Scholar]
- 7. Hildebrandt F, Benzing T, Katsanis N (2011) Ciliopathies. N Engl J Med 364: 1533–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nogales‐Cadenas R, Abascal F, Diez‐Perez J, Carazo JM, Pascual‐Montano A (2009) CentrosomeDB: a human centrosomal proteins database. Nucleic Acids Res 37: D175–D180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Andersen JS, Wilkinson CJ, Mayor T, Mortensen P, Nigg EA, Mann M (2003) Proteomic characterization of the human centrosome by protein correlation profiling. Nature 426: 570–574 [DOI] [PubMed] [Google Scholar]
- 10. Firat‐Karalar EN, Sante J, Elliott S, Stearns T (2014) Proteomic analysis of mammalian sperm cells identifies new components of the centrosome. J Cell Sci 127: 4128–4133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fu Q, Xu M, Chen X, Sheng X, Yuan Z, Liu Y, Li H, Sun Z, Yang L, Wang K et al (2017) CEP78 is mutated in a distinct type of Usher syndrome. J Med Genet 54: 190–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Namburi P, Ratnapriya R, Khateb S, Lazar CH, Kinarty Y, Obolensky A, Erdinest I, Marks‐Ohana D, Pras E, Ben‐Yosef T et al (2016) Bi‐allelic truncating mutations in CEP78, encoding centrosomal protein 78, cause cone‐rod degeneration with sensorineural hearing loss. Am J Hum Genet 99: 777–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nikopoulos K, Farinelli P, Giangreco B, Tsika C, Royer‐Bertrand B, Mbefo MK, Bedoni N, Kjellstrom U, El Zaoui I, Di Gioia SA et al (2016) Mutations in CEP78 cause cone‐rod dystrophy and hearing loss associated with primary‐cilia defects. Am J Hum Genet 99: 770–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang M, Duan T, Wang L, Tang J, Luo R, Zhang R, Kang T (2016) Low expression of centrosomal protein 78 (CEP78) is associated with poor prognosis of colorectal cancer patients. Chin J Cancer 35: 62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nesslinger NJ, Sahota RA, Stone B, Johnson K, Chima N, King C, Rasmussen D, Bishop D, Rennie PS, Gleave M et al (2007) Standard treatments induce antigen‐specific immune responses in prostate cancer. Clin Cancer Res 13: 1493–1502 [DOI] [PubMed] [Google Scholar]
- 16. Azimzadeh J, Wong ML, Downhour DM, Sanchez Alvarado A, Marshall WF (2012) Centrosome loss in the evolution of planarians. Science 335: 461–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brunk K, Zhu M, Barenz F, Kratz AS, Haselmann‐Weiss U, Antony C, Hoffmann I (2016) Cep78 is a new centriolar protein involved in Plk4‐induced centriole overduplication. J Cell Sci 129: 2713–2718 [DOI] [PubMed] [Google Scholar]
- 18. Komander D, Rape M (2012) The ubiquitin code. Annu Rev Biochem 81: 203–229 [DOI] [PubMed] [Google Scholar]
- 19. Berndsen CE, Wolberger C (2014) New insights into ubiquitin E3 ligase mechanism. Nat Struct Mol Biol 21: 301–307 [DOI] [PubMed] [Google Scholar]
- 20. Nakagawa T, Mondal K, Swanson PC (2013) VprBP (DCAF1): a promiscuous substrate recognition subunit that incorporates into both RING‐family CRL4 and HECT‐family EDD/UBR5 E3 ubiquitin ligases. BMC Mol Biol 14: 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Barbelanne M, Chiu A, Qian J, Tsang WY (2016) Opposing post‐translational modifications regulate Cep76 function to suppress centriole amplification. Oncogene 35: 5377–5387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tsang WY, Spektor A, Vijayakumar S, Bista BR, Li J, Sanchez I, Duensing S, Dynlacht BD (2009) Cep76, a centrosomal protein that specifically restrains centriole reduplication. Dev Cell 16: 649–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maddika S, Chen J (2009) Protein kinase DYRK2 is a scaffold that facilitates assembly of an E3 ligase. Nat Cell Biol 11: 409–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Becker W, Weber Y, Wetzel K, Eirmbter K, Tejedor FJ, Joost HG (1998) Sequence characteristics, subcellular localization, and substrate specificity of DYRK‐related kinases, a novel family of dual specificity protein kinases. J Biol Chem 273: 25893–25902 [DOI] [PubMed] [Google Scholar]
- 25. Belzile JP, Abrahamyan LG, Gerard FC, Rougeau N, Cohen EA (2010) Formation of mobile chromatin‐associated nuclear foci containing HIV‐1 Vpr and VPRBP is critical for the induction of G2 cell cycle arrest. PLoS Pathog 6: e1001080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chu G, Chang E (1988) Xeroderma pigmentosum group E cells lack a nuclear factor that binds to damaged DNA. Science 242: 564–567 [DOI] [PubMed] [Google Scholar]
- 27. Henderson MJ, Russell AJ, Hird S, Munoz M, Clancy JL, Lehrbach GM, Calanni ST, Jans DA, Sutherland RL, Watts CK (2002) EDD, the human hyperplastic discs protein, has a role in progesterone receptor coactivation and potential involvement in DNA damage response. J Biol Chem 277: 26468–26478 [DOI] [PubMed] [Google Scholar]
- 28. Matsushima N, Tachi N, Kuroki Y, Enkhbayar P, Osaki M, Kamiya M, Kretsinger RH (2005) Structural analysis of leucine‐rich‐repeat variants in proteins associated with human diseases. Cell Mol Life Sci 62: 2771–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Le Rouzic E, Belaidouni N, Estrabaud E, Morel M, Rain JC, Transy C, Margottin‐Goguet F (2007) HIV1 Vpr arrests the cell cycle by recruiting DCAF1/VprBP, a receptor of the Cul4‐DDB1 ubiquitin ligase. Cell Cycle 6: 182–188 [DOI] [PubMed] [Google Scholar]
- 30. Gerard FC, Yang R, Romani B, Poisson A, Belzile JP, Rougeau N, Cohen EA (2014) Defining the interactions and role of DCAF1/VPRBP in the DDB1‐cullin4A E3 ubiquitin ligase complex engaged by HIV‐1 Vpr to induce a G2 cell cycle arrest. PLoS One 9: e89195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jung HY, Wang X, Jun S, Park JI (2013) Dyrk2‐associated EDD‐DDB1‐VprBP E3 ligase inhibits telomerase by TERT degradation. J Biol Chem 288: 7252–7262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kaur M, Khan MM, Kar A, Sharma A, Saxena S (2012) CRL4‐DDB1‐VPRBP ubiquitin ligase mediates the stress triggered proteolysis of Mcm10. Nucleic Acids Res 40: 7332–7346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. D'Angiolella V, Donato V, Vijayakumar S, Saraf A, Florens L, Washburn MP, Dynlacht B, Pagano M (2010) SCF(Cyclin F) controls centrosome homeostasis and mitotic fidelity through CP110 degradation. Nature 466: 138–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li J, Kim S, Kobayashi T, Liang FX, Korzeniewski N, Duensing S, Dynlacht BD (2012) Neurl4, a novel daughter centriole protein, prevents formation of ectopic microtubule organizing centres. EMBO Rep 13: 547–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Spektor A, Tsang WY, Khoo D, Dynlacht BD (2007) Cep97 and CP110 suppress a cilia assembly program. Cell 130: 678–690 [DOI] [PubMed] [Google Scholar]
- 36. Schmidt TI, Kleylein‐Sohn J, Westendorf J, Le Clech M, Lavoie SB, Stierhof YD, Nigg EA (2009) Control of centriole length by CPAP and CP110. Curr Biol 19: 1005–1011 [DOI] [PubMed] [Google Scholar]
- 37. Kohlmaier G, Loncarek J, Meng X, McEwen BF, Mogensen MM, Spektor A, Dynlacht BD, Khodjakov A, Gonczy P (2009) Overly long centrioles and defective cell division upon excess of the SAS‐4‐related protein CPAP. Curr Biol 19: 1012–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tang CJ, Fu RH, Wu KS, Hsu WB, Tang TK (2009) CPAP is a cell‐cycle regulated protein that controls centriole length. Nat Cell Biol 11: 825–831 [DOI] [PubMed] [Google Scholar]
- 39. Tsang WY, Bossard C, Khanna H, Peranen J, Swaroop A, Malhotra V, Dynlacht BD (2008) CP110 suppresses primary cilia formation through its interaction with CEP290, a protein deficient in human ciliary disease. Dev Cell 15: 187–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li W, You L, Cooper J, Schiavon G, Pepe‐Caprio A, Zhou L, Ishii R, Giovannini M, Hanemann CO, Long SB et al (2010) Merlin/NF2 suppresses tumorigenesis by inhibiting the E3 ubiquitin ligase CRL4(DCAF1) in the nucleus. Cell 140: 477–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mund T, Lewis MJ, Maslen S, Pelham HR (2014) Peptide and small molecule inhibitors of HECT‐type ubiquitin ligases. Proc Natl Acad Sci USA 111: 16736–16741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rossi M, Rotblat B, Ansell K, Amelio I, Caraglia M, Misso G, Bernassola F, Cavasotto CN, Knight RA, Ciechanover A et al (2014) High throughput screening for inhibitors of the HECT ubiquitin E3 ligase ITCH identifies antidepressant drugs as regulators of autophagy. Cell Death Dis 5: e1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen Z, Indjeian VB, McManus M, Wang L, Dynlacht BD (2002) CP110, a cell cycle‐dependent CDK substrate, regulates centrosome duplication in human cells. Dev Cell 3: 339–350 [DOI] [PubMed] [Google Scholar]
- 44. Barbelanne M, Song J, Ahmadzai M, Tsang WY (2013) Pathogenic NPHP5 mutations impair protein interaction with Cep290, a prerequisite for ciliogenesis. Hum Mol Genet 22: 2482–2494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Barbelanne M, Hossain D, Chan DP, Peranen J, Tsang WY (2015) Nephrocystin proteins NPHP5 and Cep290 regulate BBSome integrity, ciliary trafficking and cargo delivery. Hum Mol Genet 24: 2185–2200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tsang WY, Wang L, Chen Z, Sanchez I, Dynlacht BD (2007) SCAPER, a novel cyclin A‐interacting protein that regulates cell cycle progression. J Cell Biol 178: 621–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Moudjou M, Bordes N, Paintrand M, Bornens M (1996) gamma‐Tubulin in mammalian cells: the centrosomal and the cytosolic forms. J Cell Sci 109(Pt 4): 875–887 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7