

Abstract
We recently demonstrated that incorporation of 4-amino-4-deoxy-l-arabinose (l-Ara4N) to the lipid A moiety of lipopolysaccharide (LPS) is required for transport of LPS to the outer membrane and viability of the Gram-negative bacterium Burkholderia cenocepacia. ArnT is a membrane protein catalyzing the transfer of l-Ara4N to the LPS molecule at the periplasmic face of the inner membrane, but its topology and mechanism of action are not well characterized. Here, we elucidate the topology of ArnT and identify key amino acids that likely contribute to its enzymatic function. PEGylation assays using a cysteineless version of ArnT support a model of 13 transmembrane helices and a large C-terminal region exposed to the periplasm. The same topological configuration is proposed for the Salmonella enterica serovar Typhimurium ArnT. Four highly conserved periplasmic residues in B. cenocepacia ArnT, tyrosine-43, lysine-69, arginine-254 and glutamic acid-493, were required for activity. Tyrosine-43 and lysine-69 span two highly conserved motifs, 42RYA44 and 66YFEKP70, that are found in ArnT homologues from other species. The same residues in S. enterica ArnT are also needed for function. We propose these aromatic and charged amino acids participate in either undecaprenyl phosphate-l-Ara4N substrate recognition or transfer of l-Ara4N to the LPS.
The Gram-negative opportunistic bacterium Burkholderia cenocepacia causes serious lung infections in patients with cystic fibrosis1,2. B. cenocepacia is difficult to treat because of its extraordinarily high intrinsic resistance to virtually all clinically useful antibiotics including antimicrobial peptides such as the polymyxins3,4,5. Intrinsic resistance of B. cenocepacia depends in part on its lipopolysaccharide (LPS)6,7,8. B. cenocepacia can modify its LPS lipid A moiety by incorporating 4-amino-4-deoxy-l-arabinose (l-Ara4N). The LPS inner core oligosaccharide contains a side branch d-glycero-d-talo-octo-2-ulosonic acid, which is also modified by l-Ara4N9,10. These modifications neutralize negative charges in the LPS molecule, inhibiting binding of cationic molecules11. Remarkably, l-Ara4N modification in B. cenocepacia is essential for bacterial viability12, since it is required for LPS export to the outer membrane13, and for high resistance to polymyxin B (PmB) at concentrations reaching milligram range4,7,8,13,14.
l-Ara4N biosynthesis occurs in the cytoplasm by the enzymes AraABCD. This process converts UDP-glucuronic acid into l-Ara4N linked to undecaprenyl phosphate15. Undecaprenyl phosphate-l-Ara4N is translocated across the inner membrane by the heterodimer ArnE-ArnF flippase16 and finally ArnT transfers l-Ara4N to the lipid A in a reaction occurring at the periplasmic face of the inner membrane17. Hamad et al.13 postulated that B. cenocepacia ArnT also transfers l-Ara4N to the inner core d-glycero-d-talo-octo-2-ulopyranosoic acid, but this was not directly demonstrated. Because the transfer of l-Ara4N to the LPS is essential for B. cenocepacia viability12,13, ∆arnT mutants are only viable if a suppressor mutation occurs in the lptG gene encoding a component of the LPS transport pathway, which allows export of unmodified LPS13.
Lipid A modifications requiring ArnT family proteins have been reported in Escherichia coli, Salmonella enterica serovar Typhimurium and Pseudomonas aeruginosa where LPS is modified with l-Ara4N18,19, and in Francisella novicida and two Bordetella species (B. bronchiseptica and B. pertussis) where LPS is modified with galactosamine and glucosamine, respectively20,21,22. Since LPS modifications contribute to pathogenicity and innate immunity evasion, ArnT can be considered a virulence factor in these bacteria.
ArnT is an integral membrane protein; its topology was partially characterized in S. enterica23, but little is known on functional regions and enzyme mechanism. Elucidating the topology of ArnT is critical to understand its function and developing molecules targeting l-Ara4N modification of LPS, which would abolish intrinsic bacterial resistance to antimicrobial peptides. In this study, we characterize the topology of B. cenocepacia and S. enterica ArnT. Both proteins show very similar topology, suggesting the topological arrangement in other proteins of the ArnT family is likely conserved. We propose ArnT proteins consist of thirteen transmembrane domains with two large periplasmic loops and a C-terminal segment that is exposed to the periplasm. We also identify four highly conserved periplasmic amino acids that are presumably involved in ArnT function.
Results
ArnT is a membrane protein with 13 predicted transmembrane helices
To facilitate immunodetection while exploring the ArnT topology we constructed a recombinant protein carrying a C-terminal FLAG-10xHis tag. ArnTFLAG-10xHis was expressed under the control of a rhamnose-inducible promoter in the ∆arnT-arnBC+lptGD31H suppressor strain (herein ∆arnT)13. This B. cenocepacia strain is viable but highly sensitive to PmB due to lack of l-Ara4N in the LPS13. In the presence of rhamnose, ArnTFLAG-10xHis restored ∆arnT PmB resistance to parental levels (Supplementary Figure S1A online), and was detected by immunoblot as a polypeptide with an apparent mass of 63 kDa, in agreement with its predicted mass of 66.3 kDa (Supplementary Figure S1B online). These experiments demonstrate that the FLAG-10xHis tag does not affect ArnT function and membrane localization.
Earlier work suggested that S. enterica and E. coli ArnT proteins contain 12 predicted transmembrane helices and that the C-terminus is in the cytosolic compartment17,23. However, an in silico model of B. cenocepacia ArnT topology with PolyPhobius predicted 13 transmembrane helices and a large C-terminus exposed to the periplasmic space in both S. enterica and E. coli ArnT proteins (Fig. 1). To provide experimental evidence for the predicted ArnT transmembrane topology we used the substituted cysteine accessibility method with methoxypolyethylene glycol maleimide (PEG-mal) labeling24,25. PEG-mal is a maleimide conjugate that modifies cysteines but cannot cross membranes. Reaction of a cysteine with PEG-mal results in a mass addition of ~5 kDa to the protein, which appears as a band shift by immunoblotting. PEG-mal modification using EDTA-permeabilized whole cells indicates that the cysteine has a periplasmic orientation, while PEG-mal modification under denaturing conditions (presence of SDS) suggests that the cysteine is within a transmembrane helix or faces the cytoplasm. B. cenocepacia ArnTFLAG-10xHis has two cysteine residues at positions 154 and 176, which were replaced by alanine to generate a cysteineless derivative (ArnTCysless). ArnTCysless and the single cysteine replacement derivatives (ArnTC154A and ArnTC176A) restored PmB resistance in ∆arnT at similar levels as the wild type strain (Fig. 2A), demonstrating that the native cysteines are dispensable for ArnT function. Using PEG-mal labeling, we observed that ArnTC154A and ArnTC176A were only PEGylated in detergent (2% SDS), while ArnTCysless was not PEGylated under any condition (Fig. 2B). These results suggest that Cys-154 and Cys-C176 are buried in transmembrane helices, as predicted by the in silico model (Fig. 1). Cell membrane integrity in these experiments was confirmed by co-expressing ArnTC154 and ArnTC176 with HA-SoxY, a cytoplasmic protein that is highly PEGylated24. Immunoblotting using anti-HA antibodies shows that HA-SoxY was only PEGylated under denaturing conditions (Fig. 2B), demonstrating that the bacterial membranes remained intact.
Figure 1. ArnT topological model.

The model was derived using PolyPhobius and displayed with TMRPres2D. The ArnT C-terminus is oriented towards the periplasm and the protein has 13 predicted transmembrane helices, indicated by rectangles. The residues replaced by cysteine are shown in gray circles. Functional residues are indicated by black circle. The endogenous cysteines of ArnT are shown in black squares.
Figure 2. Native cysteines in ArnT are located in transmembrane segments.

(a) Cys-less ArnT protein remains functional. Complementation of the ∆arnT-arnBC+ suppressor strain transformed with pSCrhaB2 encoding ArnTCys-less, ArnTC154A, and ArnTC176A. Cells expressing ArnTFLAG-10xHis and the vector pSCrhaB2 were used as controls. The transformants were spotted on LB supplemented with 0.4% of l-rhamnose (Rham) and 10 μg ml−1 PmB. (b) E. coli DH5α containing a plasmid encoding SoxY was transformed with plasmids expressing ArnTCys-less, ArnTC154A and ArnTC176A. Bacteria were grown to mid-exponential phase and protein expression induced with 0.2% l-arabinose. Cells were harvest and resuspend in 0.3 ml of HEPES/MgCl2 buffer. 0.1 ml of cell suspension was incubated with buffer alone or 1 mM PEG-mal with or without 2% SDS for 1h at room temperature. Reactions were quenched with 45 mM DTT, and proteins were separated by SDS-PAGE and transferred to nitrocellulose membrane. ArnT derivatives were detected with anti-FLAG and SoxY with anti-hemagglutinin serum. (c) For NEM blocking, cells were harvested and resuspended in 0.4 ml of HEPES/MgCl2 buffer, then were incubated with 5 mM NEM for 30 min to block accessible sulfhydryl groups. The pellet was washed twice with 0.5 ml of ice-cold HEPES/MgCl2 buffer and finally was PEGylated as indicated for panel b. (d) Cells were harvested and resuspended in 0.4 ml of HEPES/MgCl2 buffer. 0.1 ml of the cell suspension was incubated with 5 mM NEM for 30 min, and then was treated with 1 mM PEG-mal and 2% SDS as indicated in panel B. The position of the molecular mass marker is indicated to the left, and the position of PEGylated and non-PEGylated protein to the right.
To confirm whether Cys-154 and Cys-176 are indeed buried, PEGylation was performed in the presence or absence of N-ethylmaleimide (NEM). NEM is a small sulfhydryl-reactive molecule that crosses membranes, but cannot react with cysteines embedded within the membrane bilayer26. As a control, we used a phenylalanine-247 cysteine replacement mutant, since this residue is predicted to reside in the periplasmic side. PEGylation of the resulting protein was examined in samples pretreated with or without NEM, and also with or without SDS. This experiment showed that ArnTF247C could be PEGylated in the absence of NEM (Fig. 2C), demonstrating a periplasmic orientation for this residue. In contrast, ArnTC154A and ArnTC176A were PEGylated despite treatment with NEM, demonstrating that the single cysteine remaining in each protein (Cys-176 in ArnTC154A and Cys-154 in ArnTC176A) is inaccessible to NEM modification (Fig. 2D), as would be expected if these residues are buried in the membrane bilayer. These results support the conclusion that the native cysteine residues in ArnT are in a transmembrane helix, as predicted by the in silico topological model.
More experimental evidence for the predicted ArnT topology was obtained by introducing 38 novel cysteine replacements at various positions of ArnTCysless. As before, mutant proteins were analyzed by immunoblotting to confirm that the cysteine substitution did not affect protein stability and membrane location (Supplementary Figure S2 online). Also, the ArnT cysteine replacements restored PmB resistance in ∆arnT to wild type levels. We then analyzed the topology of amino acids predicted to reside in transmembrane segments. Cysteine residues replacing valine-106, leucine-278, valine-332, and threonine-422 resulted in ArnT proteins that could only be PEGylated in SDS irrespective of NEM pretreatment (Fig. 2D), indicating that these amino acids are embedded in the membrane bilayer, as predicted by the in silico model.
Next, we examined cysteine replacements in leucine-35, tyrosine-43, asparagine-62, lysine-69, glycine-87, asparagine-239, phenylalanine-247, arginine-254, glycine-264, arginine-380 and leucine-385. All of the resulting proteins could be PEGylated under non-denaturing conditions in whole cells, indicating that these residues are exposed to the periplasm (Fig. 3A). In contrast, proteins with cysteine replacements in residues located in predicted periplasmic hinges such as histidine-137, asparagine-139, serine-186, lysine-187, serine-321, histidine-322 and lysine-324, only reacted with PEG-mal upon SDS treatment (Fig. 3B), suggesting these residues are most likely embedded in the bilayer. However, ArnTF138C and ArnTS323C were PEGylated under non-denaturing conditions, indicating that these residues are exposed to the periplasm (Fig. 3B). To confirm this assertion we labeled total membrane preparations with and without NEM pretreatment to differentiate between solvent exposed and lipid bilayer buried residues. Membrane preparations containing proteins with cysteine replacements in histidine-137, asparagine-139, serine-186, lysine-187, serine-321, histidine-322, and lysine-324 were PEGylated in the presence of NEM, indicating that these residues are not solvent-exposed (Fig. 3C); while ArnTF138C and ArnTS323C only were PEGylated in the absence of NEM (Fig. 3C), demonstrating a periplasmic orientation for these residues. The intensity of the PEGylated band varied in some cases such as in ArnTL35C or ArnTF138C when examined by PEGylation of whole-cell preparations (Fig. 3A), and ArnTS186C or ArnTK187C when examined directly from membrane preparations (Fig. 3C). Because all the ArnT mutant proteins are equally expressed, we attributed reduced PEGylation to close proximity of the residue to the membrane bilayer border.
Figure 3. PEG-mal labeling of periplasmic and cytoplasmic ArnT amino acids.

(a, b, and d) E. coli DH5α carrying plasmids encoding ArnT cysteine replacements were grown to mid-exponential phase and protein expression induced with 0.2% l-arabinose. Cells were harvested and resuspended in 0.3 ml of HEPES/MgCl2 buffer. 0.1 ml of cell suspension was incubated with buffer alone or 1 mM PEG-mal with or without 2% SDS for 1 h at room temperature. Reactions were quenched with 45 mM DTT, and proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes, followed by immunoblot with anti-FLAG antibodies. (c) Crude membrane fractions were incubated with buffer alone or pre-treated with 5 mM NEM, after that membrane were incubated with 1 mM PEG-mal and 2% SDS for 1 h at room temperature. Reactions were quenched with 45 mM DTT. (e) Crude membrane fractions were isolated by ultracentrifugation and divided in two aliquots. One aliquot was incubated with buffer alone and the other with 1 mM PEG-mal. The reactions were quenched with 45 mM DTT and separated by SDS-PAGE. Immunoblot was probed with anti-FLAG antibody.
Cysteine replacements at phenylalanine-114, arginine-161, tryptophan-209, isoleucine-288, aspartic acid-350, and asparagine-411 resulted in proteins that were only PEGylated under prior denaturing conditions (Fig. 3D), suggesting they are on the cytosolic face of the membrane. These residues were also PEGylated in membrane preparations (Fig. 3E), as the conditions used for membrane disruption favor formation of inverted vesicles allowing exposure of cytoplasmic residues27,28. Collectively, the experimental results support the in silico predicted ArnT topology.
The C-terminus of B. cenocepacia and S. enterica ArnT proteins resides in the periplasm
According to the in silico topological model, B. cenocepacia ArnT contains an extended C-terminal segment located towards the periplasm. To investigate the orientation of the 123-amino acid soluble C-terminal region of B. cenocepacia ArnT, we introduced eight cysteine replacements at aspartic acid-439, arginine-455, tyrosine-468, histidine-483, glutamic acid-493, tryptophan-512, glutamine-530, and arginine-550. The corresponding proteins were PEGylated in whole cells under non-denaturing conditions, demonstrating that these residues are exposed to the periplasm (Fig. 4A). We also constructed a version of ArnT that was C-terminally fused to PhoA (FLAGArnTPhoA), a protein probing for periplasmic location29, which was expressed in the E. coli ∆phoA CC118 strain. Immunoblotting of total membranes with the anti-FLAG antibody revealed a polypeptide band with an apparent mass of 111.6 kDa consistent with the FLAGArnTPhoA predicted mass (Fig. 4B). The CC118 strain expressing the fusion protein produced 400 U of alkaline phosphatase activity (Fig. 4C), which was comparable to that of the positive control (CC118 expressing WzxK367-PhoA)30. Together, these experiments strongly support the conclusion that the C-terminal region of B. cenocepacia ArnT is oriented towards the periplasm.
Figure 4. The ArnT C-terminus resides in the periplasmic space.

(a) Whole cells from E. coli strain DH5α carrying plasmids encoding cysteine-substitution of the C-terminal of ArnT were treated with buffer alone or 1 mM PEG-mal with or without 2% SDS for 1 h at room temperature and quenched with 45 mM DTT. Proteins were separated by SDS-PAGE and transferred to nitrocellulose membrane for blotting with anti-FLAG to detected ArnT. (b) Immunoblot of total membranes from E. coli CC118 cells expressing FLAG-ArnT-PhoA performed with anti-FLAG. L, BLUeye prestained protein ladder. (c) Quantification of PhoA activity in CC118 E. coli expressing FLAG-ArnTPhoA. Cells were grown in LB and induced with 0.2% arabinose. Alkaline phosphatase activity was determined by measuring the rate of PNPP hydrolysis. pAH01 encodes FLAG-Wzx-K367-PhoA and pAH1801 encodes FLAG-Wzx-T242-PhoA, which were used as positive and negative controls, respectively30. The values represent the mean ± SE from three independent experiments.
In a previous report23, the C-terminal region of the S. enterica ArnT was assigned to the cytosol without experimental confirmation and despite that a periplasmic location was predicted in silico. ArnTSe has eight native cysteine residues, two of which (C148 and C149) were predicted to be in the second periplasmic loop and the remaining six cysteines embedded in transmembrane segments23. Therefore, we constructed a double replacement mutant, ArnTC148A-C149A, which was used to further introduce novel cysteines in the C-terminal domain without making a complete Cys-less version of ArnTSe. Novel cysteines replaced glutamic acid-438, aspartic acid-469, arginine-506, and glutamine-544 residues. Mutant proteins and parental ArnTSe were detectable by immunoblotting (Fig. 5A). PEGylation was performed in whole cells with and without detergent. ArnTE438C, ArnTD469C, ArnTR506C and ArnTQ544C were PEGylated in non-denaturing conditions (Fig. 5B). Since all these residues were accessible to PEG-mal we concluded that they are exposed to the periplasmic face and not to the cytosolic space as previously proposed23. As a control, the cytoplasmic HA-SoxY protein was only PEGylated under SDS treatment (Fig. 5B), ruling out loss of cell membrane integrity during whole cell PEGylation. SDS treatment also showed PEGylation of the ArnTSe derivatives under denaturing conditions (Fig. 5B, asterisk), as expected given that there were six remaining native cysteines in the protein. Importantly, the parental ArnTSe was not PEGylated in absence of SDS (Fig. 5B), indicating that cysteines 148 and 149 are not exposed to the second periplasmic loop, as it was reported23.
Figure 5. C-terminus of ArnT from S.

enterica is located in the periplasm. (a) Immunoblot of total membranes isolated from E. coli DH5α containing plasmids expressing ArnTSe cysteine replacements. The proteins were separated by SDS-PAGE and immunoblotting was performed using anti-FLAG. ArnTSe-FLAG-10xHis was used as control. C148A-C149A (CC/AA) was used as a template to generate the cysteine-substitution in the C-terminal of ArnTSe. (b) PEGylated samples of ArnTSe were prepared and analyzed as described in the legend to Fig. 2B. The positions of the molecular mass marker are indicated to the left. Asterisks indicate PEGylated ArnTSe forms. ArnTSe aggregates at the top of the western blots are attributed to the mild denaturing conditions. (c) Immunoblot of total membranes from E. coli CC118 cells expressing FLAG-ArnTSe-PhoA performed with anti-FLAG. (d) Quantification of PhoA activity in CC118 E. coli expressing FLAG-ArnTSe-PhoA. Cells were processed as described in the legend to Fig. 4C using the same positive and negative controls. The values represent the mean ± SE from three independent experiments.
Further, we investigated the location of the ArnTSe C-terminus by constructing a PhoA fusion (FLAGArnTSe-PhoA). Immunoblot of total membrane preparations reacted with anti-FLAG (Fig. 5C) identified a 110.5-kDa-polypeptide band as expected, indicating that ArnTSe is correctly fused to the PhoA protein and that is also in the membrane fraction. Lysates of strain CC118 expressing the fusion protein displayed 350 U of alkaline phosphatase activity (Fig. 5D), which was comparable to the activity of the positive control cell lysates from CC118 expressing WzxK367-PhoA30. Therefore, the ArnTSe C-terminal domain is exposed towards the periplasm, as in ArnTBc.
Identification of functional amino acids in ArnT
We examined the functionality of ArnTBc cysteine replacement mutants by determining their ability to restore PmB resistance when expressed in the B. cenocepacia ∆arnT lptG suppressor mutant (Supplementary Table S1 online). Only proteins with replacements at tyrosine-43, lysine-69, arginine-254, and glutamic acid-493 residues, all of which are exposed to the periplasmic space (Fig. 1), failed to complement PmB resistance. Multiple sequence alignment revealed that tyrosine-43, lysine-69 (first periplasmic loop), arginine-254 (second periplasmic loop), and glutamic acid-493 (C-terminal segment) residues are highly conserved in ArnT proteins from different bacteria. Also, tyrosine-43 and lysine-69 residues are part of two distinct highly conserved motifs: 42RYA44 and 66YFEKP70 (Fig. 1 and Supplementary Figure S3A online), further suggesting that the identified residues could play a functional role.
Because loss of function in ArnTY43C, ArnTK69C, ArnTR254C, and ArnTE493C could also reflect structural modifications imposed by the non-native cysteine residue, we constructed alanine replacements at the same positions. The alanine replacement mutant proteins expressed at wild type levels and they also appeared in the membrane fraction (Supplementary Figure S3B online). To investigate whether the alanine replacement alters the local conformation of the loop, each residue before alanine-43, -69, -254, and -493 in ArnTY43A, ArnTK69A, ArnTR254A, and ArnTE493A proteins was replaced by cysteine. PEGylation assays using the resulting proteins with cysteine/alanine replacements (ArnTR42C-Y43A, ArnTE68C-K69A, ArnTR253C-R254A, and ArnTD492C-E493A) showed labeling of the neighboring cysteines under non-denaturing conditions (Supplementary Figure S3C online). Since the residues next to the alanine replacements are also accessible to PEG-mal, we conclude that it would unlikely that the alanine replacements cause dramatic changes in the local conformation of the protein at these sites.
To investigate the contribution of the mutated residues to ArnT activity we evaluated the PmB sensitivity of ∆arnT expressing the alanine replacement mutant proteins. The wild type strain producing LPS with l-Ara4N was extremely resistant to PmB with a minimal inhibitory concentration (MIC) greater than 1,024 μg ml−1, while the ∆arnT MIC was 0.064 μg ml−1 (16,000-fold difference)13. MICs for ∆arnT expressing ArnTK69R, ArnTR254A and ArnTE493A ranged from 8,000-fold reduction relative to wild type, while ∆arnT expressing ArnTK69A and ArnTY43A had the same MIC as ∆arnT (Table 1), indicating that these replacements affected ArnT activity. Because alanine is a small amino acid typically used in replacements to prevent structural changes in proteins, we surmise that tyrosine-43, lysine-69, arginine-254 and glutamic acid-493 are involved in ArnT function.
Table 1. PmB MIC values of cells expressing ArnT mutant proteins unable to complement the ∆arnT-arnBC + lptG D31H (∆arnT) suppressor mutant.
| Residue | MIC (μg ml−1) | Fold difference | Presence of l-Ara4N in lipid A |
|---|---|---|---|
| ArnT | >1,024 | 1 | Yes |
| ∆arnT-arnBC+ | 0.064 | 16,000 | No |
| ArnTY43A | 0.064 | 16,000 | No |
| ArnTK69A | 0.064 | 16,000 | No |
| ArnTK69R | 0.125 | 8,192 | Yes |
| ArnTK69E | 0.094 | 10,894 | No |
| ArnTR254A | 0.125 | 8,192 | Yes |
| ArnTR254K | 0.064 | 16,000 | Yes |
| ArnTR254E | 0.064 | 16,000 | No |
| ArnTE493A | 0.125 | 8,192 | Yes |
| ArnTE493D | 128 | 8 | Yes |
| ArnTE493K | 0.064 | 16,000 | No |
MICs were determined by E-Test. The presence of l-Ara4N in the lipid A was determined by MS-MALDI-TOF using the negative ion mode, as described in Methods.
Lysine-69, arginine-254, and glutamic acid-493 are charged amino acids. To investigate whether the charge or the nature of the amino acid is important for ArnT function we replaced lysine-69 with arginine and glutamic acid, arginine-254 with lysine and glutamic acid, and glutamic acid-493 with aspartic acid and lysine. All mutant proteins were detectable in whole cells by immunoblotting (Supplementary Figure S3D online). Neither lysine-69 nor arginine-254 replaced with opposite or same-charge amino acids resulted in proteins that could restore PmB resistance at MIC values similar to the wild type strain (Table 1). In contrast, ArnT containing glutamic acid-493 replaced by aspartic acid restored PmB resistance in ∆arnT at 128 μg ml−1 (Table 1), while the opposite charge substitution, ArnTE493K, led to a MIC of 0.064 μg ml−1, identical to that of ∆arnT. Together, these data suggest that tyrosine-43 and lysine-69 are needed for ArnT function, and also that positive and negative charges at position 254 and 493, respectively, are required for ArnT activity.
The effect of the amino acid replacement mutants on ArnT function was also evaluated by examining the lipid A structure using matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry in the negative ion mode. The mass spectrum from the wild-type sample showed the presence of l-Ara4N as indicated by the ion peaks corresponding to tetra-acylated lipid A with one l-Ara4N in the monophosphate (at m/z 1496.8) and diphosphate (at m/z 1577.5) forms and penta-acylated lipid with one and two l-Ara4N (at m/z 1804.6 and m/z 1935.5, respectively) (Fig. 6A), agreeing with the reported B. cenocepacia lipid A structure13,31. As expected, these peaks were absent in the ∆arnT lipid A spectrum (Fig. 6B and Table 1). Mass spectrometry analysis of lipid A from ∆arnT expressing ArnTY43A, ArnTK69A, ArnTK69E, ArnTR254E and ArnTE493K did not reveal any peaks corresponding to glycoforms containing l-Ara4N (Table 1 and Supplementary Figure S4 online). Interestingly, l-Ara4N was present in ∆arnT-arnBC+containing, ArnTK69R, ArnTR254A, ArnTR254K, ArnTE493A, and ArnTE493D (Fig. 6C-G and Table 1). Because MALDI-TOF does not provide quantitative assessments, we interpreted the presence of l-Ara4N as a result of reduced but not abolished ArnT enzyme activity. This interpretation agrees with the hypersensitivity to PmB of the strain expressing these mutant proteins (Table 1), suggesting insufficient amount of lipid A modification to promote wild type levels of resistance to PmB. We therefore conclude that tyrosine-43, lysine-69, arginine-254 and glutamic acid-493 residues are important for ArnT function.
Figure 6. MALDI-TOF spectra of purified lipid A produced by arnT mutants.

∆arnT denotes strain MH55 (∆arnT-arnBC+ lptGS), which carries a lptG suppressor mutation (Table S1). Plasmids encoding ArnT proteins with various replacements are introduced into MH55 by triparental mating. The profiles represented were obtained using the negative ion mode. I-II Tetra-acylated lipid A with one or two phosphates molecules; III-IV, Tetra-acylated lipid A with one or two l-Ara4N molecules. V, Penta-acylated lipid A; VI-VII, Penta-acylated lipid A with one or two l-Ara4N molecules.
To better support these conclusions, we employed the conditional B. cenocepacia mutant Prha::arnT, which only grows with rhamnose (permissive condition)13. Recombinant plasmids containing ArnTBc derivatives were introduced into Prha::arnT and the resulting transformants examined for viability without rhamnose in the medium. Recombinant plasmids expressing ArnTR254A, ArnTR254K and ArnTE493D could rescue Prha::arnT growth without rhamnose (Supplementary Figure S5 online). In contrast, vector control and plasmids containing ArnTK69E, ArnTR254E, ArnTE493K and ArnTE493A, failed to rescue growth under the same condition. Bacteria containing ArnTK69A and ArnTK69R had moderate growth. These results agree with the lipid A structural analysis. Strains bearing ArnTR254A, ArnTR254K and ArnTE493D had l-Ara4N in the LPS, and they could rescue viability in absence of rhamnose. Nevertheless, the MIC value from these strains is very similar to ∆arnT-arnBC+ except for the strains with ArnTE493D, which behaves as the wild-type strain.
Together, these results indicate that ArnT with substitutions preserving the net charge of each residue were slightly functional, while substitutions reversing the charge caused loss of function. Therefore, the net charges in lysine-69 and arginine-254 are important for ArnT activity.
Tyrosine-36, lysine-62 and glutamic acid-478 are also important for S. enterica ArnT
An earlier report identified arginine-246 (corresponding arginine-254 of ArnTBc) as being functionally important for the S. enterica ArnT23. We investigated whether ArnTSe tyrosine-36, lysine-62, and glutamic acid-478 (corresponding to tyrosine-43, lysine-69, and glutamic acid-493 in ArnTBc, respectively) are also required for ArnTSe function. However, the ArnTSe-FLAG-10xHis fusion resulted in a protein that could not be detected by immunoblot. This could be due to structural constrains associated with a proline at the C-terminal end of the protein, which were resolved by replacing proline-547 by alanine without altering ArnT function32. Therefore, we constructed a P547A ArnTSe-FLAG-10xHis protein and the corresponding alanine replacements in tyrosine-36, lysine-62, and glutamic acid-478. The recombinant proteins were tested for their ability to restore PmB resistance in an E. coli ΔarnT mutant. The complementation experiment indicated that these proteins could not confer PmB resistance, suggesting that the mutated residues are also important for ArnT activity in S. enterica (Fig. 7A); the stability of these proteins was not affected as shown by immunoblotting (Fig. 7B). Therefore, tyrosine-36, lysine-62 and glutamic acid-478 play a crucial role in the function of the ArnTSe, demonstrating that the conserved amino acids in ArnTBc are also important in other ArnT family proteins.
Figure 7. S. enterica ArnT shares the same functional residues than B. cenocepacia ArnT.
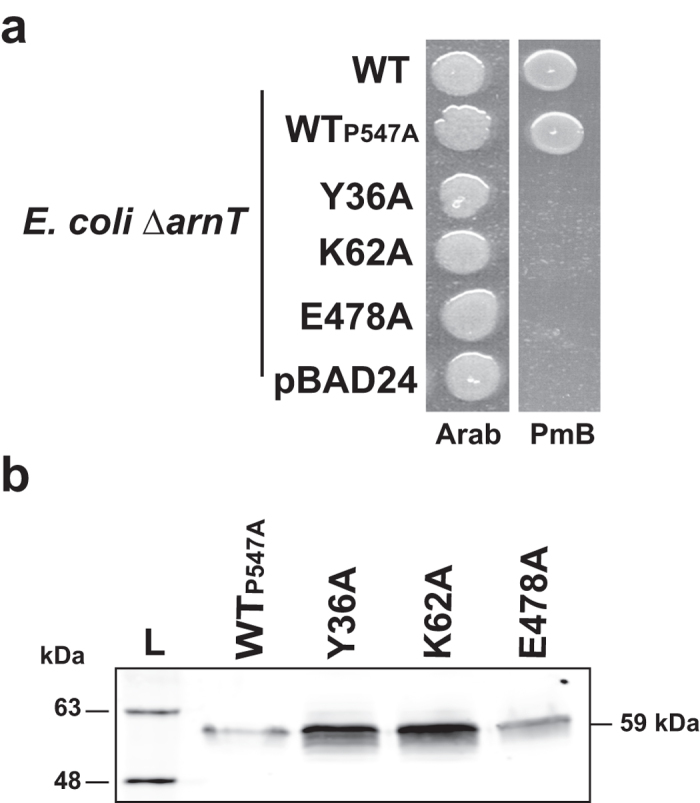
(a) Complementation of E. coli ∆arnT with functional residues replaced with alanine from ArnTSe. WT, ArnTP547A-FLAG-10xHis as a positive control; pBAD24 as a negative control. Strains were spotted in LB plate with 0.2% arabinose (Arab) or 0.2% arabinose plus PmB 2.5 μg/ml (PmB). (b) Total protein from cells expressing ArnTSe derived proteins were separated by SDS-PAGE and detected with antibody anti-FLAG. L, molecular masses of protein standards.
Discussion
Glycosylation of the lipid A phosphate groups with l-Ara4N, catalyzed by ArnT, is a strategy commonly used by bacteria to overcome the action of antimicrobial peptides such as PmB33. However, despite the growing number of ArnT homologs found in different bacteria to date very little is known on the mechanism of ArnT modification. We report in this work the membrane topology of B. cenocepacia and S. enterica ArnT proteins. Application of a topology prediction algorithm in combination with substituted cysteine scanning using PEGylation yielded an experimentally validated model for ArnTBc that suggests 13-transmembrane helices, two large periplasmic hydrophilic loops, and an extended C-terminus also located in the periplasm. Comparison of the B. cenocepacia ArnT model with an earlier model of ArnT from S. enterica reveals striking differences. Unlike ArnTBc, the proposed ArnTSe model suggested four periplasmic loops and placed the C-terminal end towards the cytosol23. Our PEGylation results do not support the earlier ArnTSe model since two adjacent cysteines at positions 148 and 149, predicted to face the periplasm, were inaccessible to PEG-mal indicating they are buried in the membrane. In the previous model23, the ArnTSe C-terminus was also assigned a cytosolic location without any experimental validation and despite bioinformatic data suggesting the opposite. In contrast, our PEGylation data and a positive alkaline phosphatase ArnT-PhoA C-terminal fusion strongly support that the C-terminal segment of ArnTSe is in the periplasm. We therefore conclude that B. cenocepacia and S. enterica ArnT proteins share similar topology, as expected for proteins of the same family performing the same function.
We also identified putative functional residues in ArnTBc that are highly conserved in ArnT proteins from different bacteria. These include tyrosine-43, lysine-69, and arginine-254. Furthermore, a negative charged amino acid is required at position 493 (glutamic acid in ArnTBc). All these putative functional residues reside on the periplasmic side of ArnT, as expected for an enzyme using substrates only available in the periplasm15,17. We propose that these residues may participate either in catalysis or binding of the undecaprenyl-l-Ara4N donor substrate. Tyrosine-43 and lysine-69 residues are within two highly conserved motifs in all ArnT family members: 42RYA44 and 66YFEKP70. The conservation of these motifs within the first periplasmic loop of ArnTBc suggest not only that they are functionally relevant but more importantly, that the members of ArnT family share similar features. Further, replacing tyrosine-43 of the RYA motif by alanine or inverting the polarity of the lysine-69 resulted in a nonfunctional protein. It is possible that the first periplasmic loop containing two highly conserved motifs could define a putative catalytic reaction center conserved in all ArnT proteins. Mutagenesis of the arginine-254 residue residing on the second periplasmic loop resulted in a protein with poor enzymatic activity. Agreeing with this conclusion, an arginine from ArnTSe corresponding to ArnTBc arginine-254 was also reported to be important for the activity of this enzyme23. Our data also suggest that the ArnTSe second periplasmic loop, between amino acids 234 and 256, is required for function, as well as a C-terminal glutamic acid-493. Replacing this residue by aspartic acid, to maintain the same charge, resulted in a protein retaining the ability to incorporate l-Ara4N to lipid A and supports the notion that the ArnT C-terminus is also involved in the overall activity of this enzyme. However, our assignment of the functional residues has to remain tentative until validation by further biochemical investigations using purified protein, substrates, and acceptor molecules.
The mechanism of transfer of l-Ara4N to LPS remains to be elucidated. Superficially, ArnT proteins are analogous to O-antigen ligases and oligosaccharyl transferases. These proteins perform the enzymatic reaction on the periplasmic side of the membrane, and involve a metal ion-independent inverting glycosyltransfer reaction using undecaprenyl-bisphosphate-linked saccharide precursors34. In contrast, bacteriophage-encoded glucosyltransferase proteins, which are responsible for the periplasmic transfer of glucose to the LPS O antigen moiety35,36, utilize a lipid phosphate-activated glucosyl residue (Und-P-glucose)37. ArnT proteins also utilize undecaprenyl-phosphate-linked saccharides, and the reaction occurs at the periplasmic site of the membrane17. Further, there is an inversion of the stereochemistry in the reaction since the substrate is Und-P-α-l-Ara4N17, but l-Ara4N is β-linked to the phosphate groups of the lipid A at 4’ and 1 positions31, suggesting ArnT proteins are also inverting glycosyltransferases. Given these similarities with other glycan ligases, it is reasonable to find that key functional residues are located on the periplasmic exposed side of ArnT.
In this work, we demonstrate that ArnT proteins from B. cenocepacia and S. enterica share topology and functional residues. In B. cenocepacia, however, l-Ara4N is added to lipid A and the d-glycero-d-talo-octo-2-ulosonic acid inner core sugar, modifications that confer extremely high resistance to PmB13. In contrast, S. enterica and E. coli ArnT transfer l-Ara4N only to lipid A and comparatively these strains present much lower levels of PmB resistance31. The topological similarities and functionally conserved residues between ArnTBc and ArnTSe suggest that both proteins have more in common that one would expect if they had different specificities. However, we cannot exclude the possibility that another region in ArnTBc is responsible for specific recognition of the Burkholderia LPS, making this protein different than that of E. coli, Salmonella, and other bacteria. Alternatively, a distinct l-Ara4N transferase could be required in B. cenocepacia to add l-Ara4N to the inner core d-glycero-d-talo-octo-2-ulosonic acid after ArnT transfers l-Ara4N to the lipid A. Further research is needed to investigate these possibilities and examine the sequential process of l-Ara4N incorporation to lipid A and inner core in detail.
In summary, this study provides experimental evidence that ArnT proteins from B. cenocepacia and S. enterica, which play key roles in remodeling LPS, share topological and functional similarities that also extend to other members of the ArnT family and provide insights for further investigation of ArnT proteins and the elucidation of their catalytic mechanism.
Methods
Bacterial strains and growth conditions
Bacterial strains and plasmids are listed in Supplementary Table S1 online. Growth medium was supplemented as needed with the following chemicals (final concentrations): 100 μg ampicillin ml−1, 30 μg chloramphenicol ml−1, 50 μg trimethoprim ml−1, 25 μg gentamicin ml−1, 10 μg PmB ml−1, 0.2% l-arabinose (w/v), and 0.2% l-rhamnose (w/v). Bacteria were grown aerobically at 37 °C in Luria-Bertani (LB) medium (Difco). For recombinant protein expression, bacteria were grown overnight in 5 mL of LB, diluted to A600 of 0.1, and incubated at 37 °C for 2 h until cultures reached A600 of 0.5, at which point l-arabinose was added and cells incubated for 3 h at 37 °C.
Construction of plasmids expressing parental and mutant ArnT proteins
A plasmid encoding ArnTFLAG-10xHis was constructed by PCR amplification of a 1.7-kb fragment using B. cenocepacia K56-2 genomic DNA and primers 6269/6270 (Supplementary Table S2 online). The fragment was digested with EcoRI and SalI and ligated into the same sites of pXR134. This plasmid is a modified pBAD24 containing an oligonucleotide sequence encoding a FLAG plus 10x-His tag, which was placed adjacent to the multiple cloning site for construction of in-frame C-terminally FLAG-10xHis-tagged proteins. The resulting plasmid, designated pFT1, expressed ArnTFLAG-10xHis under the control of the arabinose-inducible promoter. pFT1 was used as template to replace the native cysteines of ArnT (residues 154 and 176) with alanine by sequential site-direct mutagenesis using primers 6238/6239, and 6240/6241 (Supplementary Table S2 online). The plasmid encoding the cysteineless ArnTFLAG-10xHis was named pFT7. For sulfhydryl labeling experiments, novel cysteines were introduced in pFT7 by PCR using primers containing the desired mutations and Pfu AD polymerase (Stratagene). DpnI was added to PCR reactions for overnight digestion of parental plasmid DNA at 37 °C. The resulting DNA was introduced into E. coli DH5α by CaCl2 transformation. Resulting plasmids were confirmed by sequencing with primers 252, 258 and 6385 (Supplementary Table S2 online).
Alkaline phosphatase (PhoA) fusions and alkaline phosphatase assay
pFT4 and pFT15, expressing FLAGArnTBc-PhoA and FLAGArnTSe-PhoA, respectively, were constructed by PCR amplification of a 1.7-kb fragment from pFT1 and pMH494 with 6421/6716 and 7178/7182 primer pairs, respectively (Supplementary Table S2 online). The amplicons were digested with XbaI and SalI and ligated into these sites in pAH1830. The ligation mixture was introduced by transformation into E. coli CC118 cells, and transformants plated on LB-ampicillin containing 5-bromo-4-chloro-3-indolylphosphate (60 μg ml−1). Transformants were screened for blue colony phenotype denoting PhoA activity. To quantify alkaline phosphatase activity, overnight cultures were diluted 1:100 and grown for 4 h at 37 °C in LB ampicillin/0.2% arabinose, harvested, lysed, and assayed as described30.
Cloning and expression of arnT from S. enterica
The S. enterica arnT gene was isolated by amplification of a 1.6-kb fragment using pMH494 as template and primers 6475/6476. The fragment was digested with EcoRI and SalI and ligated into the same sites in pFT1 to replace arnT from B. cenocepacia by S. enterica arnT. This construct, named pFT54, expressed ArnTSe with a C-terminal FLAG-10xHis tag, and was used to introduce novel cysteines by site-directed mutagenesis. For complementation of E. coli ∆arnT, alanine replacements were introduced in pFT143 (encoding S. enterica ArnTP547A).
Topological model for ArnT
Two prediction methods were used to analyze the in silico topology of ArnT: PolyPhobious38 and TMHMM 2.039. The ArnT models were displayed using TMRPred2D40. Multiple-sequence alignments of ArnT homologues were performed with ClustalW ( http://www.ebi.ac.uk/clustalw/).
Complementation of ArnT function in B. cenocepacia
Plasmids expressing the ArnT replacement mutant proteins were subcloned into the NdeI-HindIII site of pSCrhaB2, and the recombinant constructs introduced in the ∆arnT-arnBC+suppressor strain by triparental mating41,42. The ability of each construct to restore PmB resistance indicated ArnT function. Strains were grown overnight with 0.4% (w/v) rhamnose (to induce protein expression), diluted to 10−1, 10−2, 10−3, 10−4 and 10−5 and 4 μl of each dilution plated onto LB plates supplemented with 0.4% rhamnose and 10 μg ml−1 PmB.
Membrane preparations and immunoblotting
Bacterial pellets were resuspended in 50 mM Tris-HCl, pH 8.0, with protease inhibitors and lysed at 10, 000 PSI using a cell disruptor (Constant Systems, Kennesaw, GA). Cell debris was pelleted by centrifugation at 15,000 × g for 15 min at 4 °C, and the clear supernatant was centrifuged at 40,000 × g for 30 min at 4 °C. The pellet, containing total membranes, was suspended 50 mM Tris-HCl, pH 8.0. Protein concentration was determined by the Bradford assay (Bio-Rad). Proteins were separated by SDS-PAGE on a 12% or 18% gel (if the experiment included detection of HA-SoxY) and transferred to a nitrocellulose membrane. Immunoblots were probed with anti-FLAG and anti-HA (Sigma) mouse monoclonal antibodies. Secondary anti- mouse Alexa fluor-680 IgG antibody (Invitrogen) was used to detect fluorescence using an Odyssey infrared imaging system (LI-COR Biosciences).
Sulfhydryl labeling
Cells were cultured in 20 ml of LB broth at 37 °C from an initial OD600 of 0.2 to an OD600 of 0.5 at which time arabinose was added to a final concentration of 0.2% (w/v) for induction of ArnTFLAG-10xHis protein expression, and growth continued for 3 h. Bacterial cells were harvested by centrifugation at 16,000 × g and washed with HEPES/MgCl2 buffer (50 mM HEPES, pH 6.8; 5 mM MgCl2). Cell pellets were resuspended in 0.3 ml of HEPES/MgCl2 buffer and divided in three portion: 0.1 ml of cell suspension was incubated with 1 mM methoxypolyethylene glycol maleimide (PEG-Mal) at room temperature for 1 h with 50 mM EDTA for B. cenocepacia ArnT and 5 mM EDTA for S. enterica ArnT; 0.1 ml was treated with 2% SDS prior to labeling, and 0.1 ml was incubated with buffer as control. The sulfhydryl labeling reaction was stopped with 45 mM of dithiothreitol for 10 min and mixed with 3X dye buffer (50 mM Tris-HCl [pH 6.8], 2% SDS, 10% glycerol, and 0.1% bromophenol blue), and incubated for 30 min at 45 °C. Mild denaturation conditions were used to optimize ArnT detection, as these conditions work best for membrane proteins43. The crude membrane fraction was resuspended in 50 μl of HEPES/ MgCl2 buffer supplemented with protease inhibitors. 25 μl were labeled with 1 mM PEG-Mal at room temperature for 1 h. The reaction was quenched by adding 45 mM of dithiothreitol for 10 min, and centrifuged at 16,000 × g for 1 min. Pellets were suspended in 50 μl of HEPES/MgCl2 buffer with protease inhibitor. Control samples were incubated with buffer alone. Protein was extracted and processed as described above.
Thiol-specific chemical blocking with N-ethylmaleimide (NEM)
Cell suspensions in HEPES/MgCl2 buffer (0.1 ml) were incubated with 5mM N-ethylmaleimide (NEM) for 30 min at room temperature, and centrifuged at 16,000 × g for 1 min. Pellets were washed twice with 1 ml of iced HEPES/MgCl2 buffer. The samples were suspended in 200 μl of HEPES/MgCl2 buffer, and incubated with 2% SDS and 1 mM PEG-Mal for 1 h at room temperature. Samples were quenched with 45 mM of dithiothreitol for 10 min. Protein extraction was performed as described above.
MIC determination
The MIC was determined on solid media using PmB E-test strips. Overnight cultures, were diluted to an A600 of 0.004 in sterile PBS and used to streak LB agar plates using a sterile cotton swab. E-test strips containing PmB were added and plates where incubated for 24 h at 37 °C. MIC was defined as the lowest concentration of PmB to show growth inhibition.
Lipid A isolation and mass spectrometry
L-Ara4N modifications of lipid A were identified by mass spectrometry (MS) using matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF). Cells were grown overnight in LB medium (100 ml) supplemented with 50-μg ml−1 trimethoprim and 0.4% rhamnose. Cultures were centrifuged (10,000 × g), washed twice with phosphate buffer (10 mM Na2HPO4, 1.7 mM KH2PO4) and freeze-dried. Lipid A was extracted from lyophilized cells and desalted, as previously described44. For MS analysis, dihydroxybenzoic acid (DHB) (Sigma Chemical Col, St. Louis, MO) was used as a matrix and prepared to saturation in acetonitrile: 0.1% triflouroacetic acid (1:2 v/v). Two-microliters of sample was loaded on the target, dried, and covered by 1 μl of DHB. The target was inserted in a Bruker Autoflex MALDI-TOF spectrometer. Data acquisition and analysis were performed using the Flex Analysis software.
Additional Information
How to cite this article: Tavares-Carreón, F. et al. Burkholderia cenocepacia and Salmonella enterica ArnT proteins that transfer 4-amino-4-deoxy-L-arabinose to lipopolysaccharide share membrane topology and functional amino acids. Sci. Rep. 5, 10773; doi: 10.1038/srep10773 (2015).
Supplementary Material
Acknowledgments
This work was supported grants from the Canadian Institutes of Health Research, the UK Cystic Fibrosis Trust (to M.A.V.), and COST action BM1003 “Microbial cell surface determinants of virulence as targets for new therapeutics in cystic fibrosis”. We thank Dr. M. Hamad for technical assistance, Dr. C. Klug for the gift of E. coli BL21(DE3)ΔarnT, and Dr. J. Bengoechea for assistance with MALDI-TOF spectrometry.
Footnotes
The authors declare no competing financial interests.
Author Contributions F.T-C. and M.A.V. designed all the research, analyzed the data and wrote the manuscript. F.T-C. performed the experiments. K.B.P. performed and analyzed mass spectrometry data. All authors reviewed and approved the manuscript.
References
- Govan J. R. & Deretic V. Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol Rev 60, 539–574 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang K. et al. Exploring the metabolic network of the epidemic pathogen Burkholderia cenocepacia J2315 via genome-scale reconstruction. BMC Syst Biol 5, 83 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitão J. H. et al. Variation of the antimicrobial susceptibility profiles of Burkholderia cepacia complex clonal isolates obtained from chronically infected cystic fibrosis patients: a five-year survey in the major Portuguese treatment center. Eur J Clin Microbiol Infect Dis 27, 1101–1111 (2008). [DOI] [PubMed] [Google Scholar]
- Loutet S. A. & Valvano M. A. Extreme antimicrobial peptide and polymyxin B resistance in the genus Burkholderia. Front Cell Infect Microbiol 1, 6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loutet S. A., Di Lorenzo F., Clarke C., Molinaro A. & Valvano M. A. Transcriptional responses of Burkholderia cenocepacia to polymyxin B in isogenic strains with diverse polymyxin B resistance phenotypes. BMC Genomics 12, 472 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega X., Silipo A., Saldias M. S., Bates C. C., Molinaro A. & Valvano M. A. Biosynthesis and structure of the Burkholderia cenocepacia K56-2 lipopolysaccharide core oligosaccharide: truncation of the core oligosaccharide leads to increased binding and sensitivity to polymyxin B. J Biol Chem 284, 21738–21751 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loutet S. A., Mussen L. E., Flannagan R. S. & Valvano M. A. A two-tier model of polymyxin B resistance in Burkholderia cenocepacia. Environ Microbiol Rep 3, 278–285 (2011). [DOI] [PubMed] [Google Scholar]
- Loutet S. A., Flannagan R. S., Kooi C., Sokol P. A. & Valvano M. A. A complete lipopolysaccharide inner core oligosaccharide is required for resistance of Burkholderia cenocepacia to antimicrobial peptides and bacterial survival in vivo. J Bacteriol 188, 2073–2080 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silipo A. et al. The complete structure and pro-inflammatory activity of the lipooligosaccharide of the highly epidemic and virulent gram-negative bacterium Burkholderia cenocepacia ET-12 (strain J2315). Chemistry 13, 3501–3511 (2007). [DOI] [PubMed] [Google Scholar]
- Ortega X., Silipo A., Saldías M. S., Bates C. C., Molinaro A. & Valvano M. A. Biosynthesis and structure of the Burkholderia cenocepacia K56-2 lipopolysaccharide core oligosaccharide: truncation of the core oligosaccharide leads to increased binding and sensitivity to polymyxin B. J Biol Chem 284, 21738–21751 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinion-Dubiel A. D. & Goldberg J. B. Lipopolysaccharide of Burkholderia cepacia complex. J Endotoxin Res 9, 201–213 (2003). [DOI] [PubMed] [Google Scholar]
- Ortega X. P. et al. A putative gene cluster for aminoarabinose biosynthesis is essential for Burkholderia cenocepacia viability. J Bacteriol 189, 3639–3644 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamad M. A., Di Lorenzo F., Molinaro A. & Valvano M. A. Aminoarabinose is essential for lipopolysaccharide export and intrinsic antimicrobial peptide resistance in Burkholderia cenocepacia. Mol Microbiol 85, 962–974 (2012). [DOI] [PubMed] [Google Scholar]
- El-Halfawy O. M. & Valvano M. A. Chemical communication of antibiotic resistance by a highly resistant subpopulation of bacterial cells. PLoS One 8, e68874 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raetz C. R., Reynolds C. M., Trent M. S. & Bishop R. E. Lipid A modification systems in gram-negative bacteria. Annu Rev Biochem 76, 295–329 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan A., Guan Z. & Raetz C. R. An undecaprenyl phosphate-aminoarabinose flippase required for polymyxin resistance in Escherichia coli. J Biol Chem 282, 36077–36089 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trent M. S., Ribeiro A. A., Lin S., Cotter R. J. & Raetz C. R. An inner membrane enzyme in Salmonella and Escherichia coli that transfers 4-amino-4-deoxy-L-arabinose to lipid A: induction on polymyxin-resistant mutants and role of a novel lipid-linked donor. J Biol Chem 276, 43122–43131 (2001). [DOI] [PubMed] [Google Scholar]
- Miller S. I., Ernst R. K. & Bader M. W. LPS, TLR4 and infectious disease diversity. Nat Rev Microbiol 3, 36–46 (2005). [DOI] [PubMed] [Google Scholar]
- Raetz C. R. & Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem 71, 635–700 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Ribeiro A. A., Guan Z. & Raetz C. R. Identification of undecaprenyl phosphate-beta-D-galactosamine in Francisella novicida and its function in lipid A modification. Biochemistry 48, 1162–1172 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marr N., Tirsoaga A., Blanot D., Fernandez R. & Caroff M. Glucosamine found as a substituent of both phosphate groups in Bordetella lipid A backbones: role of a BvgAS-activated ArnT ortholog. J Bacteriol 190, 4281–4290 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolin O. et al. Enzymatic modification of lipid A by ArnT protects Bordetella bronchiseptica against cationic peptides and is required for transmission. Infect Immun 82, 491–499 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impellitteri N. A., Merten J. A., Bretscher L. E. & Klug C. S. Identification of a functionally important loop in Salmonella typhimurium ArnT. Biochemistry 49, 29–35 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch S., Fritsch M. J., Buchanan G. & Palmer T. Escherichia coli TatA and TatB proteins have N-out, C-in topology in intact cells. J Biol Chem 287, 14420–14431 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasie I., Steiner-Mordoch S. & Schuldiner S. Topology determination of untagged membrane proteins. Methods Mol Biol 1033, 121–130 (2013). [DOI] [PubMed] [Google Scholar]
- Bogdanov M., Zhang W., Xie J. & Dowhan W. Transmembrane protein topology mapping by the substituted cysteine accessibility method (SCAM(TM)): application to lipid-specific membrane protein topogenesis. Methods 36, 148–171 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Ordaz A. et al. The sodium/proline transporter PutP of Helicobacter pylori. PLoS One 8, e83576 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuper C. & Jung K. CadC-mediated activation of the cadBA promoter in Escherichia coli. J Mol Microbiol Biotechnol 10, 26–39 (2005). [DOI] [PubMed] [Google Scholar]
- Haardt M. & Bremer E. Use of phoA and lacZ fusions to study the membrane topology of ProW, a component of the osmoregulated ProU transport system of Escherichia coli. J Bacteriol 178, 5370–5381 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marolda C. L. et al. Membrane topology and identification of critical amino acid residues in the Wzx O-antigen translocase from Escherichia coli O157:H4. J Bacteriol 192, 6160–6171 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silipo A. et al. Complete structural characterization of the lipid A fraction of a clinical strain of B. cepacia genomovar I lipopolysaccharide. Glycobiology 15, 561–570 (2005). [DOI] [PubMed] [Google Scholar]
- Bretscher L. E., Morrell M. T., Funk A. L. & Klug C. S. Purification and characterization of the L-Ara4N transferase protein ArnT from Salmonella typhimurium. Protein Expr Purif 46, 33–39 (2006). [DOI] [PubMed] [Google Scholar]
- Needham B. D. & Trent M. S. Fortifying the barrier: the impact of lipid A remodelling on bacterial pathogenesis. Nat Rev Microbiol 11, 467–481 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan X., Loyola D. E., Marolda C. L., Perez-Donoso J. M. & Valvano M. A. The WaaL O-antigen lipopolysaccharide ligase has features in common with metal ion-independent inverting glycosyltransferases. Glycobiology 22, 288–299 (2012). [DOI] [PubMed] [Google Scholar]
- Korres H. & Verma N. K. Topological analysis of glucosyltransferase GtrV of Shigella flexneri by a dual reporter system and identification of a unique reentrant loop. J Biol Chem 279, 22469–22476 (2004). [DOI] [PubMed] [Google Scholar]
- Korres H. & Verma N. K. Identification of essential loops and residues of glucosyltransferase V (GtrV) of Shigella flexneri. Mol Membr Biol 23, 407–419 (2006). [DOI] [PubMed] [Google Scholar]
- Nikaido K. & Nikaido H. Glucosylation of lipopolysaccharide in Salmonella: biosynthesis nof O antigen factor n12 2 . II. Structure of the lipid intermediate. J Biol Chem 246, 3912–3919 (1971). [PubMed] [Google Scholar]
- Käll L., Krogh A. & Sonnhammer E. L. An HMM posterior decoder for sequence feature prediction that includes homology information. Bioinformatics 21 (Suppl 1), i251–257 (2005). [DOI] [PubMed] [Google Scholar]
- Sonnhammer E. L., von Heijne G. & Krogh A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc Int Conf Intell Syst Mol Biol 6, 175–182 (1998). [PubMed] [Google Scholar]
- Spyropoulos I. C., Liakopoulos T. D., Bagos P. G. & Hamodrakas S. J. TMRPres2D: high quality visual representation of transmembrane protein models. Bioinformatics 20, 3258–3260 (2004). [DOI] [PubMed] [Google Scholar]
- Craig F. F., Coote J. G., Parton R., Freer J. H. & Gilmour N. J. A plasmid which can be transferred between Escherichia coli and Pasteurella haemolytica by electroporation and conjugation. J Gen Microbiol 135, 2885–2890 (1989). [DOI] [PubMed] [Google Scholar]
- Figurski D. H. & Helinski D. R. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc Natl Acad Sci U S A 76, 1648–1652 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan X. & Valvano M. A. In vitro O-antigen ligase assay. Methods Mol Biol 1022, 185–197 (2013). [DOI] [PubMed] [Google Scholar]
- El Hamidi A., Tirsoaga A., Novikov A., Hussein A. & Caroff M. Microextraction of bacterial lipid A: easy and rapid method for mass spectrometric characterization. J Lipid Res 46, 1773–1778 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.