

Abstract
Artificial antigen-presenting cells (aAPCs) have recently gained a lot of attention. They efficiently activate T cells and serve as powerful replacements for dendritic cells in cancer immunotherapy. Focusing on a specific class of polymer-based aAPCs, so-called synthetic dendritic cells (sDCs), we have investigated the importance of multivalent binding on T-cell activation. Using antibody-functionalized sDCs, we have tested the influence of polymer length and antibody density. Increasing the multivalent character of the antibody-functionalized polymer lowered the effective concentration required for T-cell activation. This was evidenced for both early and late stages of activation. The most important effect observed was the significantly prolonged activation of the stimulated T cells, indicating that multivalent sDCs sustain T-cell signaling. Our results highlight the importance of multivalency for the design of aAPCs and will ultimately allow for better mimics of natural dendritic cells that can be used as vaccines in cancer treatment.
Introduction
One important goal of cancer immunotherapy is the replacement of costly dendritic cell (DC) vaccines with synthetic variants, thereby overcoming the need of generating a customized vaccine for every individual patient.1 These synthetic variants, called artificial antigen-presenting cells (aAPCs), are designed to prime T cells against cancer-specific antigens. These aAPCs can be produced in a straightforward manner from synthetic building blocks, opening up the possibility for standardized “off-the-shelf” protocols2 and circumventing elaborate and expensive personalized medicine. Different aAPC designs have been synthesized over the last years with scaffolds varying from polymer beads,3,4 carbon nanotubes,5 liposomes,6 and many others.7 In general, the design of aAPCs is inspired by the natural DC and its interaction with the T cell. DC binding to T cells involves three main signals that are all required to fully activate the T cell: antigen-loaded major histocompatibility complexes (pMHC) of the DC bind to specific T-cell receptors (TCR; signal 1). At the same time, co-stimulatory molecules on the DC surface interact with their T-cell binding partners (signal 2). In addition to these receptor interactions, soluble factors (cytokines) are also involved in T-cell activation (signal 3). In the first stage of activation, signal 1 interactions trigger the TCR, which is prearranged in nanoclusters in the T-cell membrane (up to 20 TCRs per cluster).8−12 In the next step, triggered TCR molecules re-arrange together with signal 2 interactions, to form larger signaling microclusters containing around 20–300 TCRs.9,11,13−16 These contact areas between both cells are stabilized by a number of different adhesion molecules. After the initial stimulation, triggered microclusters move toward the so-called supramolecular adhesion complex where receptors and adhesion molecules are rearranged to form a “bulls eye” pattern of micrometer size.17 This process clearly involves the dynamic multivalent binding of many (different) binding partners.
Multivalent interactions generally form at the interface between two objects that carry multiple, complementary functionalities.18,19 The simultaneous interaction between these functionalities enhances the binding strength (avidity), sometimes by several orders of magnitude compared to the affinity of the monovalent interaction.20 This enhancement mainly originates from an increase in the effective concentration of identical binding partners. Once the first ligand is bound, the “search volume” is reduced, and the following binding events occur with a higher probability.21 We have recently introduced a new multivalent aAPC design for activating T cells: synthetic dendritic cells (sDCs).22,23 In this design, anti-CD3 antibodies (αCD3), which are known to trigger the TCR (signal 1), were bound to a semiflexible and linear polyisocyanopeptide scaffold with a length of ∼200 nm. Using these novel sDCs, T-cell activation occurred at much lower doses of antibody compared to those of freely soluble αCD3. This is a direct consequence of the unique physical properties of the polymer scaffold. Its high aspect ratio allows the efficient simultaneous binding of all αCD3 effector molecules to the T cell. At the same time, its nanometer size combined with its semiflexibility promotes the dynamic spatial rearrangement of polymer-bound effector molecules, mimicking the fluidity of the natural cell membrane and supporting receptor mobility. Coupling of additional anti-CD28 antibodies (αCD28; signal 2) to the sDC shaped the immunoresponse toward the induction of helper and killer T cells, without activating the regulatory T-cell population.23 Remarkably, this effect was only seen when both signals were bound to one and the same polyisocyanopeptide backbone, indicating that activation requires both signals to bind in close spatial proximity. These results already provided a first indication that multivalent binding of these sDCs does not only increase the binding strength of the interaction. The polymer concentrates the effector molecules in a locally confined area. It may therefore affect T-cell signaling pathways and directly influence the strength and specificity of the T-cell response.
Focusing on sDCs functionalized with only anti-CD3 antibodies (αCD3–sDC), we have now designed a series of experiments to investigate the effect and importance of multivalent binding of our sDCs. We have synthesized a library of αCD3–sDCs with different polymer lengths and αCD3 densities and investigated the influence of these parameters on T-cell activation. Incubating T cells with the αCD3–sDCs, we show that an increase in polymer length and/or effector molecule density boosts both early (Ca2+-signaling) and late (interferon γ (IFNγ) release) stages of T-cell activation and provides evidence that this effect goes beyond a simple avidity increase. A positive effect on T-cell signaling is further demonstrated after removal of the αCD3–sDCs. T-cell activation is sustained for extended periods of time (days), as confirmed by prolonged Ca2+-signaling, expression of the early activation marker CD69, and the release of IFNγ.
Results and Discussion
Synthesis of αCD3–sDCs
All polymer−αCD3 conjugates (i.e., the αCD3–sDCs) were synthesized according to previously published methods22,23 (Figure 1). The sDC scaffold is based on a water-soluble polyisocyanopeptide co-polymer bearing nonfunctional methoxy and functional azide groups. The corresponding methoxy and azide isocyanide monomers were polymerized using a nickel catalyst to obtain azide-functionalized polyisocyanopeptide polymers. The azide groups were subsequently utilized in a strain-promoted azide-alkyne cycloaddition (SPAAC) reaction with bicyclononyne-functionalized streptavidin (BCN–SAv).24 The SAv molecules allow for the binding of biotinylated αCD3 antibodies to yield the αCD3–sDCs (Figure 1a). In all experiments, the ratio between SAv and αCD3 was tuned to be 1:1.
Figure 1.

Schematic overview of the sDC library. (a) Experimental design for sDC synthesis. (b) Schematic overview of the sDCs used in this study (P1–P3) showing the corresponding polymer lengths and αCD3 densities.
Influence of αCD3–sDC Length on T-Cell Activation
Polyisocyanopeptides of different lengths were synthesized using different catalyst-to-monomer ratios during the polymerization reaction. Two polymers of different average lengths (P1′ = 175 nm and P2′ = 350 nm; azide/methoxy = 1:100) were synthesized using this strategy (Table S1, Figures S1 and S2). The density of SAv per polymer chain was determined using atomic force microscopy (AFM imaging; Table S2, Figures S1 and S3). The P1–SAv and P2–SAv conjugates possess an average density of 1 SAv molecule per 110 and 120 nm, respectively. For the synthetic protocol used, we have shown earlier that one αCD3 antibody is bound per SAv molecule.22,23 It can therefore be assumed that these values also represent the densities of αCD3 molecules on the αCD3–sDC conjugates, P1 and P2 (Figure 1b). This means that P1 carries 1–2 αCD3 molecules per polymer, whereas the total number of αCD3 molecules on P2 is ∼3.
P1 and P2 were compared with free αCD3 in a single-cell Ca2+-signaling experiment (Figure 2a). Ca2+-release from the endoplasmic reticulum is one of the earliest activation events when triggering T cells at the TCR level. The subsequent complex interplay between Ca2+-release from the endoplasmic reticulum and the calcium influx across the plasma membrane through Ca2+-release-activated Ca2+ (CRAC) channels leads to oscillations of the cytoplasmic calcium.25 These calcium oscillations, which have a direct influence on T-cell gene expression, were monitored using peripheral blood lymphocytes (PBLs) loaded with the Ca2+-sensitive fluorescent dye, Fura-2 (Table S3, Figure S4, and Movies M1 and M2).26 Determination of the number of Ca2+-signaling cells during the first hour of treatment with P1, P2, or free αCD3 (5 and 25 ng/mL) revealed that the two αCD3–sDCs as well as the free αCD3 caused a marked increase in the number of Ca2+-signaling cells (above the 30% background level of Ca2+-signaling cells observed in the absence of any stimulant27). At 0.5 ng/mL, this effect was only seen for P2 and not for P1 or free αCD3.
Figure 2.

T-cell activation using sDCs of different length and αCD3 density. (a) Fraction of activated PBLs as determined from single-cell Ca2+-signaling measurements performed during the first hour of stimulation with P1, P2, and free αCD3. (b) Relative increase in the concentration of IFNγ secreted by PBLs treated with P1, P2, and free αCD3 for 16 h. Untreated PBLs were used as a reference. (c) Fraction of activated PBLs as determined from single-cell Ca2+-signaling measurements performed during the first hour of stimulation with P3a–c, and free αCD3. (d) Relative increase in the concentration of IFNγ secreted by PBLs treated with P3a–c, and free αCD3 for 16 h. Untreated PBLs were used as a reference. For (a)–(c), the data represent the mean ± SEM of three independent experiments performed with T cells from different donors. For (d), the data represent the mean ± SEM of two independent experiments performed with T cells from different donors. The number of cells analyzed in the single-cell Ca2+-signaling experiments (a, c) is summarized in Table S4.
To probe the effect of the two αCD3–sDCs on a late and more robust event in T-cell activation, we stimulated PBLs for 16 h with P1, P2, and αCD3 and measured the release of IFNγ. Both αCD3–sDCs stimulated the production of IFNγ over a range of concentrations from 0.05 to 100 ng/mL (Figure 2b). At all concentrations tested, the effect of P2 was most pronounced. Considering that P1 and P2 possess approximately the same αCD3 density, these results suggest that the polymer length is a crucial design parameter. It increases the total number of αCD3 molecules per αCD3–sDC, thereby causing a stronger T-cell-stimulating effect.
Influence of αCD3 Density on T-Cell Activation
The previous experiment has shown that a density of one αCD3 antibody in 110–120 nm combined with a polymer length of maximally 350 nm (P2) leads to a small but clearly detectable increase in T-cell activation. We therefore decided to increase both the polymer length and the αCD3 density to investigate the multivalency effect over a larger dynamic range. On increasing the number of azide functional groups (azide/methoxy = 1:70), polymer P3′ with an average length of 400–450 nm was synthesized (Table S1, Figures S1 and S2). This polymer was then functionalized with a different number of SAv molecules per polymer, using different ratios of BCN–SAv/azide in the coupling reaction (0.5, 1, and 5 equiv of BCN–SAv). Using AFM imaging, the average SAv density on these polymer conjugates was determined to be 1 SAv molecule in every ∼130 nm (P3a–SAv, 0.5 equiv), ∼90 nm (P3b–SAv, 1 equiv), and ∼40 nm (P3c–SAv, 5 equiv) (Figure 1b; Table S2, Figures S1 and S3). Again, it was assumed that these values correspond to the density of αCD3 antibodies so that the αCD3–sDCs carry an average of 3–4 (P3a–SAv), 5 (P3b–SAv), or 10–11 (P3c–SAv) antibodies per polymer.
The single-cell Ca2+-signaling assay shows a marked increase already at the lowest tested concentration of P3c (0.005 ng/mL; Figure 2c). In sharp contrast, P3a and P3b displayed the same dose-dependency as free αCD3. Moreover, for these αCD3–sDCs, a more clear difference was observed in the IFNγ release assay. All three αCD3–sDCs were shown to be more effective than free αCD3 (Figure 2d). Most importantly, a positive correlation was observed between αCD3 density and IFNγ release over the full range of tested concentrations (0.05–50 ng/mL). At the highest αCD3 concentration of 50 ng/mL, P3a–c induced a 2.4-, 3.5-, and 6.1-fold increase of secreted IFNγ, respectively, compared to that by free αCD3. Clearly, in addition to the polymer length, the αCD3 density is an important determinant for T-cell activation by αCD3–sDCs.
Quantification of the Multivalent Enhancement Factor
The above results show that both polymer length and αCD3 density are crucial design parameters for our sDC design. Together, these parameters determine the number of interactions that can form between the polymer and the T cell. To quantify the enhancement of the multivalent binding strength, dose–response curves were established for both free αCD3 and the best performing αCD3–sDC (P3c). The dose–response curves provide the basis for determining the EC50 values and allow for calculating an enhancement factor for the multivalent interaction. To construct these dose–response curves, single-cell Ca2+-signaling experiments were performed over an extended range of αCD3 concentrations (0.001–100 ng/mL). Even though T-cell activation was more difficult to quantify in the Ca2+-signaling experiment, we have chosen this readout parameter as it corresponds to a very early activation event. We believe that an early readout parameter is more relevant for quantifying the multivalent binding strength than downstream parameters when signal amplification may have taken place.
When PBLs were treated with P3c, the smallest concentration that caused a detectable effect on the number of Ca2+-signaling cells was a factor ∼200–300-fold lower than for free αCD3 (Figure 3), which is in line with previously reported results.22 For free αCD3, an EC50 value of 16 ng/mL was found, whereas an EC50 value of 0.24 ng/mL was obtained for the multivalent P3c. This yields an enhancement factor of ∼67 for the multivalent system (Figure 3).28 This remarkable enhancement clearly indicates that multivalency is one of the key parameters responsible for the increased potency of αCD3–sDCs. It is worth mentioning that the slope of the dose–response curve usually contains additional information, for example, about positive or negative cooperativity. The sDC polymers are heterogeneous, however, when considering both their length and the αCD3 density. It is therefore highly likely that the more gradual response to increasing the concentration of P3c is a direct result of this heterogeneity. We further note that it would be interesting to increase the αCD3 density on the polymer to determine whether this multivalent system is characterized by an optimum loading. Despite several attempts, we have not been able to attach more αCD3 antibodies to the polymer, possibly due to steric hindrance.
Figure 3.
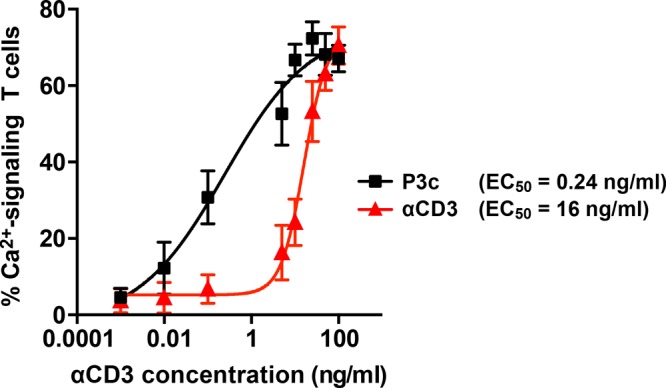
Dose–response curves for PBLs treated with P3c and free αCD3 as determined from single-cell Ca2+-signaling experiments. EC50 values were determined using a four-parameter fit. The multivalent enhancement factor is calculated by dividing the EC50 of free αCD3 by the EC50 of P3c. The data represent the mean ± SEM of three independent experiments performed with PBLs from different donors. The number of cells analyzed is summarized in Table S4.
Assuming that the enhancement factor purely characterizes the avidity increase of the multivalent interaction, the question remains whether enhanced binding of the αCD3–sDC is the only parameter that determines T-cell activation or whether T-cell signaling is also affected. The polymer linkage between several αCD3 antibodies efficiently directs these polymer-attached αCD3 antibodies to the same spatially confined area even if the overall αCD3 concentration is very low. It further enhances the probability of rebinding after dissociation (koff = 0.39 s–1)29 for individual polymer-attached αCD3 antibodies, thereby triggering a higher number of TCRs in close proximity. Considering the sequence of events occurring during T-cell activation, this may directly lower the threshold concentration for T-cell activation. A first indication for this can be obtained when re-considering the potency of the αCD3–sDCs P3a–c (Figure 2c,d) that all bind in a multivalent manner. During all experiments, the data were normalized to the αCD3 concentration so that the polymer concentration (i.e., the concentration of T-cell-stimulating entities; αCD3–sDCs) varies between the different samples. When normalizing the data with respect to the αCD3–sDC concentration, it becomes evident that a 1000-fold lower concentration of P3c is sufficient to obtain the same effect as with P3a (Figure S5). This value is considerably larger than the multivalent enhancement factor determined above and may suggest that the co-localization of a certain number of αCD3 antibodies in a small area on the cell surface is a key factor for T-cell activation. Interestingly, a total number of ∼10 αCD3 antibodies (P3c carries ∼10 antibodies) distributed over an area of several tens of nanometers matches with the predicted size of TCR nanoclusters that are preformed on the T-cell surface.8−10 It may therefore be speculated that αCD3–sDCs form a highly specific and dynamic multivalent interaction with these nanoclusters and that the T-cell fate is already determined at this very early stage of forming the initial contact with the T cell.11
Long-Term Effect of αCD3–sDC Binding on T-Cell Signaling
To investigate the effect of the αCD3–sDCs on T-cell signaling in more detail, we designed a new series of experiments to obtain information about sustained T-cell activation. Instead of measuring T-cell activation in the continuous presence of the αCD3–sDCs or free antibodies, excess stimulant was washed off after 1 h of treatment, and T-cell activation was analyzed at several time points after removal of the stimulant. This allowed for determining the long-term effect of the initial stimulation, as no free stimulant was available for binding to the T cells after the medium was replaced. To quantify the effect, we have again performed single-cell Ca2+-signaling experiments and determined the secretion of IFNγ. In addition, the expression of the surface activation marker, CD69, was measured, which is another indicator of early T-cell activation.
For the single-cell Ca2+-signaling measurements, PBLs were treated with free αCD3 or P3c (12.5 ng/mL) for 1 h. The stimulant was then removed, fresh medium was added, and the cells were incubated without a stimulant for another 15 or 23 h. Fura-2 was added in the last 20 min of this extended incubation period, and the fraction of Ca2+-signaling cells was determined during the following hour (starting at 15 or 23 h after the initial addition of the stimulant; see Figure S6 for a detailed timeline). A reference sample was imaged for 1 h in the presence of the stimulant (0 h; Figure S6). In agreement with the results presented above (Figures 2c and 3), both free αCD3 and P3c (12.5 ng/mL) readily increased the number of Ca2+-signaling T cells during the first hour of stimulation (Figure 4a). In samples treated with free αCD3, the fraction of Ca2+-signaling PBLs was significantly reduced at both poststimulation time points (Figure S7). In sharp contrast, the vast majority of P3c-treated PBLs remained active after removal of the stimulant. Even at 24 h after the initial stimulation with P3c, the fraction of Ca2+-signaling T cells was still ∼75%, indicating that the multivalent sDC causes a sustained stimulation of the intracellular pathways involved in Ca2+-responses. Negative controls involving an isotope control (mIgG2a antibody), polymers with SAv but no αCD3, and nontreated cells did not show high amounts of activated T cells before and after removal of the stimulant (Figure S7).
Figure 4.

Analysis of sustained T-cell activation. (a) Long-term Ca2+-signaling after treating PBLs with 12.5 ng/mL P3c or free αCD3 for 1 h. The first measurement (0 h) was performed directly on the microscope during 1 h of incubation with the stimulant. The other time points represent the total time of the experiment (i.e., 1 h incubation with the stimulant + incubation time after removal of the stimulant). For all experiments, the data represent the mean ± SEM of three independent experiments performed with PBLs from different donors. The number of cells analyzed is summarized in Table S5. (b) Fraction of T cells expressing CD69 after treatment with P3c or free αCD3 for 8 h. For P3c, concentrations of 1 and 50 ng/mL are shown. For αCD3, a concentration of 50 ng/mL is shown. The first measurement was performed directly before the stimulant was removed (8 h). The following time points represent the total time of the experiment. For all experiments, the data represent the mean ± SEM of three independent experiments performed with PBLs from different donors. (c) Concentration of secreted IFNγ after treating PBLs with 5 ng/mL P3c or αCD3 for 16 h. The first measurement was performed directly before the stimulant was removed (16 h). The following time points represent the total time of the experiment. Untreated PBLs were used as a reference. For all experiments, the data represent the mean ± SEM of three independent experiments performed with PBLs from different donors.
To support the results from the single-cell Ca2+-signaling experiments, FACS analysis was performed to determine the expression of the surface marker CD69. T cells were treated with P3c or free αCD3 (1, 5, and 50 ng/mL) for 8 h before placing the PBLs into a fresh medium. At the time of removal of the stimulant, higher numbers of CD69-expressing T cells were observed when the PBLs were treated with 50 ng/mL P3c than when they were treated with the same concentration of free αCD3 (Figures 4b and S8). On the basis of the multivalent enhancement factor of 67 (Figure 3), one would expect a similar level of activation for PBLs treated with 1 ng/mL of P3c or with 50 ng/mL of free αCD3. When comparing the initial time point at 8 h, the P3c-treated sample indeed contains approximately the same number of CD69-expressing cells. This amount increases during the next 40 h for the P3c-treated sample, whereas it decreases for the sample treated with free αCD3.
To further confirm that T cells were showing sustained and robust activation for an extended period of time, IFNγ release assays were performed. PBLs were treated with 5 ng/mL P3c or free αCD3 for 16 h, after which the cells were washed, and a new medium was added. The supernatant was tested for IFNγ directly before the removal of the stimulant (16 h), and a high concentration of IFNγ was determined for both treatment conditions as expected. At all subsequent time points (24, 48, 72, 96, and 120 h after the initial stimulation), a clear difference was seen between T cells treated with P3c or free αCD3 (Figure 4c). PBLs treated with free αCD3 do not seem to produce new IFNγ, and a decrease in the IFNγ level is seen over time. In contrast, PBLs stimulated with P3c produced new IFNγ. Until the 96 h time point, an approximately constant level of IFNγ was maintained before a decrease of the IFNγ concentration was observed. Taken together, these results show that sDCs stimulate T cells over much longer periods of time compared with free αCD3, and this effect is observed for both early and late T-cell activation markers.
Overall, these results show that αCD3–sDCs efficiently activate T cells, combining the benefits of multivalent binding with a spatially confined interaction with several TCRs. The equilibrium binding constant of the αCD3 antibody used is KD = 680 μM29 so that an overall increase in the multivalent binding strength is seen (enhancement factor of 67). At the same time, the antibody possesses a relatively fast kinetic off-rate, koff = 0.39 s–1 so that dissociation and immediate rebinding of individual polymer-attached αCD3 molecules is highly likely to occur while the sDC is bound to the cell surface. It has been proposed that such dynamic interactions are important factors contributing to T-cell activation.30 In addition to these thermodynamic and kinetic effects, the sDC polymers keep up to 10 individual effector molecules in close spatial proximity. It appears likely that one sDC binds within one nanocluster, and this proximity effect may have direct consequences for its signaling activity.12 Altogether, these factors ensure an efficient and long-lasting T-cell activation. Even though this appears to be the most likely mechanism, other factors such as altered TCR endocytosis and exocytosis16,31,32 or changes in membrane properties may contribute to the observed activation. Investigating these additional factors will be the subject of further study. Clearly, our sDC design is a powerful new tool that allows for studying the initial steps of T-cell activation in more detail, including the role of TCR nanoclusters. As another important next step, the αCD3 antibody will be replaced with pMHC complexes. Natural pMHC complexes bind to the TCR with a slightly higher KD but similar off-rates when compared to those of the αCD3 antibody used.33 The established sDC design principles can therefore be applied directly for the development of clinically relevant sDCs.
Conclusions
In summary, we have shown that polymer length and effector molecule density are key design parameters for the development of sDCs. These parameters have a direct effect on the valency of the sDC and, consequently, on the effective concentration required for T-cell activation. In addition to this enhancement of the binding strength, robust and sustained T-cell activation was observed that goes beyond a pure avidity effect. Our results show that the multivalent scaffold also affects T-cell signaling pathways. Co-localization of several effector molecules in the same nano- or micro-cluster leads to a long-lasting activation that cannot be achieved with nonpolymer-bound antibodies. Future studies, using natural pMHCs as effector molecules, will be directed at elucidating the mechanistic origin of this sustained T-cell response.
Experimental Section
Polymer Synthesis (P1′–P3′)
Water-soluble polyisocyanopeptides were synthesized as described using our previously published method.22 For the synthesis of P1′ and P2′, the functional azide monomer (N3) and the nonfunctional methoxy monomer (OMe) were polymerized in a 1:100 ratio. For P3′, a 1:70 N3/OMe ratio was used to increase the number of possible coupling sites. The polymer length was controlled by the amount of nickel catalyst added in the polymerization reaction. A 1:200 ratio of the catalyst/monomer was used for the synthesis of P1′. For P2′ and P3′, the catalyst/monomer ratio was 1:10 000. The molecular weight of the polymers was determined from viscosity measurements (Table S1) as described previously.22
Synthesis of Polymer–SAv Conjugates
SAv (Thermo Fisher Scientific) was functionalized with BCN–POE3-NH-C(O)CH2CH2CH2C(O)OSu (BCN–NHS; Synaffix) to couple it to the azide-groups on the polymer in a SPAAC reaction.24 The reaction was performed in borate buffer (10 mM, pH 8.5) using a 5–6-fold excess of BCN–NHS. After incubation for 1.5 hours at room temperature, the mixture was purified by ultrafiltration (10 kDa cutoff) and gel filtration (PD-10 desalting column; GE Healthcare) to remove nonreacted BCN–NHS. During purification, the buffer was exchanged to phosphate-buffered saline (PBS, pH 7.4). Mass spectrometry analysis (ESI-TOF) revealed that 1–4 BCN moieties were coupled per SAv molecule (BCN–SAv).
BCN–SAv was subsequently reacted with azide-bearing polyisocyanopeptide polymers. A 1:1 molar ratio of N3/BCN–SAv was used for the synthesis of P1–SAv and P2–SAv. N3/BCN–SAv ratios of 1:0.5, 1:1, and 1:5 were used for the synthesis of P3a–SAv, P3b–SAv, and P3c–SAv, respectively. All reactions were performed in PBS. The reaction mixtures were first incubated at room temperature for 1 h and subsequently stirred at 4 °C for 2.5 days. The resulting polymer–SAv conjugates were purified by ultrafiltration (100 kDa cutoff).
Characterization of Polymer–SAv Conjugates
The average polymer length and SAv density were determined with AFM using the method described earlier22 (Figure S1). The resulting histograms, displaying the distribution of the polymer length and the SAv distance of at least 41 individual polymers, are shown in Figures S2 and S3. The results are summarized in Table S2.
Synthesis of sDCs (P1, P2, and P3a–c)
The polymer–SAv conjugates were incubated with biotinylated, monoclonal mouse anti-human CD3 antibodies (αCD3; clone OKT3; purified in house from the hybridoma cell line) to obtain the αCD3-functionalized polymers, P1, P2, and P3a–c. The polymer–SAv conjugates were incubated with the biotinylated antibodies in a 4:1 αCD3/SAv molar ratio. As previously described,22 this 4:1 αCD3/SAv molar loading ratio yields a 1:1 binding ratio of αCD3 and SAv on the polymer backbone (αCD3–sDC).
Cell Preparation and Cell Culture
PBLs were obtained from buffy-coats of healthy individuals in accordance with institutional guidelines.22 Briefly, peripheral blood mononuclear cells were obtained from Ficoll density centrifugation. Monocytes were removed using the plastic flask adherence method. The nonadherent PBLs were then maintained at 37 °C and 5% CO2 in complete medium: RPMI-1640 (Lonza), containing 10% (v/v) fetal bovine serum (Gibco), 1% (w/v) glutamine (Lonza), and 1× antibiotic–antimycotic (Gibco).
Single-Cell Ca2+-Signaling
The fraction of PBLs responding to the αCD3–sDC treatment was determined in a single-cell Ca2+-signaling assay using the ratiometric Ca2+-indicator Fura-2. PBLs (105 cells) were loaded with 3 μM Fura-2 AM (Thermo Fisher) in complete medium for 1 h at 37 °C and 5% CO2. Fura-2-loaded PBLs were washed twice with HEPES buffered saline (20 mM HEPES pH 7.4, 115 mM NaCl, 5.4 mM KCl, 1 mM CaCl2, 0.8 mM MgCl2, 13.8 mM glucose) and allowed to adhere to poly-d-l-lysine-coated (0.05 mg/mL; Sigma-Aldrich) glass-bottom dishes (Nunc) at room temperature. Single-cell Ca2+-measurements were performed as described before.34 Dishes were placed on the stage of an inverted microscope (Axiovert 200 M; Zeiss) equipped with a temperature-controlled CO2 stage incubator (37 °C and 5% CO2) and a 63×, 1.25 NA objective (Plan NeoFluar). Fura-2 was excited at 340 and 380 nm alternatingly, using a monochromator (Polychrome IV; TILL Photonics). The emitted light was directed through a 415 DCLP dichroic mirror (Omega Optical) and a 510WB40 emission filter (Omega) onto a CoolSNAP HQ monochrome CCD camera (Roper Scientific). The camera exposure time was 30 ms, and the time between two ratio images was 2–4 s. All hardware was controlled with Metafluor 6.0 software (Universal Imaging). The images obtained were analyzed using Image-Pro Plus software (Media Cybernetics). Regions of interest (ROIs), corresponding to individual PBLs, were selected together with a cell-free ROI for background correction. For each ROI, the average pixel intensity was calculated for each excitation wavelength. After subtraction of the corresponding background value, the 340/380 nm fluorescence emission ratio was calculated as a measure of the cytosolic Ca2+-concentration. Increases in cytosolic Ca2+-concentration were identified with GraphPad Prism 5 (GraphPad Software), using as criterion that the increase in the 340 nm signal is mirrored by a decrease in the 380 nm signal.
To investigate the early effects of different αCD3–sDCs on Ca2+-signaling, a series of experiments was performed. These experiments include measurements to compare the effect of different αCD3–sDCs (experiments 1 and 2 in Table S3) and the dose dependences of P3c and freely soluble αCD3 (experiment 3 in Table S3). For these experiments, the dishes with the Fura-2-loaded T cells were placed onto the microscope, and imaging was started. The different stimulants were added 5 min after the onset of imaging. After 1 h of imaging, 1 μg/mL of ionomycin was added to test for cell viability. The number of cells analyzed for every experimental condition tested is summarized in Table S4. Typical raw data of PBLs stimulated with αCD3–sDC or free αCD3 are shown in Figure S4 and Movies M1 and M2. The movies (60× speed) show Fura-2-loaded PBLs treated with free αCD3 (Movie M1) or with the sDC-αCD3 P3c (Movie M2). Each movie shows 5 min of baseline imaging and 1 h of imaging in the presence of the stimulant. The last 5 min of each movie show the cells in the presence of ionomycin. The movies were captured with a frame rate of 0.25 s–1. The data show that PBLs display different patterns of Ca2+-signaling. In this study, we restricted ourselves to determining the fraction of Ca2+-signaling cells.
To establish the long-term effect of P3c on single-cell Ca2+-signaling, PBLs were treated with the respective stimulant for 1 h, followed by incubation without the stimulant for an extended period of time. The following conditions were tested: untreated, P3c, free αCD3, P3–mIgG2a (isotype control; 40 nm mIgG2a spacing), and P3c–SAv (lacking αCD3). In more detail, PBLs (105 cells) were incubated with the respective stimulant (12.5 ng/mL) for 1 h at 37 °C and 5% CO2. At the end of this stimulation period, the cells were washed with RPMI-1640 medium lacking the serum and the antibiotics to remove the stimulant. The cells were resuspended in a fresh complete medium and maintained at 37 °C and 5% CO2 for another 15 or 23 h (Figure S6). During the last 20 min of this poststimulation period, the cells were loaded with Fura-2. The Fura-2-loaded cells were washed twice with PBS + 1% BSA (PBA) and imaged under the microscope for 1 h. Cell viability was assessed at the end of the measurement as described above (treatment with ionomycin for 5 min). To be able to compare these poststimulation results with the previously obtained data, a “0 h” time point was taken at which the PBLs (105 cells) were first loaded with Fura-2 for 20 min and imaged exactly as described above.
Enzyme-Linked Immunosorbent Assay (ELISA; IFNγ Secretion)
To investigate the effect of the different αCD3–sDCs on IFNγ secretion, PBLs (105 cells/well) were seeded in 96-well plates and treated with P1, P2, and P3a–c or free antibodies (αCD3) at different concentrations (0.05, 0.5, 5, 50, and 100 ng/mL) for 16 h at 37 °C and 5% CO2. To determine the effect of the polymer length, the treatment variables were P1, P2, and free αCD3 along with an untreated control. The experiment was performed with PBLs from three different donors, each measured in duplicate. P3a–c were used to investigate the effect of the αCD3 density (again using free αCD3 and untreated cells as a control). This experiment was performed with PBLs from two different donors (measured in duplicate). The concentration of the secreted IFNγ was determined using a sandwich ELISA as described in our earlier publications.22,23 Briefly, 96-well plates (Nunc Immunomodules) were coated with a mouse anti-human IFNγ antibody (Thermo Fisher). After incubation at 4 °C overnight, the plates were washed and blocked with PBS/Tween (0.05%) and PBS + 1% BSA (PBA), respectively. IFNγ standards (Thermo Fisher) and supernatants (from treated and untreated cells) were added into the respective wells and incubated for 1 h at room temperature. After washing 3× with PBA, the concentration of IFNγ was detected using a biotinylated mouse anti-human IFNγ antibody (Thermo Fisher) and a SAv–horseradish peroxidase (HRP) conjugate (Life Technologies). HRP activity was detected using tetramethyl benzidine (TMB; Sigma-Aldrich). Absorption at 450 nm was measured using an iMark Microplate Reader (Bio-Rad).
To measure the long-term effect of the αCD3–sDC treatment, PBLs (105 cells/well) were seeded in 96-well plates and treated with the respective stimulant at 5 ng/mL. The cells were incubated with the stimulant for 16 h at 37 °C and 5% CO2. After this incubation time, the cells were washed with RPMI-1640 medium lacking the serum and the antibiotics. They were then resuspended in a fresh complete medium with serum to observe the activation state of the cells after the stimulant was removed. The IFNγ concentration was determined directly before the medium was removed (16 h) and at the following time points after the initial stimulation: 24, 48, 72, 96, and 120 h.
Flow Cytometry (CD69 Expression)
To measure the long-term effect of the αCD3–sDC treatment on CD69 expression, PBLs (105 cells/well) were seeded in 96-well plates and treated with the respective stimulant (1, 5, and 50 ng/mL). The cells were incubated with the stimulant for 8 h at 37 °C and 5% CO2. After this incubation time, a sample was taken for analysis. The remaining cells were washed with RPMI-1640 medium lacking the serum and the antibiotics. They were then resuspended in a fresh complete medium with serum to observe the activation state of the cells after the stimulant was removed. The cell suspensions were collected at the respective time points (24, 48, 72, 96, and 120 h after the initial stimulation) and used for flow cytometric analysis (CyAn ADP; Beckman Coulter) following the same methodology as described in our previous publications.22,23
Briefly, PBLs (treated or untreated) were washed twice with PBA to remove unbound sDCs or antibodies. PBLs were stained with antibodies specific for the CD4/8+ T-cell subpopulations (APC-labeled mouse anti-human CD4/8 mAb; T-cell marker; BD Pharmingen) and with PE-labeled mouse anti-human CD69 (eBioSciences). After 1 h of incubation, the cells were again washed twice with PBA before performing flow cytometric analysis. The data obtained were analyzed using FlowJo ver. 9.2 Software (TreeStar Inc.). The gating strategy is shown in Figure S9.
Acknowledgments
This work was supported by grants from the Dutch Cancer Society (grants KUN2006-3699 and KUN2009-4402), the Dutch government to the Netherlands Institute for Regenerative Medicine (NIRM, grant FES0908), the European Research Council (ERC; grant ERC-2010-AdG269019, C.G.F.), the Netherlands Organization for Scientific Research (NWO; Spinoza award 2006, C.G.F; VICI grant 700.56.444, A.E.R.; VIDI grant 700.58.430, K.G.B.), and NanoNext (grants 7A.06 and 3D.12).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.6b00436.
Author Present Address
School of Pharmacy & Health Professions, Creighton University, 2500 California Plaza, Omaha, Nebraska 68178, United States (S.M.).
Author Present Address
Department of Biomedical Engineering and Institute for Complex Molecular Systems, Laboratory of Immunoengineering, Eindhoven University of Technology, De Zaale, 5612 AP Eindhoven, The Netherlands (J.T.).
Author Present Address
Mechano(bio)chemistry, Max Planck Institute of Colloids and Interfaces, Science Park Potsdam-Golm, 14424 Potsdam, Germany (K.G.B.).
Author Contributions
∥ R.H. and S.M. contributed equally. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Banchereau J.; Steinman R. M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Eggermont L. J.; Paulis L. E.; Tel J.; Figdor C. G. Towards efficient cancer immunotherapy: advances in developing artificial antigen-presenting cells. Trends Biotechnol. 2014, 32, 456–465. 10.1016/j.tibtech.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy N. M.; Mossoba M. E.; Steinberg S. M.; Fellowes V.; Yan X. Y.; Hakim F. T.; Babb R. R.; Avila D.; Gea-Banacloche J.; Sportes C.; Levine B. L.; June C. H.; Khuu H. M.; Carpenter A. E.; Krumlauf M. C.; Dwyer A. J.; Gress R. E.; Fowler D. H.; Bishop M. R. Phase I trial of adoptive cell transfer with mixed-profile type-I/type-II allogeneic T cells for metastatic breast cancer. Clin. Cancer Res. 2011, 17, 6878–6887. 10.1158/1078-0432.CCR-11-1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlie N. K.; LeFever A. V.; Siebenlist R. E.; Levine B. L.; June C. H.; Lum L. G. T cells coactivated with immobilized anti-CD3 and anti-CD28 as potential immunotherapy for cancer. J. Immunother. 1999, 22, 336–345. 10.1097/00002371-199907000-00007. [DOI] [PubMed] [Google Scholar]
- Fadel T. R.; Sharp F. A.; Vudattu N.; Ragheb R.; Garyu J.; Kim D.; Hong E.; Li N.; Haller G. L.; Pfefferle L. D.; Justesen S.; Herold K. C.; Fahmy T. M. A carbon nanotube-polymer composite for T-cell therapy. Nat. Nanotechnol. 2014, 9, 639–647. 10.1038/nnano.2014.154. [DOI] [PubMed] [Google Scholar]
- Prakken B.; Wauben M.; Genini D.; Samodal R.; Barnett J.; Mendivil A.; Leoni L.; Albani S. Artificial antigen-presenting cells as a tool to exploit the immune ‘synapse’. Nat. Med. 2000, 6, 1406–1410. 10.1038/82231. [DOI] [PubMed] [Google Scholar]
- van der Weijden J.; Paulis L. E.; Verdoes M.; van Hest J. C. M.; Figdor C. G. The right touch: design of artificial antigen-presenting cells to stimulate the immune system. Chem. Sci. 2014, 5, 3355–3367. 10.1039/C4SC01112K. [DOI] [Google Scholar]
- Lillemeier B. F.; Mortelmaier M. A.; Forstner M. B.; Huppa J. B.; Groves J. T.; Davis M. M. TCR and Lat are expressed on separate protein islands on T cell membranes and concatenate during activation. Nat. Immunol. 2010, 11, 90–96. 10.1038/ni.1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schamel W. W. A.; Alarcón B. Organization of the resting TCR in nanoscale oligomers. Immunol. Rev. 2013, 251, 13–20. 10.1111/imr.12019. [DOI] [PubMed] [Google Scholar]
- Dinic J.; Riehl A.; Adler J.; Parmryd I. The T cell receptor resides in ordered plasma membrane nanodomains that aggregate upon patching of the receptor. Sci. Rep. 2015, 5, 10082 10.1038/srep10082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nika K.; Acuto O. Membrane nanodomains in T-cell antigen receptor signalling. Essays Biochem. 2015, 57, 165–175. 10.1042/bse0570165. [DOI] [PubMed] [Google Scholar]
- Pageon S. V.; Tabarin T.; Yamamoto Y.; Ma Y.; Nicovich P. R.; Bridgeman J. S.; Cohnen A.; Benzing C.; Gao Y.; Crowther M. D.; Tungatt K.; Dolton G.; Sewell A. K.; Price D. A.; Acuto O.; Parton R. G.; Gooding J. J.; Rossy J.; Rossjohn J.; Gaus K. Functional role of T-cell receptor nanoclusters in signal initiation and antigen discrimination. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, E5454–E5463. 10.1073/pnas.1607436113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma R.; Campi G.; Yokosuka T.; Saito T.; Dustin M. L. T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity 2006, 25, 117–127. 10.1016/j.immuni.2006.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokosuka T.; Saito T. Dynamic regulation of T-cell costimulation through TCR–CD28 microclusters. Immunol. Rev. 2009, 229, 27–40. 10.1111/j.1600-065X.2009.00779.x. [DOI] [PubMed] [Google Scholar]
- Choudhuri K.; Dustin M. L. Signaling microdomains in T cells. FEBS Lett. 2010, 584, 4823–4831. 10.1016/j.febslet.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto-Tane A.; Saito T. Dynamic Regulation of TCR–Microclusters and the Microsynapse for T Cell Activation. Front. Immunol. 2016, 7, 255 10.3389/fimmu.2016.00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fooksman D. R.; Vardhana S.; Vasiliver-Shamis G.; Liese J.; Blair D. A.; Waite J.; Sacristán C.; Victora G. D.; Zanin-Zhorov A.; Dustin M. L. Functional Anatomy of T Cell Activation and Synapse Formation. Annu. Rev. Immunol. 2010, 28, 79–105. 10.1146/annurev-immunol-030409-101308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder A.; Huskens J.; Reinhoudt D. N. Multivalency in supramolecular chemistry and nanofabrication. Org. Biomol. Chem. 2004, 2, 3409–3424. 10.1039/b413971b. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy V. M.; Estroff L. A.; Whitesides G. M.. Multivalency in Ligand Design. In Fragment-Based Approaches in Drug Discovery; Jahnke W.; Erlanson D. A., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2006; Vol. 34, pp 11–53. [Google Scholar]
- Kitov P. I.; Bundle D. R. On the Nature of the Multivalency Effect: A Thermodynamic Model. J. Am. Chem. Soc. 2003, 125, 16271–16284. 10.1021/ja038223n. [DOI] [PubMed] [Google Scholar]
- Huskens J. Multivalent interactions at interfaces. Curr. Opin. Chem. Biol. 2006, 10, 537–543. 10.1016/j.cbpa.2006.09.007. [DOI] [PubMed] [Google Scholar]
- Mandal S.; Eksteen-Akeroyd Z. H.; Jacobs M. J.; Hammink R.; Koepf M.; Lambeck A. J. A.; van Hest J. C. M.; Wilson C. J.; Blank K.; Figdor C. G.; Rowan A. E. Therapeutic nanoworms: towards novel synthetic dendritic cells for immunotherapy. Chem. Sci. 2013, 4, 4168–4174. 10.1039/c3sc51399h. [DOI] [Google Scholar]
- Mandal S.; Hammink R.; Tel J.; Eksteen-Akeroyd Z. H.; Rowan A. E.; Blank K.; Figdor C. G. Polymer-Based Synthetic Dendritic Cells for Tailoring Robust and Multifunctional T Cell Responses. ACS Chem. Biol. 2015, 10, 485–492. 10.1021/cb500455g. [DOI] [PubMed] [Google Scholar]
- Dommerholt J.; Schmidt S.; Temming R.; Hendriks L. J. A.; Rutjes F. P. J. T.; van Hest J. C. M.; Lefeber D. J.; Friedl P.; van Delft F. L. Readily Accessible Bicyclononynes for Bioorthogonal Labeling and Three-Dimensional Imaging of Living Cells. Angew. Chem., Int. Ed. Engl. 2010, 49, 9422–9425. 10.1002/anie.201003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph N.; Reicher B.; Barda-Saad M. The calcium feedback loop and T cell activation: How cytoskeleton networks control intracellular calcium flux. Biochim. Biophys. Acta, Biomembr. 2014, 1838, 557–568. 10.1016/j.bbamem.2013.07.009. [DOI] [PubMed] [Google Scholar]
- Williams D. A.; Fogarty K. E.; Tsien R. Y.; Fay F. S. Calcium gradients in single smooth muscle cells revealed by the digital imaging microscope using Fura-2. Nature 1985, 318, 558–561. 10.1038/318558a0. [DOI] [PubMed] [Google Scholar]
- Donnadieu E.; Bismuth G.; Trautmann A. The intracellular Ca2+ concentration optimal for T cell activation is quite different after ionomycin or CD3 stimulation. Pfluegers Arch. 1995, 429, 546–554. 10.1007/BF00704160. [DOI] [PubMed] [Google Scholar]
- Montet X.; Funovics M.; Montet-Abou K.; Weissleder R.; Josephson L. Multivalent Effects of RGD Peptides Obtained by Nanoparticle Display. J. Med. Chem. 2006, 49, 6087–6093. 10.1021/jm060515m. [DOI] [PubMed] [Google Scholar]
- Kjer-Nielsen L.; Dunstone M. A.; Kostenko L.; Ely L. K.; Beddoe T.; Mifsud N. A.; Purcell A. W.; Brooks A. G.; McCluskey J.; Rossjohn J. Crystal structure of the human T cell receptor CD3εγ heterodimer complexed to the therapeutic mAb OKT3. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 7675–7680. 10.1073/pnas.0402295101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dushek O.; van der Merwe P. A. An induced rebinding model of antigen discrimination. Trends Immunol. 2014, 35, 153–158. 10.1016/j.it.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths G. M.; Tsun A.; Stinchcombe J. C. The immunological synapse: a focal point for endocytosis and exocytosis. J. Cell Biol. 2010, 189, 399–406. 10.1083/jcb.201002027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhuri K.; Llodra J.; Roth E. W.; Tsai J.; Gordo S.; Wucherpfennig K. W.; Kam L. C.; Stokes D. L.; Dustin M. L. Polarized release of T-cell-receptor-enriched microvesicles at the immunological synapse. Nature 2014, 507, 118–123. 10.1038/nature12951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone J. D.; Chervin A. S.; Kranz D. M. T-cell receptor binding affinities and kinetics: impact on T-cell activity and specificity. Immunology 2009, 126, 165–176. 10.1111/j.1365-2567.2008.03015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visch H.-J.; Koopman W. J. H.; Zeegers D.; van Emst-de Vries S. E.; van Kuppeveld F. J. M.; van den Heuvel L. W. P. J.; Smeitink J. A. M.; Willems P. H. G. M. Ca2+-mobilizing agonists increase mitochondrial ATP production to accelerate cytosolic Ca2+ removal: aberrations in human complex I deficiency. Am. J. Physiol. 2006, 291, C308–16. 10.1152/ajpcell.00561.2005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.