

Abstract
Natriuretic peptide receptor-A (NPR-A), also known as guanylyl cyclase-A, is a transmembrane receptor guanylyl cyclase that is activated by the cardiac hormones atrial natriuretic peptide and B-type natriuretic peptide. Although ligand-dependent NPR-A degradation (also known as down-regulation) is widely acknowledged in human and animal models of volume overload, down-regulation in cultured cells is controversial. Here, we examined the effect of ANP exposure on cellular NPR-A levels as a function of time. Relative receptor concentrations were estimated using guanylyl cyclase and immunoblot assays in a wide variety of cell lines that endogenously or exogenously expressed low or high numbers of receptors. ANP exposures of 1 h markedly reduced hormone-dependent but not detergent-dependent guanylyl cyclase activities in membranes from exposed cells. However, 1-h ANP exposures did not significantly reduce NPR-A concentrations in any cell line. In contrast, exposures of greater than 1 h reduced receptor concentrations in a time-dependent manner. The time required for half of the receptors to be degraded (t1/2) in primary bovine aortic endothelial and immortalized HeLa cells was approximately 8 h. In contrast, a 24-h exposure of ANP to 293T cells stably overexpressing NPR-A caused less than half of the receptors to be degraded. To our knowledge, this is the first report to directly measure NPR-A down-regulation in endogenously expressing cells. We conclude that down-regulation is a universal property of NPR-A but is relatively slow and varies with receptor expression levels and cell type.
NPR-A is degraded in response to long-term but not short-term ligand exposure, an issue that was previously controversial.
Atrial natriuretic peptide (ANP) and B-type NP (BNP) are endogenous hormones that are essential for cardiovascular homeostasis (1, 2, 3). In response to cardiomyocyte stretch, ANP and BNP are released into the circulation from the heart, where they bind receptors in target tissues like the kidneys, heart, adrenal gland, endothelium, and vasculature. NP receptor-A (NPR-A), or guanylyl cyclase-A, is the primary signaling receptor for ANP and BNP. It is a transmembrane receptor guanylyl cyclase with intracellular regulatory, dimerization, and catalytic domains (4). Activation of NPR-A leads to the synthesis of the intracellular second messenger cyclic (c)GMP, which mediates the vast majority of NP effects (1, 5). ANP and BNP also bind the NP clearance receptor (NPR-C), which mediates intracellular NP degradation and may reduce cellular cAMP concentrations (6, 7, 8).
Circulating ANP and BNP levels are dramatically elevated in congestive heart failure (9). Initially, these cardiac peptides stimulate compensatory hemodynamic functions, but over time, NP-dependent cardiac unloading effects wane despite continued elevation of serum ANP and BNP concentrations (10). One explanation for the blunted response is accelerated NPR-A degradation resulting from ligand-dependent receptor degradation. Recent studies using 125I-NP binding or antibody-mediated receptor detection indicate that NPR-A levels are reduced in tissues from heart-failed animals or patients compared with nonfailed subjects (11, 12, 13, 14).
Whether NPR-A is degraded in response to ligand binding in cell culture systems is controversial. 125I-ANP binding studies conducted by Pandey (15) and Pandey et al. (16) suggested that ANP-NPR-A complexes rapidly internalize after ligand exposure and that a portion of the internalized population is recycled to the plasma membrane and another portion is degraded. In contrast, 125I-ANP binding studies by Maack and co-workers (17) indicated that NPR-A is a constitutively membrane-resident protein that does not internalize in the presence or absence of ANP. Instead of intracellular lysosomal-mediated receptor-ligand dissociation, the Maack group concluded that the termination of the interaction between ANP and NPR-A results from a rapid, temperature-dependent, dissociation that occurs at the cell surface (17, 18). Consistent with these findings, Jewett et al. (19) reported that ANP binding to NPR-A in 293 cells causes a transition from high to low affinity binding within 15 min at 4 C that is not associated with receptor loss from the cell surface. Similarly, we found that 80% of 125I-ANP bound to NPR-A overexpressed in 293T cells was released intact into culture medium after 10 min at 37 C (20). Additionally, we reported that NPR-A stably expressed in 293 cells was down-regulated in a manner positively associated with time of ANP exposure up to 4 h (21). However, a more recent study from our group found that NPR-A expressed in a related but different 293 cell line (293T) was not detectably down-regulated by a 14-h ANP exposure (20).
The effect of ANP on NPR-A down-regulation in cell culture is decidedly unclear. Furthermore, we recently reported two confounding observations. First, NPR-A is not down-regulated by ligand exposure in 293T cells (20), and second, NPR-A is down-regulated in a mouse model of congestive heart failure (12, 13). Because ANP and BNP are highly elevated in congestive heart failure and NPR-A mRNA levels are not decreased in failing hearts (22, 23), we investigated the effect of prolonged ANP incubations on NPR-A levels in four cell lines using guanylyl cyclase assays and immunoblot analysis with antibodies against natural intracellular and artificial extracellular receptor epitopes. Primary, immortalized, and overexpressing cell lines were studied to develop a consistent understanding of the problem. We specifically avoided 125I-ANP binding studies due to the previously described interpretation issues associated with changing binding affinities, difficulties in removing prior bound ANP from receptors (prior receptor occupation), and the common scenario of cells expressing two ANP receptors (NPR-A and NPR-C). We found that ANP unequivocally stimulates NPR-A degradation in all cell types examined by a process that occurs over hours, not minutes.
Materials and Methods
Materials
Rat ANP, cycloheximide, and microcystin-LR were purchased from Sigma-Aldrich (St. Louis, MO). Cyclic GMP RIA kit was from PerkinElmer (Waltham, MA).
Cell culture
Bovine aortic endothelial cells (bAEC) were purchased from Lonza (Walkersville, MD) and maintained in endothelial cell growth medium (Lonza). Stably transfected 293T NPR-A cells were grown as previously described (20). CHO-pCEP6 cells were a generous gift from Kathy Griendling (Emory University, Atlanta, GA) and were maintained in DMEM containing 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 500 μg/ml hygromycin B, and 20 μg/ml l-proline.
FLAG-NPR-A construct
Rat NPR-A was excised from pCMV3-GC-A (24) by digestion with NotI and BglII and subcloned into the same sites in the multiple cloning region of pFLAG-CMV1 (Sigma-Aldrich), which contains a FLAG epitope-inserted C-terminal to the Met-preprotrypsin coding sequence to generate pFLAG-NPR-A-CMV1. The resulting plasmid generates an amino-terminal FLAG-tagged receptor after cleavage of the signal sequence.
Small interfering RNA (siRNA) knock down of NPR-A
Silencer glyceraldehyde-3-phosphate dehydrogenase siRNA and siRNAs targeting human NPR-A (NPR1) [no. 143349: sense, 5′-CCCAGAUAAUCCCGAGUACtt-3′; antisense, 5′-GUACUCGGGAUUAUCUGGGtc-3′; no. 143350: sense, 5′-GCAUAUUAUAAGGGCAACCtt-3′; antisense, 5′-GGUUGCCCUUAUAAUAUGCtg-3′; and no. 143351: sense, 5′-CCGUAAACGCAUUGAGCUGtt-3′; antisense, 5′-CAGCUCAAUGCGUUUACGGtt-3′] were from Ambion (Austin, TX); 30 nmol siRNA were transfected into HeLa cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Cells were incubated for 48–72 h posttransfection before analysis.
Membrane preparation
Cells were washed at 4 C with PBS and scraped off the plate in the presence of phosphatase inhibitor buffer containing 25 mm HEPES (pH 7.4), 50 mm NaCl, 20% glycerol, 50 mm NaF, 2 mm EDTA, 0.5 μm microcystin-LR, and 1× Roche complete protease inhibitors. Suspended cells were sonicated for 1–2 sec and centrifuged at 20,000 × g for 10 min at 4 C. The supernatant was aspirated, and the pellet was resuspended in the same buffer. Total protein concentrations were determined by the Bradford method.
Guanylyl cyclase assay
Twenty microliters of membranes were assayed for guanylyl cyclase activity in the presence of 1 mm ATP and 5 mm MgCl2 (basal stimulation), 1 mm ATP, 5 mm MgCl2 and 1 μm rat ANP (hormone stimulation), or 1% Triton X-100 and 5 mm MnCl2 (detergent stimulation). The reaction was initiated by adding 55 μl of prewarmed cocktail containing 25 mm HEPES (pH 7.4), 50 mm NaCl, 0.1% BSA, 0.5 mm 3-isobutyl-1-methylxanthine, 1 mm GTP, 5 mm creatine phosphate, 0.1 μg/μl creatine kinase, 1 μm EDTA, and 1 μm microcystin-LR. Reactions were performed at 37 C for 3 min and stopped with 400 μl of ice-cold 50 mm sodium acetate solution containing 5 mm EDTA.
Immunoprecipitation and immunoblotting
Cells were solubilized in 1 ml of ice-cold modified radioimmunoprecipitation assay buffer containing 50 mm Tris (pH 7.5), 100 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 10 mm NaH2PO4, 50 mm NaF, 2 mm EDTA, 0.5 μm microcystin-LR, and 1× Roche complete protease inhibitors. To reduce nonspecific binding, solubilized cells were incubated at 4 C with 50 μl protein A (RepliGen, Waltham, MA) for at least 30 min. NPR-A was immunoprecipitated overnight at 4 C using 2 μl of polyclonal rabbit 6325 antiserum, which recognizes the last 17 C-terminal amino acids of rat NPR-A, and 50 μl protein A. The FLAG epitope was immunoprecipitated with 40 μl anti-FLAG M2 affinity gel (Sigma-Aldrich) overnight at 4 C. Immunocomplexes were washed three times with 1 ml of ice-cold radioimmunoprecipitation assay buffer, fractionated by SDS-PAGE, and transferred to polyvinylidene fluoride membrane.
Primary antibodies for immunoblot analysis were polyclonal rabbit 6325 (1:2500) to NPR-A (25) and monoclonal anti-FLAG M2 (1:5000; Sigma-Aldrich) to the FLAG epitope. Secondary antibodies were peroxidase-conjugated donkey antirabbit IgG (1:20,000; GE Healthcare; Buckinghamshire, UK), peroxidase-conjugated sheep antimouse IgG (1:20,000; GE Healthcare), or goat antirabbit IRDye 680 (1:10,000; LI-COR Biosciences; Lincoln, NE).
Whole-cell cGMP elevation assay
The 293 cells, grown on poly-d-lysine-coated 24-well plates, were transfected using a calcium phosphate protocol. The day of the assay, the cells were incubated in serum-free medium for at least 4 h. Medium was aspirated, and the cells were incubated for 20 min at 37 C in DMEM containing 20 mm HEPES (pH 7.4) and 0.5 mm 3-isobutyl-1-methylxanthine. This medium was then replaced with the same medium containing 1 μm rat ANP, a 1:2000 dilution of anti-FLAG M2 antibody, or both 1 μm rat ANP and a 1:2000 dilution of anti-FLAG M2 antibody and stimulated for 5 min. The reaction was terminated by aspirating the medium and adding 1 ml of ice-cold 80% ethanol. One hundred and fifty microliters of the resulting supernatant were dried in a centrifugal vacuum concentrator and analyzed for cGMP content by RIA.
Data analysis
Immunoblots were quantified on either a GS-700 Imaging Densitometer with Molecular Analyst software (Bio-Rad, Hercules, CA) or an Odyssey Infrared Imaging System (LI-COR Biosciences). Microsoft Excel was used for statistical analysis of the data; statistical significance was determined by paired t tests as indicated in the figure legends.
Results
ANP-dependent degradation of endogenous NPR-A
To examine whether NPR-A is down-regulated in response to prolonged ligand exposure in cultured cells, initial experiments were conducted in primary bAEC. These cells express detectable levels of endogenous NPR-A and are more likely to exhibit physiologic regulation than immortalized or transfected cells. Exposure of bAEC to saturating levels of ANP (200 nm) for 1 h reduced ANP-dependent guanylyl cyclase activity by 75% (Fig. 1A). To determine whether the reduced cyclase activity was explained by decreased receptor protein levels, cellular NPR-A levels from cells exposed to ANP for increasing periods of time were determined by immunoblot analysis (Fig. 1B). A characteristic doublet detection pattern for NPR-A was observed, which consists of the more prominent upper completely glycosylated and phosphorylated band and lower incompletely glycosylated and unphosphorylated band (26). Importantly, immunoblot signals obtained from cells incubated with or without ANP for 1 h were not markedly different, indicating that decreases in receptor protein levels do not explain the reduced ANP-dependent guanylyl cyclase activity. Previous studies indicate that NPR-A dephosphorylation is tightly correlated with early losses in ANP-dependent guanylyl cyclase activities in 293 cells and likely explains the initial activity declines in the bAEC as well (21, 24, 26, 27). Exposure of the bAEC to ANP for 4, 8, or 20 h resulted in additional loss of ANP-dependent cyclase activity. Immunoblot analysis indicated that the fully processed form of NPR-A (upper band) declined in a time-dependent manner, whereas the lower incompletely processed form (lower band) was unaffected by ligand exposure (Fig. 1B). By 8 and 20 h, mature NPR-A protein levels were reduced by approximately 57 and 85%, respectively. To our knowledge, this is the first report to directly demonstrate large time-dependent reductions in NPR-A protein and guanylyl cyclase levels in response to ANP exposure in primary cells.
Fig. 1.
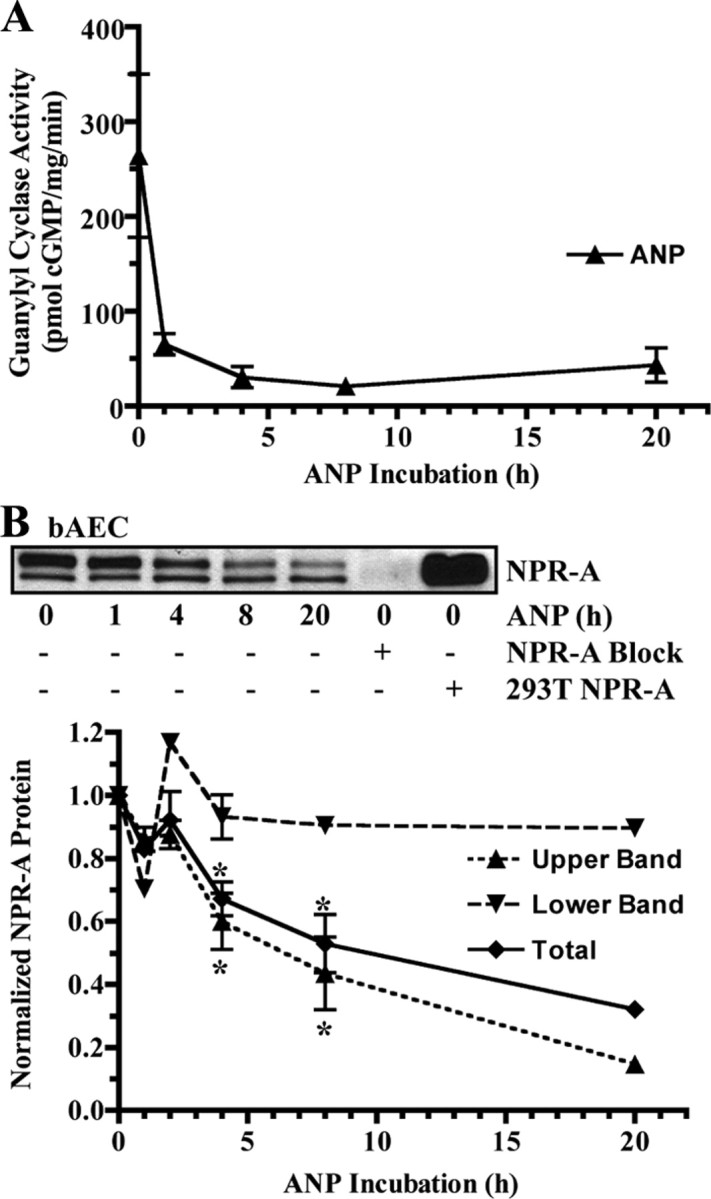
Endogenous NPR-A is down-regulated by ANP in primary bAEC. Cells were incubated with 200 nm ANP for the indicated periods of time at 37 C. A, Membranes were prepared and assayed for hormone-dependent guanylyl cyclase activity and plotted as a function of time in the presence of ANP. The data are representative of at least three separate experiments. B, In a parallel experiment, NPR-A was immunoprecipitated, fractionated by SDS-PAGE, electroblotted, and detected by immunoblot. NPR-A isolated from 293T cells stably expressing NPR-A was used as a positive control. A synthetic NPR-A blocking peptide was used as a negative control to block NPR-A antibody/antigen binding. NPR-A protein levels from three independent immunoblots were quantitated, normalized, and presented in graphical form as mean ± sem or range, where n = 3 (0, 4, and 8 h), n = 2 (1 and 2 h), or n = 1 (20 h). Statistical significance was determined by a paired t test, where *, P < 0.05.
ANP-dependent down-regulation of endogenous NPR-A was also examined in immortalized human cervical HeLa cells. To focus on protein degradation in the absence of new protein synthesis, these cells were incubated with 10 μg/ml cycloheximide during the ANP exposure periods. Cycloheximide clearly blocked protein synthesis, because the lower migrating precursor form of NPR-A was not present in immunoblots from cells incubated with the protein synthesis inhibitor (Fig. 2B). As was observed in the primary bAEC, exposure to 200 nm ANP for 1 h markedly reduced hormone-dependent guanylyl cyclase activity without affecting NPR-A protein levels (Fig. 2, A and B). However, exposure to hormone for 2, 4, 8, or 12 h further reduced ANP-dependent guanylyl cyclase activity, and the reductions were consistent with decreased NPR-A protein levels as revealed by immunoblot analysis.
Fig. 2.
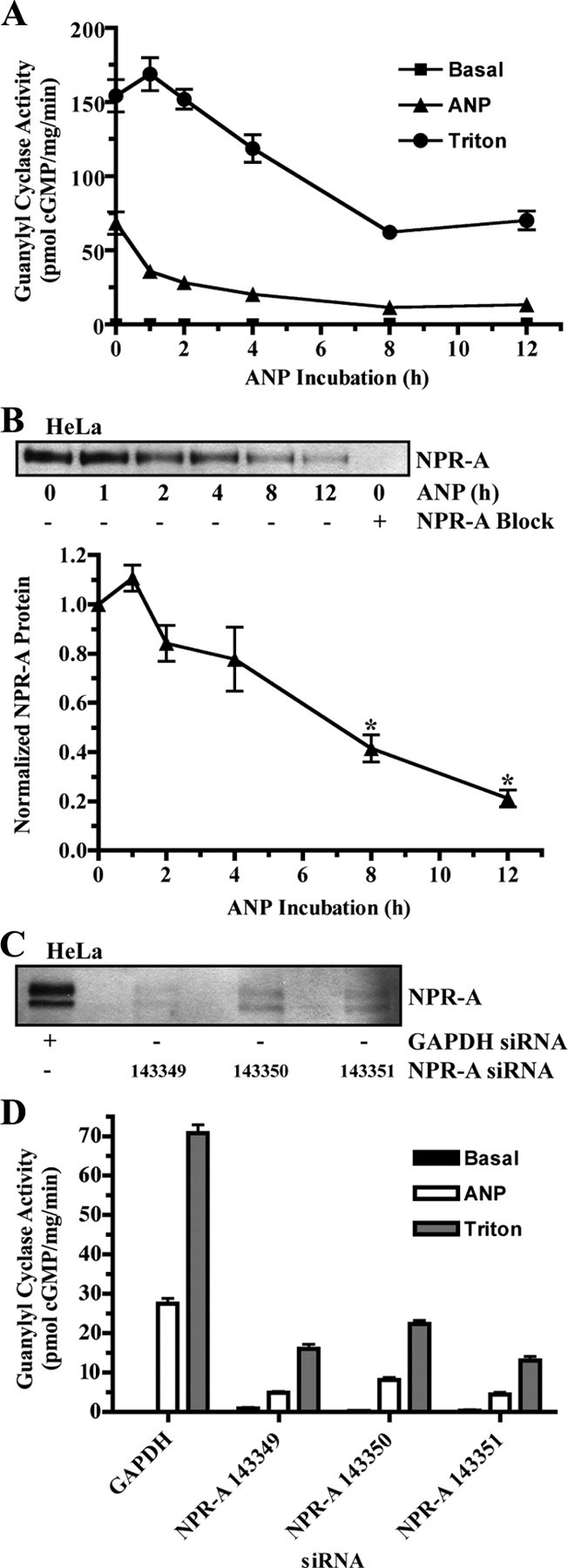
Endogenous NPR-A is down-regulated by ANP in immortalized human cervical HeLa cells. A and B, Cells were incubated with 200 nm ANP for the indicated periods of time at 37 C. Cycloheximide (10 μg/ml) was also added to the medium to block protein synthesis. A, Membranes were prepared and assayed for guanylyl cyclase activity after basal, ANP, or Triton X-100 stimulation. Activity levels were plotted as a function of time in the presence of ANP. Data points are represented as mean ± sem, where n = 3. B, In a parallel experiment, NPR-A was immunoprecipitated and detected by immunoblot. NPR-A isolated from 293T cells stably expressing NPR-A was used as a positive control. A synthetic NPR-A blocking peptide was used as a negative control to block NPR-A antibody/antigen binding. NPR-A protein levels from four independent immunoblots were quantitated, normalized, and presented in graphical form as mean ± sem, where n = 4. Statistical significance was determined by a paired t test, where *, P < 0.005. C and D, Cells were transfected with siRNA against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or NPR-A. C, NPR-A was immunoprecipitated and detected by immunoblot to verify the NPR-A knock down. D, Membranes were prepared and assayed for guanylyl cyclase activity after a 3-min basal, ANP, or Triton X-100 stimulation. Data points are represented as mean ± sem, where n = 3.
In both the bAEC and HeLa cells, about half of the protein was lost after exposure to ANP for 8 h. In the experiment with the HeLa cells, we also measured guanylyl cyclase activity in the presence of 1% Triton X-100 and manganese as the divalent metal cofactor. These nonphysiologic conditions activate the enzyme in a hormone- and phosphorylation-independent manner. Hence, they are a reliable indicator of total NPR-A protein levels (28). Guanylyl cyclase activity determined in the presence of detergent declined gradually and paralleled the diminishing immunoblot signal (Fig. 2, A and B). To verify that NPR-A alone was responsible for the ANP- and Triton X-100-dependent guanylyl cyclase activities, guanylyl cyclase activity was measured after a 3-min stimulation in HeLa cells transfected with siRNA against NPR-A mRNA to reduce NPR-A protein. Three separate siRNAs targeting different regions of NPR-A produced similar results (Fig. 2, C and D). In Fig. 2C, note that the lower immature band of NPR-A is present, because no cycloheximide was added in this experiment. Both ANP-dependent and ANP-independent guanylyl cyclase activities were reduced by approximately 80% in cells expressing NPR-A-specific siRNA, which indicates that NPR-A is responsible for the vast majority of guanylyl cyclase activity in these cells (Fig. 2D). Thus, two independent assays, detergent-dependent guanylyl cyclase activities and immunoblot detection, indicate that prolonged ANP exposure causes NPR-A degradation in HeLa cells.
ANP-dependent degradation of NPR-A in overexpressing cells
Previously, our laboratory reported that NPR-A was not internalized or degraded in response to ANP in 293T cells overexpressing NPR-A (20). Because we observed endogenous NPR-A down-regulation in a mouse model of congestive heart failure (12, 13) and in ANP-exposed “regular” 293 (21), bAEC, and HeLa cells, we reexamined the effect of prolonged ANP exposure on NPR-A stably expressed in 293T cells (Fig. 3). However, to optimize our ability to detect ANP-dependent degradation in the 293T NPR-A cells, several modifications were made to the original down-regulation protocol (20). Specifically, the time of incubation in the presence of ANP was extended, new protein synthesis was blocked with cycloheximide, and fresh ANP was added several times throughout the incubation period to replenish ANP levels resulting from possible peptide proteolysis. Under these conditions, ANP-dependent degradation of NPR-A in 293T cells was observed. However, the rate and extent of NPR-A degradation was markedly reduced compared with degradation observed in the bAEC and HeLa cells. Visually, NPR-A protein levels were not obviously reduced until after 16 h of ANP exposure. Quantification of these data indicated that NPR-A protein levels were reduced by 17, 38, and 48% after being exposed to ANP for 8, 16, or 24 h, respectively (Fig. 3, bottom).
Fig. 3.

ANP stimulates NPR-A down-regulation in 293T cells overexpressing NPR-A. Cells were incubated with 10 μg/ml cycloheximide and with or without 1 μm ANP for indicated periods of time. ANP was refreshed every 4 h. Top, NPR-A levels were detected by sequential immunoprecipitation-immunoblotting as described in Fig. 1. Bottom, Data from the immunoblot were quantitated and presented in graphical form as a mean and range, where n = 2. Error bars are within the symbols. 293T NPR-A data are representative of three similar experiments.
The antibody used in the previous experiments recognizes the last 17 amino acids of NPR-A. Thus, it is possible that ANP exposure causes cleavage of this small peptide from the receptor without causing complete inactivating proteolysis of NPR-A. To rule out this possibility, we engineered a NPR-A construct with an amino-terminal FLAG antibody binding epitope. ANP stimulated cGMP elevations in 293 cells transfected with a plasmid encoding FLAG-NPR-A similarly to cells transfected with the wild-type receptor (Fig. 4A). Simultaneous additions of ANP and anti-FLAG M2 antibody caused a comparable increase in cGMP levels, and addition of the anti-FLAG antibody alone did not stimulate cGMP production. Thus, FLAG-NPR-A is expressed at the cell surface and neither the FLAG epitope nor its accompanying antibody stimulated or hindered receptor activation.
Fig. 4.
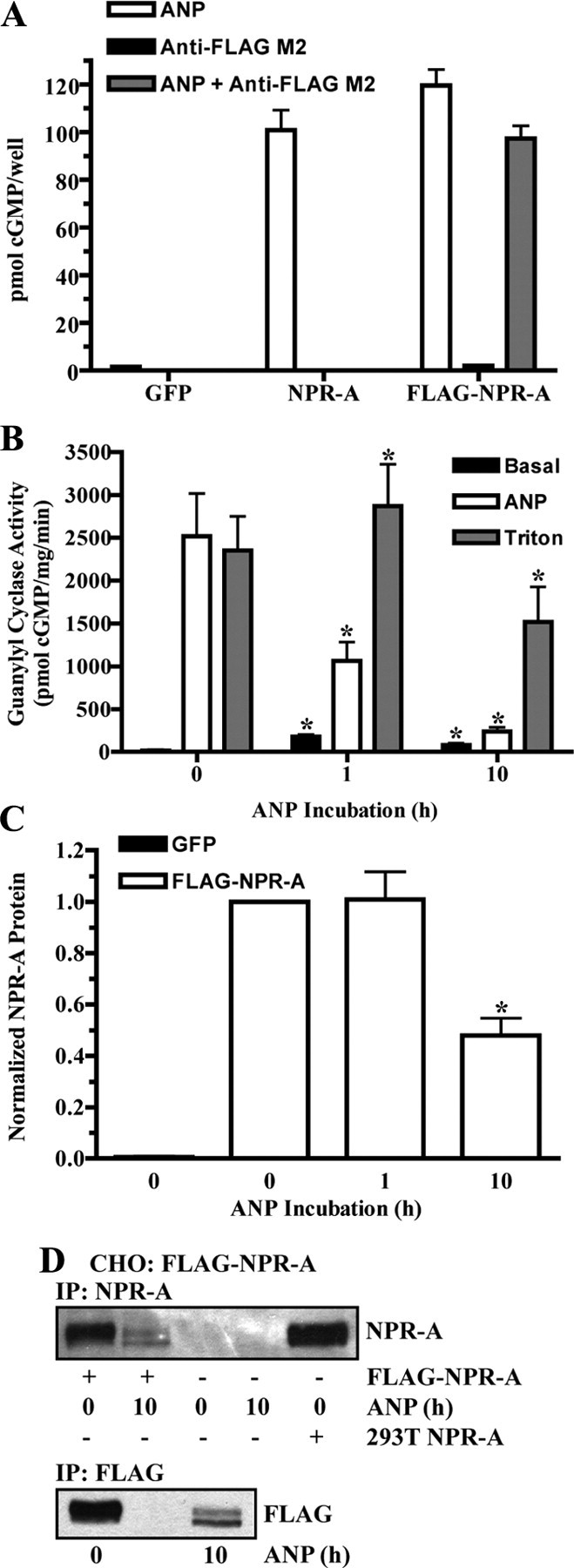
ANP stimulates FLAG-NPR-A down-regulation. A, 293 cells transiently transfected with GFP, NPR-A (wild type), or FLAG-NPR-A were stimulated with 1 μm ANP for 3 min, and cGMP levels were measured. FLAG-NPR-A transfected cells were also stimulated with anti-FLAG M2 antibody (1:2000) or both ANP and anti-FLAG M2 antibody. Data points are represented as mean ± sem, where n = 8. B–D, CHO cells were transiently transfected with or without FLAG-NPR-A and incubated with 10 μg/ml cycloheximide in the absence or presence of 200 nm ANP. B, Membranes were prepared and assayed for guanylyl cyclase activity after basal, ANP, or Triton X-100 stimulation. Data points are represented as mean ± sem, where n = 6. C, FLAG-NPR-A was immunoprecipitated with antibody to the FLAG epitope and detected by immunoblot using antibody to NPR-A. Protein levels were quantitated, normalized, and presented in graphical form as a mean ± sem, where n = 4. D, FLAG-NPR-A was immunoprecipitated and detected by immunoblot using antibody to both NPR-A (top) and the FLAG epitope (bottom). NPR-A isolated from 293T NPR-A cells was used as a positive control. Statistical significance was determined by a paired t test (vs. 0 h ANP), where *, P < 0.01 (B) and P < 0.005 (C).
To determine whether FLAG-NPR-A undergoes ANP-dependent degradation similarly to the wild-type receptor, Chinese hamster ovary (CHO) cells were transfected with FLAG-NPR-A and incubated with cycloheximide in the absence or presence of ANP. Consistent with results from the bAEC and HeLa cells, ANP-stimulated guanylyl cyclase activity dropped 58 and 90% after 1 and 10 h, respectively, of ANP exposure compared with unexposed cells (Fig. 4B). Triton X-100-dependent guanylyl cyclase activity was only reduced after the 10-h ANP incubation (Fig. 4B). FLAG-NPR-A protein levels in CHO cells were not reduced after 1-h ANP incubation; however, a 10-h incubation with ligand decreased protein levels by 52% (Fig. 4C). In contrast to the experiments conducted in the HeLa cells, cycloheximide treatment did not prevent the detection of the faster migrating NPR-A species. However, consistent with the HeLa cell studies, ANP exposure only reduced the mature form (upper band) of NPR-A. No endogenous NPR-A was detected in either GFP-transfected or GFP-untransfected CHO cells (Fig. 4, C and D). Importantly, detection of FLAG-NPR-A was determined by reactivity to an epitope at the amino terminus (Fig. 4D, bottom) and carboxyl terminus (Fig. 4D, top) of the receptor. Thus, loss of antibody reactivity to both the amino and carboxyl termini of NPR-A is consistent with ANP exposure stimulating the complete degradation of NPR-A.
Discussion
Here, we show that prolonged ANP exposure results in a time-dependent decrease in NPR-A concentration in a variety of cell culture models, including 293T cells. The decreased receptor levels result from increased degradation, not reduced synthesis, because cycloheximide blocked new NPR-A synthesis without affecting NPR-A degradation rate as measured by immunoblot or Triton X-100-dependent guanylyl cyclase activities. The fact that only the upper band but not the lower band of NPR-A is degraded is also consistent with an mRNA-independent mechanism. It is unknown why the incompletely processed form of NPR-A is not degraded, but it may be because this form is not on the cell surface, and is therefore, unable to bind ANP. Thus, data from primary, immortalized, and transfected cells are consistent and in agreement with the in vivo observations for ANP-dependent NPR-A down-regulation. In other words, prolonged cellular exposure to ANP stimulates NPR-A degradation.
The reason why we did not observe significant ANP-dependent NPR-A degradation in our previous studies with 293T cells may result from the slow rate of degradation in these cells (Fig. 3). However, it is also noteworthy that ANP was frequently added to the cell culture medium to ensure constant NP concentrations, and cycloheximide was added to inhibit new protein synthesis in the present but not previous study. In addition, the previous report examined ANP-dependent degradation of cell-surface NPR-A after 0, 10, 60, or 840 min of ANP exposure. Although endogenous NPR-A in bAEC and HeLa cells degraded more rapidly than NPR-A in the 293T NPR-A cells, even in the bAEC and HeLa cells, there was virtually no loss of receptor protein levels after 1 h of ligand exposure. After 16 h of ANP exposure, we observed only a 38% reduction in NPR-A protein levels in the 293T NPR-A cells. Thus, a slight reduction in NPR-A levels at the 14-h time point in the previous study was not detected. It should be noted that the internalization and degradation of the epidermal growth factor receptor is reduced in cells expressing high concentrations of receptors (29), and the 293T cell line used in the previous study expressed very high numbers of receptor (∼106 receptors/cell) (20), which is consistent with reduced trafficking due to saturation of the basal endocytic machinery. However, we cannot rule out the possibility that the majority of NPR-A is not on the cell surface and, therefore, is unable to be regulated by ligand binding in these cells.
An increased rate of NPR-A internalization is one explanation for ANP-induced NPR-A degradation. The internalization of NPR-A in response to ligand binding has been controversial and limited to 125I-ANP binding studies, which track the receptor via the 125I-ANP-NPR-A complex. A disadvantage of this approach is that previously added unlabeled ANP can block subsequent binding of 125I-ANP via a process known as prior receptor occupation (30). In addition, 125I-ANP binds NPR-C, which is not only the most widely and abundantly distributed NPR, but is also constitutively internalized (7). Regardless of these limitations, we note that the Pandey group used ligand binding assays to suggest that ANP binding stimulates NPR-A down-regulation as early as 1986 (31), and now, the majority of long-term exposure data confirm this hypothesis. However, similarly to the Maack group (17, 18), and unlike the results from Pandey et al. (16), we failed to observe significant NPR-A degradation within the first hour of ANP exposure. A caveat to this interpretation is that our studies measured reductions in the total cellular pool of NPR-A, and if the amount of NPR-A that is degraded at the cell surface is low compared with the total cellular receptor population, then changes in extracellular NPR-A levels may not be detectable with our methods.
In conclusion, prolonged ANP exposure stimulates NPR-A degradation in all cellular systems tested. Future studies will investigate how ANP exposure affects NPR-A trafficking and degradation.
Acknowledgments
We thank Dedra Fagan for constructing the pFLAG-NPR-A-CMV1 plasmid.
Footnotes
This work was supported by American Heart Association Grants 0815607G (to D.R.F.) and 0950053G (to L.R.P.).
Disclosure Summary: The authors have nothing to disclose.
First Published Online April 9, 2010
Abbreviations: ANP, Atrial natriuretic peptide; bAEC, bovine aortic endothelial cells; BNP, B-type NP; c, cyclic; CHO, Chinese hamster ovary; LR, lysine-arginine; NPR-A, NP receptor-A; NPR-C, NP clearance receptor; siRNA, small interfering RNA.
References
- 1.Potter LR, Yoder AR, Flora DR, Antos LK, Dickey DM 2009. Natriuretic peptides: their structures, receptors, physiologic functions and therapeutic applications. Handb Exp Pharmacol 191:341–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuhn M 2003. Structure, regulation, and function of mammalian membrane guanylyl cyclase receptors, with a focus on guanylyl cyclase-A. Circ Res 93:700–709 [DOI] [PubMed] [Google Scholar]
- 3.Garbers DL, Chrisman TD, Wiegn P, Katafuchi T, Albanesi JP, Bielinski V, Barylko B, Redfield MM, Burnett Jr JC 2006. Membrane guanylyl cyclase receptors: an update. Trends Endocrinol Metab 17:251–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Potter LR, Hunter T 2001. Guanylyl cyclase-linked natriuretic peptide receptors: structure and regulation. J Biol Chem 276:6057–6060 [DOI] [PubMed] [Google Scholar]
- 5.Potter LR, Abbey-Hosch S, Dickey DM 2006. Natriuretic peptides, their receptors, and cyclic guanosine monophosphate-dependent signaling functions. Endocr Rev 27:47–72 [DOI] [PubMed] [Google Scholar]
- 6.Matsukawa N, Grzesik WJ, Takahashi N, Pandey KN, Pang S, Yamauchi M, Smithies O 1999. The natriuretic peptide clearance receptor locally modulates the physiological effects of the natriuretic peptide system. Proc Natl Acad Sci USA 96:7403–7408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nussenzveig DR, Lewicki JA, Maack T 1990. Cellular mechanisms of the clearance function of type C receptors of atrial natriuretic factor. J Biol Chem 265:20952–20958 [PubMed] [Google Scholar]
- 8.Anand-Srivastava MB 1992. Enhanced expression of inhibitory guanine nucleotide regulatory protein in spontaneously hypertensive rats. Relationship to adenylate cyclase inhibition. Biochem J 288(Pt 1):79–85 [DOI] [PMC free article] [PubMed]
- 9.Ruskoaho H 2003. Cardiac hormones as diagnostic tools in heart failure. Endocr Rev 24:341–356 [DOI] [PubMed] [Google Scholar]
- 10.Cody RJ, Atlas SA, Laragh JH, Kubo SH, Covit AB, Ryman KS, Shaknovich A, Pondolfino K, Clark M, Camargo MJ, Scarborough RM, Lewicki JA 1986. Atrial natriuretic factor in normal subjects and heart failure patients. Plasma levels and renal, hormonal, and hemodynamic responses to peptide infusion. J Clin Invest 78:1362–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsutamoto T, Kanamori T, Morigami N, Sugimoto Y, Yamaoka O, Kinoshita M 1993. Possibility of downregulation of atrial natriuretic peptide receptor coupled to guanylate cyclase in peripheral vascular beds of patients with chronic severe heart failure. Circulation 87:70–75 [DOI] [PubMed] [Google Scholar]
- 12.Bryan PM, Xu X, Dickey DM, Chen Y, Potter LR 2007. Renal hyporesponsiveness to atrial natriuretic peptide in congestive heart failure results from reduced atrial natriuretic peptide receptor concentrations. Am J Physiol Renal Physiol 292:F1636–F1644 [DOI] [PubMed]
- 13.Dickey DM, Flora DR, Bryan PM, Xu X, Chen Y, Potter LR 2007. Differential regulation of membrane guanylyl cyclases in congestive heart failure: natriuretic peptide receptor (NPR)-B, Not NPR-A, is the predominant natriuretic peptide receptor in the failing heart. Endocrinology 148:3518–3522 [DOI] [PubMed] [Google Scholar]
- 14.Singh G, Kuc RE, Maguire JJ, Fidock M, Davenport AP 2006. Novel snake venom ligand dendroaspis natriuretic peptide is selective for natriuretic peptide receptor-A in human heart: downregulation of natriuretic peptide receptor-A in heart failure. Circ Res 99:183–190 [DOI] [PubMed] [Google Scholar]
- 15.Pandey KN 1993. Stoichiometric analysis of internalization, recycling, and redistribution of photoaffinity-labeled guanylate cyclase/atrial natriuretic factor receptors in cultured murine Leydig tumor cells. J Biol Chem 268:4382–4390 [PubMed] [Google Scholar]
- 16.Pandey KN, Nguyen HT, Sharma GD, Shi SJ, Kriegel AM 2002. Ligand-regulated internalization, trafficking, and down-regulation of guanylyl cyclase/atrial natriuretic peptide receptor-A in human embryonic kidney 293 cells. J Biol Chem 277:4618–4627 [DOI] [PubMed] [Google Scholar]
- 17.Koh GY, Nussenzveig DR, Okolicany J, Price DA, Maack T 1992. Dynamics of atrial natriuretic factor-guanylate cyclase receptors and receptor-ligand complexes in cultured glomerular mesangial and renomedullary interstitial cells. J Biol Chem 267:11987–11994 [PubMed] [Google Scholar]
- 18.Vieira MA, Gao M, Nikonova LN, Maack T 2001. Molecular and cellular physiology of the dissociation of atrial natriuretic peptide from guanylyl cyclase a receptors. J Biol Chem 276:36438–36445 [DOI] [PubMed] [Google Scholar]
- 19.Jewett JR, Koller KJ, Goeddel DV, Lowe DG 1993. Hormonal induction of low affinity receptor guanylyl cyclase. EMBO J 12:769–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fan D, Bryan PM, Antos LK, Potthast RJ, Potter LR 2005. Down-regulation does not mediate natriuretic peptide-dependent desensitization of natriuretic peptide receptor (NPR)-A or NPR-B: guanylyl cyclase-linked natriuretic peptide receptors do not internalize. Mol Pharmacol 67:174–183 [DOI] [PubMed] [Google Scholar]
- 21.Potter LR, Hunter T 1999. A constitutively “phosphorylated” guanylyl cyclase-linked atrial natriuretic peptide receptor mutant is resistant to desensitization. Mol Biol Cell 10:1811–1820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown LA, Nunez DJ, Wilkins MR 1993. Differential regulation of natriuretic peptide receptor messenger RNAs during the development of cardiac hypertrophy in the rat. J Clin Invest 92:2702–2712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Christoffersen TE, Aplin M, Strom CC, Sheikh SP, Skott O, Busk PK, Haunso S, Nielsen LB 2006. Increased natriuretic peptide receptor A and C gene expression in rats with pressure-overload cardiac hypertrophy. Am J Physiol Heart Circ Physiol 290:H1635–H1641 [DOI] [PubMed]
- 24.Potter LR, Garbers DL 1992. Dephosphorylation of the guanylyl cyclase-A receptor causes desensitization. J Biol Chem 267:14531–14534 [PubMed] [Google Scholar]
- 25.Abbey SE, Potter LR 2002. Vasopressin-dependent inhibition of the C-type natriuretic peptide receptor, NPR-B/GC-B, requires elevated intracellular calcium concentrations. J Biol Chem 277:42423–42430 [DOI] [PubMed] [Google Scholar]
- 26.Koller KJ, Lipari MT, Goeddel DV 1993. Proper glycosylation and phosphorylation of the type A natriuretic peptide receptor are required for hormone-stimulated guanylyl cyclase activity. J Biol Chem 268:5997–6003 [PubMed] [Google Scholar]
- 27.Joubert S, Labrecque J, De Léan A 2001. Reduced activity of the NPR-A kinase triggers dephosphorylation and homologous desensitization of the receptor. Biochemistry 40:11096–11105 [DOI] [PubMed] [Google Scholar]
- 28.Potter LR, Hunter T 1998. Phosphorylation of the kinase homology domain is essential for activation of the A-type natriuretic peptide receptor. Mol Cell Biol 18:2164–2172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sorkin A, Goh LK 2008. Endocytosis and intracellular trafficking of ErbBs. Exp Cell Res 314:3093–3106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schiffrin EL, Turgeon A, Tremblay J, Deslongchamps M 1991. Effects of ANP, angiotensin, vasopressin, and endothelin on ANP receptors in cultured rat vascular smooth muscle cells. Am J Physiol 260:H58–H65 [DOI] [PubMed]
- 31.Pandey KN, Inagami T, Misono KS 1986. Atrial natriuretic factor receptor on cultured Leydig tumor cells: ligand binding and photoaffinity labeling. Biochemistry 25:8467–8472 [DOI] [PubMed] [Google Scholar]