

Abstract
Itaconic acid is an important metabolite produced by macrophages after stimulation with LPS. The role of itaconate in the inflammatory cascade is unclear. Here we used [13C]itaconate and dimethyl [13C]itaconate (DMI) to probe itaconate metabolism, and find that [13C]DMI is not metabolized to itaconate. [13C]Itaconate in the cell culture medium leads to elevated intracellular levels of unlabeled succinate, with no evidence of intracellular uptake. The goal of this study is to encourage the development of effective pro-drug strategies to increase the intracellular levels of itaconate, which will enable more conclusive analysis of its action on macrophages and other cell and tissue types.
Keywords: inflammation, isotopic tracer, macrophage, mass spectrometry (MS), metabolomics, dimethyl-itaconate, itaconic acid
Introduction
Itaconic acid is a dicarboxylic acid polar metabolite originally characterized in Aspergillus terreus, but is also produced in mammalian cells (1). After LPS stimulation, itaconic acid is secreted by macrophages, where it can inhibit bacterial cell growth (1). In macrophages, itaconate synthesis is catalyzed by the immune-responsive gene 1 (IRG1) protein, which mediates the decarboxylation of cis-aconitate to itaconate (2). Metabolomic and fluxomic analysis of LPS-stimulated macrophages demonstrated reduced Isocitrate dehydrogenase-1 (IDH1) expression and increased IRG1 expression, resulting in a diversion of citrate from the TCA cycle2 toward itaconate production (3). This metabolic remodeling results in glutamate serving as the anaplerotic substrate to maintain or elevate succinate levels (3). Moreover, elevated succinate acts as an inflammatory signal that induces secretion of IL-1β and stabilization of HIF-1α (4). Based on the similarity between itaconate (methylene succinic acid) and succinate, recent investigations have focused on the link between itaconate synthesis and succinate accumulation. Two complementary studies reported that itaconate inhibits succinate dehydrogenase and drives succinate accumulation (5, 6). Studying the role of itaconate requires either lowering its intracellular concentration by using IRG1 knock-out mouse models (5, 6) or increasing its intracellular concentration by using either itaconate (5) or a “cell-permeable” analog, dimethyl itaconate (DMI) (6). However, there is no direct evidence that itaconate or DMI can cross cell membranes and increase intracellular itaconate. Without direct evidence of intracellular delivery, it remains unclear whether itaconate-mediated metabolic and inflammatory effects are induced by increasing intracellular itaconate or by an extracellular mechanism.
Here we synthesized isotopically labeled [13C]itaconate and dimethyl [13C]itaconate ([13C]DMI) to directly profile itaconate metabolism and uptake (Fig. 1). This analysis suggests that exogenous itaconate is not taken up into cells and [13C]DMI is not metabolized into [13C]itaconate in bone marrow-derived macrophages. We also report that [13C]itaconate in the cell culture medium leads to elevated intracellular levels of unlabeled succinate, yet there is no evidence of intracellular uptake. Overall, this study highlights current limitations in intracellular itaconate delivery, and emphasizes the development of effective pro-drug strategies to conclusively define its action on macrophages and other cell and tissue types.
FIGURE 1.

Synthesis of [13C]DMI from [13C5]itaconic acid. Shown is an overlay of the mass spectra of unlabeled (black) and labeled (red) DMI showing the +5 mass shift in the parent [M+H]+ and its fragment.
Results and Discussion
Itaconate is a highly polar metabolite that is unlikely to cross into cells without the presence of an active transporter. Because no transporter has been identified, it remains unclear whether the addition of exogenous itaconate can affect intracellular itaconate levels. To test whether exogenous itaconate can enter cells, we treated RAW-264.7 cells with 10 mm itaconate neutralized with sodium hydroxide (pH 7). Cells were incubated for 2–3 h, and then rinsed and quenched. To reduce background signal from residual extracellular and cell surface-associated metabolites, cells were washed either once or twice with 150 mm ammonium acetate before freezing in liquid nitrogen as described previously (7). The extracted metabolites were then analyzed by HILIC-LC/MS. The levels of citrate and succinate did not change significantly following the second wash. Conversely, itaconate levels fell more than 50% after the second wash, suggesting that much of the cell-associated itaconate signal is due to nonspecific association (Fig. 2A). Thus, analysis of cellular uptake is prone to misleading results, because any itaconate adsorbed to the cell surface would contribute to the measured cellular uptake.
FIGURE 2.
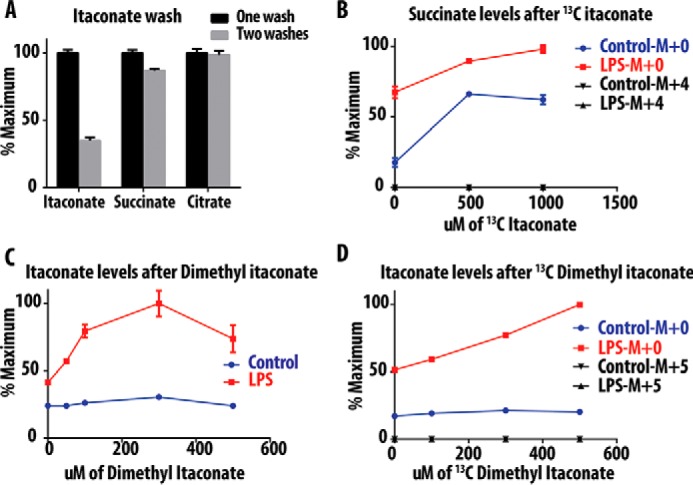
Itaconate and succinate levels after treatment with dimethyl itaconate. A, levels of different metabolites in RAW cells after incubation with itaconic acid and subjecting plates to one or two wash steps. B, levels of succinate in mice BMDM after treatment with serial concentrations of [13C]itaconic acid. C, levels of itaconate in mice BMDM after treatment with serial concentration of DMI. D, levels of itaconate in mice BMDM after treatment with serial concentration of [13C]DMI. Data indicate S.E.
As reported previously (5), we also found that the addition of exogenous itaconate elevates succinate levels in RAW cells and bone marrow-derived macrophage (BMDM) cells (data not shown). To evaluate whether this effect is caused by itaconate conversion to succinate, we treated mouse BMDM with increasing concentrations of [13C]itaconate (0–1000 μm). Similarly, the addition of [13C]itaconate increased levels of succinate in both LPS-stimulated and unstimulated cells. However, the accumulated succinate lacked the M+4 label, and thus cannot be the product of [13C]itaconate metabolism (Fig. 2B). Based on these results, itaconate is not metabolized by cells to form succinate.
Intracellular delivery of polar carboxylate metabolites is typically achieved by synthesis of methyl or ethyl ester analogues, as has been done for succinate (8, 9) and glutamate (10). This pro-drug strategy relies on methyl or ethyl ester hydrolysis by intracellular esterases, leaving the unmasked polar metabolite trapped inside cells. Methyl ester derivatives of DMI have been used to presumably deliver intracellular itaconate; however, the conversion of DMI to itaconate has not been demonstrated (6). To address this possibility, BMDM cells were incubated with vehicle or LPS along with a range of DMI concentrations, followed by LC-MS quantitation of intracellular itaconate (Fig. 2C). DMI treatment had no effect on itaconate levels in vehicle-treated cells (Fig. 2C). In contrast, LPS-stimulated cells revealed a dose-dependent increase in itaconate levels when incubated with increasing concentrations of DMI (Fig. 2C). From these data, we concluded either that DMI potentiates LPS-mediated de novo itaconate synthesis or that LPS stimulates cellular esterases to accelerate DMI methyl ester hydrolysis.
To trace the intracellular fate of DMI, we synthesized isotopically labeled dimethyl [13C]itaconate, which, after esterase hydrolysis, would release the M+5 isotopomer of [13C]itaconate (see “Materials and Methods”). Vehicle-treated and LPS-stimulated BMDM macrophages were incubated with increasing concentrations of [13C]DMI. Again, only LPS-treated cells demonstrated a dose-dependent increase in unlabeled (M+0) itaconate (Fig. 2D). However, there was no measurable 13C-labeled (M+5) itaconate in either vehicle-treated or LPS-treated cells, suggesting either that DMI is not converted to itaconate or that the resulting itaconate is quickly excreted or further metabolized, without intracellular accumulation (Fig. 2D).
Based on these findings, we conclude that exogenous DMI is not directly metabolized to itaconate, but instead somehow potentiates the effects of LPS activation to increase itaconate biosynthesis. How cells achieve alteration in metabolism, including amplification of itaconate and succinate production, following itaconate or DMI treatment will require further investigation. The findings here appear to reveal an alternative mode of dimethyl itaconate action. Importantly, although itaconate is not very electrophilic, methyl esterification is predicted to activate the electrophilic acrylate. Once introduced to cells, DMI could potentially alkylate active site of cysteine residues or alter redox homeostasis. Thus, we agree with previous studies (5, 6) that treatment of cells with either itaconate or DMI has a broad effect on cellular metabolism. However, these metabolic effects do not appear to be due to accumulation of intracellular itaconate, and could be the result of electrophilic stress and covalent inactivation of select metabolic enzymes. Alternatively, itaconate or DMI could bind to surface receptors, such as succinate receptor 1 (SUCNR1/GPR91), which is expressed on macrophages (11), or an as yet unknown receptor to modulate macrophage activation, and thus act entirely extracellularly. It is tempting to speculate that accumulation of extracellular itaconate, which may occur following macrophage death, could provide an amplification signal to enhance the inflammatory activity of local macrophages. Indeed, a recent study has found that succinate release into the extracellular fluid in a model of antigen-induced arthritis can increase macrophage production of IL-β, in a GPR91-dependent manner (12).
Finally, suitable pro-drugs are needed to manipulate intracellular itaconate levels to probe its exact role in the mechanism of macrophage activation and stimulation. Future pro-drug strategies to introduce itaconate into cells should avoid enhancing the electrophilicity of the conjugated acrylate, and focus on selective esterification or amidation of the distal carboxylate.
Materials and Methods
Dimethyl Itaconate and Itaconic Acid
Dimethyl itaconate and itaconic acid were purchased from Sigma (part numbers 109533 and I29204). [13C5]Itaconic acid was synthesized by the Metabolite Standards Synthesis Core at SRI International, arranged through the National Institutes of Health Common Fund's Metabolomics Initiative (13).
[13C5]Itaconic acid dimethyl ester ([13C]DMI) was synthesized by the following procedure. A magnetic stir bar was added to a 10-ml dry round-bottom flask. Next, 5 ml of methanol was added, the flask was cooled to 0 °C on ice, and 1 ml of thionyl chloride was slowly added while stirring. [13C5]Itaconic acid (22 mg, 0.17 mmol) was dissolved in 1 ml of methanol, and then added dropwise to the mixture under a positive pressure of nitrogen for 1 h. The reaction was then warmed to room temperature and stirred overnight. TLC analysis indicated the consumption of starting materials. The solvent was gently removed by rotary evaporation, and the residue was directly purified by flash chromatography eluted with MeOH in dichloromethane (0–5%). Pure fractions of [13C5]itaconic acid dimethyl ester were collected (20 mg, 73%). The product was confirmed by NMR. Using 1H NMR (401 MHz, CDCl3): δ 6.75 − 6.00 (m, 1H), 5.98 − 5.36 (m, 1H), 3.75 (dd, J = 3.9, 1.0 Hz, 3H), 3.68 (dd, J = 3.9, 1.0 Hz, 3H), 3.32 (ddt, J = 130.2, 8.4, 4.7 Hz, 2H), while for 13C NMR (101 MHz, CDCl3) δ 171.28 (dt, J = 58.7, 3.3 Hz), 166.76 (dd, J = 71.8, 3.2 Hz), 133.74 (tdd, J = 72.1, 46.6, 3.0 Hz), 131.05 − 126.04 (m), 37.62 (dddd, J = 58.7, 46.7, 3.3, 1.8 Hz). Product was also confirmed by high resolution mass spectrometry (Fig. 1).
[13C5]Itaconic acid was synthesized by pyrolyzing [13C6]citric acid to [13C5]itaconic anhydride followed by hydrolysis to give [13C5]itaconic acid with an isotopic purity of 99%.3
Bone Marrow-derived Macrophages
Bone marrow-derived macrophages were derived from bone marrow of C57/BL mice and cultured with 30% L-Cell-enriched medium for 6 days prior to the experiment. Macrophage stimulation was performed in RPMI containing 10 mm glucose, 2 mm l-glutamine, 100 units/ml penicillin/streptomycin, and 10% FBS overnight using LPS (20 ng/ml) and IFN-γ (50 ng/ml). RAW 264.7 cells were obtained from the ATCC and cultured until a maximum passage number of 10. RAW cells were grown in DMEM supplied with 100 units/ml penicillin/streptomycin and 10% FBS.
Targeted Metabolite Profiling by LC-MS
Polar metabolites were separated by hydrophilic interaction chromatography using a Luna NH2 column and detected using 6520 Agilent Q-TOF (Santa Clara, CA) as described previously (14–17). Metabolites were identified by accurate mass and by matching their retention time with standards.
Author Contributions
M. E., C. R. E., K. A. G., B. R. M., and C. F. B conceived and designed the experiments. L. L. O and M. J. T synthesized and provided the [13C]itaconate. C. T. M. B. T and B. R. M. synthesized and provided the [13C]DMI. M. E and C. E. performed the experiments. All authors contributed to the manuscript writing
Acknowledgments
We thank Amrita Joshi for macrophage isolation and Hashim Motiwala for helpful discussions (University of Michigan). The Michigan Nutrition Obesity Research Center is funded by National Institutes of Health Grant P30DK089503, the Michigan Diabetes Research Center is funded by National Institutes of Health Grant P30DK020572, and the Michigan Regional Comprehensive Metabolomics Resource Core is supported by National Institutes of Health Grant U24DK097153.
This work was supported by NHLBI, National Institutes of Health Contract Number HHSN268201300022C. This work was also supported by National Institutes of Health Grants DP2GM114848 (to B. R. M.); F31EB019320 (to C. T. M. B. T.); and K08DK102357 (to K. A. G.) The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
L. Jong, L. Olson, and J. J. Tanga, manuscript in review.
- TCA cycle
- tricarboxylic acid cycle
- DMI
- dimethyl itaconate
- HILIC
- hydrophilic interaction liquid chromatography
- BMDM
- bone marrow-derived macrophage.
References
- 1. Strelko C. L., Lu W., Dufort F. J., Seyfried T. N., Chiles T. C., Rabinowitz J. D., and Roberts M. F. (2011) Itaconic acid is a mammalian metabolite induced during macrophage activation. J. Am. Chem. Soc. 133, 16386–16389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Michelucci A., Cordes T., Ghelfi J., Pailot A., Reiling N., Goldmann O., Binz T., Wegner A., Tallam A., Rausell A., Buttini M., Linster C. L., Medina E., Balling R., and Hiller K. (2013) Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc. Natl. Acad. Sci. U.S.A. 110, 7820–7825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jha A. K., Huang S. C., Sergushichev A., Lampropoulou V., Ivanova Y., Loginicheva E., Chmielewski K., Stewart K. M., Ashall J., Everts B., Pearce E. J., Driggers E. M., and Artyomov M. N. (2015) Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42, 419–430 [DOI] [PubMed] [Google Scholar]
- 4. Tannahill G. M., Curtis A. M., Adamik J., Palsson-McDermott E. M., McGettrick A. F., Goel G., Frezza C., Bernard N. J., Kelly B., Foley N. H., Zheng L., Gardet A., Tong Z., Jany S. S., Corr S. C., et al. (2013) Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496, 238–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cordes T., Wallace M., Michelucci A., Divakaruni A. S., Sapcariu S. C., Sousa C., Koseki H., Cabrales P., Murphy A. N., Hiller K., and Metallo C. M. (2016) Immunoresponsive gene 1 and itaconate inhibit succinate dehydrogenase to modulate intracellular succinate levels. J. Biol. Chem. 291, 14274–14284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lampropoulou V., Sergushichev A., Bambouskova M., Nair S., Vincent E. E., Loginicheva E., Cervantes-Barragan L., Ma X., Huang S. C., Griss T., Weinheimer C. J., Khader S., Randolph G. J., Pearce E. J., Jones R. G., et al. (2016) Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. 24, 158–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lorenz M. A., Burant C. F., and Kennedy R. T. (2011) Reducing time and increasing sensitivity in sample preparation for adherent mammalian cell metabolomics. Anal. Chem. 83, 3406–3414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. MacDonald M. J., Fahien L. A., Mertz R. J., and Rana R. S. (1989) Effect of esters of succinic acid and other citric acid cycle intermediates on insulin release and inositol phosphate formation by pancreatic islets. Arch. Biochem. Biophys. 269, 400–406 [DOI] [PubMed] [Google Scholar]
- 9. Mills E. L., Kelly B., Logan A., Costa A. S., Varma M., Bryant C. E., Tourlomousis P., Däbritz J. H., Gottlieb E., Latorre I., Corr S. C., McManus G., Ryan D., Jacobs H. T., Szibor M., et al. (2016) Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167, 457–470.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maechler P., and Wollheim C. B. (1999) Mitochondrial glutamate acts as a messenger in glucose-induced insulin exocytosis. Nature 402, 685–689 [DOI] [PubMed] [Google Scholar]
- 11. Rubic T., Lametschwandtner G., Jost S., Hinteregger S., Kund J., Carballido-Perrig N., Schwärzler C., Junt T., Voshol H., Meingassner J. G., Mao X., Werner G., Rot A., and Carballido J. M. (2008) Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat. Immunol. 9, 1261–1269 [DOI] [PubMed] [Google Scholar]
- 12. Littlewood-Evans A., Sarret S., Apfel V., Loesle P., Dawson J., Zhang J., Muller A., Tigani B., Kneuer R., Patel S., Valeaux S., Gommermann N., Rubic-Schneider T., Junt T., and Carballido J. M. (2016) GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J. Exp. Med. 213, 1655–1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sud M., Fahy E., Cotter D., Azam K., Vadivelu I., Burant C., Edison A., Fiehn O., Higashi R., Nair K. S., Sumner S., and Subramaniam S. (2016) Metabolomics Workbench: an international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Res. 44, D463–D470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. El Azzouny M., Longacre M. J., Ansari I. U., Kennedy R. T., Burant C. F., and MacDonald M. J. (2016) Knockdown of ATP citrate lyase in pancreatic beta cells does not inhibit insulin secretion or glucose flux and implicates the acetoacetate pathway in insulin secretion. Mol. Metab. 5, 980–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. El-Azzouny M., Evans C. R., Treutelaar M. K., Kennedy R. T., and Burant C. F. (2014) Increased glucose metabolism and glycerolipid formation by fatty acids and GPR40 receptor signaling underlies the fatty acid potentiation of insulin secretion. J. Biol. Chem. 289, 13575–13588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. ElAzzouny M. A., Evans C. R., Burant C. F., and Kennedy R. T. (2015) Metabolomics analysis reveals that AICAR affects glycerolipid, ceramide and nucleotide synthesis pathways in INS-1 Cells. PLoS ONE 10, e0129029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lorenz M. A., El Azzouny M. A., Kennedy R. T., and Burant C. F. (2013) Metabolome response to glucose in the β-cell line INS-1 832/13. J. Biol. Chem. 288, 10923–10935 [DOI] [PMC free article] [PubMed] [Google Scholar]