

Abstract
The retrovirus integrase (IN) inserts the viral cDNA into the host DNA genome. Atomic structures of five different retrovirus INs complexed with their respective viral DNA or branched viral/target DNA substrates have indicated these intasomes are composed of IN subunits ranging from tetramers, to octamers, or to hexadecamers. IN precursors are monomers, dimers, or tetramers in solution. But how intasome assembly is controlled remains unclear. Therefore, we sought to unravel the functional mechanisms in different intasomes. We produced kinetically stabilized Rous sarcoma virus (RSV) intasomes with human immunodeficiency virus type 1 strand transfer inhibitors that interact simultaneously with IN and viral DNA within intasomes. We examined the ability of RSV IN dimers to assemble two viral DNA molecules into intasomes containing IN tetramers in contrast to one possessing IN octamers. We observed that the last 18 residues of the C terminus (“tail” region) of IN (residues 1–286) determined whether an IN tetramer or octamer assembled with viral DNA. A series of truncations of the tail region indicated that these 18 residues are critical for the assembly of an intasome containing IN octamers but not for an intasome containing IN tetramers. The C-terminally truncated IN (residues 1–269) produced an intasome that contained tetramers but failed to produce an intasome with octamers. Both intasomes have similar catalytic activities. The results suggest a high degree of plasticity for functional multimerization and reveal a critical role of the C-terminal tail region of IN in higher order oligomerization of intasomes, potentially informing future strategies to prevent retroviral integration.
Keywords: DNA-protein interaction, integrase, protein purification, retrovirus, structure-function, Rous sarcoma virus integrase, intasome, integrase oligomerization, size exclusion chromatography, strand transfer inhibitors
Introduction
The retrovirus integrase (IN)3 is responsible for the concerted integration of linear viral cDNA ends into the host DNA genome and has multiple auxiliary roles including reverse transcription and virus particle maturation (for reviews, see Refs. 1–4). Reverse transcription of the viral RNA in virus-infected cells results in the cytoplasmic preintegration complex (PIC). The PIC is transported into the nucleus where IN inserts the viral DNA ends into the host genome. To understand the structure and function of IN binding at the viral DNA ends, the crystal structure of the spumaretrovirus prototype foamy virus (PFV) intasome containing recombinant IN and two molecules of viral DNA (19 bp in length) was determined (5). PFV IN is monomeric in solution and assembles into a tetrameric structure complexed with either blunt-ended or 3′-OH recessed viral DNA ends, producing a soluble and stable synaptic complex termed the intasome (5, 6). PFV IN assembled onto blunt-ended viral DNA ends can undergo a 3′-OH processing reaction, releasing a dinucleotide in the presence of divalent metal ions (7). This primed 3′-OH recessed viral DNA is now positioned to be covalently inserted into a bendable target DNA sequence that produces the strand transfer complex (STC) (8–10).
Atomic structures of other assembled IN-DNA complexes of alpharetrovirus and betaretrovirus were also accomplished (9, 11). Purified virus-derived and recombinant avian retrovirus INs (12–15) as well as the recombinant mouse mammary tumor virus (MMTV) IN are dimeric in solution (11). In contrast to the tetrameric IN in the PFV intasome and STC, the crystal structure of the Rous sarcoma virus (RSV) STC revealed that an octamer of IN was complexed with a branched viral/target DNA substrate that represents the product produced by the concerted integration reaction (9). Cryo-EM analysis of the MMTV intasome with two viral DNA molecules also revealed IN as an octameric structure (11). These above structural studies of retrovirus IN-viral DNA complexes suggest divergent evolution even though all retrovirus INs contain three canonical domains necessary for integration, the N-terminal (NTD), the catalytic core (CCD), and the C-terminal (CTD) domains (16) (Fig. 1). All three domains are necessary for concerted integration.
FIGURE 1.
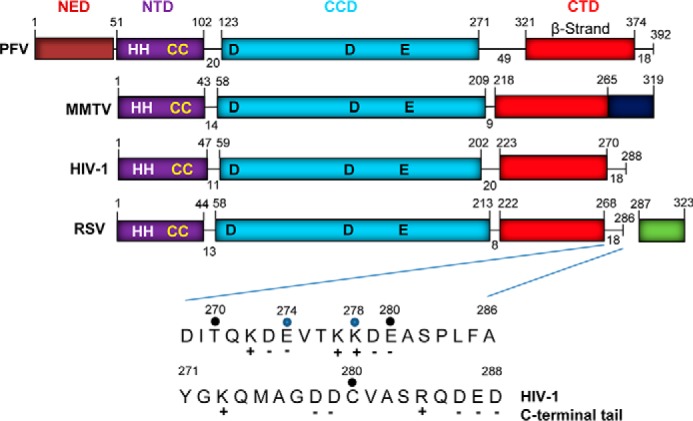
Retrovirus IN domain organizations and the tail region of RSV IN. The N-terminal extension domain (NED), NTD, CCD, and CTD of PFV, MMTV, HIV-1, and RSV INs are shown in different colors. The β-strand-rich region (red) in the CTD is identified. The last 18 residues comprising the tail region of PFV, HIV-1, and RSV INs are identified at their very C termini. Residues 266–319 of MMTV IN (dark blue) are shown from the terminal residue of the β-strand region of the SH3 domain (11). WT RSV IN (amino acids 1–286) and the cleaved 37-amino acid fragment (amino acids 287–323) (green) from the polymerase polyprotein are shown. IN truncations at Glu-274 and Lys-278 are identified by blue dots. Residue Ser-282 is partially phosphorylated in RSV particles (20). Charged residues are indicated. The sequence of the HIV-1 IN tail is also shown.
The domain organizations of IN found in four different retroviruses are shown in Fig. 1. All of the INs have similar size and functional NTD, CCD, and CTD domains, whereas PFV IN possesses an additional N-terminal extension domain that binds viral DNA (5). The linkers between the domains are similar in size except for PFV IN that possesses larger size linkers. These INs except recombinant MMTV IN possess a short “tail” region of 18 residues at their very C terminus adjacent to highly conserved β-strand-rich region of the SH3 domain fold in the CTD (Fig. 1). The tail region of MMTV is 54 residues. For RSV IN, the viral protease processes a precursor polymerase protein, producing wild type (WT) IN that terminates at residue 286, which includes the 18 residues of the tail region (17, 18). The cleaved 37-amino acid polypeptide, whose function is unknown, is not essential for virus replication (19). Approximately 5–6 residues at the C-terminal end of RSV IN (amino acids 1–286) are also not essential for replication (20, 21). There are also 10–12 residues at the very C terminus of the tail region of human immunodeficiency virus type 1 (HIV-1) IN (amino acids 1–288) that are not critical for virus replication but nonetheless enhance the functions of IN with increasing efficiency in accordance with its length (22, 23). The structure of MMTV IN in virus particles and whether there is additional proteolytic cleavage of IN to produce a shorter tail region and a separate cleaved protein fragment are unknown.
Biochemical and biophysical studies have suggested that lentivirus HIV-1 IN is tetrameric in the presence of viral DNA or a branched viral/target DNA product, the result of concerted integration (24–27). Protein-protein cross-linking studies of isolated HIV-1 IN-DNA complexes by agarose gel electrophoresis or visualized by atomic force microscopy suggest the presence of an IN tetramer in these complexes. Recombinant HIV-1 IN forms a variety of multimeric states from monomers to octamers in solution (28–32). Very recently intasome and STC atomic structures with IN from HIV-1 (33) and maedi-visna virus (ovine lentivirus) (34) were determined by cryo-EM to be tetrameric to hexadecameric in nature.
PFV (10) and RSV (9) INs form insoluble complexes in low salt buffers with a branched viral/target DNA substrate that mimics the product of concerted integration. These insoluble complexes can be dialyzed in higher salt buffers, which produces soluble and stable STCs for crystallographic analysis. Using a similar procedure, recombinant MMTV IN complexed with viral DNA was analyzed by cryo-EM (11). Without going through the first insoluble IN-DNA complex formation step used in the above studies, a C-terminally truncated RSV IN (amino acids 1–269) is directly capable of producing a soluble IN-viral DNA complex in the presence of HIV-1 strand transfer inhibitors (STIs) that stabilize the intasome (14). STIs act as interfacial inhibitors that trap kinetically stabilized intasomes (14, 35) by binding to protein-DNA interfaces (5, 36). The RSV intasome contains a tetramer of IN determined by absolute molecular mass measurements using size exclusion chromatography-multiangle light scattering (SEC-MALS) analysis. C-terminally truncated RSV INs (residues 1–269 or 1–270) possess concerted integration activity using 3′-OH recessed viral DNA substrates (9, 14, 15), form IN tetrameric structures in STI-trapped intasomes with 3′-OH recessed viral DNA substrates (18–22 bp) (14), and promote STC assembly containing an octameric IN structure (9).
The crystal structure of RSV STC assembled with IN (residues 1–270) shows a pair of IN dimers engaging the two viral DNA ends for catalysis; these are designated “proximal” subunits (9). Another pair of IN dimers bridge between the viral DNA molecules and help capture target DNA; these are termed “distal” subunits. The CTDs of both the proximal and distal IN dimers make direct contacts with the viral DNA. The CCD-CTD configuration for the proximal IN dimers (9) is comparable with that of the DNA-free IN dimers in its native configuration primed for viral DNA binding (15, 37). The CCD-CTD configuration for the distal RSV IN dimers deviates from the canonical conformation, allowing the distal IN dimers to fit into the RSV STC without making steric clashes with the proximal IN subunits or the 5′-overhang of the viral DNA strand (9).
What controls the assembly of different IN-DNA complexes under different solution conditions? The assembly mechanisms for these complexes are controlled by the structure of the DNA substrates and/or by specific regions of IN. The CTD of WT RSV IN(1–286), which extends from residues 222 to 286, contains a β-strand-rich region of the SH3 domain fold (residues 222–268) and a tail region of 18 residues that are flexible (Fig. 1) (15, 37). The tail region has not been resolved by X-ray structural studies for any retrovirus IN by itself (15, 37–39) or in the presence of viral DNA or branched viral/target DNA substrates (5, 8, 10, 33, 34). Previous results suggested that the RSV IN tail region may affect assembly of the intasome using a viral DNA substrate (14).
Our current study shows that two RSV proximal IN dimers (amino acids 1–269) assemble onto two viral DNA ends at 4 °C to produce an STI-trapped intasome containing IN tetramers. Increasing the temperature for assembly to 18 °C and the length of the tail region of different IN constructs containing residues 1–274 and 1–278 and WT IN(1–286) without and with a single point mutation (S282D) (Fig. 1) allows the formation of intasomes containing IN octamers. RSV IN(1–278) possesses the optimal length for assembly of the octameric IN complex. IN(1–269) failed to produce the octameric IN structure with viral DNA. The results suggest that the tail region of RSV IN plays a key role in assembly of the intasome containing an octameric IN structure, possibly by making further contacts with viral DNA and other IN subunits. In contrast, all of the above RSV IN constructs as well as WT IN containing an additional attached 37 residues at the very C terminus, termed IN(1–323), assemble as an octamer on a branched viral/target DNA substrate, producing the STC. Our results suggest that the tail region of RSV IN plays a critical role for assembly of the intasome containing only viral DNA complexed with IN octamers.
Results
Different Sizes of Assembled RSV Intasomes Are Observed upon Increasing the Length of IN Tail Region beyond the β-Sheet
The assembly of STI-trapped RSV intasome with IN(1–269), 18R gain-of-function (G) U3, and an STI (MK-2048) (40) occurs rapidly at 4 °C, and the major complex is stable under these assembly conditions for at least 120 h (Fig. 2A) (14). SEC-MALS analysis established the absolute molecular mass of this major complex as 151,000 ± 2,000 Da, suggesting the presence of an IN tetramer complexed with two viral DNA molecules (14). The appearance of a minor hump on the slower migrating slope of the tetrameric intasome suggested the presence of another intasome species. Increasing the length of RSV IN by 5 residues from 1–269 to 1–274 under the same assembly conditions resulted in the slow conversion of the tetrameric RSV intasome into an independent IN-DNA complex that migrated with the apparent molecular mass of ∼269,000 Da, consistent with an octameric intasome (Fig. 2B). The conversion of intasomes containing IN tetramers to this slower eluting IN-DNA complex was not complete in this assembly buffer at 4 °C. Similar results were obtained using a 19R GU3 substrate. There was no evidence of other larger size IN-DNA complexes produced under these conditions.
FIGURE 2.
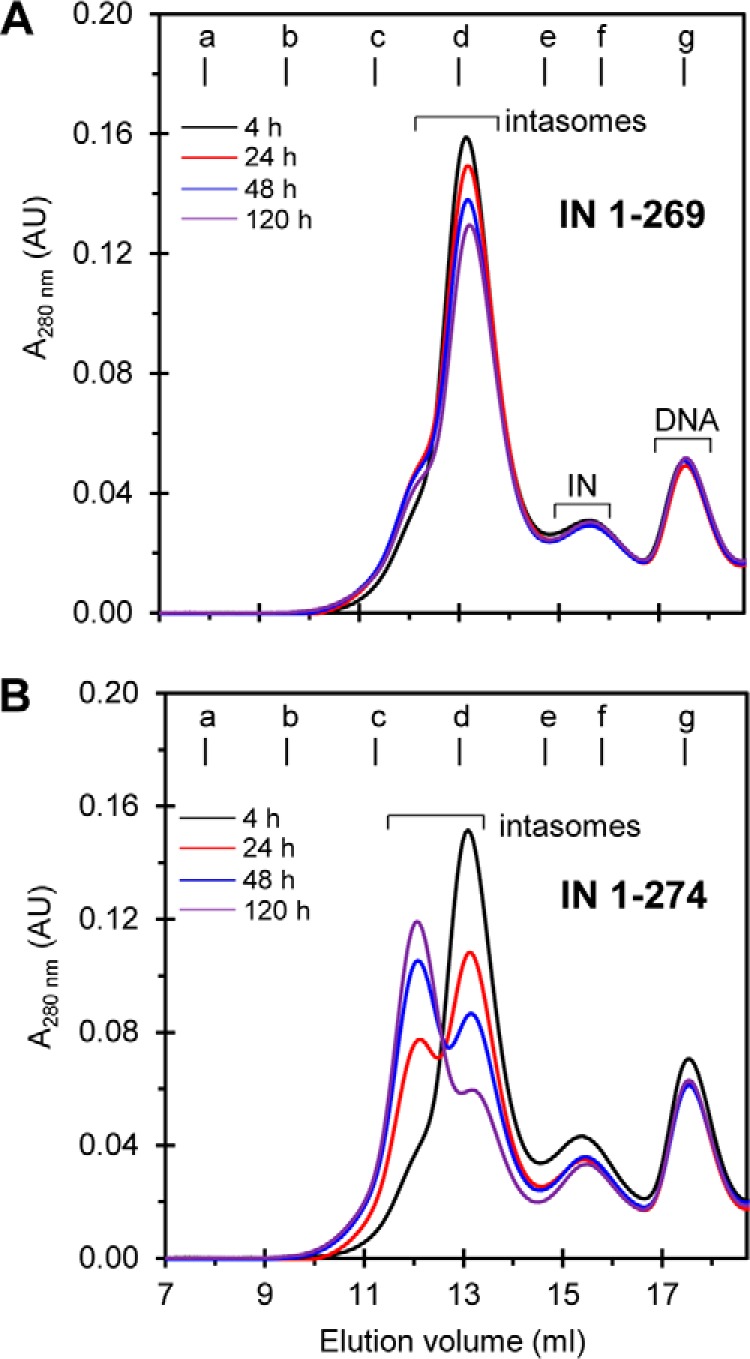
RSV intasome containing an IN tetramer is converted to an intasome containing an IN octamer by C-terminal truncated IN(1–274) but not by IN(1–269). Standard assembly conditions were utilized to produce STI-trapped intasomes with RSV IN(1–269) (A) and IN(1–274) (B) with 18R GU3 in the presence of MK-2048 at 4 °C for extended time periods as indicated. The samples were analyzed by SEC using a Superdex 200 (10/300) column at 4 °C for each time point. With RSV IN(1–269) (A), the tetrameric IN form of the trapped intasome was predominantly observed even after extended times of incubation (120 h). With RSV IN(1–274) (B), a mixture of octameric and tetrameric intasome structures was observed after 24 h in contrast to tetrameric intasome only at 4 h of incubation. After 24 h, the ratio of trapped intasomes with IN octamers to intasomes with IN tetramers increased further with time of assembly. Molecular mass standards labeled from a to g indicate the elution volumes for the void volume and 670, 443, 158, 66, 44, and 17 kDa, respectively. Elution volume (ml) and absorbance at 280 nm are indicated. The void volume is 8.1 ml, corresponding to >700 kDa. AU, arbitrary units.
Length of Tail Region and Temperature Accelerate Conversion of Tetrameric RSV Intasome to an Octameric Intasome
The tail region of IN was extended from 1–274 to 1–278 to address whether this increased length enhanced the assembly of the octameric intasome. Also, we addressed whether increasing the temperature from 4 to 18 °C during assembly would accelerate the conversion of the tetrameric intasome to the octameric IN form. Incubation of IN(1–278) at 18 °C for 4 h with 18R GU3 produced both forms of intasomes, whereas further incubation to 24 h essentially converted all of the transient tetrameric intasome to the most stable octameric structure (Fig. 3A). Incubation up to 72 h slightly increased the yield of the octameric intasome. Note that all of the oligonucleotide (ODN) was essentially utilized after 24 h with IN(1–278). At 18 °C, IN(1–274) also converted the tetrameric intasome to the octameric intasome in a less efficient manner, and not all of IN and ODN were utilized even upon longer incubation to 72 h (Fig. 3B). RSV IN(1–269) was capable of producing the tetrameric intasome in a less efficient manner upon incubation at 18 °C and produced minor larger size complexes (Fig. 3C). The results demonstrated that extension of IN to 1–278 or 1–274 from 1–269 increased the efficiency of IN to produce the octameric intasome at 18 °C and importantly prevented nonspecific aggregation of IN-DNA complexes.
FIGURE 3.
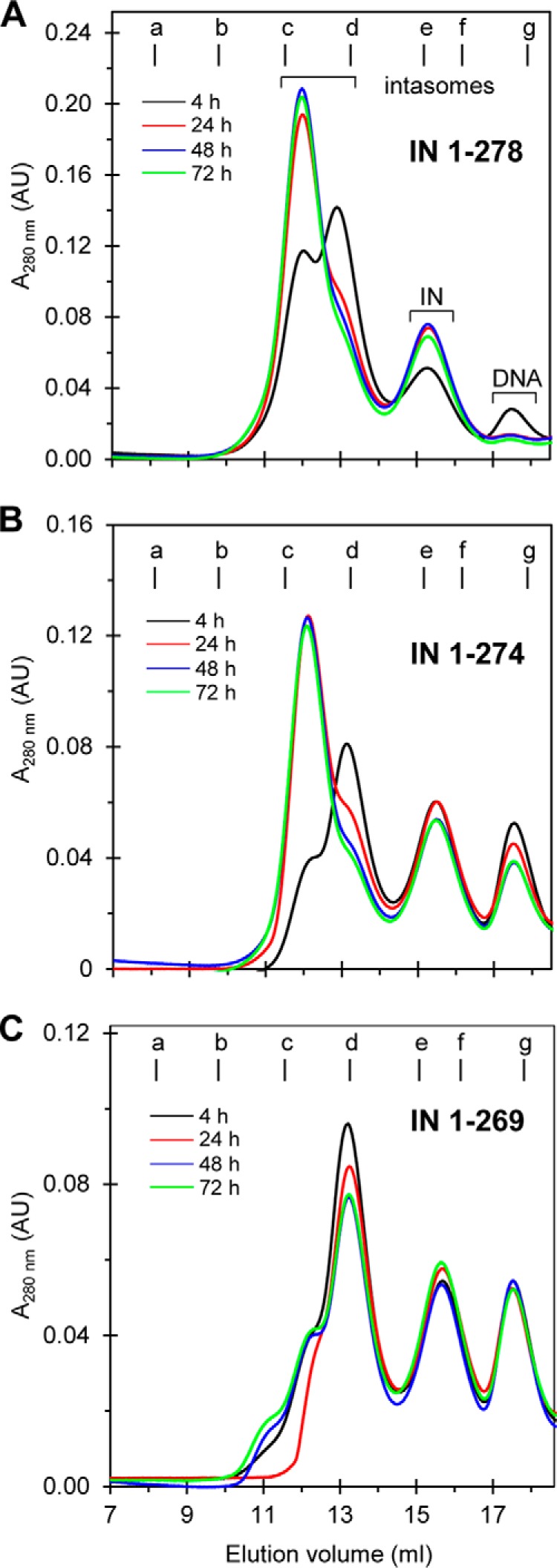
Extension of C-terminal tail region of RSV IN beyond the β-sheet region accelerates the formation of intasomes containing IN octamers at 18 °C. Intasomes were assembled with either RSV IN(1–278) (A), IN(1–274) (B), or IN(1–269) (C) with 18R GU3 DNA in the presence of MK-2048 at 18 °C for the indicated time periods. Standard assembly conditions were used for all proteins. Samples were analyzed by SEC at 4 °C. IN(1–278) and IN(1–274) produced predominantly STI-trapped intasomes containing IN octamers after 24 h and later. With IN(1–269), extended incubation at 18 °C resulted in a minor quantity of octameric STI-trapped intasome and another species. Elution volume (ml) and absorbance at 280 nm are indicated. Molecular mass standards labeled from a to g indicate the elution volumes for the void volume and 670, 443, 158, 66, 44, and 17 kDa, respectively. The void volume is 8.1 ml, corresponding to >700 kDa. AU, arbitrary units.
SEC-MALS Analysis of Octameric RSV Intasome and STC
The absolute molecular mass of the RSV intasome assembled with IN(1–278) was determined. The assembly was for 48 h at 18 °C using standard IN, 18R GU3, and MK-2048 concentrations. The SEC-MALS analysis was performed at 22 °C. The mass of the STI-trapped intasome was 257,000 ± 8,000 Da (n = 4), which correlates with the presence of IN octamers and two viral DNA molecules (Fig. 4A). For comparison, the STC produced with IN(1–278) and the branched viral/target DNA substrate was assembled for 24 h at 4 °C. The mass of this assembled STC was 252,000 ± 9,000 Da (n = 5) (Fig. 4B), similar to that previously reported for the STC using IN(1–269) (9). The mass of purified dimeric IN(1–278) was 59,000 ± 2,000 Da (n = 2). WT RSV IN(1–286), IN(1–269), and IN(1–274) are also dimers (14).
FIGURE 4.

SEC-MALS analysis of octameric intasome and STC assembled by RSV IN(1–278). The STI-trapped RSV octameric intasome was formed with IN(1–278) and 18R GU3 at 18 °C for 48 h. The intasome was subjected to Superdex 200 (10/300) analysis in 20 mm HEPES, 100 mm ammonium sulfate, 200 mm NaCl, 5% glycerol, and 1 mm TCEP, pH 7.5, at 22 °C (A). The RSV STC was formed with IN(1–278) and a branched viral/target DNA substrate at 4 °C for 24 h and analyzed by SEC in 20 mm HEPES, 1000 mm NaCl, 5% glycerol, and 1 mm TCEP, pH 7.5, at 22 °C (B). The absolute molecular mass was determined by light scattering using Wyatt in-line detectors. The mass profile for the complexes is shown in red across each peak. In both panels, the left y axis and right y axis indicate absorbance at 280 nm and molar mass, respectively. The molecular mass of IN(1–278) intasome was 257,000 ± 8,000 Da. The STC produced with IN(1–278) showed a molar mass of 252,000 ± 9,000 Da. Elution volume (ml) and absorbance at 280 nm are indicated. The void volume is 8.1 ml, corresponding to >700 kDa. AU, arbitrary units.
For comparison, we also examined the ability of IN(1–323) to assemble both the intasome and STC. The intasome was assembled in the presence of MK-2048 and 18R GU3 for 48 h at 18 °C under standard conditions. The molecular mass of the RSV intasome produced with IN(1–323) was 293,000 ± 3,000 Da (n = 3), and that of the STC was 272,000 ± 20,000 Da (n = 3). The mass of purified dimeric IN(1–323) was 66,000 ± 2,400 Da (n = 3). In summary, these above measurements of different RSV intasomes and STCs produced by different lengths of IN molecules are consistent with an octameric IN structure associated with each complex.
Specificity for Assembly of Octameric RSV Intasome with IN(1–278)
We determined what components are required for assembly of intasomes containing IN octamers. RSV IN(1–278) and 18R GU3 were incubated with and without MK-2048 at 18 °C for 24 h. The octameric intasome was produced as usual with the STI, whereas there were minimal quantities of intasomes formed in the absence of MK-2048 (Fig. 5). The octameric intasome was also efficiently produced by IN(1–278) in the presence of MK-0536 or dolutegravir but not elvitegravir (data not shown); these HIV-1 IN STIs with the exception of elvitegravir effectively inhibit RSV IN concerted integration (14). No intasomes were produced with a 21R-NSP ODN containing nonspecific DNA sequences using IN(1–278) in the presence of MK-2048 (Fig. 5). Similar STI and viral DNA requirements to produce the RSV intasome containing two proximal IN dimers using IN(1–269) were observed previously (14).
FIGURE 5.
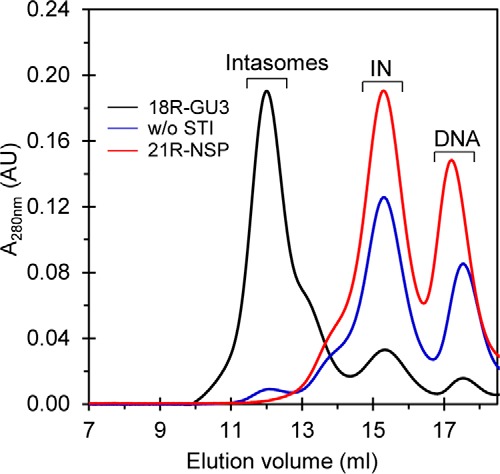
Formation of STI-trapped intasomes with IN octamers requires viral DNA and STI. RSV IN(1–278) was used with 18R GU3 in the presence (black line) and absence (w/o STI) (blue line) of MK-2048 to assemble intasomes under standard conditions at 18 °C for 48 h. A parallel assembly with MK-2048 using a 21R-NSP ODN with nonspecific DNA sequences (red line) was also analyzed. Elution volume (ml) and absorbance at 280 nm are indicated. AU, arbitrary units.
STI binding to the intasome requires 3′-OH processing of the dinucleotide from the catalytic DNA strand by IN prior to displacement of the terminal 3′-deoxyadenosine from the active site by the STI (5, 7). The STI is in equilibrium with the assembled intasomes (14). We investigated whether intasome assembly occurred with blunt-ended 20B GU3 in the presence of MK-2048 at 18 °C for 72 h under standard assembly conditions. Only IN(1–278) and IN(1–274) produced ∼5–10% of the normal quantities of intasomes as compared with using the 3′-OH recessed GU3 DNA substrate (data not shown), suggesting that the proper IN-DNA interface for MK-2048 missing in blunt-ended DNA is important for stabilizing the intasome.
RSV IN 1–278 Achieved the Highest Efficiency for Producing Octameric Intasomes
A direct quantitative comparison among RSV IN(1–274), IN(1–278), and WT IN(1–286) to produce the intasome containing octamers of IN using standard assembly conditions was performed (Fig. 6). Following incubation at 18 °C for 48 h each, the samples were analyzed by SEC. The efficiency of IN(1–278) to produce the intasome containing IN octamers was ∼30% higher than observed with IN(1–274) and IN(1–286). Note again that all of the DNA substrate was incorporated into the intasome using IN(1–278) and not with the other two proteins, suggesting that IN(1–278) has the highest affinity for 18R GU3. Similar results were obtained in this same series of experiments when the samples were analyzed after 24 h (data not shown). The results show that the most efficient formation of the octameric intasome occurs at 18 °C with IN(1–278). This result suggests residues from 270 to 278 in RSV IN preferentially play a critical role in assembling the octameric intasome.
FIGURE 6.
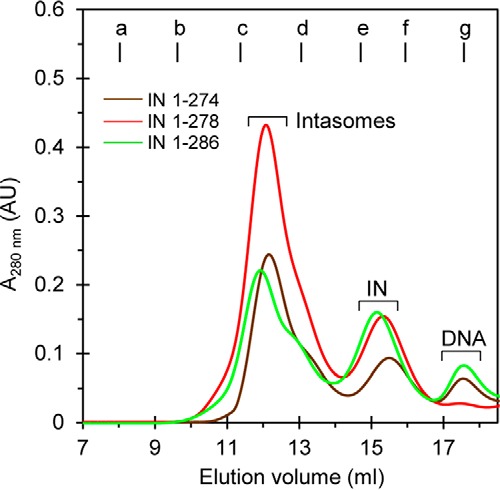
RSV IN(1–278) is most efficient for producing intasomes. Intasomes with IN octamers were assembled under standard conditions at 18 °C for 48 h with IN(1–274), IN(1–278), or WT IN(1–286). Samples (500 μl each) were analyzed by SEC, and the profiles are shown. Molecular mass standards labeled from a to g indicate the elution volumes for the void volume and 670, 443, 158, 66, 44, and 17 kDa, respectively. Elution volume (ml) and absorbance at 280 nm are indicated. The void volume is 8.1 ml, corresponding to >700 kDa. AU, arbitrary units.
Close Proximity of Viral DNA Ends in Intasomes
The ability of STIs to trap the RSV (14) and HIV-1 (26, 35) intasomes containing a tetramer of WT IN suggests close proximity (from ∼10 to 100 Å) of the 5′-ends of the two viral DNA molecules within the complex as measured by fluorescence resonance energy transfer (FRET) (41). We determined that this is also the case with the RSV tetrameric intasome produced by IN(1–269) and the octameric intasome produced by IN(1–278) (Fig. 7). The 5′-terminal nucleotide (A) of a 2-bp overhang of the non-catalytic strand of 20R GU3 was labeled with either Cy3 (donor) or Cy5 (acceptor) fluorophore. Each assembled intasome mixture was diluted to 0.5 μm DNA in the assembly buffer containing 1 μm MK-2048 prior to FRET measurements at 10 °C (Fig. 7). FRET was also observed with assembly mixtures that were diluted to 0.1 μm DNA (data not shown). The results suggest that both STI-trapped intasomes maintain the positions of the 5′-ends of two viral DNA molecules in close proximity.
FIGURE 7.
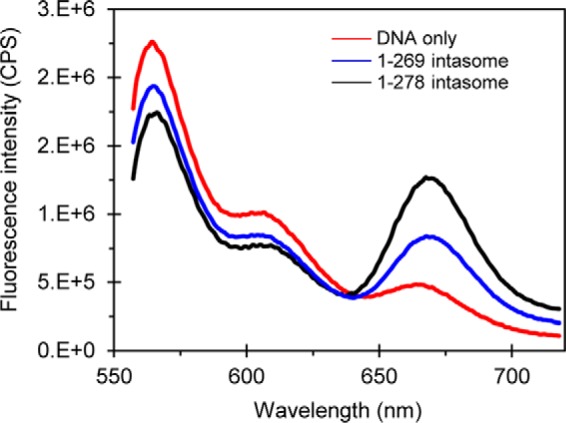
FRET analysis of RSV intasomes. Fluorescence emission spectra of assembled intasomes containing Cy3- and Cy5-labeled 20R GU3 with RSV IN(1–269) (blue line) and IN(1–278) (black line) are shown. The control sample containing only Cy3 and Cy5 DNA and buffer is shown (red line). The quenched emission of Cy3 and simultaneously sensitized emission of Cy5 are observed for both assembled intasomes. CPS, counts per second.
Further Mapping of the Tail Region Residues That Participate in Assembly of Octameric Intasome
We determined whether WT RSV IN(1–286) without and with a S282D mutation and IN(1–323), which extended an additional 37 residues past the terminal Ala (Fig. 1), were also capable of producing the octameric intasome. Standard assembly conditions were used. The yield and assembly efficiency were nearly the same for WT and IN(1–323) after 24 h at 18 °C, whereas IN S282D was the least efficient (Fig. 8, A, B, and C, respectively). In addition, the transition of the tetrameric intasome to the octameric form was slow at 4 °C with these IN constructs, similar to that observed with IN(1–274) and IN(1–278) (data not shown).
FIGURE 8.
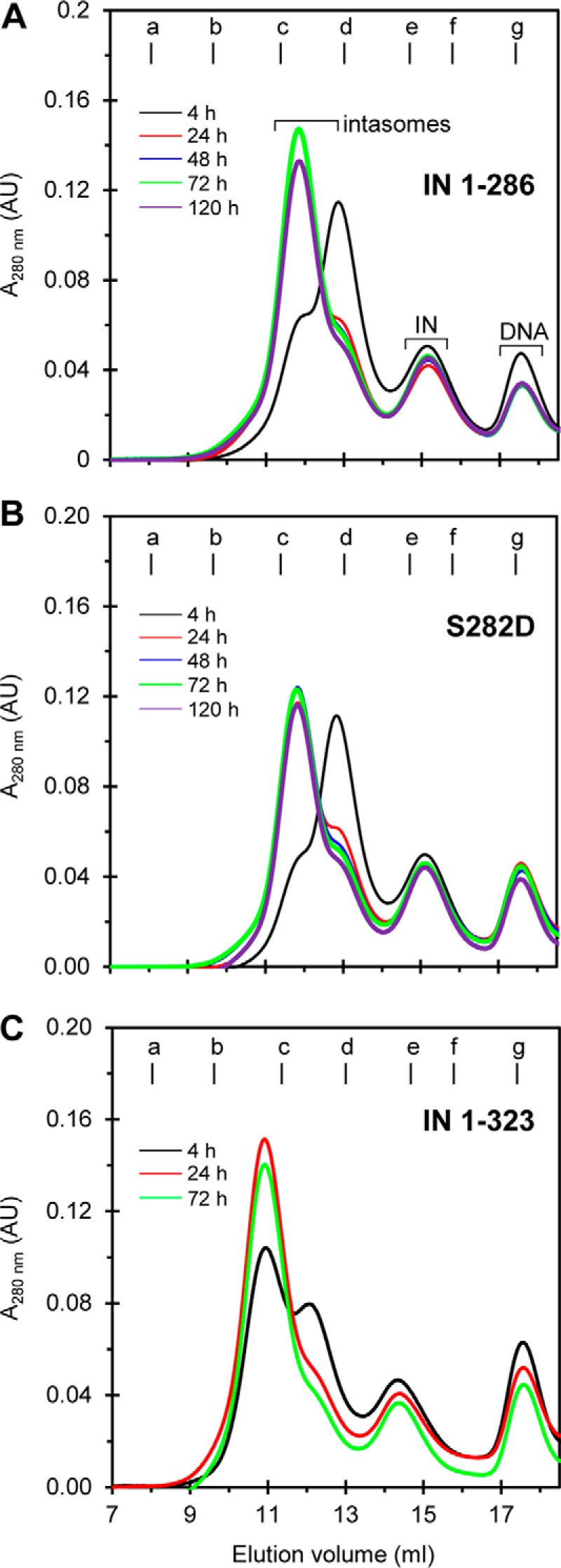
RSV octameric intasomes were assembled by IN with different extended tail regions. WT IN(1–286) was assembled with GU3 in the presence of MK-2048 under standard conditions at 18 °C for various time periods and subsequently analyzed by SEC at 4 °C (A). These same conditions were used to analyze IN(1–286) that contained a S282D mutation (B). The same conditions as above were used for IN(1–323) (C). Elution volume (ml) and absorbance at 280 nm are indicated. Molecular mass standards labeled from a to g indicate the elution volumes for the void volume and 670, 443, 158, 66, 44, and 17 kDa, respectively. The void volume is 8.1 ml, corresponding to >700 kDa. AU, arbitrary units.
Functional Analysis-coupled Concerted Reaction Using Blunt-ended GU3 Substrate
To further investigate other possible functions of the tail region besides affecting the assembly of the RSV intasome, we investigated whether the tail region beyond the β-sheet region affects the ability of IN to promote concerted integration using blunt-ended and 3′-OH recessed 18R GU3 substrates. The 3′-OH processing activity of retrovirus IN lags behind the more active strand transfer activity. As shown previously, IN(1–269) appeared to have diminished concerted integration activity relative to IN(1–274) and IN(1–286) using the GU3 blunt-ended substrate but not with 3′-OH recessed GU3 substrate at 37 °C (Fig. 9) (14). Using initial rates of concerted integration activities at 5 and 10 min, we determined that IN(1–269) had less activity than IN(1–274), IN(1–278), and IN(1–286) using blunt-ended substrate for concerted integration (Fig. 9A). This effect was also observed somewhat after 10 min but not later (Fig. 9B). IN(1–286) containing the S282D mutation displayed similar lagging kinetics as IN(1–269), whereas IN(1–323) displayed diminished capabilities throughout (Fig. 9). The concerted integration activity of these IN constructs using 18R GU3 were essentially equivalent except for IN(1–323) under the same conditions (Fig. 9, C and D). The results suggest that the tail region may facilitate stabilization of the complex for coupling the 3′-OH processing reaction of blunt-ended DNA by IN for concerted integration activity. All of the IN constructs were inhibited by STIs (data not shown) as shown previously (14).
FIGURE 9.

Initial rate of coupled concerted integration using blunt-ended GU3 substrate was controlled by length of tail region. The concerted integration activity of each RSV IN construct was determined with a blunt-ended 20B GU3 substrate (A). Strand transfer was carried out with each respective IN (top) (2 μm) and DNA (1 μm) at 37 °C for 5, 10, and 15 min. Products were deproteinized and run on a 1.8% agarose gel. Lane 1, marked “M,” contains the molecular size markers (Promega kb ladder). Lane 20, marked “C,” does not contain any IN. Concerted products were quantified by a Typhoon 9500 laser scanner, and the -fold difference in activity relative to IN(1–269) was plotted (B). The colors of each IN are indicated. The standard deviation (error bars) was determined from at least three independent experiments. C and D are the same as A and B except 3′-OH recessed 18R GU3 was used. CHS, circular half-site; S.C., supercoiled; O.C., open circular.
We had shown that WT RSV IN(1–286), IN(1–274), and IN(1–269) had equivalent 3′-OH processing activities at 37 °C for 30 min using 32P-labeled blunt-ended 4.6-kbp viral DNA (14). Similar results were obtained with these same IN constructs including IN(1–278) and IN S282D but not IN(1–323), which had significantly lower processing activity (Table 1), explaining the very low activity of IN(1–323) in integration of blunt-ended DNA substrate. IN(1–278) was slightly faster at 5 min of incubation, whereas all were essentially equivalent at 30 min.
TABLE 1.
The 3′-OH processing activities of RSV INs
The 3′-OH processing activity of the RSV IN constructs was determined as described earlier (15) in the presence of Mg2+ at 37 °C for the indicated times. The standard deviation was determined from at least three independent experiments.
| Activity at 5 min | Activity at 30 min | |
|---|---|---|
| % | % | |
| RSV IN(1–269) | 18 ± 1 | 39 ± 5 |
| RSV IN(1–274) | 16 ± 2 | 39 ± 2 |
| RSV IN(1–278) | 24 ± 2 | 44 ± 3 |
| RSV IN S282D | 20 ± 1 | 43 ± 5 |
| RSV IN(1–286) | 21 ± 2 | 43 ± 4 |
| RSV IN(1–323) | 1 ± 0.1 | 4 ± 1 |
Role of Viral/Target DNA Sequences in Formation of the RSV STC
PFV, MMTV, and RSV INs are capable of forming insoluble IN-DNA complexes under low salt conditions. When these insoluble complexes are dialyzed against higher salt buffers they produce either a PFV tetrameric STC (10), an RSV octameric STC (9), or an MMTV octameric intasome for atomic resolution studies (11). PFV IN is capable of directly producing a soluble tetrameric intasome without first producing insoluble IN-DNA complexes (5, 7, 8). Likewise, RSV IN is capable of directly producing a soluble STI-trapped intasome that contains an IN tetramer (14). Using SEC analysis, a direct comparison of these two different assembly methods using RSV IN(1–269) and a branched viral/target DNA substrate (Fig. 10A, see inset) was performed in the absence of STIs. Both methods are essentially equivalent for producing soluble RSV STC (Fig. 10A) that contains an octameric IN structure (9).
FIGURE 10.

Assembly of RSV IN with a branched viral/target DNA substrate to produce the STC. The direct assembly method (dark line) to produce the soluble STC was compared with the dialysis method that causes precipitation of IN-DNA complexes followed by resolubilization in high salt buffer (red line) (A). Complexes were produced with RSV IN(1–269) and a 42-bp STC DNA substrate and analyzed by SEC. A schematic of the branched viral/target DNA substrate is shown (inset). The target DNA sequence in the substrate is blue, viral DNA is black, and 6-bp host site duplication sequences are red. B, six different IN constructs represented by different colored lines were used to produce the STC by the direct assembly method and analyzed by SEC. Representative SEC profiles are shown. The change in elution volume for each sample corresponded to the size of the RSV IN construct. Molecular mass standards labeled from a to g indicate the elution volumes for the void volume and 670, 443, 158, 66, 44, and 17 kDa, respectively. Elution volume and absorbance at 280 nm are indicated. The void volume is 8.1 ml, corresponding to >700 kDa. AU, arbitrary units.
We wanted to determine whether all of the RSV IN constructs were capable of forming the STC in contrast to the inability of only IN(1–269) to produce the octameric intasome containing only viral DNA (Figs. 2A and 3C) (14). Using the direct assembly method, WT RSV IN and all of the modified C-terminal IN constructs essentially produce nearly equivalent quantities of soluble STCs at 4 °C under identical assembly conditions (Fig. 10B). The data suggest that both viral and target DNA sequences play key roles in forming the RSV STC (9), and its assembly is independent of the C-terminal tail. In summary, our studies suggest that residues particularly between 269 and 278 of IN as well as the entire 18-residue tail region of IN(1–286) play a critical role in prompting the assembly of the octameric intasome containing 3′-OH recessed GU3 DNA.
Discussion
The specific functions for the tail region extending beyond the β-strand-rich region of the SH3 domain fold in the CTD of RSV IN or similar tail regions of HIV-1 and PFV INs have not been determined (Fig. 1). Our C-terminal RSV IN truncation studies suggest that these 18 residues are critical for assembly of the STI-trapped intasome containing an octameric IN structure with 18R GU3 DNA but not for the intasome containing only a tetrameric IN structure (14). The assembly conditions for the most efficiently produced and stable RSV intasome containing the octameric IN structure is with IN(1–278) at 18 °C (Fig. 6), suggesting that an optimal kinetic threshold for this specific assembly is met with the viral DNA substrate.
The two proximal IN dimers engage the viral DNA ends for catalysis, whereas the distal IN dimers bridge between the two GU3 DNA molecules and engage the target DNA substrate (9). Almost all of the IN dimers can produce the tetrameric and octameric RSV intasome in the presence of different STIs (Figs. 2, 3, and 6). Only IN(1–269) is not capable of producing the octameric intasome in the presence of MK-2048 (Figs. 2A and 3C), whereas IN(1–278) and IN(1–274) (Figs. 2B and 3), IN S282D, and IN(1–286) (Fig. 8) are fully capable of assembling the octameric RSV intasome at 18 °C. FRET results demonstrated that both IN(1–269) and IN(1–278) produced intasomes that maintain the viral DNA ends in close proximity (Fig. 7), also previously demonstrated with WT RSV IN without STI present (41). The presence of an STI in an intasome formed with HIV-1 IN (26) or PFV IN (5) does not drastically alter its structure. An alternative possibility of the tetramer consisting of two DNA molecules bound to a proximal and distal dimer was also considered. However, this possibility was ruled out as proximal and distal dimers cannot simultaneously take the native (and DNA-binding compatible) conformation (15). An IN dimer can bind the viral DNA but cannot dimerize unless they swap NTDs to form the core tetramer of the intasome (9). The results suggest the additional tail residues in IN(1–278) are necessary and sufficient for interacting with viral DNA and/or other residues for assembly of the stable octameric intasome without target DNA being present.
Functional insights into how the 18-residue length of the tail region in HIV-1 IN affects the replication and integration properties of HIV-1 IN mutants in cell culture and their biochemical properties in vitro have been investigated (22, 23). Single amino acid truncations of the CTD, starting from residues 265 to 277 of HIV-1, revealed several interesting results (23). Truncated “tail-less” CTD HIV-1 IN(1–269) mutant accommodated approximately half of the WT integration frequencies without and with viral complementation in vivo. Truncations from residues 273 to 282 in IN mutants produced near WT HIV-1 replication capabilities. The results suggested that the increased length of the tail region stabilizes the viral DNA ends for 3′-OH processing in vivo, thus promoting concerted integration (23). Similar results were obtained using other sets of C-terminal HIV-1 IN truncations (22). Significantly, the tail region fragment (residues 271–288) of HIV-1 IN cross-linked to viral and target DNA sequences with a hybrid dumbbell DNA substrate used to investigate disintegration activity (42). These HIV-1 IN studies suggest that the tail region is not critical for replication but enhances IN functions with increasing efficacy with its length.
The C terminus on WT IN(1–286) in avian retroviruses is produced by viral protease p15 cleavage of a polymerase precursor, and the cleaved 37-amino acid fragment is not essential for virus replication (Fig. 1) (12, 17, 19, 43–45). Further natural partial proteolytic processing of the IN tail region results in approximately 5–6 residues being removed from its C terminus, which does not grossly affect replication of the virus; partial phosphorylation of RSV IN at Ser-282 in vivo (Fig. 1) prevents proteolytic processing of the tail region at the very C terminus (20, 21, 43). Mutagenesis studies both in vitro and in vivo of the β-strand region of the SH3 domain in RSV IN showed that this region is essential for virus replication and concerted integration (13, 15, 20, 46, 47). These results suggest that both HIV-1 and RSV INs can tolerate partial removal of their tail regions without affecting reverse transcription and integration in vivo. There are no apparent amino acid sequence or charge similarities between the tail regions of PFV, HIV-1, and RSV INs (Fig. 1).
The efficiency for assembling the RSV octameric intasome at 18 °C increased with the length of the tail region to residue 278 and was directly related to the complete disappearance of the GU3 substrate in the assembly mixture in the presence of IN(1–278) (Figs. 3 and 6). Increasing or decreasing the length of the tail region of IN(1–278) hindered the efficient use of the 18R GU3 substrate for assembly (Fig. 6). Further insights into the functions of the tail region of RSV IN were observed. We addressed whether residues beyond Ile-269 increased the ability of IN to couple 3′-OH processing of blunt-ended DNA to promote concerted integration in vitro similarly to what was observed with HIV-1 IN in vivo (23). Increasing the length from IN(1–269) to either IN(1–274) or IN(1–278) also increased the initial rate of concerted integration activity several times in the presence of 20B GU3 substrate (Fig. 9A). This initial faster rate essentially disappeared after 15-min incubation at 37 °C for catalysis in comparison with IN(1–286). It is noteworthy that IN(1–286) containing the S282D mutation also had an initial slower rate of concerted integration activity relative to WT IN (Fig. 9), which is consistent with the slower replication rate of the virus containing this mutation relative to RSV (20). Lastly, the attachment of the 37-amino acid fragment (not essential for replication) to WT IN(1–286) to produce IN(1–323) effectively prevented IN(1–323) to promote concerted integration activity with blunt-ended viral DNA (Fig. 9). Apparently, the positioning of the blunt ends of the GU3 are not correct in this intasome produced by IN(1–323) to allow proper positioning of these ends for coupling 3′-OH processing and concerted integration. The 3′-OH processing of the 32P-labeled blunt-ended 4.6-kbp DNA by IN(1–323) is also defective (Table 1). The results suggest that observed proteolytic processing of polymerase polyprotein in RSV particles to WT IN(1–286) is essential for correct assembly of the intasome and for concerted integration activity in vivo.
DNase I footprint protection studies using virion-purified avian myeloblastosis virus or recombinant RSV IN(1–286) with larger size DNA substrates (∼1 kbp) demonstrated that IN is capable of fully protecting the GU3 or WT U3 non-catalytic DNA strand to ∼20th nucleotide from the DNA end, and this protection is directly coupled to concerted integration activity catalyzed by the intasome (46, 48, 49). IN(1–269) produces the tetrameric intasome (14), whereas the addition of 5 residues or 9 residues to IN(1–269) culminates in the formation of the octameric intasome (Fig. 3). This suggests that the C-terminal tail interacts with viral DNA as well as with adjacent IN molecules to stabilize the octamer.
What are the mechanisms responsible for assembly of the RSV STC and the two different STI-trapped intasomes containing either a tetrameric or octameric IN structure? The branched viral/target DNA is a substrate for RSV IN(1–270) to assemble the STC containing the octameric IN structure for crystallization (9) and formation of the STC in solution (Fig. 10). There are multiple protein-DNA and protein-protein contacts that play various roles in the RSV STC structure. All of the INs in this current study can utilize the branched viral/target DNA substrate (Fig. 10B) to assemble the STC containing IN octamers, suggesting that both viral and target DNA sequences play roles in these assembly processes.
The atomic structure of IN in the intasome without the target DNA is probably similar to what is observed in the RSV STC including the CTD interactions of the distal IN dimers with viral DNA (9). The RSV octameric STC structure modeled with the C-terminal tails (residues 270–286) suggests important roles of the different tail regions for assembly and stability of the octameric intasome (Fig. 11). The positioning of the C-terminal tails (residues 270–286) in the STC complex viewed from the viral DNA side is shown in Fig. 11A. Six of the total eight subunits, from both proximal and distal IN dimers, have their well ordered CTD residue Ile-269 (9) in close proximity with the viral DNA, and subsequently the C-terminal tails potentially have extensive interactions with the viral DNA molecules. Rotating the view of Fig. 11A for 180° in Fig. 11B (shows the view from the target DNA side) demonstrates that the two inner subunits of distal dimers C and G chains have no interactions with DNA molecules. Instead, the three neighboring IN subunits form a pocket where each C-terminal tail fits snugly into the pocket. Fig. 11C shows the side view of the assembled STC and the fitting of the C-terminal tail of C chain (inner subunit of a distal dimer) in the pocket. Fig. 11, D and E, show the detailed conformation of C-terminal tails of D chain (outer subunit of a distal dimer) and C chain with three lysine residues emphasized. There are numerous other potential interactions like the Nζ atom of Lys-277 forming a salt bridge with Asp-7 of the E chain (Fig. 11E). All of the interchain interactions could facilitate the assembly of the octameric complex by incorporating IN subunits into the tetrameric complex. Our model offers the opportunity to investigate the role of various residues in the assembly of both the tetrameric and the octameric intasome.
FIGURE 11.

Hypothetical model of RSV octameric STC with C-terminal residues 270–286. A, view from the viral DNA side. Color coding is as follows: A chain, green; B chain, cyan; C chain, warm pink; D chain, yellow; E chain, light pink; F chain, gray; G chain, sky blue; H chain, orange. Proximal IN dimers include A/B and E/F dimers. Distal IN dimers include C/D and G/H. For the proximal subunits, the A and E chains perform catalysis, whereas the B and F chains are the outer subunits. Viral DNA has interactions with C-terminal tail residues of A, B, D, E, F, and G chains. B, 180° rotation of A viewing from the target DNA side. The C-terminal tail of the C chain fit snuggly in the pocket formed by A, B, D, and E chains. C, side view of C-terminal tails of the C chain in the pocket. D, close-up view of C-terminal tails of the D chain in proximity of viral DNA chain. The positively charged Lys-272, Lys-277, and Lys-278 are identified as blue residues. E, close-up view of the C-terminal tail of the C chain in the pocket formed by A, B, D, and E chains.
In conclusion, our studies suggest that residues between 269 and 278 of RSV IN in the 18-residue tail region of IN(1–286) play a critical role in prompting the assembly of the STI-trapped octameric intasome containing viral DNA. Both HIV-1 and RSV IN biological data support a functional role for the tail region, although atomic structural information is lacking to support a complete understanding. The differential observations to assemble the tetrameric and octameric intasomes between IN(1–269) and IN(1–278), respectively, suggest critical roles of residues 270–278 in recruitment of distal IN dimers. Assembly of the two proximal IN dimers with viral DNA is presumably the first step in the cytoplasmic PIC of infected cells, paralleling the assembly of the intasome in vitro. These observations suggest that a tetramer of IN is the basic catalytic unit for concerted integration for PFV, HIV-1, and RSV INs supported by biological, structural, and biochemical data. The auxiliary role or indeed a possible transient role performed by the tail region of the two proximal and distal IN dimers may provide additional stabilization to the octameric intasome and thus provide a platform for target binding (9). The observations that HIV-1 (50) and murine leukemia virus (51) INs protect the very termini as well as extended regions of the viral DNA ends to ∼200 bp in the cytoplasmic PIC of virus-infected cells suggest that further oligomerization properties of IN are to be discovered.
Experimental Procedures
Production and Purification of RSV IN Constructs
WT RSV Prague A IN (amino acids 1–286) was produced as described (52). C-terminal IN truncation mutants are designated by numbers from the N terminus to the terminating amino acid in the construct. C-terminal truncations 1–269, 1–274, 1–278, and full-length IN containing a single point mutation (S282D) were produced by site-directed mutagenesis of WT RSV IN. The addition of 37 residues to RSV IN resulted in IN(1–323), whose sequence was derived from the polymerase sequence of RSV PrA and PrC strains (53, 54). Negatively charged residues Asp and Glu comprise 27% of these shared protein sequences in this 37-residue fragment. The sequence of this 37-amino acid protein is GISDWIPWEDEQEGLQGETASNKQERPGEDTLAANES. The IN clones were verified by DNA sequence analysis.
RSV IN constructs were expressed in Escherichia coli BL21 (DE3)pLysS, purified, and concentrated to 10–30 mg/ml using Amicon Ultra-15 centrifugal filters as described previously (14). All purified INs were free of contaminating DNA endonuclease activities using supercoiled DNA as substrate.
Concerted Integration and 3′-OH Processing Assays
The concerted integration assay using blunt-ended and 3′-OH recessed ODN viral DNA substrates with RSV IN was described previously (14). The concentrations of IN and the viral ODNs were 2 and 1 μm, respectively. The strand transfer products were separated on a 1.8% agarose gel, stained with SYBR Gold (Invitrogen), and analyzed by a Typhoon 9500 laser scanner (GE Healthcare).
The 3′-OH processing of 32P-labeled blunt-ended viral 4.6-kbp DNA at 37 °C was described previously (15). The time of incubation was varied as indicated. Concentrations of the various purified RSV INs and DNA in the assay mixture were 20 and 0.5 nm, respectively.
Viral DNA and Viral DNA/Target ODNs for Assembly of IN-DNA Complexes
Double-stranded blunt-ended and 3′-OH recessed ODNs containing RSV GU3 long terminal repeat sequences from 18 to 22 nucleotides in length were synthesized by Integrated DNA Technologies (14). The DNA substrates were recessed by 2 nucleotides on the catalytic strand and designated with an R or were blunt-ended and designated with a B. The identified length of the ODN denotes the non-catalytic strand. The sequences were as follows: 18R, 5′-ATTGCATAAGACAACA-3′ and 5′-AATGTTGTCTTATGCAAT-3′; 20B, 5′-GTATTGCATAAGACAACATT-3′ and 5′-AATGTTGTCTTATGCAATAC-3′. The following 3′-OH recessed ODN with nonspecific sequences was also prepared: 21R-NSP, 5′-GCAATGATACCGCGAGACC-3′ and 5′-TGGGTCTCGCGGTATCATTGC-3′.
The 5′-end on the non-catalytic strand of GU3 was also labeled with either Cy5 or Cy3 and synthesized by Integrated DNA Technologies. The RSV branched viral/target DNA substrate mimicking the product of concerted integration was used for the assembly of the STC (9).
Assembly Protocols for Intasomes and STC
There are two methods for assembly of stable RSV IN-DNA complexes. In the first method, intasomes produced with viral DNA require the presence of STIs to form kinetically “trapped” intasomes (14). The standard direct assembly buffer was 20 mm HEPES, pH 7.5, 100 mm ammonium sulfate, 100 mm NaCl, 1 m non-detergent sulfobetaine-201, 10% dimethyl sulfoxide (DMSO), 10% glycerol, and 1 mm tris(2-carboxyethyl)phosphine (TCEP). IN (as monomers), 3′-OH recessed DNA ODN, and STI concentrations were generally set at 45, 15, and 125 μm, respectively, unless otherwise indicated. After addition of DNA to the assembly mixture and subsequently IN, the samples were incubated at 4 or 18 °C for various times as indicated. The STIs used were raltegravir, MK-2048, MK-0536, dolutegravir, and elvitegravir. The direct assembly condition to produce the RSV STC using a branched viral/target DNA substrate was the same as described above for the intasomes except the NaCl concentration was 0.4 m, a 42-bp STC DNA substrate was used (15 μm) (9), and no STIs were present.
The second method to assemble the RSV STC involved the precipitation of RSV IN with the branched viral/target DNA substrate under low salt buffer conditions and subsequent dialysis against high salt buffer to produce a stable and soluble STC as described (9). The high salt buffer for dialysis was 20 mm HEPES, pH 7.5, 0.75 m NaCl, 20% glycerol, and 1 mm TCEP.
Size Exclusion Chromatography and SEC-MALS Analysis
SEC using Superdex 200 (10/300) (GE Healthcare) and molecular mass standards were described previously (14). The SEC buffer for analysis of the RSV intasome was 20 mm HEPES, pH 7.5, 100 mm ammonium sulfate, 200 mm NaCl, 5% glycerol, and 1 mm TCEP. The SEC buffer for analysis of the RSV STC was the same except 1 m NaCl was used. Chromatography was at 4 °C, and UV absorption was monitored at 280 nm. Generally, 250 μl of sample was injected into the column. SEC-MALS analysis to determine absolute molecular weights was as described (9, 14) except the column and buffers were kept at room temperature (22 °C).
Steady-state Fluorescence Measurements
RSV IN (45 μm), fluorophore-labeled ODNs (15 μm), and MK-2048 (125 μm) were used to assemble intasomes for FRET measurements as described earlier (26, 41). Equimolar quantities (7.5 μm each) of Cy3 (donor)- and Cy5 (acceptor)-labeled 20R GU3 were used in the assembly mixture without IN or with IN (IN(1–269) or IN(1–278)) at 4 or 18 °C, respectively, for 24–48 h. To avoid inner filter effects, the assembled mixture was diluted to 0.1 or 0.5 μm DNA with assembly buffer containing 1 μm MK-2048. The samples were analyzed within 10 min of dilution. Fluorescence spectra were collected using a Fluoromax-3 (Jobin Yvon, Inc., Edison, NJ) spectrofluorometer at 10 °C with a temperature-regulated cell holder. The samples were excited with 550-nm wavelength (donor excitation), and emission spectra were collected from 555 to 720 nm for quenching of Cy3 (peak at ∼565 nm) and sensitized emission of Cy5 (peak at ∼668 nm). All spectra were corrected for buffer and instrument.
Modeling
The starting model was the X-ray structure of the RSV octameric STC (Protein Data Bank code 5EJK) (9) in which the nicks in the target DNA strands were sealed. The nucleotides previously missing in the X-ray crystallographic coordinates due to disorder were modeled based on non-crystallographic symmetry-related DNA atoms, and the full-length DNA molecules used in crystallization were built using the MOE program (55). The conformation of C-terminal residues 270–286 was modeled using MODELLER (56). The resulting coordinates were then hydrated, and 0.15 m NaCl was added to a box with dimensions of 160, 156, and 175 Å in the x, y, and z directions, respectively. The system underwent 5,000 cycles of minimization. The molecular dynamics was performed under 310 K for 50,000 cycles using NAMD2 (57). Figures were prepared using PyMOL software.
Author Contributions
K. K. P., S. B., and D. P. G. designed the studies and analyzed the results. K. K. P. and S. B. performed the experiments. K. S. and H. A. constructed and analyzed the model for the C-terminal tail in the intasome. D. P. G. supervised the project and wrote the manuscript with input from all authors. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Merck & Co. and GlaxoSmithKline for HIV-1 inhibitors. Computer resources were provided by the Basic Sciences Computing Laboratory of the University of Minnesota Supercomputing Institute.
This work was supported by National Institutes of Health Grants AI127196 (to D. P. G.) and GM118047 (to H. A.) and by Saint Louis University (to D. P. G.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- IN
- integrase
- RSV
- Rous sarcoma virus
- HIV-1
- human immunodeficiency virus type 1
- PFV
- prototype foamy virus
- MMTV
- mouse mammary tumor virus
- ODN
- oligonucleotide
- PIC
- preintegration complex
- STC
- strand transfer complex
- STI
- strand transfer inhibitor
- G
- gain-of-function
- B
- blunt
- R
- recessed
- SEC
- size exclusion chromatography
- MALS
- multiangle light scattering
- NSP
- nonspecific
- NTD
- N-terminal domain
- CCD
- catalytic core domain
- CTD
- C-terminal domain
- SH3
- Src homology 3
- TCEP
- tris(2-carboxyethyl)phosphine.
References
- 1. Lesbats P., Engelman A. N., and Cherepanov P. (2016) Retroviral DNA integration. Chem. Rev. 16, 12730–12757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Grandgenett D. P., Pandey K. K., Bera S., and Aihara H. (2015) Multifunctional facets of retrovirus integrase. World J. Biol. Chem. 6, 83–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Andrake M. D., and Skalka A. M. (2015) Retroviral integrase: then and now. Annu. Rev. Virol. 2, 241–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Craigie R., and Bushman F. D. (2012) HIV DNA integration. Cold Spring Harb. Perspect. Med. 2, a006890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hare S., Gupta S. S., Valkov E., Engelman A., and Cherepanov P. (2010) Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 464, 232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gupta K., Curtis J. E., Krueger S., Hwang Y., Cherepanov P., Bushman F. D., and Van Duyne G. D. (2012) Solution conformations of prototype foamy virus integrase and its stable synaptic complex with U5 viral DNA. Structure 20, 1918–1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hare S., Maertens G. N., and Cherepanov P. (2012) 3′-Processing and strand transfer catalysed by retroviral integrase in crystallo. EMBO J. 31, 3020–3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Maertens G. N., Hare S., and Cherepanov P. (2010) The mechanism of retroviral integration from X-ray structures of its key intermediates. Nature 468, 326–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yin Z., Shi K., Banerjee S., Pandey K. K., Bera S., Grandgenett D. P., and Aihara H. (2016) Crystal structure of the Rous sarcoma virus intasome. Nature 530, 362–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yin Z., Lapkouski M., Yang W., and Craigie R. (2012) Assembly of prototype foamy virus strand transfer complexes on product DNA bypassing catalysis of integration. Protein Sci. 21, 1849–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ballandras-Colas A., Brown M., Cook N. J., Dewdney T. G., Demeler B., Cherepanov P., Lyumkis D., and Engelman A. N. (2016) Cryo-EM reveals a novel octameric integrase structure for betaretroviral intasome function. Nature 530, 358–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grandgenett D. P., Vora A. C., and Schiff R. D. (1978) A 32,000-dalton nucleic acid-binding protein from avian retravirus cores possesses DNA endonuclease activity. Virology 89, 119–132 [DOI] [PubMed] [Google Scholar]
- 13. Bojja R. S., Andrake M. D., Weigand S., Merkel G., Yarychkivska O., Henderson A., Kummerling M., and Skalka A. M. (2011) Architecture of a full-length retroviral integrase monomer and dimer, revealed by small angle X-ray scattering and chemical cross-linking. J. Biol. Chem. 286, 17047–17059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pandey K. K., Bera S., Korolev S., Campbell M., Yin Z., Aihara H., and Grandgenett D. P. (2014) Rous sarcoma virus synaptic complex capable of concerted integration is kinetically trapped by human immunodeficiency virus integrase strand transfer inhibitors. J. Biol. Chem. 289, 19648–19658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shi K., Pandey K. K., Bera S., Vora A. C., Grandgenett D. P., and Aihara H. (2013) A possible role for the asymmetric C-terminal domain dimer of Rous sarcoma virus integrase in viral DNA binding. PLoS One 8, e56892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Skalka A. M. (2014) Retroviral DNA transposition: themes and variations. Microbiol. Spectr. 2, 1–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Grandgenett D., Quinn T., Hippenmeyer P. J., and Oroszlan S. (1985) Structural characterization of the avian retrovirus reverse transcriptase and endonuclease domains. J. Biol. Chem. 260, 8243–8249 [PubMed] [Google Scholar]
- 18. Hippenmeyer P. J., and Grandgenett D. P. (1984) Requirement of the avian retrovirus pp32 DNA binding protein domain for replication. Virology 137, 358–370 [DOI] [PubMed] [Google Scholar]
- 19. Alexander F., Leis J., Soltis D. A., Crowl R. M., Danho W., Poonian M. S., Pan Y. C., and Skalka A. M. (1987) Proteolytic processing of avian sarcoma and leukosis viruses pol-endo recombinant proteins reveals another pol gene domain. J. Virol. 61, 534–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Horton R., Mumm S. R., and Grandgenett D. P. (1991) Phosphorylation of the avian retrovirus integration protein and proteolytic processing of its carboxyl terminus. J. Virol. 65, 1141–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mumm S. R., Horton R., and Grandgenett D. P. (1992) v-Src enhances phosphorylation at Ser-282 of the Rous sarcoma virus integrase. J. Virol. 66, 1995–1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dar M. J., Monel B., Krishnan L., Shun M. C., Di Nunzio F., Helland D. E., and Engelman A. (2009) Biochemical and virological analysis of the 18-residue C-terminal tail of HIV-1 integrase. Retrovirology 6, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mohammed K. D., Topper M. B., and Muesing M. A. (2011) Sequential deletion of the integrase (Gag-Pol) carboxyl-terminus reveals distinct phenotypic classes of defective HIV-1. J. Virol. 85, 4654–4666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li M., and Craigie R. (2005) Processing of viral DNA ends channels the HIV-1 integration reaction to concerted integration. J. Biol. Chem. 280, 29334–29339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li M., and Craigie R. (2009) Nucleoprotein complex intermediates in HIV-1 integration. Methods 47, 237–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bera S., Pandey K. K., Vora A. C., and Grandgenett D. P. (2009) Molecular interactions between HIV-1 integrase and the two viral DNA ends within the synaptic complex that mediates concerted integration. J. Mol. Biol. 389, 183–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kotova S., Li M., Dimitriadis E. K., and Craigie R. (2010) Nucleoprotein intermediates in HIV-1 DNA integration visualized by atomic force microscopy. J. Mol. Biol. 399, 491–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Engelman A., Bushman F. D., and Craigie R. (1993) Identification of discrete functional domains of HIV-1 integrase and their organization within an active multimeric complex. EMBO J. 12, 3269–3275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Faure A., Calmels C., Desjobert C., Castroviejo M., Caumont-Sarcos A., Tarrago-Litvak L., Litvak S., and Parissi V. (2005) HIV-1 integrase crosslinked oligomers are active in vitro. Nucleic Acids Res. 33, 977–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cherepanov P., Maertens G., Proost P., Devreese B., Van Beeumen J., Engelborghs Y., De Clercq E., and Debyser Z. (2003) HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J. Biol. Chem. 278, 372–381 [DOI] [PubMed] [Google Scholar]
- 31. Pandey K. K., Bera S., and Grandgenett D. P. (2011) The HIV-1 integrase monomer induces a specific interaction with LTR DNA for concerted integration. Biochemistry 50, 9788–9796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee S. P., Xiao J., Knutson J. R., Lewis M. S., and Han M. K. (1997) Zn2+ promotes the self-association of human immunodeficiency virus type-1 integrase in vitro. Biochemistry 36, 173–180 [DOI] [PubMed] [Google Scholar]
- 33. Passos D. O., Li M., Yang R., Rebensburg S. V., Ghirlando R., Jeon Y., Shkriabai N., Kvaratskhelia M., Craigie R., and Lyumkis D. (2017) Cryo-EM structures and atomic model of the HIV-1 strand transfer complex intasome. Science 355, 89–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ballandras-Colas A., Maskell D. P., Serrao E., Locke J., Swuec P., Jónsson S. R., Kotecha A., Cook N. J., Pye V. E., Taylor I. A., Andrésdóttir V., Engelman A. N., Costa A., and Cherepanov P. (2017) A supramolecular assembly mediates lentiviral DNA integration. Science 355, 93–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pandey K. K., Bera S., Vora A. C., and Grandgenett D. P. (2010) Physical trapping of HIV-1 synaptic complex by different structural classes of integrase strand transfer inhibitors. Biochemistry 49, 8376–8387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pommier Y., Kiselev E., and Marchand C. (2015) Interfacial inhibitors. Bioorg. Med. Chem. Lett. 25, 3961–3965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang Z. N., Mueser T. C., Bushman F. D., and Hyde C. C. (2000) Crystal structure of an active two-domain derivative of Rous sarcoma virus integrase. J. Mol. Biol. 296, 535–548 [DOI] [PubMed] [Google Scholar]
- 38. Chen J. C., Krucinski J., Miercke L. J., Finer-Moore J. S., Tang A. H., Leavitt A. D., and Stroud R. M. (2000) Crystal structure of the HIV-1 integrase catalytic core and C-terminal domains: a model for viral DNA binding. Proc. Natl. Acad. Sci. U.S.A. 97, 8233–8238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen Z., Yan Y., Munshi S., Li Y., Zugay-Murphy J., Xu B., Witmer M., Felock P., Wolfe A., Sardana V., Emini E. A., Hazuda D., and Kuo L. C. (2000) X-ray structure of simian immunodeficiency virus integrase containing the core and C-terminal domain (residues 50–293)—an initial glance of the viral DNA binding platform. J. Mol. Biol. 296, 521–533 [DOI] [PubMed] [Google Scholar]
- 40. Grobler J., McKemma P. M., Ly S., Stillmock K. A., Bahnck C. M., Danovich R. M., Dornadula G., Hazuda D., and Miller M. D. (2009) HIV integrase inhibitor dissociation rates correlate with efficacy in vitro. Antivir. Ther. 14, Suppl. 1, A27 [Google Scholar]
- 41. Bera S., Vora A. C., Chiu R., Heyduk T., and Grandgenett D. P. (2005) Synaptic complex formation of two retrovirus DNA attachment sites by integrase: a fluorescence energy transfer study. Biochemistry 44, 15106–15114 [DOI] [PubMed] [Google Scholar]
- 42. Heuer T. S., and Brown P. O. (1997) Mapping features of HIV-1 integrase near selected sites on viral and target DNA molecules in an active enzyme-DNA complex by photo-cross-linking. Biochemistry 36, 10655–10665 [DOI] [PubMed] [Google Scholar]
- 43. Horton R., Mumm S., and Grandgenett D. P. (1988) Avian retrovirus pp32 DNA endonuclease is phosphorylated on Ser in the carboxyl-terminal region. J. Virol. 62, 2067–2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Katz R. A., and Skalka A. M. (1988) A C-terminal domain in the avian sarcoma-leukosis virus pol gene product is not essential for viral replication. J. Virol. 62, 528–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Katz R. A., Kotler M., and Skalka A. M. (1988) cis-Acting intron mutations that affect the efficiency of avian retroviral RNA splicing: implication for mechanisms of control. J. Virol. 62, 2686–2695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chiu R., and Grandgenett D. P. (2003) Molecular and genetic determinants of Rous sarcoma virus integrase for concerted DNA integration. J. Virol. 77, 6482–6492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Moreau K., Faure C., Violot S., Verdier G., and Ronfort C. (2003) Mutations in the C-terminal domain of ALSV (avian leukemia and sarcoma viruses) integrase alter the concerted DNA integration process in vitro. Eur. J. Biochem. 270, 4426–4438 [DOI] [PubMed] [Google Scholar]
- 48. Vora A., and Grandgenett D. P. (2001) DNase protection analysis of retrovirus integrase at the viral DNA ends for full-site integration in vitro. J. Virol. 75, 3556–3567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vora A., Bera S., and Grandgenett D. (2004) Structural organization of avian retrovirus integrase in assembled intasomes mediating full-site integration. J. Biol. Chem. 279, 18670–18678 [DOI] [PubMed] [Google Scholar]
- 50. Chen H., Wei S. Q., and Engelman A. (1999) Multiple integrase functions are required to form the native structure of the human immunodeficiency virus type I intasome. J. Biol. Chem. 274, 17358–17364 [DOI] [PubMed] [Google Scholar]
- 51. Wei S. Q., Mizuuchi K., and Craigie R. (1998) Footprints on the viral DNA ends in Moloney murine leukemia virus preintegration complexes reflect a specific association with integrase. Proc. Natl. Acad. Sci. U.S.A. 95, 10535–10540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McCord M., Stahl S. J., Mueser T. C., Hyde C. C., Vora A. C., and Grandgenett D. P. (1998) Purification of recombinant Rous sarcoma virus integrase possessing physical and catalytic properties similar to virion-derived integrase. Protein Expr. Purif. 14, 167–177 [DOI] [PubMed] [Google Scholar]
- 53. Schwartz D. E., Tizard R., and Gilbert W. (1983) Nucleotide sequence of Rous sarcoma virus. Cell 32, 853–869 [DOI] [PubMed] [Google Scholar]
- 54. Mumm S. R., and Grandgenett D. P. (1991) Defining nucleic acid-binding properties of avian retrovirus integrase by deletion analysis. J. Virol. 65, 1160–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chemical Computing Group Inc. (2015) Molecular Operating Environment (MOE), Chemical Computing Group Inc., Montreal, Quebec, Canada [Google Scholar]
- 56. Sali A., and Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 57. Kale L., Skeel R., Bhandarkar M., Brunner R., Gursoy A., Krawetz N., Phillips J., Shinozaki A., Varadarajan K., and Schulten K. (1999) NAMD2: greater scalability for parallel molecular dynamics. J. Comput. Phys. 151, 283–312 [Google Scholar]