

Abstract
Patients with lung cancers harboring anaplastic lymphoma kinase (ALK) gene fusions benefit from treatment with ALK kinase inhibitors but acquired resistance inevitably arises. A better understanding of proximal ALK signaling mechanisms may identify sensitizers to ALK inhibitors that disrupt the balance between pro-survival and pro-apoptotic effector signals. Using affinity purification coupled with mass spectrometry in an ALK fusion lung cancer cell line (H3122), we generated an ALK signaling network and investigated signaling activity using tyrosine phosphoproteomics. We identified a network of 464 proteins composed of subnetworks with differential response to ALK inhibitors. A small hairpin RNA screen targeting 407 proteins in this network revealed 64 and 9 proteins whose loss sensitized cells to crizotinib and alectinib, respectively. Among these, knocking down fibroblast growth factor receptor substrate 2 (FRS2) or coiled-coil and C2 domain-containing protein 1A (CC2D1A, both scaffolding proteins, sensitized multiple ALK fusion cell lines to the ALK inhibitors crizotinib and alectinib. Collectively, our data provides a resource that enhances our understanding of signaling and drug resistance networks consequent to ALK fusions, and identifies potential targets to improve the efficacy of ALK inhibitors in patients.
Introduction
Mutations or gene rearrangements of key receptor tyrosine kinases (RTKs) confer oncogenic function by disrupting the balance between downstream pro-survival and pro-apoptotic signaling pathways (1). Direct analysis and modeling support the idea that oncogene inhibition by kinase inhibitors leads to a temporal imbalance in these signals whereby pro-apoptotic signals outweigh pro-survival signals (2). For example, pro-survival signals from the kinases ERK and AKT, regulated by the epidermal growth factor receptor (EGFR), degrade more quickly in response to EGFR-targeted tyrosine kinase inhibitors (TKIs) than pro-apoptotic signals from the mitogen-activated protein kinase (MAPK) p38, leading to cell death (1). Changes in downstream signaling that alter the decay rates of survival signals can alter the aggregate survival and death signaling, resulting in changes in tumor cell survival and ultimately tumor growth or regression (2). This model implies that the molecular network circuitry that lies between the oncogene and the distal pro-survival or pro-apoptotic signals could play an important role in affecting the temporal relationships and the ultimate cell decision in response to kinase inhibitors directed against a driver oncogene. This has potential clinical relevance in developing strategies to thwart residual disease in oncogene-driven cancers and eliminate “persister” cells that give rise to overt disease recurrence (3–5).
Downstream of RTKs is a complex network of kinases, phosphatases, adaptor proteins, and negative regulators that tune survival signals emanating from RTKs. A protein network centered on EGFR using literature knowledge identified sub-networks of proteins that influenced sensitivity to EGFR-targeting agents and led to rational combinations that enhanced responses to EGFR antagonists (6). Similarly, an experimentally generated protein network using mass spectrometry-based proteomics centered on mutant EGFR in lung cancer cells was shown to harbor sub-network proteins that affect cell survival (7). Determining the functional relevance of each component in the balance of pro-survival and pro-death signals, as well as tuning responses to kinase inhibition, is complicated by complexity of the network architecture and protein expression levels of each component. Simple signaling models along with mathematical modeling have demonstrated that combination effects of hitting two proteins can be non-obvious and is a manifestation of the topology or circuitry of the signaling network (8). The existence of feedback modules can further drive uncertainly as to the role of particular combination therapies. Counterintuitive results can be observed based on which nodes are inhibited and how the nodes are organized in a network. For these reasons, focused experiments that assess removal of each node within a complex system may be necessary to fully understand their effects.
We hypothesized that an RTK-centered protein network could identify sub-network proteins that affect responses to a kinase inhibitor directed against RTK. We hypothesized that a natural area to hunt for such sub-networks would be in the proximal signaling machinery used by RTK to transduce downstream signaling, by virtue of its ability to shape downstream imbalances between pro-survival and pro-apoptotic signals. To test this idea, we explored cells harboring a fusion of the gene encoding anaplastic lymphoma kinase (ALK) with that encoding echinoderm microtubule associated protein-like 4 (EML4). This EML4-ALK rearrangement occurs in approximately 4% to 5% of lung cancer patients, and these patients derive some initial benefit from treatment with ALK TKIs (9–11). However, primary resistance and acquired resistance attenuate the curative potential of ALK TKIs and are thus major hurdles in ALK-directed therapy (12, 13). One resistance mechanism is secondary ALK domain mutations, which in some cases can be overcome by newer generation ALK TKIs that have activity against secondary mutations (12, 14) (15). A second resistance mechanism class involves bypass signaling mechanisms, such as activation of other RTKs, including EGFR and insulin-like growth factor 1 receptor (IGFR) (16–18). Preclinical results suggest that co-targeting bypass targets, such as heat shock protein HSP 90 (HSP90) (19), EGFR (18), or IGF-1R (16), can overcome ALK TKI resistance driven through these mechanisms.
To enrich for these ALK TKI sensitizer proteins, we used (i) affinity purification combined with mass spectrometry to identify ALK protein complexes in ALK-rearranged and ALK TKI-sensitive lung cancer cells, and (ii) tyrosine phosphoproteomics to further identify both direct and indirect ALK signaling substrates. Mass spectrometry-based quantitative proteomics provides a powerful approach for characterizing the phosphotyrosine (pTyr) proteome, identifying targetable signaling, and decoding the composition of protein complexes. Integration of these two datasets enabled us to create an ALK signaling network or interactome from which to identify with RNA interference analysis key proteins regulating the balance between pro-survival and pro-apoptotic signals (Fig. 1). In the end, our findings revealed new insights into the EML4-ALK signaling network, the mechanisms of ALK TKIs, and the identities of a number of targets that sensitize ALK rearranged lung cancer cells to ALK TKI.
Figure 1. Schematic of the EML4-ALK centric signaling network strategy.
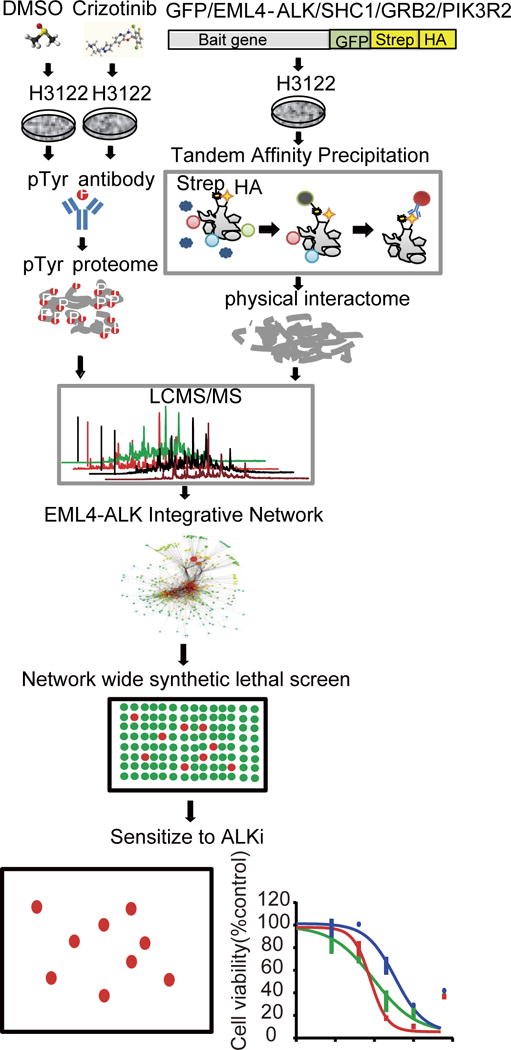
Phosphoproteomics and tandem affinity precipitation approaches were applied to profile the global Tyr phosphoproteome, characterize the perturbation of the Tyr phosphoproteome by ALK TKIs, and uncover EML4-ALK protein complexes in EML4-ALK-driven NSCLC cells. All identified Tyr-phosphorylated proteins and physically interacting proteins were integrated into an EML4-ALK interactome, which informed a global EML4-ALK signaling network. A knowledge-based pathway analysis combined with TKI effects and an unbiased network-wide synthetic lethal screen were used to identify sensitizers to ALK TKI.
Results
Identification of proximal EML4-ALK protein complexes
To gain insight into key protein interactions of EML4-ALK, we set out to dissect complexes of EML4-ALK and three known interacting partners and adaptor proteins (Src homology 2 domain-containing transforming protein 1 (SHC1), growth factor receptor bound protein 2 (GRB2), and phosphoinositide-3-kinase regulatory subunit 2 (PIK3R2), which form complexes with ALK fusion proteins in various cellular contexts (20–23). A recent study highlighting the importance of ALK-driven mitogen-activated protein kinase (MAPK) signaling indirectly suggests an important role for adaptor proteins, such as GRB2 and SHC1, that couple RTKs to MAPK signaling (24). We first examined the effect of loss-of-function of SHC1 or GRB2 on H3122 and STE1 cell viability using real-time analysis technology (Fig. 2A). Both H3122 and STE1 cells harbor an EML4-ALK fusion and are sensitive to ALK TKI. Our results show that siRNA-mediated knockdown of ALK inhibits proliferation in both H3122 and STE1 cells, as expected. GRB2 knockdown resulted in the strongest suppression on cell viability compared to ALK knockdown in both cells. Knockdown of ALK, and to a lesser extent GRB2, reduced the downstream phosphorylation of ERK (Supplemental Figure S1). SHC1 knockdown had more modest effects compared with that of GRB2, but with stronger effects seen in STE1 compared with H3122 cells. These results indicate an important functional role for the EML4-ALK-SHC1-GRB2 complex in these ALK-rearranged lung cancer cells. Because ALK inhibitors decrease AKT phosphorylation, we also examined the effect of the loss of PIK3R2 on H3122 cell viability and found that knockdown of PIK3R2 reduced cell viability (Fig. 2A) (25). Next, we experimentally determined protein complexes of EML4-ALK and created a physical map (or “interactome”) of protein complexes. Currently reported ALK protein-protein interactions come from fusion of nucleophosmin with anaplastic lymphoma kinase (NPM-ALK), full-length ALK, or EML4, but to our knowledge no studies have performed experiments in EML4-ALK-rearranged lung cancer cells (20, 21). We genetically tagged four bait proteins (EM4-ALK, SHC1, GRB2, and PIK3R2) with Strep and HA sequences and then expressed them in H3122 cells using retroviruses. Green fluorescent protein (GFP) was also tagged as the negative control. Two-step pulldown against two tags was subsequently used to isolate the protein complexes, which were then identified with mass spectrometry (7). We identified 84, 96, 64, and 62 unique proteins from EML4-ALK, SHC1, GRB2, and PIK3R2 pulldowns, respectively, in H3122 cells. Results from the individual pulldowns were merged into a physical EML4-ALK bait-prey network containing 169 unique proteins (Fig. 2B). In addition to SHC1, GRB2, and PIK3R2, six other proteins (heat shock protein family D (Hsp60) member 1 (HSPD1), signal transducer and activator of transcription 3 (STAT3), heat shock protein 90kDa alpha family class A member 1 (HSP90AA1), lymphoid-restricted membrane protein (LRMP), tubulin beta class I (TUBB), and ubiquitin A-52 residue ribosomal protein fusion product 1(UBA52)) previously reported to interact with ALK were identified in our experiment and are common binding partners of ALK (21, 26, 27). We next determined how crizotinib altered the ALK complex. H3122 cells containing Strep-HA tagged EML4-ALK were treated with and without crizotinib, and the EML4-ALK complexes were isolated with Strep pulldown and profiled with MS. We found that crizotinib dissociated both SHC1 and GRB2 from the ALK complex consistent with their role in mediating ALK signaling (Fig. 2C).
Figure 2. Physical EML4-ALK protein-protein interactome.
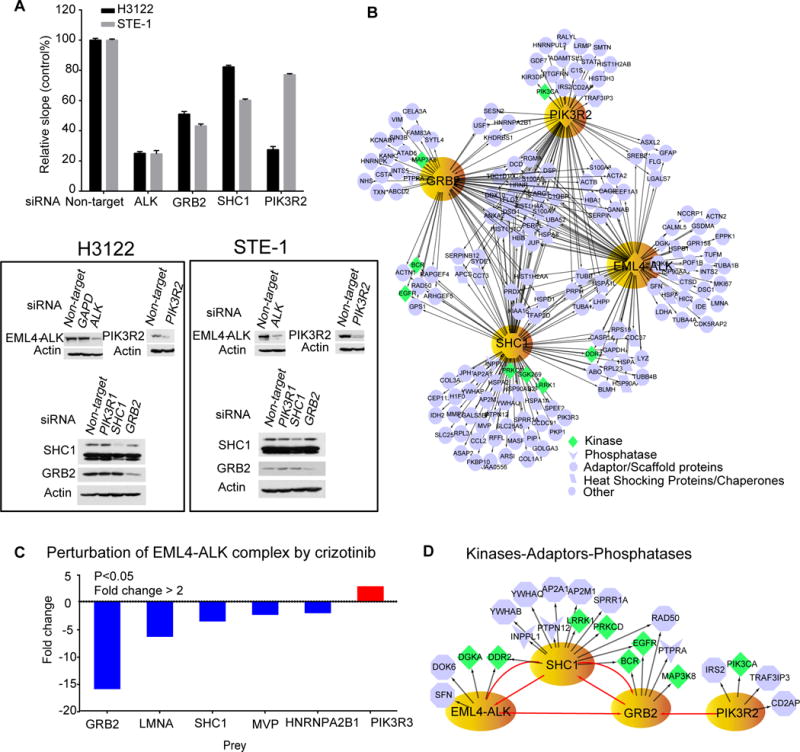
(A) Functional association of selected bait proteins with EML4-ALK signaling. H3122 and STE1 cell viability (relative slope 1/hour of index curve) was analyzed by real time cellular analysis after treating with control siRNA or siRNA targeting ALK, SHC1, GRB2, and PIK3R2. The cell index was recorded every 15 min over 120 hours. Data are means ± SD of 3 experiments. Knock down of each protein was confirmed with Western blot. Blots are representative of 3 experiments. (B) Protein complex network constructed from tandem affinity precipitation for GFP and LC MS/MS in H3122 cells expressing strep- and HA-tagged GFP-EML4-ALK, SHC1, GRB2 and PIK3R2 and visualized in Cytoscape 2.8.3. Bait proteins are indicated in yellow. Data represents two experiments, each with two biological replicates for each bait protein and each biological replicate run in a technical replicate. To remove non-specific protein interactions, prey proteins identified from GFP pulldowns were subtracted from pulldown results from each bait. (C) Fold change in the abundance of EML4-ALK complex in response to crizotinib (3 hours) in H3122 cells, assessed with one-step Strep pulldown and profiled by LC MS/MS. Changes in the abundance of binding partners of EML4-ALK were quantified by comparing peak area of their representative unmodified peptides with or without crizotinib treatment. Data represents a single experiment with biological replicate for both DMSO or crizotinib and each biological replicate run in a technical replicate. A t-test was used to compare EML4-ALK prey abundance between untreated and treated samples (D) Kinase-phosphatase-adaptor subnetwork was extracted from the entire physical interactome. Red lines indicate interactions identified with the four bait proteins.
The physical EML4-ALK interactome is composed of different protein types such as kinases, phosphatases, and adaptors, which are critical elements for kinase signaling. We focused our attention on these core elements and extracted kinases, adaptors, and phosphatases from EML4-ALK physical network and created a smaller subnetwork using PhosphoSitePlus (28). Examination of this smaller subnetwork identified potential insertion points for other signaling proteins to modulate ALK driven signaling events (Fig. 2D). We found EGFR bound to GRB2, suggesting how EGFR signaling can control this network and drive resistance to ALK TKI by facilitating EGFR:GRB2 signaling complexes. Interestingly, we found discoidin domain receptor tyrosine kinase 2 (DDR2), a receptor tyrosine kinase whose ligand is collagen, bound to both EML4-ALK and SHC1 baits. This may indicate a role for collagen signaling in affecting ALK signaling networks and ALK TKI sensitivity. Protein kinase C delta (PKC-δ), previously found to play a role in ALK TKI resistance, was found in complex with SHC1 (13). We also found insulin receptor substrate 2 (IRS2) bound to PIK3R2, suggesting an insertion point for IGF signaling that can drive ALK TKI resistance (16). Finally, we identified two phosphatases, protein tyrosine phosphatase, non-receptor type 12 (PTPN12) and inositol polyphosphate phosphatase like 1 (INPPL1) that could be involved in dephosphorylation of ALK signaling substrates. PTPN12 has been reported to interact with and inhibit other kinases, including MAPK (29), protein tyrosine kinase 2 (PTK2) (29), ERB-B2 receptor tyrosine kinase 2 (ERBB2) (30), EGFR (30), hepatocyte growth factor receptor (MET) (31), activated CDC42 kinase 1 (ACK1) (31), and the SRC family nonreceptor tyrosine kinase LCK (31). INPPL1 was reported to interact with MET (32), EGFR (33), and phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) (34). In summary, the physical interactome and visualization of smaller subnetworks identified known mediators of ALK TKI sensitivity, suggesting potential complexes that could be associated with ALK TKI resistance.
Characterizing basal EML4-ALK tyrosine phosphoproteome and crizotinib-induced perturbations
One limitation of this group of experiments is that tandem affinity purification followed by mass spectrometry may miss more distal changes in signaling, including ALK kinase substrates and changes induced by ALK inhibition. To address this limitation, we characterized tyrosine phosphorylation in EML4-ALK-rearranged and ALK TKI-sensitive H3122 cells using a mass spectrometry-based quantitative phosphoproteomic approach. Briefly, H3122 cells were exposed to crizotinib or DMSO for 3 hours and then tyrosine phosphorylated peptides were enriched with an antibody specifically recoginizing phosphorylated tyrosine residues and detected by liquid chromatography tandem mass chromatography (LC MS/MS) (35). We identified a total of 487 unique pTyr sites assigned to 310 unique phosphoproteins (table S1). Phosphorylation of 68 unique pTyr sites on 48 unique proteins decreased with crizotinib, whereas phosphorylation of 69 unique pTyr sites on 58 unique proteins increased with crizotinib (P<0.05, |fold change| >1.5) (Fig. 3A). These results are consistent with our studies in EGFR mutant lung cancer cells where we also identified increased pTyr peptides following EGFR TKI (35). We identified several pTyr proteins previously linked to ALK signaling pathways in non-EML4-ALK cell contexts. These included receptor-type tyrosine-protein phosphatase eta (PTPRJ) and protein tyrosine phosphatase, non-receptor type 6 (PTPN6) (31, 36), which function as phosphatases and dephosphorylate ALK to decrease its activity. This group also included the p85 subunit of PI3K (37), STAT3 (38), P130CAS (39), fibroblast growth factor receptor substrate 2 (FRS2) (40), bifunctional purine biosynthesis protein PURH (PUR9) (41), and mitogen-activated protein kinase 1 (MAPK1) (21), which have been previously identified as ALK substrates. Our analysis found that phosphorylation of STAT3 (pTyr705, −3.76-fold), FRS2 (pTyr349, −7.58-fold), MAPK1 (pTyr187, −2.65-fold), and PTPN6 (pTyr 536, 2.34-fold) are reduced by crizotinib in H3122 cells (table S1), indicating that these proteins are common components of EML4-ALK signaling.
Figure 3. Phosphoproteomics characterizes the response of H3122 cells to crizotinib.
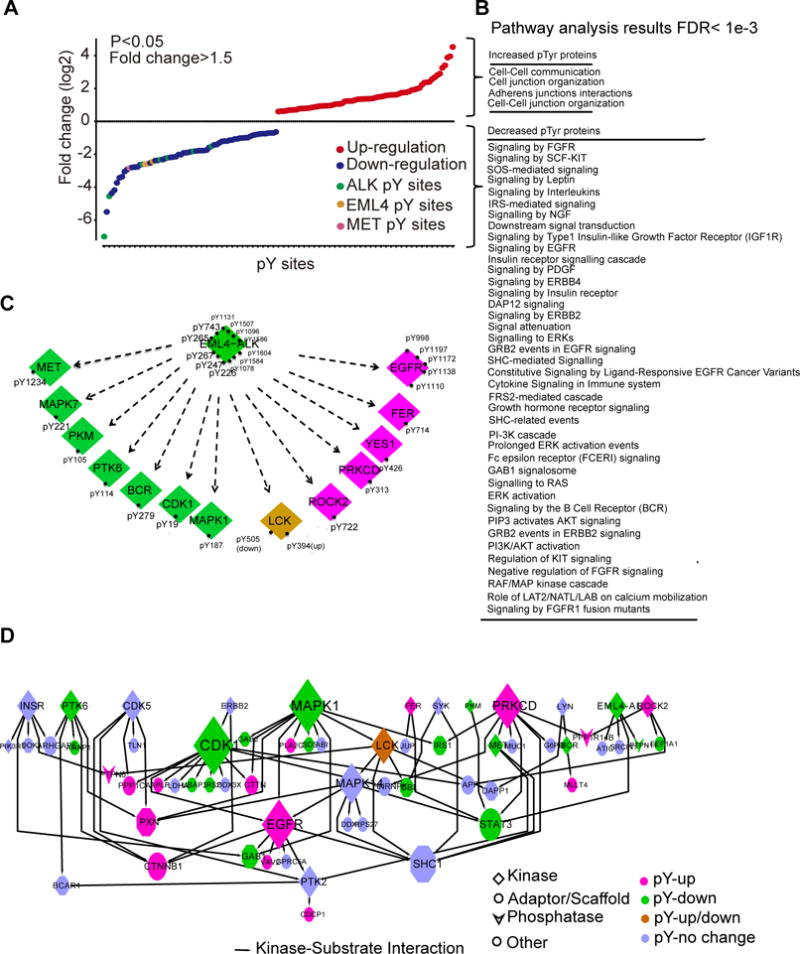
(A) Fold-change in the abundance of tyrosine-phosphorylated (pTyr) peptides in H3122 cells treated with crizotinib (1 μM, 3 hours), relative to those treated with DMSO, was quantified using label-free, peak area. Data are representative of a single experiment with biological replicates for DMSO, crizotinib 100 nM, and crizotinib 1000 nM treated samples with each run in a technical replicate. A two-sample t-test was performed to compare differential phosphorylation for each tyrosine between the control and treatment group treated at 1000 nM (B) Pathways in which crizotinib increased or decreased the Tyr-phosphorylation of proteins found in (A) in H3122 cells (A). (C) Kinases and pTyr sites regulated by crizotinib in cells described in (A). (D) Kinase-substrate interactions among pTyr proteins identified in (A) was extracted from PhosphositePlus (http://www.phosphosite.org/homeAction.action), with the node size representing the number of substrates, and colors representing changes by crizotinib.
We next used pathway analysis tools to annotate signaling pathways regulated by crizotinib. We identified in total 119 signaling pathways overrepresented by the entire pTyr dataset (FDR <0.001, N>5; table S2). Next, we examined pathways enhanced or diminished by crizotinib treatment. As expected, many signaling pathways annotated to kinase signaling were inhibited by crizotinib, including signaling associated with RTK signaling as well as signaling from key adaptor proteins, such as IRS, GRB2, SHC, FRS2, and GRB2 associated binding protein 1 (GAB1) (Fig. 3B). Interestingly, crizotinib treatment was associated with increased abundance of phosphopeptides associated with cell junction pathway signaling, including members of the catenin signaling complex (catenin beta 1(CTNNB1), cortactin (CTTN), catenin delta 1(CTNND1)) and other proteins such as PXN, rho-associated, coiled-coil containing protein kinase 2 (ROCK2), vav guanine nucleotide exchange factor 2 (VAV2), integrin subunit beta 1 (ITGB1), tight junction protein 1 (TJP1), and myeloid/lymphoid or mixed-lineage leukemia; translocated to, 4 (MLLT4).
Next, we examined alterations in pTyr peptide abundance of kinases affected by crizotinib using our pTyr dataset. Of 44 identified tyrosine phosphorylated kinases, phosphorylation of 14 kinases was affected by crizotinib (Fig. 3C). These effects included reduced EML4-ALK, MAPK1, and MET phosphorylation. Altered changes in MET are expected since crizotinib has activity as a MET kinase inhibitor. To associate changes in kinase phosphorylation with changes in substrate phosphorylation, we extracted evidence on kinase-substrate pairs from PhosphositePlus (42). Kinases affected by crizotinib, including EML4-ALK, MET, cyclin-dependent kinase 1 (CDK1), protein tyrosine kinase 6 (PTK6), MAPK1, PKC-δ, EGFR, ROCK2, breakpoint cluster region protein (BCR), pyruvate kinase, muscle (PKM), the tyrosine protein kinase FER (FPS/FES-related), and the tyrosine protein kinase LCK, have known pTyr protein substrates (Fig. 3D), suggesting that they have important roles in ALK signaling in these cells and hence are potential co-target candidates for combination therapy. Adaptive resistance to kinase inhibitors, as evidenced by increased tyrosine-specific phosphorylation by TKI, can point to co-targeting strategies to thwart these adaptive changes (35). Our phosphoproteomics results show that phosphorylation of both EGFR and protein kinase C-δ (PKC-δ) was enhanced by crizotinib. Co-targeting both ALK and EGFR has resulted in enhanced ALK TKI therapeutic efficacy (43), and studies in human samples have identified increased EGFR phosphorylation as a potential mechanism of ALK inhibitor resistance (44). Recently, PKC-δ signaling was shown to confer resistance to ALK TKI alone; co-targeting of PKC and ALK has been shown to synergistically eliminate ALK-rearranged lung cancer cells (13). PKC-δ directly binds and phosphorylates its known substrates, including phosphatases protein phosphatase 1 regulatory inhibitor subunit 14B (PPP1R14B) (45), ADAM metallopeptidase domain 9 (ADAM9) (46), the adaptor protein TJP1 (47), and the kinases EGFR (48) and LCK (49), as well as CUB domain containing protein 1(CDCP1) (50), heat shock protein 90kDa alpha family class B member 1 (HSP90AB1) and cytosolic phospholipase A2 (PLA2G4A) (51)]. We found that crizotinib increased the phosphorylation of both PKC-δ and its substrates, including EGFR, LCK, PTPN6, PPP1R14B, TJP1, CDCP1, phospholipase A2 group 4A (PLA2G4A), ADAM9, and HSP90AB1 (fig. S2). These results suggest that ALK inhibition by crizotinib is associated with an adaptive change whereby PKC becomes activated and phosphorylates a set of substrate proteins. This may explain how gain of function of PKC signaling may drive resistance to ALK inhibitors (13).
The ALK interactome identifies kinase inhibitors that sensitize cells to ALK TKI
To fully understand EML4-ALK function and signaling, we constructed an EML4-ALK-centric protein-protein interactome network by integrating all known literature reporting protein-protein interactions between our experimentally identified pTyr proteins and our experimentally derived bait-prey interactions. This network, which contains a total of 464 proteins and 4443 interactions (Data file S1), enabled us to track the signaling from EML4-ALK to downstream substrates and biological output involved in EML4-ALK-dependent signaling. We hypothesized that this interactome could provide a strategy to sensitize cells to ALK TKI and overcome resistance in some circumstances by selectively targeting proteins on central nodes/edges of the network (52). To make biological inferences from our EML4-ALK integrative network, we performed signaling pathway analysis on the entire network and identified a spectrum of overrepresented signaling pathways using Reactome FI (P<0.001, FDR<0.001, N≥6) (table S2) (http://apps.cytoscape.org/apps/reactomefiplugin). Our results confirm known associations with PI3K (53), IGF-1R (16), MAPK (53), and EGFR (54) signaling pathways. Comparison of the entire network with functional protein groups reveals that the “kinome,” defined as all known human kinases, represents major signaling pathways of the entire network (54% overlap) and pTyr protein group (64% overlap) (fig. S3). We found that 77% and 89% of pathways represented by crizotinib increased and decreased pTyr proteins are covered by the kinome (fig. S4).
To enrich key kinases and signaling pathways and define their perturbations by crizotinib, we first generated three subnetworks through filtering pTyr peptides corresponding to kinases perturbed by crizotinib from the entire ALK interactome. In general, we wanted to identify protein interaction modules that were increased, decreased, or unaffected by crizotinib. Our goal was to cluster these three subnetworks to identify functional modules and predict co-targeting kinases and signaling pathways, which potentially sensitize cells to ALK TKI (Fig. 4A). We identified four protein modules, all found in our experimental data, that were decreased by crizotinib. We identified eight kinases (EML4-ALK, CDK1, mitogen-activated protein kinase 7 (MAPK7), PKM, PTK6, BCR, and MET) that were in clusters inhibited by crizotinib. We also identified a module corresponding to reduce STAT3 activity (38). We identified modules corresponding to histone deacetylase (HDAC), proteasome, IRS, and FGFR as being negatively regulated by crizotinib (Fig. 4A).
Figure 4. Generating EML4-ALK integrative network to identify ALK TKI sensitizers.
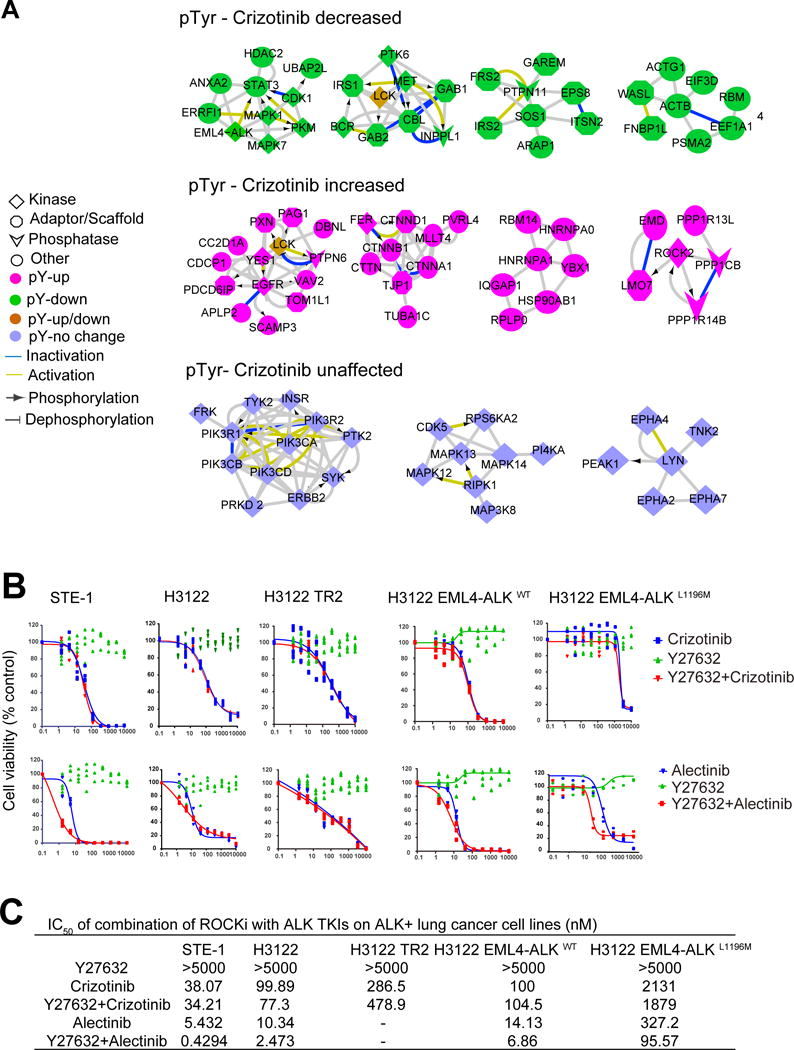
(A) Integrative EML4-ALK protein-protein network (Data File S1) was generated by integrating (i) the physical EML4-ALK interactome identified in Fig. 2B and (ii) all pTyr protein results from H3122 cells. Clustering analysis was conducted to enrich functional protein modules on pTyr peptide abundance that was decreased, increased or unaffected by crizotinib. (B) Cell viability of drug-sensitive cell lines (STE1, H3122, H3122 EML4-ALKWT) and drug-resistant cell lines (H3122 TR2 and H3122 EML4-ALKL1196M) treated with ALK TKI (crizotinib or alectinib), ROCK inhibitor (ROCKi; Y27632), or combinations thereof (crizotinib or alectinib+Y27632). Data represents three - four data points for each drug concentration in a representative experiment for each cell line and drug combination(C) Mean IC50 values from treatments in (B) were calculated with Prism6. No IC50 value was obtainable from H3122 TR2 cells (“-”) because the drug dosage graphs of single-agent alectinib, and its combination with Y27632 did not produce “S” curves.
In contrast, we identified four protein modules that their tyrosine phosphorylations were increased by crizotinib and five kinases (tyrosine-protein kinase YES, EGFR, LCK, FER, and ROCK2) in complexes increased by crizotinib (Fig. 4A). Based on this observation of the ROCK cluster, we next tested the ability of a ROCK inhibitor, Y27632, to sensitize cells to ALK TKI. We tested parenteral and ALK TKI sensitive H3122 and STE1 cells with both crizotinib and alectinib, as well as two ALK TKI resistant lines, one by virtue of a gatekeeper L1196M mutation (H3122- EML4-ALKL1196M) and the other through a gain of EGFR signaling (H3122-TR2) (55). The L1196M mutation in ALK has been clinically identified as a major cause of acquired drug resistance and cell lines harboring this mutant represent an ideal model to examine the ability of drugs to overcome ALK TKI resistance (12, 56). We generated a H3122 EML4-ALKL1196M cell line by cloning the gate-keeper mutation L1196M of EML4-ALK into parenteral H3122 cells, which results in resistance to crizotinib but sensitivity to alectinib (fig. S5) (14). Exposure of cells to Y27632 had no effect on cell viability across any of the cell lines, and neither did the combination of Y27632 with crizotinib; however, combining Y27632 with alectinib reduced the IC50 in multiple cell lines (H3122, STE1), including H3122 EML4-ALKL1196M crizotinib resistant cells (Figure 4, B and C). We examined ROCK2 protein abundance in three ALK rearranged cells (H3122, STE1, and H2228) and H3122-derived resistant lines and found that ROCK2 was abundant in all tested cell lines (fig. S6A). We found no difference in ROCK2 abundance between the sensitive and resistant H3122 pair. Lastly, we also identified HSP90 grouped into another crizotinib increased module. Our results confirmed the direct binding of HSP90 and cell division cycle 37 (CDC37) with ALK in complex (56, 57). Functionally, inhibiting HSP90 with AUY 922 or knocking down CDC37 decreased H3122 cell viability (fig. S6, B and C).
ALK interactome-wide synthetic lethal screening identifies ALK TKI sensitizers
Although pathway analysis is powerful in mining biologic insights from well-annotated proteins, many important network proteins are still not assigned into existing signaling pathways, and thus their functional role in sensitizing to ALK TKI could be missed. To identify new ALK TKI sensitizers, we used a shRNA library screen to systemically examine the combination effects of loss-of-function for every node of the entire EML4-ALK integrative network with ALK inhibitors, including crizotinib and alectinib, on H3122 cell viability. Alectinib is a second-generation ALK TKI that can overcome EML4-ALK gatekeeper mutant resistance and hence was used to test the ability of loss of function of network components to sensitize resistance (14). A total of 2,029 shRNA constructs targeting 407 proteins from the interactome were screened (table S3). We required five shRNA for each gene and thus 59 proteins were not included in this analysis. The IC20, IC50 and IC80 doses of crizotinib and alectinib in H3122 cells were determined using GLO assays. Cells containing the shRNA library were treated with these doses, and the synergic effect of loss-of-function of each component of the EML4-ALK network with ALK TKIs was evaluated to identify ALK TKI sensitizers (Fig. 5A). The synergic effect of loss-of-function of each component of EML4-ALK network with ALK TKIs on H3122 cell viability was evaluated to identify ALK TKI sensitizers.
Figure 5. shRNA library screen to identify the ALK TKI sensitizers.
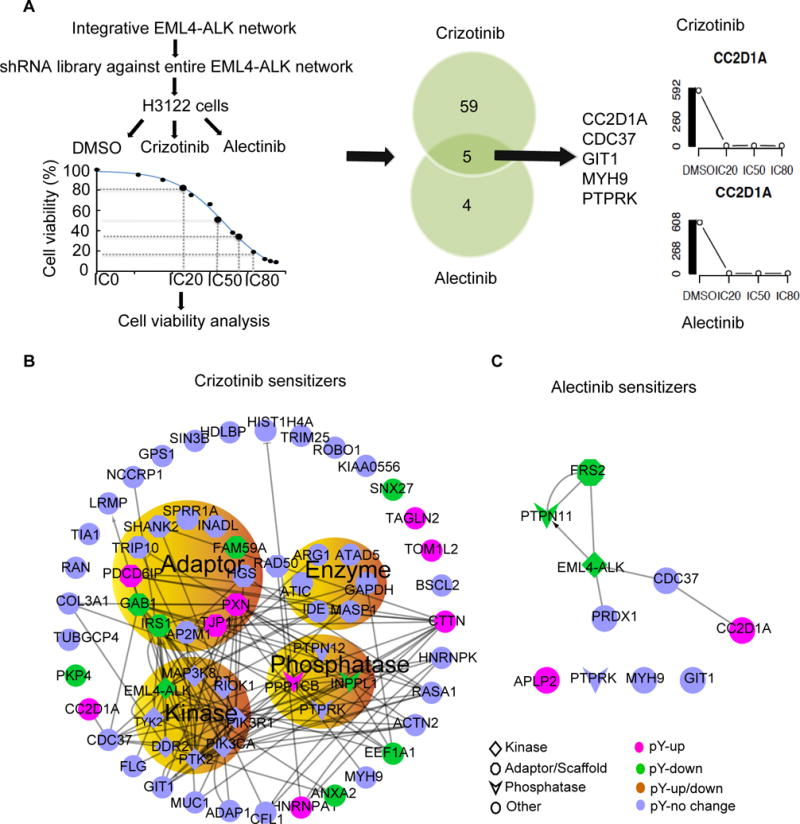
(A) Schematic of the shRNA library screen for ALK TKI sensitizers (left), with a representative Venn diagram of shRNAs that sensitized cells to crizotinib, alectinib or both (middle), and representative viability assays for one of these targets, CC2D1A (right). Data represents results from a single experiment, where five unique shRNA were targeted to each gene, and each drug/shRNA effect was assayed in five replicates (B and C) Interactions among the proteins that sensitized cells to crizotinib (B) and alectinib (C) were extracted from the entire EML4-ALK interaction network. The protein types and pTyr change induced by crizotinib are highlighted.
Our results show the synergistic effect between loss-of-function of 64 proteins with crizotinib including 8 kinases, 12 adaptors, 7 enzymes, and 4 phosphatases (Fig. 5B and table S4). Of these 64 hits, four are known from previous literature to be related to ALK signaling (ALK phosphorylates PTK2 (FAK1) (58), phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha isoform (PIK3CA) (37), bifunctional purine biosynthesis protein PURH (ATIC)(41), and LRMP (26, 59) and increases their activities) (Figure 5b). In addition, another 7 hits were in proteins whose pTyr was reduced by crizotinib in our data [GAB1, insulin receptor substrate 1 (IRS1) (16), annexin A2 (ANXA2), eukaryotic translation elongation factor 1 alpha 1 (EEF1A1), sorting nexin family member 27 (SNX27), GRB2-associated and regulator of MAPK protein 1 (FAM59A), and plakophilin 4 (PKP4)] (Fig. 5B). These results suggest that co-targeting these proteins along the ALK axis can enhance sensitivity to crizotinib. Interestingly, the kinase COT, encoded by the MAP3K8 gene, was found in our analysis and also identified to drive resistance to ALK inhibitors in a gain of function screen (13). Conversely, we identified 9 hits corresponding to proteins whose phosphorylation was increased by crizotinib, namely coiled-coil and C2 domain containing 1A (CC2D1A), TJP1, heterogeneous nuclear ribonucleoprotein A1 (HNRNPA1), Ser/Thr protein phosphatase catalytic subunit, beta isozyme (PPP1CB), paxillin (PXN), CTTN, target of MYB1-like 2 membrane trafficking protein (TOM1L2), transgelin 2 (TAGLN2), and programmed cell death 6-interacting protein (PDCD6IP) (Fig. 5B). Because they are not inhibited by crizotinib, these proteins may represent bypass signaling mechanisms and could also represent adaptive resistance changes induced by loss of negative feedback.
We found that the loss of function (through knockdown) of 9 proteins [protein tyrosine phosphatase receptor type K (PTPRK), amyloid beta (A4) precursor-like protein 2 (APLP2), peroxiredoxin 1 (PRDX1), CC2D1A, FRS2, CDC37, G protein-coupled receptor kinase interacting ArfGAP 1 (GIT1), myosin heavy chain 9 (MYH9) and protein tyrosine phosphatase, non-receptor type 11 (PTPN11)] sensitized H3122 cells to alectinib (Fig. 5C). We focused on FRS2, a kinase scaffold protein typically associated with fibroblast growth factor signaling. Previous studies found that ALK physically binds to and activates FRS2 (40). Our quantitative phosphoproteomics analysis found that crizotinib decreased phosphorylation of FRS2 (table S1). Together with the results from our shRNA screen, these results suggest that FRS2 is directly regulated by ALK in these cells and is necessary for ALK-driven survival. In addition, overexpression of another FRS family member, FRS3, was previously shown to drive resistance to ALK inhibitors (13). To validate the shRNA screen, we used two individual hairpins to deplete FRS2 and assessed the ability of FRS2 depletion to potentiate ALK TKI sensitivity. Our results verified that FRS2 depletion sensitizes H3122 cells to both crizotinib and alectinib (Fig. 6A). Decreased mRNA expression and protein abundance of FRS2 was verified by reverse transcription polymerase chain reaction (RT-PCR) and Western blotting, respectively (Fig. 6B). That depletion of FRS2 by shRNA enhanced the sensitivity of H3122 cells to crizotinib was also confirmed by real-time cell analysis (fig. S7). Combining FRS2 knockdown with alectinib decreased viability in crizotinib-resistant H3122 EML4-ALKL1196M cells (Fig. 6A). Next, we examined both FRS2 tyrosine phosphorylation and total protein abundance in multiple ALK-fusion cell lines, including H3122 parent, H3122 TR2, H3122 EML4-ALKwild-type, H3122 EML4-ALKL1196M, H2228, and STE1 cells. We found that FRS2 was present in all the examined cell lines, with STE1 cells appearing to have the highest abundance of total and phosphorylated FRS2 (fig. S8A). We further evaluated the change of phosphorylation and total protein abundance of FRS2 in response to crizotinib treatment in a time-course manner. We found that crizotinib reduced the abundance of Tyrosine-phosphorylated FRS2 over the same time course as it did to the abundance of Tyr-phosphorylated ALK in H3122 cells (fig. S8B). Together, our loss of function studies along with previously reported gain of function studies support that FRS2-mediated signaling is a key adaptor hub for ALK-rearranged lung cancer.
Figure 6. Validating knock-down of FRS2 and CC2D1A as ALK TKI sensitizers.
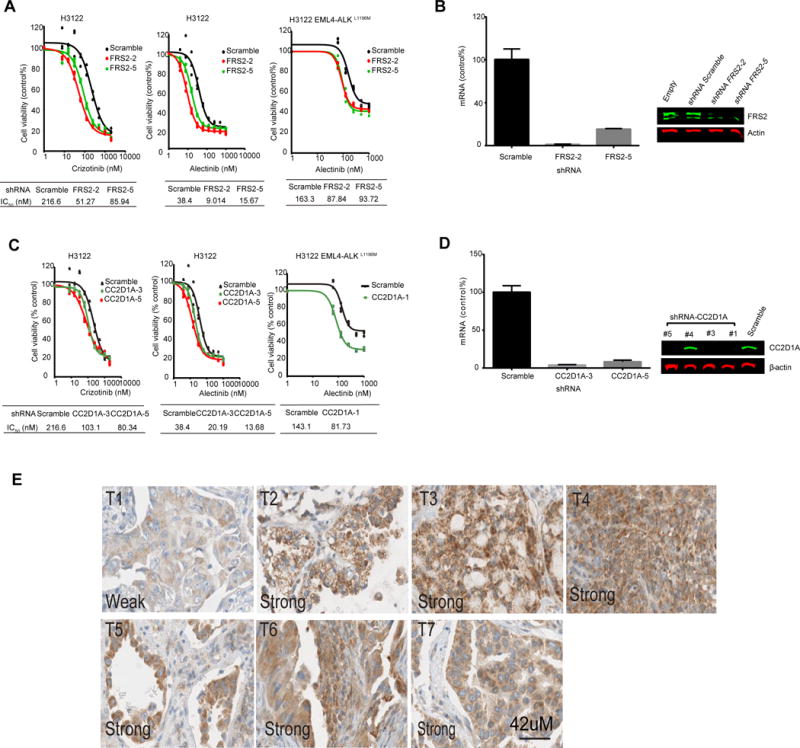
(A) Cell viability in H3122 or H3122 EML4-ALKL1196M cell cultures transfected with two shRNAs targeting FRS2 and treated with crizotinib or alectinib. (B) Quantitative RT-PCR (left) and Western blotting (right) to assess FRS2 knockdown by shRNA in H3122 cells. (C) Cell viability in H3122 or H3122 EML4-ALKL1196M cell cultures transfected with shRNA targeting CC2D1A and treated with to crizotinib or alectinib. (D) Quantitative RT-PCR (left) and Western blotting (right) to assess CC2D1A knockdown by shRNA in H3122 cells. RT-PCR data in (B) and (D) are means ± S.D. from a single experiment each with triplicate datapoints. Western blots are representative of two experiments. (E) Immunohistochemistry staining for CC2D1A in a panel of ALK-positive human lung cancer tumor tissues. Shown are representative sections from 7 patients.
Loss of coiled-coil and C2 domain-containing protein 1A (CC2D1A) sensitizes cells to ALK TKIs
Knockdown of five proteins (CC2D1A, GIT1, PTPRK, CDC37, and MYH9) sensitized H3122 cells to both crizotinib and alectinib. CC2D1A, also known as AKT-kinase-interacting-protein-1,) is a receptor-selective scaffold protein that modulates diverse signaling pathways and determines the selectivity of receptor kinases (60, 61). We found that inhibition of ALK increased the phosphorylation of CC2D1A at pTyr 207 (table S1). The exact biological role of pTyr207 of CC2D1A remains unclear. Our loss-of-function screen indicated that knockdown of CC2D1A sensitizes H3122 cells to both crizotinib and alectinib (Fig. 5). To verify these shRNA library screen results, we used two individual hairpins to knockdown CC2D1A and assessed the ability of CC2D1A knockdown increase ALK TKI sensitivity. Our results verified that shRNA-mediated depletion of CC2D1A sensitized H3122 cells to both crizotinib and alectinib (Fig. 6C). Decreased mRNA and protein abundance were verified by RT-PCR and Western blotting (Fig. 6D). Using a third shRNA against CC2D1A, we found that depletion of CC2D1A also sensitized H3122 EML4-ALKL1196M cells to alectinib(Fig. 6C). We next examined the expression of CC2D1A in seven distinct EML4-ALK rearranged human lung cancer tissues using immunohistochemistry. We assessed both the staining intensity and percentage of tumors staining for CC2D1A and combined these into a singular measurement. Six of the 7 EML4-ALK-positive tissues had strong and broad staining—and thus high abundance—of CC2D1A, whereas the one tissue had weak abundance of CC2D1A (Fig. 6E). Though limited to a small number of samples, this suggests that CC2D1A might be coexpressed with EML4-ALK. Together our results indicate that targeting CC2D1A may enhance ALK TKI efficacy in patients.
Discussion
We set out to determine if an experimentally derived signaling network or interactome centered on ALK in ALK-rearranged lung cancer cells could identify sensitizers to ALK TKI. We hypothesized that this interactome could be involved in modulating the balance of pro-survival and pro-apoptotic signals in aggregate and could identify targets of potential therapeutic value that may shift the survival/apoptosis signal balance. The interactome would be highly relevant to acquired drug resistance and could enable modeling or other hypotheses based on genetic alterations or epigenetic alterations in interactome components. From a clinical point of view, targeting sensitizers could convert a limited partial response into a deeper partial response or even complete response and thus improve patient outcomes. Upfront targeting could be important in elimination of “persister” cells and help reduce minimal residual disease that can lead to overt drug resistance and tumor progression. Better understanding of key sensitizers could also be involved in predicting the degree of response to ALK TKI prior to onset of therapy, by identifying those patients likely to have deep and durable responses from those with weaker and more transient responses requiring initial upfront combination therapy. Future experiments using results generated here are necessary to answer these questions.
We devised a strategy combining tyrosine phosphoproteomics, tandem affinity precipitation, and interactome-wide RNA interference screening to physically and functionally characterize an EML4-ALK signaling network or interactome. Our EML4-ALK interactome provides ALK biologists and clinicians with an important information source for further study. One advantage of the EML4-ALK interactome in this study is that it integrated both relatively static physical protein-protein interactions and transient phosphotyrosine events based on experimental observations and as such is not limited to prior literature-based interactions. Our data reveal that only a small portion (18.9%) of pathways enriched from the entire network are associated with ALK TKI (crizotinib) sensitivity, indicating that the majority (81.2%) of pathways are ALK TKI-independent and could be potential bypass signaling pathways of ALK TKIs. These pathways are possible co-targeting candidates for developing combined therapy with ALK TKIs. Furthermore, our results give mechanic insights into EML4-ALK signaling and crizotinib action linking the perturbation of pTyr phosphoproteome by crizotinib and hits identified by unbiased loss-of-function analysis to create an integrative EML4-ALK network. We have pinpointed 18 functional targets from clustering analysis of the ALK network, and some of these were experimentally shown to decrease H3122 EML4-ALKL1196M cell viability. For instance, crizotinib increased the phosphorylation of paxillin, which is a known substrate of the kinase SRC. Co-targeting ALK and SRC signaling has been shown to overcome ALK inhibitor resistance in patient-derived lung cancer cells (62). We recognize that a limit of our studies is the single time point used for the phosphoproteomic analysis, thus dynamics and mechanisms of signaling changes remain unclear. For example, changes in tyrosine phosphorylation of paxillin may represent rebound changes following initial inhibition or truly represent immediate gain of tyrosine phosphorylation following ALK inhibition.
Our interactome-wide unbiased shRNA library synthetic lethal screen revealed novel protein sensitizers that could not be obtained from screening of inhibitors and knowledge-based network analysis. For instance, there are only 28% total crizotinib sensitizers whose phosphorylation is regulated by crizotinib, whereas phosphorylation of the 72% crizotinib sensitizers identified from shRNA library screen were not regulated by crizotinib. One advantage of using the interactome to direct the shRNA functional screen is that it increases the likelihood of identifying sensitizers through a focused and deep analysis of the shRNA barcodes. Of note, we observed that despite aiming for an IC20, IC50, and IC80 dose for each drug, there was significantly more cell death at a given dose in the alectinib-treated shRNA library cells than the crizotinib-treated cells. Given that the robustness of synthetic lethality screens relies on representation of each shRNA in multiple cells, a substantial decrease in cell number can bring about a bottleneck, reducing representation of many shRNAs below the threshold needed to achieve significance. This likely accounts for the discrepancy between the number of synergistic shRNA’s between crizotinib and alectinib. We note here that this type of technical issue is much more likely to bias the experiment toward false negative results and that the hits that were found for both drugs represent relevant biology. Thus, the failure for a gene that was a hit in the crizotinib screen to score as synthetic lethal in the alectinib screen should not indicate that this gene was a false positive in the crizotinib screen.
In addition to kinases, many adaptors, phosphatases, enzymes, and other types of proteins were recruited into various macromolecular signaling complexes, which could not be predicted by pathway analysis, but could be important in altering the balance of pro-survival and pro-death effector signals downstream of EML4-ALK. FRS2 and CC2D1A are representative examples. Whereas other studies have found that ALK physically interacts with FRS2 in some contexts and increases its activity, our results show that FRS2 knockdown enhances the effects of both crizotinib and alectinib, thus demonstrating its role as a sensitizer to ALK TKI therapy (40). Although adaptor proteins such as FRS2 were traditionally felt to be poor candidates for therapeutic development, newer chemistry approaches, such as through phthalimide conjugation, may challenge this mindset (63). Our study also demonstrates CC2D1A as a sensitizer to ALK TKI therapy. Although CC2D1A is reportedly a scaffold the PI3K–AKT signaling pathway, we observed no effect of knockdown CC2D1A on either Thr308 or Ser473 phosphorylation of AKT. Further studies are necessary to delineate the exact mechanisms of sensitization, as CC2D1A may regulate the EGFR signaling pathway (60) or the NF-κB cascade (64). Our analysis of tumor tissues from patients with EML4-ALK rearrangements shows a distribution of CC2D1A protein expression, suggesting that this could be a modulator of tumor cell intrinsic sensitivity to ALK TKI. Further studies using larger numbers of ALK-rearranged lung cancers with known responses to ALK TKIs are needed to assess how the expression of FRS2 or CC2D1A may relate to the extent of clinical response. Lastly, we found that the knockdown of a number of phosphatases sensitized cells to ALK TKI, including PTPN12, PTPRK, PP1CB, and INPPL1. This result is interesting in light of previous studies implicating that phosphatase signaling affected the pro-survival and pro-death signal balance driven by oncogenes (1). Further experiments targeting phosphatases identified in our experiments are needed to see how they may disrupt the balance between pro-survival and pro-death signaling.
In conclusion, this proteome level and RNA interference sensitization perspective provides a valuable resource for identifying other resistance mechanisms and co-targeting strategies for ALK rearranged lung cancers. Further studies on biological mechanisms of other components within EML4-ALK interactome created in this study may identify more combinational therapeutic strategies to either improve the ALK-based therapy efficacy or overcome ALK TKI resistance. Biomarker approaches to discern key facets of the interactome may identify patients likely to derive more benefit from upfront combination therapy as opposed to single use of ALK inhibitors.
Materials and Methods
Materials
Materials used included iodoacetamide, dithiothreitol, 1 M triethylammonium bicarbonate (TEAB), protease inhibitor cocktail, HA antibody conjugated agarose, Polybrene (Sigma-Aldrich, St. Louis, MO); trypsin (Promega Corp., Madison, WI); formic acid (HCOOH) (MERCK, Darmstadt, Germany); Strep-Tactin sepharose (IBA TAGnologies, Gottingen, Germany); D-biotin (Alfa Aesar, Karlsruhe, Germany); micro Bio-Spin chromatography columns (Bio-Rad, Hercules, CA); Gateway LR Clonase II Enzyme Mix Kit, fetal bovine serum, and Lipofectamine 2000 (Invitrogen, Carlsbad, CA).
Cell lines and culture
Human NSCLC H3122 cell lines were provided by Dr. William Pao (Vanderbilt-Ingram Cancer Center, Vanderbilt University, Nashville, TN) and confirmed to be free of Mycoplasma contamination. Cells were cultured in 10% FBS-RPMI (Invitrogen) medium at 37°C and 5% CO2 in a humidified environment, digested with Accutase (Innovative Cell Technologies, Inc), and separated into single cells by passing through BD Falcon Cell Strainer (Sigma-Aldrich). Phoenix HEK293 cells used for producing lentivirus were obtained from ATCC (Manassas, VA). Crizotinib (PF 2341066) and NVP AUY-922, were purchased from ChemieTek (Indianapolis, IN). Alectinib and Y27632 were purchased from Selleck Chemicals.
Stable crizotinib-resistant cell lines
Oncogenic fusion gene EML4-ALK variant 1 wild type or point mutant at L1196M was cloned into the retroviral pBabe-puro backbone (kindly provided by Dr. Johannes Heuckmann, Universität zu Köln, Köln, Germany). The construction plasmids were co-transfected with VSV-g vector to Phoenix cells and cultured at 32°C for 48 hours to make the retroviral virus. Retroviral viruses carrying targeting genes were harvested, followed by infection of H3122 cells with retroviral virus containing target genes. Positive cell clones were selected with 2 μg/ml of puromycin for a continuous 2 weeks to remove cells. Resistance of stable cell lines to crizotinib was verified using CellTiter-Glo® Luminescent (Promega) cell viability assay.
Tyrosine phosphoproteome extraction
Because only two original EML4-ALK-positive patient NSCLC cell lines were originally available (H3122 (variant1) and H2228 (variant 3a/b)), with the former demonstrating sensitivity to crizotinib and clear mechanism to create the acquired resistant cell model to ALK TKIs, but the latter not demonstrating resistant mechanisms and no resistant cell model available, we selected H3122 cells for our experiments. pTyr phosphoproteomes were purified, applying PhosphoScan kit (P-Tyr-100) (Cell Signaling Technology) according to the manufacturer’s recommendations. Briefly, 2 × 108 cells were lysed in urea buffer; extracted proteins (40–80 mg) were then reduced by dithiothreitol, alkylated by iodoacetamide, and then digested by trypsin. Peptide mixture was isolated from lysate using Sep-Pak C18 columns and then lyophilized. Phosphorylated peptides were dissolved in Immunoaffinity buffer and enriched using immobilized antibody specific for phosphotyrosine, and eluted with acid. Sample volumes were then reduced to 20 μL by vacuum centrifugation (Speedvac) for LC-MS/MS analysis.
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) and peptide assignment
After in-gel digestion (for TAP experiment) or in-solution digestion (for phosphoproteomics experiment), peptides were analyzed using nanoflow liquid chromatography (U3000, Dionex, Sunnyvale, CA) coupled to an electrospray hybrid ion trap mass spectrometer (LTQ-Orbitrap, Thermo, San Jose, CA). Samples were first loaded onto a precolumn (5 × 300-mm internal diameter packed with C18 reversed phase resin, 5 mm, 100 Å) and washed for 8 minutes with aqueous 2% acetonitrile and 0.04% trifluoroacetic acid. The trapped peptides were eluted onto the analytical column (C18, 75 μm internal diameter × 15 cm; Pepmap 100, Dionex, Sunnyvale, CA). The 60-minute gradient delivered at 300 nl/min was programmed as 95% solvent A (2% acetonitrile + 0.1% formic acid) for 8 minutes; solvent B (90% acetonitrile + 0.1% formic acid) was ramped from 5% to 50% over 35 minutes and then from 50% to 90% in 1 minute and held at 90% for 5 minutes, followed by ramping down from 90% to 5% in 1 minute and re-equilibration for 10 minutes. After each survey scan, 5 tandem mass spectra were collected in a data-dependent manner. The MS scans were performed in Orbitrap to obtain accurate peptide mass measurement, and the MS/MS scans were performed in linear ion trap using 60-second exclusion for previously sampled peptide peaks.
For peptide sequence assignment, Sequest, and Mascot searches were performed against human entries in the UniProt database. Two missed tryptic cleavages were allowed, and the precursor mass tolerance was set to 1.08 Da (to accommodate incorrect selection of the monoisotopic peak). MS/MS mass tolerance was 0.8 Da. Dynamic modifications included carbamidomethylation (Cys), oxidation (Met), deamidation (Asn, Gln) and phosphorylation (Ser, Thr, Tyr). To accurately identify phosphorylation sites, we integrated database search results from both Sequest and Mascot into Scaffold (www.proteomesoftware.com) and took multiple parameters (SCAFFOLD peptide probability, XCorr, DeltaCN, Mascot ion score, E-value) into consideration. The following limits were used to establish data quality: 80% peptide probability, 10 ppm fragment error, 40 Mascot score, and XCorr for 2+ >2.5, XCorr for 3+ >3, and DeltaCN >0.1. Peptides could be identified by either database search alone as long as the quality metrics were exceeded. Finally, phosphorylation site assignment was manually validated from the raw data using published methods.
Quantification of tyrosine phosphorylated peptides using extracted ion chromatograms
To quantify the perturbation of pTyr proteome change response to ALK TKI treatment, the integrated peak areas for each pTyr peptide were calculated from extracted ion chromatograms (EIC) using QuanBrowser from Xcalibur 2.0. These values were restricted by m/z (±0.02) and retention time (60 s). Other parameters were Genesis peak integration using smoothing point 9.0, S/N threshold 0.5, and peak detection minimum peak height (S/N) 3.0. The masses and isotopic peak patterns of the target peptides were manually inspected to ensure proper sequence assignment and to verify peak quality. Peak area values of all precursors from all samples were merged to one spreadsheet using PeakAreaSummary software, which was developed in-house (http://proteome.moffitt.org/proteomics/). PeakAreaSummary is an Excel add-in using VBA. It first calculates the sum of peak areas for all the precursors in the same sample with the same m/z and retention time (using the same delta values as mentioned above) and then merges the peak area values from all the samples to get one value for any precursor.
Pre-processing to identify unique phosphorylated tyrosine (pY) sites
After quantification, 628 phosphorylated tyrosine sites were identified. An in-house algorithm, implemented to identify unique phosphorylated tyrosine (pY) site, was used to remove redundant sites, merge peptides containing missed cleavages by using protein ID, peptide sequence, and phosphorylation site index (i.e. amino acid residue number), and quantify peak areas. When it is only identifiable to the level of pairs of pYs, then the independent unit for analysis is the unique pY pair (instead of single site). Phosphopeptides produced with missed cleavages or fragments of the same phosphopeptides were merged. A total of 487 unique phosphotyrosine units (pYs or pY pairs) were identified.
Because of their detection and signal stability across samples, the CDC2-pY15 peptides were used for normalizing the peak areas across all 12 samples (6 biological samples with technical duplicates) fig. S9. The normalized quantities across samples are shown in fig. S10.
Reproducibility between technical replicates for each pY was high and therefore included in the analyses. Averages of technical replicates from 6 biological samples were used in the analyses. Data were analyzed in log2 scale prior to parametric analyses and for the ease of interpretation. A two-sample t-test was performed to compare differential phosphorylation for each tyrosine between the control and treatment group treated at 1000 nM. Because follow-up siRNA screening was planned, multiple comparisons were not adjusted. P-value of less than 0.05 and 1.5-fold change was used to measure the change of phosphotyrosines.
SH tandem-affinity purification
To dissect the EML4-ALK physical protein-protein interaction network, we isolated and identified components of protein complexes of EML4-ALK V1, SHC1, GRB2, and PIK3R2 coupling TAP with MS analysis. GFP was used as the control. cDNA for EML4-ALK V1 was provided by Dr. William Pao (Vanderbilt University, Nashville, TN). The design of PCR primers, amplification, and pENTR TOPO cloning of EML4-ALK V1, SHC1, GRB2, and PIK3R2 were performed as previously described (7). Briefly, EML4-ALK V1, SHC1, GRB2, P85B, and GFP were separately inserted into pfMSCV-C-SH IRES GFP gateway vector from pENTR TOPO vector using Gateway LR Clonase II Enzyme Mix Kit. The retroviral expression clone was verified by DNA sequencing using an Applied Biosystems 3130 × 1 Genetic analyzer (HITACHI) with data analyses performed using Lasergene software V7.2. Phoenix HEK293 cells were obtained from ATCC (Manassas, VA) and grown in DMEM medium containing 10% fetal bovine serum (FBS). On day 1, 8 × 105 Phoenix cells per well were seeded in 6-well plates. On day 2, cells were transfected with 3 μg VSV-G and 5 μg retroviral plasmids using Lipofectamine 2000. Six hours after transfection, the supernatant was replaced with 2 mL DMEM-20% FBS, and the cells were incubated in a 5% CO2 incubator at 32°C for 48 hours. The supernatant (viruses) was collected by centrifugation at 4°C and either stored at −80°C or used immediately to infect the target cells. H3122 cells were maintained in RPMI 1640 medium supplemented with 10% FBS. For retroviral transduction, 2 × 105 cells per well were seeded in 6-well plates. After overnight incubation, cells were infected with 800 μL of the virus supernatant plus 6 μg/mL Polybrene for 24 hours and then supplemented with 4 mL media per well. Cells were grown continuously until cell sorting. One week after infection, GFP-positive cells were sorted by FACSVantage (BD Biosciences, San Diego, CA). GFP positivity and HA expression were assessed by flow cytometry and immunoblot, respectively, before expanding the cells to 10 × 15-cm dishes. When approximately 90% confluent, the EML4-ALK V1, SHC1, GRB2, and PIK3R2-tagged cells were washed with ice-cold PBS containing 1 mM sodium orthovanadate and scraped with a cell lifter on ice. Five of 150mm dishes of each EML4-ALK, SHC1, GRB2, and PIKR2 and GFP-expressing H3122 cell pellets (each consisting of 5 × 15-cm dishes) were collected in 15-mL conical tubes by centrifugation at 120 × g at 4°C for 5 minutes and lysed in TNN-HS buffer (50 mM HEPES, pH 8.0, 150 mM NaCl, 5 mM EDTA, 0.5% NP-40, 50 mM NaF, 1.5 mM Na3VO4, 1.0 mM PMSF, and protease inhibitor cocktail). Insoluble material was removed by centrifugation at 14000× g for 15 minutes at 4°C. We transferred 200 μL of StrepTactin sepharose (400 μL slurry/pulldown) to a 14-mL dust-free Falcon tube and washed with 2 × 1 mL TNN-HS buffer. The lysates (extracted from 5 × 15-cm plates) were added to the washed StrepTactin sepharose and rotated for 20 minutes at 4°C. The sepharose beads and supernatant were transferred to a spin column and gravity drained. The sepharose was washed with 4 × 1 mL TNN-HS buffer, and the bound proteins were eluted with 3 × 300 μL freshly prepared 2.5 mM D-biotin in TNN-HS buffer into a fresh dust-free 1.5-mL Eppendorf tube. One hundred microliters of HA antibody conjugated agarose beads (200 μL slurry/pulldown) were added to the biotin elute and rotated for 1 hour at 4°C. The samples were centrifuged at 200× g for 1 minute at 4°C, and the supernatant was removed. The HA antibody conjugated beads were suspended in 1 mL TNN-HS buffer, and the washed beads and buffer loaded into a fresh dust-free Biospin column and gravity drained. The HA antibody conjugated agarose was sequentially washed with 3 × 1 mL TNN-HS buffer and 2 × 1 mL TNN-HS buffer consisting of only HEPES, NaCl, and EDTA. Retained proteins were eluted from the column directly into a glass HPLC vial using 500 μL of 100 mM HCOOH and immediately neutralized with 125 μL of 1 M TEAB. Two hundred microliters were removed for immunoblot analysis. The remaining samples were desalted by loading into SDS-PAGE gels and running short-time electrophoresis; the bands were then sliced out from gel and cut into small pieces. Regular in-gel digestion with trypsin was then performed, and the peptides were extracted from gel pieces with 40% of acetonitrile in 0.1% TFA solution. Resulting peptides were submitted to LC MS/MS and database to identify the proteins.
To evaluate the effects of crizotinib on EML4-ALK complexes, we utilized a one step STREP pulldown, instead of tandem affinity purification of STREP and HA. The peak intensity of each protein was calculated by MaxQuant. A t-test was used to determine changes induced by crizotinib as compared to DMSO control treated samples.
Generating EML4-ALK integrative protein-protein network
To facilitate the application of various protein-protein interaction analysis software and databases, all identified proteins from phosphoproteomics and TAP experiments were first converted to uniform Gene name, UniProt name, and UniProt access number using g:Convert Gene ID Converter (65), UniProtKB, and PhosphoSitePlus (28). To maximize the known protein-protein interactions among identified tyrosine phosphorylated proteins and EML4-ALK physical interact proteins, four reciprocal tools including PSICQUIC (66), MetaCore, PhosphositePlus, and PhosphoPoint(67) were used to retrieve protein-protein interactions from existing databases. We first loaded all identified protein UniProtKB access numbers to PSICQUIC Universal Client Service (V 0.31), a plug-in of Cytoscape (V2.8.3), allowing access to 31 registered protein-protein databases and more than 151 million binary interactions. The query mode was set as GET_BY_QUERY. We next loaded gene names of all identified proteins to MetaCore, another tool for protein-protein annotation that provides detailed reference and specific statement for each protein-protein interaction. The effect categories of MetaCore, including active, inactive, and unspecific, are especially meaningful for phosphoproteins. Analyzed networks and canonical pathways were selected as the network building algorithm, the maximum number of nodes in a network was set as 50, and functional and binding interactions were used for network building. All mechanisms and effects were selected. All protein-protein interactions obtained from PSICQUIC and MetaCore and kinase-substrate interactions from PhosphoSitePlus and PhosphoPoint were merged in Cytoscape 2.8.3 to generate a union protein-protein interaction network. Contaminated non-experimental nodes were removed. Because of different formats and protein-protein interaction names from different search engines, this network still contains some repeated interactions between protein-protein pairs. To refine the network to unique interactions for each protein-protein pair, we exported all interactions together with edge attribute to Excel file and manually checked each of the protein-protein interactions and removed all redundant interactions. To ensure that each protein-protein interaction was unique, we followed this policy: 1) if there are similar interactions between the same protein pair, the more clearly stated and evidenced with literature one was kept; 2) if reverse effects (for example, active and inactive) were found between the sample protein pair, we kept both interactions; 3) if there are kinase-substrate interaction and other types of interactions found between the same protein pair, keep KSI only; and 4) similar interaction statements were combined to make one interaction. The refined protein-protein interactions were merged with bait-prey interactions from TAP experiments in Cytoscape to generate the EML4-ALK integrated protein network.
Bioinformatics analysis to mine biological implications embedded in EML4-ALK integrative network
After constructing the EML4-ALK integrated protein network, our next goal was to mine the biological implication of EML4-ALK network to cancer mechanism and discover potential new drug targets for therapy. To facilitate understanding the function of proteins in the EML4-ALK integrated network, we annotated the entire network proteins using PhosphoSitePlus® (PSP) and assistance from Cell Signaling Technology to 14 protein types: kinase, phosphatase, GAB, GEF, adaptor/scaffold, enzyme, chaperone, vesicle, motility polarity chemotaxis, transcription factor, ligase, heat shock proteins, cytoskeletal proteins, and other.
To globally understand the functional hints embedded in the network, we simplified the network through creating a series of subnetworks according to different functional attributes including phosphorylation changes, drug response, and kinase proteins. Also, inteactions among first neighbor nodes for selected proteins were extracted to create the subnetwork; these subnetworks allow the network-wide understanding of the function of certain key proteins or protein group.
To identify key players from complicated networks, we performed cluster analysis on networks and subnetworks using Clust&See (68), a Cytoscape 2.8.3 plug-in. The graph clustering algorithm TFit (iterated Transfer-Fusion) was selected to cluster the networks due to its ability to gain accurate modules from multi-levels. Modules containing less than 5 nodes were filtered out.
Pathway analysis was conducted on network and each subnetwork using Reactome FI. The FDR value cut off was set as 0.001 to obtain significantly enriched pathways.
Network statistics analysis
Subnetworks in pairs according to different conditions were compared using NetworkAnalyzer plug-in (69). All comparison analyses of different group lists were conducted and visualized with Venny 2. 0.2 (Oliveros, J.C. http://bioinfogp.cnb.csic.es/tools/venny/index.html).
Cell viability analysis
CellTiter-Glo® Luminescent (Promega) cell viability assay was used to examine the effects of TKI and RNAi alone and in combination with ALK TKI on cell viability of H3122 parent and ALK TKI-resistant cell lines following the manufacturer’s instructions. In general, cells were seeded to 96-well plates and grown in RPMI 1640 medium containing 10% FBS at 37°C. After 24 hours of cell attachment, the inhibitors targeting the selected component of EML4-ALK integrated network were added in a series of concentrations. To determine appropriate concentration scope for each drug, we started all drugs from maximum 10 μM and sequentially diluted them in 1:5. According to screen results, we adjusted the maximum concentration and dilution ratio for some inhibitors to ensure most data points were located within the “S” curve. After 72-hour incubation, CellTiter-Glo was added and the results were read with SpectraMax M5 (Molecular Device). The IC50 values for each inhibitor were calculated using GraphPad Prism 6 software.
Lethal screening using shRNA library
To further validate the functions of the EML4-ALK integrated network, we investigated the effect of loss of function of each of the whole network proteins on cell viability with or without ALK inhibitor treatment. We first built a shRNA sublibrary by selecting 5 shRNA clone IDs for each of the 407 genes from the Sigma-Aldrich Website (http://www.sigmaaldrich.com/united-states.html) and the target sequence from the Broad Institute Website (http://www.broadinstitute.org/rnai/public/clone/search). The detailed shRNA library screen procedure is described below.
Preparation of viral media and shRNA-containing cell lines
293FT cells were transfected using Turbofect (ThermoScientific) with a library containing TRC HIV-based (pLKO.1 and pLKO.2) lentiviral vectors, and viral medium was harvested as described previously (70). We plated106 H3122 wild-type cells in 10-cm cell culture dishes, allowing adherence for 24 hours. The cells were inoculated with shRNA viral media for 72 hours, split upon confluency into experimental plates, and allowed to grow for another 24 hours. The cells were treated with a 1:10000 dilution of DMSO or crizotinib or alectinib at IC20, IC50, or IC80 dose levels, in replicates of 3 plates per dose. After 72 hours, drug treatment media were replaced with fresh media, and the cells were allowed to grow for an additional 72 hours. Cells were removed using Accutase (Cell Innovative Technologies), pelleted, and stored at −20°C.
PCR and HiSeq
DNA was extracted from a cell pellet using a DNEasy Blood and Tissue Kit (Qiagen). DNA (500 ng) was added to a 50-μL PCR reaction using the Phusion system (New England Biolabs) and custom indexing primers, and reactions were performed in duplicate for a total of 1000 ng per sample. The PCR reaction sequence was as follows: 98°C for 1 minute, 35 cycles of 98°C for 10 seconds, 60°C for 20 seconds, and 72°C for 30 seconds, followed by 72°C for 5 minutes. We used 10 μL of each sample to check for appropriate bands on a 2% agarose gel. We pooled 5 μL of each sample into a single tube, concentrated using the Wizard Gel and PCR Clean-up System (Promega) and eluted in 50 μL. The eluent was then loaded on a Pippen Prep 2% gel (Sage Science) for targeted size selection. Pippen Prep parameters were set to elute DNA between 250 and 400 ng. The size-selected eluent was then run on an Illumina HiSeq2500 with dual-indexing.
Search for significant gene hits
We employed the Bioinformatics for Next Generation Sequencing! (BiNGS!) algorithm for analyzing and interpreting functional genetic screen by deep sequencing data(71). In brief, a preprocessing step filtered out erroneous and low-quality reads. Filtered reads were mapped against the shRNA reference library (2 days after infection) using Bowtie(72). Output from this step is a P × N matrix, where P and N represent shRNA counts and samples, respectively. shRNAs were also filtered where the median raw count in the concentration 1 (e.g. IC20) is greater than the maximum raw count in the concentration 2 (e.g. IC50) if the shRNA is enriched in the concentration 1, and vice versa. We then employed edgeR (73) to compute the differentially represented shRNAs. We then computed the false discovery rate (q-value) for these shRNAs, and carried out meta-analysis by combining q-values for all shRNAs representing the same gene using weighted Z-transformation approach (74). For each gene, we computed a weighted p-value P(wZ), and we used this P(wZ) to sort the shRNA hits. We performed pair-wise comparisons for each concentrations, and grouped the genes into three classes using the following rules based on the P(wZ) obtained from each gene (similar to the classification rules in Marcotte et al 2012 (75)). In this study, we considered genes, when deleted by shRNAs, induced cell death in all dosages (IC20, IC50 and IC80) as candidate hits. The counts of gene hits were significantly lower from Vehicle to IC20 and IC80 (E-value < 2), maximum difference is Vehicle to IC20 among all possible combinations, and no significant changes from IC20 to IC50 and IC50 to IC80. This analysis approach is similar to our previously published research (76).
shRNA library screen results validation
To validate the results from shRNA library screening and examine their ability to sensitize ALK TKI-resistant cells, we constructed 5 hairpins of each CC2D1A and FRS2 into lentivirus in H239FT cells and then infected H3122, H3122 EML4-ALKL1196M, and STE-1 cells. The infected cells were selected with 10 μg/ml puromycin for more than 10 days. The shRNA-positive cells were seeded into 96-well plates, series-diluted crizotinib and alectinib were added, and GLO assay was performed to measure the combined effects of shRNA knockdown with ALK TKI on sensitive and resistant cell viability at 72 hours. At the same time, cells were seeded into 10-cm dishes and treated with DMSO, 1 μM crizotinib, and 1 μM alectinib for 3 hours. Cell pellets were collected for further signaling and mechanism analyses with both RT-PCR and Western blotting.
Western blot analysis
Cell lysates were separated by SDS-PAGE, and proteins were electrotransferred to nitrocellulose membranes. Regular ECL western blot was used in this study. Basically, members were blocked with 5% milk in TBS for 1 hour, incubated with primary antibodies recognizing target proteins at 4°C overnight, washed membranes with TTBS for 5 minutes three times, incubated membranes with ECL HRP-conjugated second antibodies at room temperature for 1 hour, washed membranes with TTBS three times for 5 minutes, added super ECL and read the results. Another infrared based western blotting method was used in this study too. Simply, membranes, blocked with Odyssey Blocking Buffer (LI-COR Biosciences) for 1 hour at room temperature, were washed for 5 minutes three times with TBS/T. Membranes were incubated with primary antibody in blocking buffer with gentle agitation overnight at 4°C and then washed three times for 5 minutes each with TBS/T. Incubated membranes were fluorescently labeled with secondary antibody (1 μL IRDye 680 goat mouse source antibody and 1 μL IRDye 680 goat rabbit source antibody (1:10000)) in blocking buffer, with gentle rotation for 1 hour at room temperature. Membranes were washed with TBS/T for 5 minutes three times and rinsed with PBS to remove residual Tween 20. The Odyssey Infrared Imaging System quantified the 700- and 800-nm channel images and Odyssey V3.0 to calculate the intensities at 800 nm; these values were normalized to corresponding β-actin results (700 nm).
Quantitative RT-PCR
Cells containing scramble and target shRNA hairpins were washed with pre-cold PBS. Cell pellets were then collected. RNA was extracted using RNeasy Mini Kit (Qiagen). Purified RNA was reverse transcribed to cDNA using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). TaqMan® (Hs00183614_m1 FRS2 and Hs00214594_m1 CC2D1A FAM, Applied Biosystems) PCR reactions were conducted in 96-well Optical Reaction Plate with Barcode (code 128) on ABI 7900HT Fast Real Time PCR (Applied Biosystems) instruments. 18s rRNA (VIC-MGB 4319413E1202056, Applied Biosystems) was used to normalize all samples. Gene expression was quantified by cycle threshold (CT) method, where expression was determined by comparing normalized CT of samples containing target gene shRNA hairpin to CT of samples containing control shRNA scramble hairpin.
Dynamic cell viability analysis using real-time cell assay (RTCA)
The xCELLigence RTCA technology provides an accurate platform for noninvasive detection of cell viability (77). For cell viability analysis, effects of siRNA or drugs on cell viability were monitored by the dynamic, impedance-based xCELLigence system (Roche Applied Science, Germany) in a real-time, label-free manner. In general, H3122 cells were transfected with siRNA or treated with drugs as described previously in this paper and then seeded at a density of 8000 cells/well into E-Plate 16 (ACEA Biosciences, Inc., San Diego, CA) containing 100 μL RPMI 1640 medium per well in 96-well plates supplemented with 10% FBS. The cell index was derived from measured cell-electrode impedance that correlates with cells viability. Non-targeted siRNA and DMSO were used as control of RNAi and drug treatment, respectively. Data were collected every 10 minutes, exported into an Excel file after analyses with RTCA software (version 1.2), and plotted in Graphpad Prism 6.0 software. The slope/hour of cell index across all experimental times was used to monitor the cell viability.
TMA and immunohistochemistry analysis
A patient-derived tumor microarray (TMA) was created in the Moffitt Tissue Procurement Core Facility (Lung 6.2 TMA) under the auspices of an IRB-approved protocol. No protected health information related to the tissue included in the TMA was revealed to the study team. Donor paraffin blocks were obtained through biopsy or surgical resection specimens. All tumors were tested as standard of care prior to ALK TKI therapy and used the Vysis FISH probe. The Lung 6.2 TMA contained the following tissues: 8 EML4-ALK-rearranged cases (7 with triplicate cores and 1 with a single core); 6 non-ALK-rearranged NSCLC cases (all 6 with triplicate cores); malignant tissue including breast, colon, diffuse large B-cell lymphoma, glioma, hepatocellular carcinoma, osteosarcoma, ovarian carcinoma, prostate, and renal cell carcinoma (all with single cores); and normal tissue controls including placenta, spleen, stomach, and tonsil (all with single cores). All TMAs were sectioned at 5 μm, placed on charged slides, and baked for 60 minutes at 60°C.
Slides containing TMA and positive and negative controls were immunohistochemically stained using a Ventana Discovery XT automated system (Ventana Medical Systems, Tucson, AZ) following manufacturer’s instructions. Briefly, slides were deparaffinized with EZ Prep solution (Ventana) on the automated system. The rabbit primary antibody recognizing CC2D1A (HPA005436, Sigma, St. Louis, MO) was diluted with Dako antibody diluent (Carpenteria, CA) at 1:200 and incubated with TMA for 60 minutes. Slides were then incubated with Ventana OmniMap rabbit source secondary antibody for 8 minutes. The Ventana ChromoMap kit was used to detect the staining. Slides were also then counterstained with hematoxylin. Slides were then dehydrated and coverslipped as per normal laboratory protocol. Stained tissue micro arrays were scanned using the Aperio™ ScanScope XT (Aperio, Vista, California) with a 20x/0.8NA objective lens via Basler tri-linear-array detection. Images were viewed with ImageScope version 12.1.0.5029 (Aperio, Vista, California) and snapshot images from select cores were extracted to tif file format.
Stained TMA slides were scored as previously described (60) with slight modification. Staining intensity was graded as 0 (negative), 1 (weak), and 2 (strong); percentage of positive cells examined was scored as 0 (<5%), 1 (5–50%), and 2 (>50%). The two scores were multiplied, and the immunoreactive score (values from 0–4) was determined: 0 as negative, values 1–2 as weakly positive, and values 4 as strongly positive. The average immunoreactive score for EML4-ALK were calculated using Prism6 software.
Supplementary Material
Acknowledgments
We thank W. Pao (Roche, Basel) for providing H3122 cell lines, C. Lovly (Vanderbilt University) for providing STE1 cell lines, J. Tanizaki (Dana Farber Cancer Institute) for providing H3122/TR2 cells, J. Heuckmann and R. Thomas (Universität zu Köln, Köln, Germany) for providing plasmid constructs of EML4-ALK LL96M, and P. V. Hornbeck (Cell Signaling Technology) for his courtesy annotating protein types for all experimental identified proteins in this study using PhosphoSitePlus (www.phosphosite.org). We thank R. Hamilton (Moffitt Cancer Center) for editorial assistance. We thank F. Kinose (Moffitt Cancer Center) for help with cell culture. We also thank the Moffitt Analytic Microscopy Core Facility for scanning the TMA slides.
Funding: Moffitt Analytic Microscopy Core Facility is supported by the National Cancer Institute (NCI) as a Cancer Center Support Grant (P30-CA076292). This project was supported by a grant from Moffitt Cancer Center SPORE in Lung Cancer (P50-CA119997), Moffitt Lung Cancer Center of Excellence, Colorado Lung Cancer SPORE in Lung Cancer (P50-CA058187) and R01 (R01-CA157850) awarded by NCI, National Institutes of Health (NIH). Moffitt Proteomics is supported in part by the NCI(P30-CA076292) as a Cancer Center Support Grant and the Moffitt Foundation.
Footnotes
Author contributions: G. Z. and E. B. H. conceived the study. G. Z, H. S., J.K., A.I.R., Y.A.C., X. H. Z., L. X. S., R. Z. L., B. F., Y. B., J. K., A.C.T., J. D., and E.B.H. contributed to the methodology. G.Z, H. S., J.K., A.I.R., Y. A. C., X. H. Z., L. X. S. Y.B., R. Z. L., and B. F. performed the data acquisition and analysis. G.Z. and E. B. H. wrote the manuscript and G.Z, H. S., Y.A.C., B. F., J. K., A.C.T., J. D., and E.B.H. revised the manuscript. J. K., J.D. and E.B.H. provided the funding support through grants noted below. J.K., J.D. and E.B.H. provided resources. J.K., A.C.T., J.D. and E.B.H. supervised the study.
Competing interests: The authors have no conflicts of interest to disclose.
Data and materials availability: The mass spectrometry data are deposited to the ProteomeXchange Consortium via PRIDE partner repository with the dataset identifier PXD004935 and 10.6019/PXD004935.
References and Notes
- 1.Sharma SV, et al. A common signaling cascade may underlie “addiction” to the Src, BCR-ABL, and EGF receptor oncogenes. Cancer cell. 2006 Nov;10:425. doi: 10.1016/j.ccr.2006.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tran PT, et al. Survival and death signals can predict tumor response to therapy after oncogene inactivation. Sci Transl Med. 2011 Oct 5;3:103ra99. doi: 10.1126/scitranslmed.3002018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramirez M, et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nature communications. 2016;7:10690. doi: 10.1038/ncomms10690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hata AN, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nature medicine. 2016 Mar;22:262. doi: 10.1038/nm.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bivona TG, Doebele RC. A framework for understanding and targeting residual disease in oncogene-driven solid cancers. Nature medicine. 2016 May 5;22:472. doi: 10.1038/nm.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Astsaturov I, et al. Synthetic lethal screen of an EGFR-centered network to improve targeted therapies. Science signaling. 2010;3:ra67. doi: 10.1126/scisignal.2001083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li J, et al. Perturbation of the mutated EGFR interactome identifies vulnerabilities and resistance mechanisms. Molecular systems biology. 2013;9:705. doi: 10.1038/msb.2013.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fitzgerald JB, Schoeberl B, Nielsen UB, Sorger PK. Systems biology and combination therapy in the quest for clinical efficacy. Nat Chem Biol. 2006 Sep;2:458. doi: 10.1038/nchembio817. [DOI] [PubMed] [Google Scholar]
- 9.Soda M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007 Aug 2;448:561. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 10.Kwak EL, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. The New England journal of medicine. 2010 Oct 28;363:1693. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shaw AT, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2009 Sep 10;27:4247. doi: 10.1200/JCO.2009.22.6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi YL, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. The New England journal of medicine. 2010 Oct 28;363:1734. doi: 10.1056/NEJMoa1007478. [DOI] [PubMed] [Google Scholar]
- 13.Wilson FH, et al. A functional landscape of resistance to ALK inhibition in lung cancer. Cancer Cell. 2015 Mar 9;27:397. doi: 10.1016/j.ccell.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sakamoto H, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer cell. 2011 May 17;19:679. doi: 10.1016/j.ccr.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 15.Shaw AT, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. The New England journal of medicine. 2014 Mar 27;370:1189. doi: 10.1056/NEJMoa1311107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lovly CM, et al. Rationale for co-targeting IGF-1R and ALK in ALK fusion-positive lung cancer. Nat Med. 2014 Sep;20:1027. doi: 10.1038/nm.3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kimura M, et al. Analysis of ERBB ligand-induced resistance mechanism to crizotinib by primary culture of lung adenocarcinoma with EML4-ALK fusion gene. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer. 2015 Mar;10:527. doi: 10.1097/JTO.0000000000000381. [DOI] [PubMed] [Google Scholar]
- 18.Katayama R, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci Transl Med. 2012 Feb 8;4:120ra17. doi: 10.1126/scitranslmed.3003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sang J, et al. Targeted inhibition of the molecular chaperone Hsp90 overcomes ALK inhibitor resistance in non-small cell lung cancer. Cancer discovery. 2013 Apr;3:430. doi: 10.1158/2159-8290.CD-12-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu F, Wang P, Young LC, Lai R, Li L. Proteome-wide identification of novel binding partners to the oncogenic fusion gene protein, NPM-ALK, using tandem affinity purification and mass spectrometry. The American journal of pathology. 2009 Feb;174:361. doi: 10.2353/ajpath.2009.080521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crockett DK, Lin Z, Elenitoba-Johnson KS, Lim MS. Identification of NPM-ALK interacting proteins by tandem mass spectrometry. Oncogene. 2004 Apr 8;23:2617. doi: 10.1038/sj.onc.1207398. [DOI] [PubMed] [Google Scholar]
- 22.Riera L, et al. Involvement of Grb2 adaptor protein in nucleophosmin-anaplastic lymphoma kinase (NPM-ALK)-mediated signaling and anaplastic large cell lymphoma growth. The Journal of biological chemistry. 2010 Aug 20;285:26441. doi: 10.1074/jbc.M110.116327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bai RY, et al. Nucleophosmin-anaplastic lymphoma kinase associated with anaplastic large-cell lymphoma activates the phosphatidylinositol 3-kinase/Akt antiapoptotic signaling pathway. Blood. 2000 Dec 15;96:4319. [PubMed] [Google Scholar]
- 24.Hrustanovic G, et al. RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK-positive lung cancer. Nat Med. 2015 Sep;21:1038. doi: 10.1038/nm.3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koivunen JP, et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res. 2008 Jul 1;14:4275. doi: 10.1158/1078-0432.CCR-08-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lai AC, Tsai CF, Hsu CC, Sun YN, Chen YJ. Complementary Fe(3+)- and Ti(4+)-immobilized metal ion affinity chromatography for purification of acidic and basic phosphopeptides. Rapid communications in mass spectrometry: RCM. 2012 Sep 30;26:2186. doi: 10.1002/rcm.6327. [DOI] [PubMed] [Google Scholar]
- 27.Chatr-Aryamontri A, et al. The BioGRID interaction database: 2015 update. Nucleic acids research. 2015 Jan;43:D470. doi: 10.1093/nar/gku1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hornbeck PV, et al. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic acids research. 2012 Jan;40:D261. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng Y, et al. Ras-induced and extracellular signal-regulated kinase 1 and 2 phosphorylation-dependent isomerization of protein tyrosine phosphatase (PTP)-PEST by PIN1 promotes FAK dephosphorylation by PTP-PEST. Molecular and cellular biology. 2011 Nov;31:4258. doi: 10.1128/MCB.05547-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun T, et al. Activation of multiple proto-oncogenic tyrosine kinases in breast cancer via loss of the PTPN12 phosphatase. Cell. 2011 Mar 4;144:703. doi: 10.1016/j.cell.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barr AJ, et al. Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell. 2009 Jan 23;136:352. doi: 10.1016/j.cell.2008.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koch A, Mancini A, El Bounkari O, Tamura T. The SH2-domian-containing inositol 5-phosphatase (SHIP)-2 binds to c-Met directly via tyrosine residue 1356 and involves hepatocyte growth factor (HGF)-induced lamellipodium formation, cell scattering and cell spreading. Oncogene. 2005 May 12;24:3436. doi: 10.1038/sj.onc.1208558. [DOI] [PubMed] [Google Scholar]
- 33.Pesesse X, et al. The Src homology 2 domain containing inositol 5-phosphatase SHIP2 is recruited to the epidermal growth factor (EGF) receptor and dephosphorylates phosphatidylinositol 3,4,5-trisphosphate in EGF-stimulated COS-7 cells. The Journal of biological chemistry. 2001 Jul 27;276:28348. doi: 10.1074/jbc.M103537200. [DOI] [PubMed] [Google Scholar]
- 34.Backers K, Blero D, Paternotte N, Zhang J, Erneux C. The termination of PI3K signalling by SHIP1 and SHIP2 inositol 5-phosphatases. Advances in enzyme regulation. 2003;43:15. doi: 10.1016/s0065-2571(02)00043-2. [DOI] [PubMed] [Google Scholar]
- 35.Yoshida T, et al. Tyrosine phosphoproteomics identifies both codrivers and cotargeting strategies for T790M-related EGFR-TKI resistance in non-small cell lung cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014 Aug 1;20:4059. doi: 10.1158/1078-0432.CCR-13-1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Honorat JF, Ragab A, Lamant L, Delsol G, Ragab-Thomas J. SHP1 tyrosine phosphatase negatively regulates NPM-ALK tyrosine kinase signaling. Blood. 2006 May 15;107:4130. doi: 10.1182/blood-2005-06-2421. [DOI] [PubMed] [Google Scholar]
- 37.Polgar D, et al. Truncated ALK derived from chromosomal translocation t(2;5)(p23;q35) binds to the SH3 domain of p85-PI3K. Mutation research. 2005 Feb 15;570:9. doi: 10.1016/j.mrfmmm.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 38.Sattu K, et al. Phosphoproteomic analysis of anaplastic lymphoma kinase (ALK) downstream signaling pathways identifies signal transducer and activator of transcription 3 as a functional target of activated ALK in neuroblastoma cells. The FEBS journal. 2013 Nov;280:5269. doi: 10.1111/febs.12453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ambrogio C, et al. p130Cas mediates the transforming properties of the anaplastic lymphoma kinase. Blood. 2005 Dec 1;106:3907. doi: 10.1182/blood-2005-03-1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Degoutin J, Vigny M, Gouzi JY. ALK activation induces Shc and FRS2 recruitment: Signaling and phenotypic outcomes in PC12 cells differentiation. FEBS letters. 2007 Feb 20;581:727. doi: 10.1016/j.febslet.2007.01.039. [DOI] [PubMed] [Google Scholar]
- 41.Boccalatte FE, et al. The enzymatic activity of 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase/IMP cyclohydrolase is enhanced by NPM-ALK: new insights in ALK-mediated pathogenesis and the treatment of ALCL. Blood. 2009 Mar 19;113:2776. doi: 10.1182/blood-2008-06-161018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hornbeck PV, et al. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic acids research. 2015 Jan;43:D512. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shaozhang Z, et al. Detection of EML4-ALK fusion genes in non-small cell lung cancer patients with clinical features associated with EGFR mutations. Genes, chromosomes & cancer. 2012 Oct;51:925. doi: 10.1002/gcc.21976. [DOI] [PubMed] [Google Scholar]
- 44.Katayama R, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci Transl Med. 2012 Feb 8;4:120ra17. doi: 10.1126/scitranslmed.3003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eto M, Karginov A, Brautigan DL. A novel phosphoprotein inhibitor of protein type-1 phosphatase holoenzymes. Biochemistry. 1999 Dec 21;38:16952. doi: 10.1021/bi992030o. [DOI] [PubMed] [Google Scholar]
- 46.Izumi Y, et al. A metalloprotease-disintegrin, MDC9/meltrin-gamma/ADAM9 and PKCdelta are involved in TPA-induced ectodomain shedding of membrane-anchored heparin-binding EGF-like growth factor. The EMBO journal. 1998 Dec 15;17:7260. doi: 10.1093/emboj/17.24.7260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim YA, et al. Role of PKCbetaII and PKCdelta in blood-brain barrier permeability during aglycemic hypoxia. Neuroscience letters. 2010 Jan 14;468:254. doi: 10.1016/j.neulet.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 48.Hsieh HL, et al. Thrombin induces EGF receptor expression and cell proliferation via a PKC(delta)/c-Src-dependent pathway in vascular smooth muscle cells. Arteriosclerosis, thrombosis, and vascular biology. 2009 Oct;29:1594. doi: 10.1161/ATVBAHA.109.185801. [DOI] [PubMed] [Google Scholar]
- 49.Garofalo T, et al. A novel mechanism of CD4 down-modulation induced by monosialoganglioside GM3. Involvement of serine phosphorylation and protein kinase c delta translocation. The Journal of biological chemistry. 1998 Dec 25;273:35153. doi: 10.1074/jbc.273.52.35153. [DOI] [PubMed] [Google Scholar]
- 50.He Y, et al. Proteolysis-induced N-terminal ectodomain shedding of the integral membrane glycoprotein CUB domain-containing protein 1 (CDCP1) is accompanied by tyrosine phosphorylation of its C-terminal domain and recruitment of Src and PKCdelta. The Journal of biological chemistry. 2010 Aug 20;285:26162. doi: 10.1074/jbc.M109.096453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cho SH, You HJ, Woo CH, Yoo YJ, Kim JH. Rac and protein kinase C-delta regulate ERKs and cytosolic phospholipase A2 in FcepsilonRI signaling to cysteinyl leukotriene synthesis in mast cells. Journal of immunology. 2004 Jul 1;173:624. doi: 10.4049/jimmunol.173.1.624. [DOI] [PubMed] [Google Scholar]
- 52.Csermely P, Korcsmaros T, Kiss HJ, London G, Nussinov R. Structure and dynamics of molecular networks: a novel paradigm of drug discovery: a comprehensive review. Pharmacology & therapeutics. 2013 Jun;138:333. doi: 10.1016/j.pharmthera.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen Z, et al. Inhibition of ALK, PI3K/MEK, and HSP90 in murine lung adenocarcinoma induced by EML4-ALK fusion oncogene. Cancer research. 2010 Dec 1;70:9827. doi: 10.1158/0008-5472.CAN-10-1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miyanaga A, et al. Activity of EGFR-tyrosine kinase and ALK inhibitors for EML4-ALK-rearranged non-small-cell lung cancer harbored coexisting EGFR mutation. BMC cancer. 2013;13:262. doi: 10.1186/1471-2407-13-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tanizaki J, et al. Activation of HER family signaling as a mechanism of acquired resistance to ALK inhibitors in EML4-ALK-positive non-small cell lung cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012 Nov 15;18:6219. doi: 10.1158/1078-0432.CCR-12-0392. [DOI] [PubMed] [Google Scholar]
- 56.Tanimoto A, et al. Receptor ligand-triggered resistance to alectinib and its circumvention by Hsp90 inhibition in EML4-ALK lung cancer cells. Oncotarget. 2014 Jul 15;5:4920. doi: 10.18632/oncotarget.2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Taipale M, et al. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell. 2012 Aug 31;150:987. doi: 10.1016/j.cell.2012.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schreiber TB, Mausbacher N, Keri G, Cox J, Daub H. An integrated phosphoproteomics work flow reveals extensive network regulation in early lysophosphatidic acid signaling. Molecular & cellular proteomics: MCP. 2010 Jun;9:1047. doi: 10.1074/mcp.M900486-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Blouin J, Roby P, Arcand M, Beaudet L, Lipari F. Catalytic specificity of human protein tyrosine kinases revealed by Peptide substrate profiling. Current chemical genomics. 2011;5:115. doi: 10.2174/1875397301105010115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yamada T, et al. Akt kinase-interacting protein1, a novel therapeutic target for lung cancer with EGFR-activating and gatekeeper mutations. Oncogene. 2013 Sep 12;32:4427. doi: 10.1038/onc.2012.446. [DOI] [PubMed] [Google Scholar]
- 61.Yamada T, et al. Akt Kinase-Interacting Protein 1 Signals through CREB to Drive Diffuse Malignant Mesothelioma. Cancer research. 2015 Oct 1;75:4188. doi: 10.1158/0008-5472.CAN-15-0858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Crystal AS, et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science. 2014 Dec 19;346:1480. doi: 10.1126/science.1254721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Winter GE, et al. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015 Jun 19;348:1376. doi: 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Basel-Vanagaite L, et al. The CC2D1A, a member of a new gene family with C2 domains, is involved in autosomal recessive non-syndromic mental retardation. Journal of medical genetics. 2006 Mar;43:203. doi: 10.1136/jmg.2005.035709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reimand J, Arak T, Vilo J. g:Profiler–a web server for functional interpretation of gene lists (2011 update) Nucleic acids research. Jul;39:W307. doi: 10.1093/nar/gkr378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aranda B, et al. PSICQUIC and PSISCORE: accessing and scoring molecular interactions. Nature methods. 2011 Jul;8:528. doi: 10.1038/nmeth.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang CY, et al. PhosphoPOINT: a comprehensive human kinase interactome and phospho-protein database. Bioinformatics. 2008 Aug 15;24:i14. doi: 10.1093/bioinformatics/btn297. [DOI] [PubMed] [Google Scholar]
- 68.Spinelli L, et al. Clust&See: a Cytoscape plugin for the identification, visualization and manipulation of network clusters. Bio Systems. 2013 Aug;113:91. doi: 10.1016/j.biosystems.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 69.Assenov Y, Ramirez F, Schelhorn SE, Lengauer T, Albrecht M. Computing topological parameters of biological networks. Bioinformatics. 2008 Jan 15;24:282. doi: 10.1093/bioinformatics/btm554. [DOI] [PubMed] [Google Scholar]
- 70.Casas-Selves M, et al. Tankyrase and the canonical Wnt pathway protect lung cancer cells from EGFR inhibition. Cancer research. 2012 Aug 15;72:4154. doi: 10.1158/0008-5472.CAN-11-2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim J, Tan AC. BiNGS!SL-seq: a bioinformatics pipeline for the analysis and interpretation of deep sequencing genome-wide synthetic lethal screen. Methods in molecular biology. 2012;802:389. doi: 10.1007/978-1-61779-400-1_26. [DOI] [PubMed] [Google Scholar]
- 72.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010 Jan 1;26:139. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Whitlock MC. Combining probability from independent tests: the weighted Z-method is superior to Fisher’s approach. Journal of evolutionary biology. 2005 Sep;18:1368. doi: 10.1111/j.1420-9101.2005.00917.x. [DOI] [PubMed] [Google Scholar]
- 75.Marcotte R, et al. Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer discovery. 2012 Feb;2:172. doi: 10.1158/2159-8290.CD-11-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim J, et al. Bioinformatics-driven discovery of rational combination for overcoming EGFR-mutant lung cancer resistance to EGFR therapy. Bioinformatics. 2014 Sep 1;30:2393. doi: 10.1093/bioinformatics/btu323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Limame R, et al. Comparative analysis of dynamic cell viability, migration and invasion assessments by novel real-time technology and classic endpoint assays. PloS one. 2012;7:e46536. doi: 10.1371/journal.pone.0046536. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.