

Abstract
HIV-1 CA capsid protein possesses intrinsic conformational flexibility, which is essential for its assembly into conical capsids and interactions with host factors. CA is dynamic in the assembled capsid, and residues in functionally important regions of the protein undergo motions spanning many decades of timescales. Chemical shift anisotropy (CSA) tensors, recorded in magic-angle-spinning NMR experiments, provide direct residue-specific probes of motions on nano- to microsecond timescales. We combined NMR, MD, and Density-Functional-Theory calculations, to gain quantitative understanding of internal backbone dynamics in CA assemblies, and found that the dynamically averaged 15N CSA tensors calculated by this joined protocol are in remarkable agreement with experiment. Thus, quantitative atomic-level understanding of the relationships between CSA tensors, local backbone structure and motions in CA assemblies is achieved, demonstrating the power of integrating NMR experimental data and theory for characterizing atomic-resolution dynamics in biological systems.
Keywords: magic-angle spinning NMR, HIV-1 capsid, CA protein assemblies, HIV-AIDS, conformational dynamics, chemical shift anisotropy, quantum mechanics/molecular mechanics
Graphical Abstract
INTRODUCTION
HIV-1 capsids, assembled from ~1,500 copies of the capsid (CA) protein, are an internal part of mature virions (Figure 1).1,2 Conical in shape, capsids enclose the viral genome together with several proteins that are essential for viral replication.3 During HIV-1 entry, intact capsids are released into the cytoplasm of the host cell, followed by disassembly (uncoating), which takes place in a tightly orchestrated fashion. The net result of these events is the integration of the reverse transcribed viral DNA into the host genome, necessary for viral replication.4,5
Figure 1.

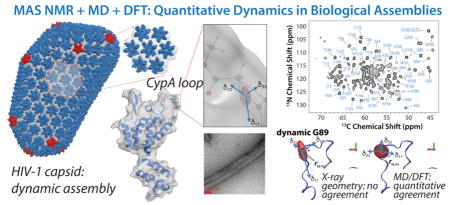
a) All-atom model of mature HIV-1 capsid determined by an integrated cryo-ET, solution NMR, and MD approach (PDBID: 3J3Y 6). Hexameric (predominant) and pentameric (minor) building blocks are shown in blue and red, respectively. b) Hexamer of hexamers (HOH), a predominant structural element of the capsid. c) 3D structure of an HIV-1 CA monomer (PDB file 3NTE). d) Illustration of a backbone 15NH CSA tensor and its orientation in the protein molecular frame, e) Transmission electron microscopy (TEM) image of tubular assemblies of CA HXB2. f) A 2D NCA spectrum of tubular assemblies of U-13C, 15N CA, recorded at 21.1 T and the MAS frequency of 14 kHz.
Capsids are pleomorphic comprised of varied number of copies of CA protein structural blocks, with variable curvature in the capsid shell and overall appearance. Pleomorphism is directly related to the conformational plasticity of the CA protein.7 Remarkably, in the assembled state, CA is dynamic over a range of timescales from nano- to milliseconds.6,8–10 Specifically, motions in the linker, which connects the CA’s N- and C-terminal domains (NTD and CTD) occur on the milliseconds timescale, and are directly linked to capsid’s ability to assemble into varied morphologies.8 These motions, coupled with pH-dependent electrostatic interactions may contribute to assembly control, involving a molecular switch.8
In the host cells, capsid interacts with host proteins. These interactions can either facilitate or inhibit the HIV-1 replication. One such protein is Cyclophilin A (CypA), which is required for the virus to be active in certain cell types.11,12 CypA interacts with the capsid by binding to the CypA loop located in CA’s NTD, and mutants in the CypA loop can escape from CypA dependence. For instance, the A92E and G94D CA escape mutants are fully infectious when CypA/CA interactions are inhibited, while in the presence of CypA these mutants retain only ~10% of infectivity. It has been proposed that CypA plays a role in stabilizing the capsid through a novel binding interface, where one CypA molecule bridges two CA hexamers, along the highest-curvature direction.13
CA/CypA interactions modulate capsid dynamics, and the virus takes advantage of this effect to escape from the CypA dependence. Specifically, CypA binding attenuates the motions of the CypA loop and also induces allosteric conformational changes in other regions of the protein. Remarkably, the dynamic behavior A92E and G94D CA mutants closely resembles that of the CA/CypA assembly, and dynamic allosteric regulation mechanism was proposed to explain the capsid’s escape from CypA dependence.10
Internal dynamics of CA also play a role in HIV-1 maturation. Initially, during virus budding from the host cell, the Gag polyprotein, which contains the main structural proteins of HIV (MA, CA, MA, NC, and p6), is a single polypeptide chain. During maturation, Gag is proteolytically cleaved into the individual domains by the HIV protease in an orderly, stepwise fashion. The final step is the cleavage of a 14-residue spacer peptide 1 (SP1) from the CA-SP1 maturation intermediate to release CA, which in turn organizes into the conical cores characteristic of mature virions. In CA-SP1, the SP1 peptide is highly flexible and its presence affects the mobility of distal residues, such as those in the CypA loop.14 Clearly, dynamic regulation is an important ingredient in the final maturation step, and the above-described studies highlight the importance of understanding residue-specific conformational dynamics in HIV-1 capsid assemblies, given the direct relationship between dynamics and virus function.
Previously, we analyzed 1H-15N/1H-13C/15N-13C dipolar interactions as well as resonance intensities and linewidths, demonstrating the power of combining MAS NMR and MD simulations for determining the motional modes of the individual residues. Here, we focus on 15N chemical shift anisotropy (CSA) tensors as sensitive probes of nano- to microsecond dynamics in HIV-1 capsid assemblies. We integrate quantum mechanics/molecular mechanics (QM/MM) calculations of magnetic shielding anisotropy, using Density Functional Theory (DFT), with MD simulations and MAS NMR to gain quantitative insights into the effects of dynamic averaging on CSA tensors. We show that the experimental and calculated 15N CSA tensors are in remarkable agreement, including those for dynamic residues, such as in the CypA-binding loop, and that dynamic averaging of chemical shifts has to be taken into account. The integrated approach described here is broadly applicable to any system and is particularly important in NMR-based structure determination of dynamic regions in protein assemblies.
RESULTS AND DISCUSSION
Experimental NMR parameters
Tubular assemblies of HIV-1 CA yield highly resolved MAS NMR spectra (Figure 1), and permitted assignments 13Cα, 13C° and 15N chemical shifts for 222, 188, and 210 residues, respectively. Backbone 15N CSA tensors for individual residues were derived from a 3D R1425-symmetry based RNCSA experiment.15,16 The experimental 15N, 13Cα, and 13C° isotropic chemical shifts, δiso are plotted vs. residue number in Figure 2 and 3, and reduced anisotropy parameters in Fig. 3d as well as CSA lineshapes are shown in Fig. 3e and Fig. S1 (Supplementary Information).
Figure 2.

The backbone isotropic chemical shift parameters in tubular assemblies of CA, plotted as a function of the residue number: (a) 13Cα δiso and (b) 13C° δiso. The parameters are shown as follows: isotropic chemical shifts recorded from NMR experiments, black; calculated by MD/DFT, red; calculated by DFT from a single X-ray structure, blue; calculated by ShiftX, orange. The grey rectangles denote the loop regions of CA protein.
Figure 3.

The backbone 15NH chemical shift parameters in tubular assemblies of CA, plotted as a function of the residue number: δiso (a–c) and δσ (d). The parameters are shown as follows: isotropic chemical shifts or reduced chemical shift anisotropy recorded from 3D RNCSA NMR experiments, black; calculated by MD/DFT, red; calculated by DFT from a single X-ray structure, blue; calculated by ShiftX, orange. The error bars are shown on the experimental δσ values. The grey rectangles denote the loop regions of CA protein, (e) Experimental (solid black lines) and simulated (dashed red lines) 15NH RNCSA lineshapes plotted for selected CA residues; the best fit CSA parameters are indicated above each spectrum.
It is well known from solution NMR studies that isotropic chemical shifts are modulated by dynamics,17 and MD simulations in conjunction with empirical ShiftX-based calculations were reported to explain the dynamic averaging of δiso in RNaseH.18 Here, for CA assemblies, large variation in 15N, 13Cα, and 13C° δiso is seen for the different residues, and the extent of variability is generally greater for the loop vs. the helical regions of CA (Fig. S2, Supplementary). However, no obvious trends that would suggest the presence of motions in specific regions of the protein can be noted. In contrast, the experimental reduced anisotropy parameters of the 15N CSA tensors, δσ, are dramatically different along the CA sequence, see Fig. 3d. Most notably, the residues in CypA loop exhibit low δσ values for A88, G89, A92, and G94 of 50.4, 40.3, 70.9, and 59.0 ppm, respectively (the corresponding rigid-limit values are 96.0, 95.3, 102.6, and 92.8 ppm, based on a DFT calculation using X-ray structure geometry; PDBID: 3NTE). While it is known that differences in the secondary structure and local interactions (hydrogen bonding and solvent interactions) affect the 15N CSA tensors in proteins,19,20 the dramatic reduction of δσ observed for the residues in the CypA loop residues, compared to the rigid value limits, cannot be explained by a structural effect alone. Greatly attenuated δσ values were also observed for several other residues, primarily in loop regions of both, the NTD and CTD.
We previously demonstrated via a combined MAS NMR and MD simulations approach, using 1H-15N/13Cα dipolar tensors, that residues in loop regions are mobile, with those in the CypA loop unusually flexible on the nano- to microseconds timescale.10 Therefore, we hypothesized that these internal motions may cause partial averaging of the backbone CSA tensors, similar to the dynamic effects on CSA tensors observed in other proteins.21,22 This notion was examined in the present study.
Understanding the Mechanism of Dynamic CSA Averaging by Integrating NMR, MD, and QM/MM Calculations
To test the hypothesis that dynamics influence CSA tensors in CA assemblies in a major way, we computed CSA tensors using a QM/MM approach, with the quantum region being treated at the DFT level. In parallel we also calculated isotropic shifts by ShiftX.23 These calculations were carried out for a monomer extracted from the CA X-ray structure (PDBID: 3NTE) as well as for individual CA substructures, using the reported 100-ns MD trajectories.10
As evidenced from the data in Fig. 2 and Fig. 3, the ShiftX-based predictions of isotropic chemical shifts agree well with the NMR experiments, because these calculations, by nature of their database driven properties, take into account dynamic averaging of isotropic shifts for mobile regions, as reported previously by several groups.24–27 On the other hand, DFT-calculated values based on the CA X-ray structure are not in agreement with experiment, neither for the reduced anisotropy parameters nor the isotropic shifts. For example, the experimental 15N δσ vary over a large range, from 40.4 ppm to 106.3 ppm, while the calculated 15N δσ exhibit only small variations (99.0 ± 15.4 ppm). RMSD values from DFT calculations are 6.2, 4.4, and 12.6 ppm for 15N δσ, 13C δiso, and 15N δσ, respectively. Furthermore, no correlation with secondary structure is seen and residues in CypA-binding loop were not predicted to have uniquely attenuated anisotropy. Likewise, the calculated isotropic 15N, 13Cα, and 13C° shifts do not reproduce the experimental parameters.
In contrast, the dynamically averaged 15N CSA tensors computed by MD/DFT are generally in remarkably good agreement with experiment, as shown in Figures 3–4. Importantly, the dramatic reduction in 15N δσ values for the CypA-loop residues that was observed experimentally is captured in the computations, indicating that this decrease is caused by internal loop dynamics rather than by structural effects. This finding is consistent with our prior results from dipolar tensors.10 In addition, the extent of CSA averaging increases for the residues in the middle of the CypA loop, where the largest motional amplitudes and extensive sampling of conformational space is expected.10 An outlier in that regard is G89, for which the computational results suggest a considerably smaller 15N δσ (23.75 ppm) than the experimentally observed value (40.3 ppm). While such a deviation in δσ values is generally considered to be acceptable for quantum mechanical calculations, it may also be the result of insufficient sampling, due to the length of the MD trajectory.
Figure 4.

(a–c) The backbone 15NH principal components of the CSA tensors, δii, in tubular assemblies of CA, plotted as a function of the residue number. The experimental values obtained from 3D RNCSA are shown in black; the values calculated by MD/DFT are in red. (d–i) The MD-DFT calculated CSA tensor parameters, δσ, δiso, and δii, plotted vs. the corresponding experimental parameters. The parameters for both NTD and CTD are displayed in (d–f); those for NTD only are shown in (g–i).
The overall agreement between the experimental and MD/DFT computed δiso is poorer than that for δσ, see Fig. 2–3 and 4e. Considerable scatter is noted, and the slope of 0.77 and offset of 21.6 ppm are significantly greater than those reported for previous DFT calculations of 15N chemical shifts for 20 structurally characterized proteins.28 To gain further insights into the effect of dynamic averaging on the 15N CSA tensors we analyzed their experimental and computed principal components, δ11, δ22, and δ33. These are plotted vs. the residue number in Fig. 4a–c. The experimental and calculated δii correlate well, see Fig. 4f: the slope is 0.98, and a negligible offset of −2.6 ppm is seen (1.3% of the entire range of δii values). The largest scatter for any principal component is observed for δ33 (Fig. S3, Supplementary), suggesting that reorientation of the CSA tensor due to motion affects most prominently the principal component perpendicular to the amide plane (Fig. 1d). These results highlight the importance of measuring CSA tensors, not only isotropic shifts, for systems that possess internal dynamics.
Another interesting observation in this study relates to the finding that better agreement between the experiment and calculations is seen for NTD residues, compared to the CTD. This is evident from the data depicted in Fig. 4, both, for δiso and δσ. We observed a similar effect in our studies of 1H-15N/13Cα dipolar tensors, where alignments were carried out for the individual domains in the calculations in order to eliminate overall translational and rotational motions.10 The NTD and CTD are connected by a flexible linker, which is mobile on the slow-microsecond to millisecond timescales.8 CTD residues are more rigid according to the experimental CSA parameters than those computed by MD/QM/MM. Since MD/QM/MM calculations were conducted on monomeric CA, the intermolecular CTD-CTD interactions, which are essential for the capsid assembly and stability,6 could not be captured, rendering the CTD in the MD/QM/MM calculations more flexible than in tubular assemblies studied by MAS NMR.
To understand how the CSA tensors are affected by the reorientation of the corresponding residues during local motions, we examined the computed principal components of the 15N CSA tensors, δii, and the isotropic shifts, δiso, for each residue along the MD trajectory. In Fig. S3 Supplementary) we illustrated the results for two residues: G89 in the tip of the CypA-binding loop, which is highly mobile, and the more rigid E98. Interestingly, the variations in the principal CSA components are relatively modest, and independent of whether the corresponding residue is dynamic or static. However, as shown in Fig. 5, for the dynamic G89 very large changes in the Euler angles (α, β, γ) are observed, associated with different orientations of the CSA tensors in the individual substructures in the MD trajectory. These kinds of motions are expected to attenuate the observed CSA tensors (see an illustration for a spin model in Fig. S4. Supplementary). Conversely, for the static E98 these variations are much smaller. The polar angles θ, which relate each δii to the N-H bond vector, on the other hand, are similar for both static and dynamic residues, see Fig. 5. This indicates that motions exert larger effects on the relative orientations of the individual CSA tensors, while the individual principal components of the diagonal CSA tensor are affected less. This is vividly seen when the individual components of a non-diagonal CSA tensor, δij (where i/j=x,y,z) are plotted in the molecular fixed frame (MFF) representation (Fig. S5, Supplementary), with variations predominantly being caused by the changing Euler angles. (MFF here coincides with the lab frame since only one protein molecule is considered in calculations.) The dynamic averaging of the 15N CSA tensors is illustrated in Fig. 6a–f for representative CypA-loop residues. The differences between mobile and rigid residues are also apparent when comparing experimental δiso values with those computed by MD/DFT and MD/ShiftX (Fig. S2, Supplementary). Not surprisingly, loop residues that are mobile exhibit broad distributions, while residues in helices possess narrow chemical shift ranges. The corresponding residue-specific distributions computed by MD/ShiftX (Fig. S6, Supplementary) reveal that the width of the CS distribution correlates with the conformational space sampled by each residue in the course of MD trajectory. This result is consistent with our prior findings for dipolar tensors10 and with solution-NMR studies of RNaseH.18
Figure 5.

(a) The amplitudes and orientations of backbone 15NH CSA tensors for G89 (left) and E98 (right) in HIV-1 CA protein with respect to the molecular frame, for the individual structures along the MD trajectory at t = 0, 1, 2, and 3 ns (from top to bottom, respectively). (b–d) Plots of the relative angles between the principal components of the 15N CSA tensor, δii, and the N-H bond, rN-H. (e–g) Euler angles for the backbone 5NH CSA tensors in the molecular frame. The CSA parameters and relative tensor orientations were calculated by QM/MM (at the DFT level) for the individual structures along the MD trajectory as a function of time, as shown for G89 (black triangles) and E98 (red circles).
Figure 6.

Left two panels: The plots of the amplitudes and orientations of the backbone 15NH CSA tensors in HIV-1 CA, in the molecular frame, calculated by QM/MM (at the DFT level) for the starting structure (left) and averaged along the MD trajectory (right). Right panel: probability distributions for Euler angles α (black), β (blue), and γ (red) over the course of MD trajectory. The plots are shown for selected CA residues: (a) H84, (b) H87, (c) G89, (d) 191, (e) A92, and (f) E98.
Given the computationally expensive nature of QM/MM CSA tensor calculations, it is important to evaluate whether the number of MD frames and the sampling intervals, which are required to accurately represent the accessible conformational space for each residue, are sufficient. Such analysis was carried out for the highly mobile G89 and rigid E98 residues (Fig. S7, Supplementary). Interestingly, for both residues we find that averaging either 100 or 200 frames from a 100-ns trajectory (1- or 0.5-ns sampling interval, respectively) yields similar δσ values (±2%), essentially identical within experimental error. Averaging 100 frames from the first 10 ns of the MD trajectory yields essentially identical δσ values for the static E98, while a difference of 5.4 ppm is observed for the dynamic G89. Therefore, it appears that longer MD trajectories are required to reach more quantitative agreement with experiment.
To rule out that details of the particular DFT methodology used in the current calculations for G89 biased the resulting CSA parameters, we applied three commonly used density functionals: B3LYP, OLYP, and 03LYP. All three yield δσ values to within 4%, i.e. vary less than the difference between the experimental and the calculated values, confirming that all three functionals are indistinguishable in outcome (Fig. S8, Supplementary).
Overall, the differences observed here between computations and experiment for certain residues highlight the importance of long trajectories and the development of MD protocols that enhance sampling of biologically relevant states. With computational cost decreasing rapidly, they should no longer be an impediment to calculate dynamically averaged CSA calculations by quantum mechanical methods. Such calculations are feasible and represent the method of choice for a rigorous analysis of the structural vs. dynamic contributions to chemical shifts, as demonstrated here.
CONCLUSIONS AND FUTURE OUTLOOK
Residue-specific 15N, 13C, and 1H CSA tensors are readily accessible using contemporary multidimensional RNCSA methods,15,16,29 even for large protein assemblies. These experiments provide uniquely rich information about the geometry, electronic structure, and dynamics. 15N CSA tensors provide a direct and sensitive probe of internal residue backbone dynamics occurring on timescales of nano- to microseconds. For tubular assemblies of the HIV-1 CA protein, investigated here, residues in the mobile CypA-binding loop exhibit dramatic decreases in 15N chemical shift anisotropy. The reduced anisotropy parameters, δσ, unequivocally report on the presence of motions, unlike isotropic chemical shifts, where dynamic effects are not immediately obvious. Knowledge of dynamic regions in proteins and assemblies thereof is invaluable for accurate interpretation of chemical shifts in terms of geometric structure. The integrated analysis of combined experimental MAS NMR data, MD simulations as well as QM methods for calculating chemical shifts in proteins using Density Functional Theory, provides rich and in-depth atomic-level information on the inter-relatedness of internal motions and local structure. Here we show that the dynamic averaging of the CSA parameters results from Euler angle changes rather than variations in the principal components of the CSA tensor. This finding has important implications for the analysis of dynamics by solution NMR relaxation methods, where the shift anisotropy is taken as a constant.30 The integrated MAS NMR and MD/DFT approach developed and illustrated here is a powerful tool for atomic-resolution characterization of structure and dynamics of biological assemblies.
MATERIALS AND METHODS
Protein expression, purification and NMR sample preparation were performed as described previously31. CA was expressed in E. coli Rosetta 2 (DE3), cultured in Luria-Bertani or modified minimal media, after induction with 0.4 mM IPTG at 23 °C for 16h. The purification was performed with cation exchange column chromatography with 0–1 M NaCI gradient in a buffer containing 25 mM sodium phosphate pH 5.8, 1 mM DTT, and 0.02% NaN3. Uniform 15N- and 13C-labeling was performed using 15NH4CI and 13C6-glucose as sole nitrogen and carbon sources. Tubular assemblies of CA protein were prepared from 30 mg/mL protein solution in 25 mM phosphate buffer (pH5.5) containing 1M NaCI. The solution was incubated at 37 °C for 1 h and stored at 4 °C for subsequent NMR experiments. The incubated CA sample was pelleted by centrifugation at 6,000 G for 15 minutes, the supernatant was removed, and 15 mg (0.59 mmoles) of full-length tubular WT CA assemblies were packed into a Varian 3.2 mm MAS rotor. The rotor was sealed using an upper spacer and a top spinner.
MAS NMR experiments were carried out on a 21.1 T ultra-wide bore spectrometer outfitted with a 3.2 mm HXY Enhanced-Design Low-E MAS probe designed and built at NHMFL.32 Larmor frequencies were 899.1 MHz (1H), 226.1 MHz (13C), and 91.1 MHz (15N). The MAS frequency was set at 14 kHz for all experiments, and was controlled to within ± 5 Hz by a Bruker MAS controller. KBr was used as temperature sensor, and the actual temperature of the sample was maintained to within 4 ± 0.1 °C using the Bruker temperature controller. 13C and 15N chemical shifts were referenced with respect to the external standards adamantane and NH4CI, respectively. In 15N RNCSA 3D experiment, the typical 90° pulse lengths were 2.5 μs (1H), 3 μs (13C), and 5 μs (15N), and the contact time of 1H-15N cross polarization (CP) was 1.0 ms. 1H-15N CP employed a linear amplitude ramp on the 1H channel with the center Hartmann-Hahn matched to the first spinning side band. The band-selective magnetization transfer from 15N to 13Cα was realized through a 4.5 ms SPECIFIC-CP with a tangent amplitude ramp on the 15N channel (49 kHz rf field center) and a constant rf field on the 13C channel (35 kHz). High-power 1H continuous wave (CW) decoupling (110 kHz) was applied during the SPECIFIC-CP period, and SPINAL-64 decoupling (100 kHz) was applied during the direct (t3) and indirect (t2) acquisition periods. R1425-based symmetry sequence was used to reintroduce 15N CSA interaction during the t1 evolution period, and the phase-alternated (±5Π/14 or ± 64.29°) rf field irradiation (49 kHz) was applied on the 15N channel. Simultaneous Π pulses were applied on the 13C channel at the center of every two rotor periods to decouple 15N-13C dipolar interactions, and 1H CW was applied to decouple 1H-15N dipolar interactions.
Numerical simulations of 15N RNCSA lineshapes were performed with the SIMPSON software package, versions 2.0. To produce a powder average, 320 pairs of {α, β} angles generated according to the REPULSION algorithm and 16 γ angles (resulting in a total of 5,120 angle triplets) were used for all simulations. Haberlen-Mehring-Spiess convention was followed to define the CSA parameters in the present work, where the isotropic chemical shift δiso, the reduced anisotropic chemical shift δσ, and the asymmetry parameter η, are related to the principal elements δii (i = 1, 2 or 3) of the chemical shift tensor according to |δ11 - δiso| ≥ |δ33 - δiso| ≥ |δ22 - δiso|, δiso = ⅓ (δ11+δ22+δ33), δσ = δ11 - δiso, and . For extracting the best-fit 15N CSA NMR parameters (δσ, η), all of the experimental and processing parameters (i.e., Larmor frequency, MAS frequency, RF field strength, number of t1 points, finite pulse lengths, zero-filling, line broadening, etc.) were taken into account in the simulations. With the attainable sensitivity and resolution, a total of 76 non-overlapping 15N CSA lineshapes were extracted and fitted.
Molecular dynamics simulations of the wild type CA monomer (HXB2 polymorph, PDB ID 3NTE) were performed using ACEMD as reported previously.10 Ions in close proximity to the protein were placed using the CIONIZE routine in VMD, bulk waters and Na/CI ions were then added using VMD33,34, setting the total concentration of NaCI to 1M. The system was then equilibrated using NAMD 2.9 with the CHARMM36FF at 300 K for 100 ns.
QM/MM calculations of magnetic shielding anisotropy tensors were carried out in Gaussian09,35 and automated fragmentation quantum mechanics/molecular mechanics approach (AF-QM/MM) from AFNMR package25 of ShiftS was used to divide the entire protein system into nonoverlapping fragments, in which QM region included a fragment within 4 or 3 Å from the target residue (without or with protons included, respectively). MM region included the rest residues beyond the core and the buffer regions. AF-QM/MM was performed to generate individual spin system for each target residue. In this work, the DFT calculations of the NMR calculations were performed by the GIAO method using the OLYP density functional and the TZVP basis set, and all of the calculated 15N magnetic shieldings were referenced to the isotropic shielding constant of 237.8 ppm, as reported previously for the computations at the same level of theory in Ubiquitin (PDB ID: 1D3Z); the calculated 13C magnetic shieldings were referenced to the isotropic shielding constant of 190.8 ppm, according to the RMSD-based referencing method36.
Averaged chemical shift tensor calculations were performed by combining QM/MM calculations with MD simulations. A total of 100 frames were extracted from first 10 ns from the 100 ns MD trajectory. For each frame, the AF-QM/MM was performed to generate individual spin system for each target residue. For each target residue, we performed the 100 QM/MM calculations using Gaussion09, and the backbone 15N chemical shielding tensors for the target residue were extracted from each calculation, as follows:
| (1) |
where i denotes the index of the frame extracted from the MD trajectory.
The magnetic shielding anisotropy tensor is expressed in its principal axes system (PAS) as
| (2) |
The dynamically averaged 15N chemical shielding tensor for each residue was then obtained by linear averaging of its nine elements extracted from the QM/MM calculations for each of the 100 individual substructures in the MD trajectory,
| (3) |
where N denotes the number of total frames taken for QM/MM calculations for the residue of interest; in the present work N=100. The averaged 15N chemical shielding tensors were referenced to the isotropic shielding constant of 237.8 ppm to obtain the motion-averaged chemical shift tensor, 〉δ〈, and the principal components, δii, for each target residue.
To obtain the time-dependent orientation of the chemical shielding tensors in the laboratory frame, for each target residue, rotations in Cartesian space were used to express the orientation relative to the stationary set of axes in each frame. These were parameterized by three Euler angles α, β, and γ, according to the standard notation:37
| (4) |
where R denotes the corresponding rotation matrix, expressed as
| (5) |
Chemical shift prediction ShiftX was also used to calculate the dynamically averaged 15N isotropic chemical shifts for the individual 5000 frames during the 100 ns MD trajectory.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (NIH Grants P50 GM082251, P41 GM104601, and P50 GM103297 from NIGMS). We acknowledge the support of the National Science Foundation (NSF Grant CHE0959496) for the acquisition of the 850 MHz NMR spectrometer at the University of Delaware and of the National Institutes of Health (NIH Grants P30GM103519 and P30GM110758) for the support of core instrumentation infrastructure at the University of Delaware. This research is part of the Blue Waters sustained-petascale computing project supported by NSF (PRAC Award ACI-1440026). A portion of this work was performed at the National High Magnetic Field Laboratory, which is supported by National Science Foundation Cooperative Agreement No. DMR-1157490 and the State of Florida.
Footnotes
Experimental and simulated 15N CSA lineshapes; distribution plots for isotropic chemical shifts; principal components of 15N CSA tensor for selected CA residues; simulated rigid and motionally reduced 15N CSA line shapes; individual components of 15N CSA tensor for selected CA residues; probability distribution plots of 15N isotropic chemical shifts calculated by Shiftx based on MD simulation; Euler angles of the 15N CSA tensors for G89 and E98 residues of CA calculated from MD/DFT with different sampling schedules, and with different functionals. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Briggs JAG, Simon MN, Gross I, Krausslich HG, Fuller SD, Vogt VM, Johnson MC. Nat Struct Mol Biol. 2004;11:672–675. doi: 10.1038/nsmb785. [DOI] [PubMed] [Google Scholar]
- 2.Sundquist WI, Krausslich HG. Csh Perspect Med. 2012;2 doi: 10.1101/cshperspect.a006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu K, Heng X, Summers MF. J Mol Biol. 2011;410:609–633. doi: 10.1016/j.jmb.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aiken C. Curr Opin Hiv Aids. 2006;1:194–199. doi: 10.1097/01.COH.0000221591.11294.c1. [DOI] [PubMed] [Google Scholar]
- 5.Freed EO. Nat Rev Microbiol. 2015;13:484–496. doi: 10.1038/nrmicro3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao GP, Perilla JR, Yufenyuy EL, Meng X, Chen B, Ning JY, Ahn J, Gronenborn AM, Schulten K, Aiken C, Zhang PJ. Nature. 2013;497:643–646. doi: 10.1038/nature12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ganser BK, Li S, Klishko VY, Finch JT, Sundquist WI. Science. 1999;283:80–83. doi: 10.1126/science.283.5398.80. [DOI] [PubMed] [Google Scholar]
- 8.Byeon IJL, Hou GJ, Han Y, Suiter CL, Ahn J, Jung J, Byeon CH, Gronenborn AM, Polenova T. J Am Chem Soc. 2012;134:6455–6466. doi: 10.1021/ja300937v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bayro MJ, Chen B, Yau WM, Tycko R. J Mol Biol. 2014;426:1109–1127. doi: 10.1016/j.jmb.2013.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu MM, Hou GJ, Zhang HL, Suiter CL, Ahn J, Byeon IJL, Perilla JR, Langmead CJ, Hung I, Gor’kov PL, Gan ZH, Brey W, Aiken C, Zhang PJ, Schulten K, Gronenborn AM, Polenova T. P Natl Acad Sci USA. 2015;112:14617–14622. doi: 10.1073/pnas.1516920112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hatziioannou T, Perez-Caballero D, Cowan S, Bieniasz PD. J Virol. 2005;79:176–183. doi: 10.1128/JVI.79.1.176-183.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Y, Kar AK, Sodroski J. J Virol. 2009;83:10951–10962. doi: 10.1128/JVI.00682-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu C, Perilla JR, Ning JY, Lu MM, Hou GJ, Ramalho R, Himes BA, Zhao GP, Bedwell GJ, Byeon IJ, Ahn J, Gronenborn AM, Prevelige PE, Rousso I, Aiken C, Polenova T, Schulten K, Zhang PJ. Nat Commun. 2016;7 doi: 10.1038/ncomms10714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han Y, Hou GJ, Suiter CL, Ahn J, Byeon IJL, Lipton AS, Burton S, Hung I, Gor’kov PL, Gan ZH, Brey W, Rice D, Gronenborn AM, Polenova T. J Am Chem Soc. 2013;135:17793–17803. doi: 10.1021/ja406907h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hou G, Paramasivam S, Byeon IJL, Gronenborn AM, Polenova T. Phys Chem Chem Phys. 2010;12:14873–14883. doi: 10.1039/c0cp00795a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hou GJ, Byeon IJL, Ahn J, Gronenborn AM, Polenova T. J Chem Phys. 2012;137 doi: 10.1063/1.4754149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guerry P, Mollica L, Blackledge M. Chemphyschem. 2013;14:3046–3058. doi: 10.1002/cphc.201300377. [DOI] [PubMed] [Google Scholar]
- 18.Robustelli P, Stafford KA, Palmer AG., 3rd J Am Chem Soc. 2012;134:6365–74. doi: 10.1021/ja300265w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saito H, Ando I, Ramamoorthy A. Prog Nucl Mag Res Sp. 2010;57:181–228. doi: 10.1016/j.pnmrs.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wylie BJ, Franks WT, Rienstra CM. J Phys Chem B. 2006;110:10926–10936. doi: 10.1021/jp060507h. [DOI] [PubMed] [Google Scholar]
- 21.Yang J, Tasayco ML, Polenova T. J Am Chem Soc. 2008;130:5798–5807. doi: 10.1021/ja711304e. [DOI] [PubMed] [Google Scholar]
- 22.Paluch P, Pawlak T, Jeziorna A, Trebosc J, Hou GJ, Vega AJ, Amoureux JP, Dracinsky M, Polenova T, Potrzebowski MJ. Phys Chem Chem Phys. 2015;17:28789–28801. doi: 10.1039/c5cp04475h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han B, Liu YF, Ginzinger SW, Wishart DS. J Biomol Nmr. 2011;50:43–57. doi: 10.1007/s10858-011-9478-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen Y, Bax A. J Biomol Nmr. 2010;48:13–22. doi: 10.1007/s10858-010-9433-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swails J, Zhu T, He X, Case DA. J Biomol Nmr. 2015;63:125–39. doi: 10.1007/s10858-015-9970-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen Y, Lange O, Delaglio F, Rossi P, Aramini JM, Liu G, Eletsky A, Wu Y, Singarapu KK, Lemak A, Ignatchenko A, Arrowsmith CH, Szyperski T, Montelione GT, Baker D, Bax A. Proc Natl Acad Sci U S A. 2008;105:4685–90. doi: 10.1073/pnas.0800256105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mollica L, Baias M, Lewandowski JR, Wylie BJ, Sperling LJ, Rienstra CM, Emsley L, Blackledge M. J Phys Chem Lett. 2012;3:3657–62. doi: 10.1021/jz3016233. [DOI] [PubMed] [Google Scholar]
- 28.Xu XP, Case DA. Biopolymers. 2002;65:408–423. doi: 10.1002/bip.10276. [DOI] [PubMed] [Google Scholar]
- 29.Hou G, Paramasivam S, Yan S, Polenova T, Vega AJ. J Am Chem Soc. 2013;135:1358–68. doi: 10.1021/ja3084972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palmer AG. Annu Rev Bioph Biom. 2001;30:129–155. doi: 10.1146/annurev.biophys.30.1.129. [DOI] [PubMed] [Google Scholar]
- 31.Han Y, Ahn J, Concel J, Byeon IJL, Gronenborn AM, Yang J, Polenova T. J Am Chem Soc. 2010;132:1976–1987. doi: 10.1021/ja908687k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gor’kov PL, Chekmenev EY, Li CG, Cotten M, Buffy JJ, Traaseth NJ, Veglia G, Brey WW. J Magn Reson. 2007;185:77–93. doi: 10.1016/j.jmr.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 33.Humphrey W, Dalke A, Schulten K. J Mol Graph. 1996;14:33–8. 27–8. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 34.Bak M, Schultz R, Vosegaard T, Nielsen NC. J Magn Reson. 2002;154:28–45. doi: 10.1006/jmre.2001.2454. [DOI] [PubMed] [Google Scholar]
- 35.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Wallingford CT: 2009. [Google Scholar]
- 36.Salager E, Day GM, Stein RS, Pickard CJ, Elena B, Emsley L. J Am Chem Soc. 2010;132:2564–6. doi: 10.1021/ja909449k. [DOI] [PubMed] [Google Scholar]
- 37.Edmonds AR. Angular momentum in quantum mechanics; Princeton University Press; 1960. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.