

Abstract
Colon cancer is one of the most common cancers. In this study, we isolated a lignan [(−)-(2R,3R)-1,4-O-diferuloylsecoisolariciresinol, DFS] from Alnus japonica (Betulaceae) and investigated its biological activity and mechanism of action on colon cancer. DFS reduced the viability of colon cancer cells and induced cell cycle arrest. DFS also suppressed β-catenin nuclear translocation and β-catenin target gene expression through a reduction in FoxM1 protein. To assess the mechanism of the action of DFS, we investigated the effect of DFS on endogenous and exogenous FoxM1 protein degradation in colon cancer cells. DFS-induced FoxM1 protein degradation was suppressed by lysosomal inhibitors, chloroquine and bafilomycin A1, but not by knock-down of proteasomal proteins. The mechanism of DFS for FoxM1 degradation is lysosomal dependent, which was not reported before. Furthermore, we found that FoxM1 degradation was partially lysosomal-dependent under normal conditions. These observations indicate that DFS from A. japonica suppresses colon cancer cell proliferation by reducing β-catenin nuclear translocation. DFS induces lysosomal-dependent FoxM1 protein degradation. This is the first report on the lysosomal degradation of FoxM1 by a small molecule. DFS may be useful in treating cancers that feature the elevated expression of FoxM1.
The Wnt/β-catenin signaling pathway plays a primary role in cellular differentiation and proliferation. Beta-catenin forms a complex with APC/Axin/GSK3β and is degraded by the proteasome under Wnt-free conditions. However, the Wnt/β-catenin pathway is constitutively activated in most sporadic and hereditary colorectal tumors caused by mutations in Wnt/β-catenin pathway-related molecules, such as adenomatous polyposis coli (APC) and β-catenin1. Aberrantly activated β-catenin increases nuclear translocation of other oncogenes2,3 and binds to T-cell factor/lymphoid enhancer factor transcription factors to promote expression of target genes, such as cyclin D1, survivin, and c-Myc, which play key roles in cellular differentiation and proliferation4,5. Thus, aberrantly activated Wnt/β-catenin signaling is regarded as a target for the chemoprevention and treatment of colorectal cancer.
FoxM1 is a member of the Forkhead box transcription factor family. The varied biological activities of FoxM1, include regulation of cellular proliferation, DNA damage repair, angiogenesis, apoptosis, and tumorigenesis6. From the early stage of tumor development to later metastasis, FoxM1 expression is highly elevated in a variety of cancers6,7. Elevation in FoxM1 levels promotes cancer initiation and maintenance through regulation of the progression of cancer cell cycle and proliferation6,7. For example, elevation in FoxM1 levels promotes development and proliferation of colon adenocarcinomas in vivo, and depletion of FoxM1 reduces colon cancer cell growth in vitro8. Furthermore, FoxM1 also increases the transcriptional activity of other oncogenes to promote tumorigenesis. For example, FoxM1 interacts with β-catenin and promotes the nuclear translocation of β-catenin to promote glioma stem cell self-renewal and tumorigenesis through increased expression of β-catenin target genes3. Therefore, targeting FoxM1 is a good strategy for chemoprevention and treatment of colorectal cancer.
Alnus japonica (Betulaceae) grows in the low mountainous areas of Korea, northeast China, and Japan. It has been used in traditional oriental medicine to treat fever, hemorrhage, diarrhea, and alcoholism. Recent studies have shown that Alnus japonica has various phytochemicals, such as diarylheptanoids, triterpenoids, and flavonoids9,10,11,12,13,14. In this study, we isolated a lignan [(−)-(2R,3R)-1,4-O-diferuloylsecoisolariciresinol, DFS] from Alnus japonica and explored its activity against colon cancer. DFS was first reported by Nomura and Tokoroyama15 and its cytotoxic action against several cancer cell types has been described16,17,18. Presently, we describe the ability of DFS to block β-catenin nuclear translocation through the lysosomal-dependent degradation of FoxM1 protein.
Results
DFS suppresses the β-catenin pathway
TOPFlash and FOPFlash reporter cell lines were used to test the effects of DFS (Fig. 1A) on the Wnt/β-catenin pathway. Treatment with Wnt3a-conditioned media (CM) significantly increased TOPFlash activity, and treatment with DFS suppressed Wnt3a-induced TOPFlash activity in a dose-dependent manner (Fig. 1B). To test whether GSK-3β is involved in the inhibition of β-catenin transcription, we treated HEK293 cells with LiCl as an inhibitor of GSK-3β14. DFS suppressed LiCl-induced TOPFlash activity in a dose-dependent manner (Fig. 1C). These data indicate that DFS suppresses the β-catenin pathway in a GSK-3β-independent manner.
Figure 1. DFS suppresses the β-catenin signaling pathway.

The structure of DFS (A). TOPFlash or FOPFlash reporter expressed HEK293 cells were treated with the indicated concentrations of DFS in the presence of Wnt3a (B) or LiCl (C) for 16 h and TOPFlash activity was measured. SW480 and HCT116 colon cancer cells were transiently transfected with the TOPFlash plasmid and treated with the indicated concentrations of DFS for 16 h, TOPFlash activity was measured (D).
Next, we tested the ability of DFS to suppress the Wnt/β-catenin pathway in colon cancer cells. SW480 and HCT116 cells (adenomatous polyposis [APC] mutated or β-catenin mutated, respectively) were transiently transfected with the TOPFlash plasmid and treated with DFS to assess luciferase activity. DFS significantly suppressed TOPFlash activity in both colon cancer cell types with an IC50 value of 7.68 μM and 7.17 μM, respectively (Fig. 1D). These data indicate that DFS suppresses the β-catenin pathway in both APC mutated and β-catenin mutated colon cancer cells.
DFS suppresses colon cancer cell proliferation and induces cell death
As abnormal activation of the Wnt/β-catenin pathway is the main cause of colon cancer cell proliferation19, we tested the inhibitory potential of DFS on the growth of colon cancer cells using the MTT and cell cycle assays. DFS reduced cell viability of SW480 and HCT116 (Fig. 2A) colon cancer cells in a dose-dependent manner. In the case of normal cells, such as colon CCD-18Co and mouse embryonic fibroblast (MEF), DFS displayed only marginal cytotoxicity at a high dose of 25 μM (Fig. 2A). Cell cycle analysis showed that DFS significantly induced G1 phase arrest in both SW480 and HCT116 colon cancer cells at 24 h, and induced cell death at 48 h (Fig. 2B,C). Taken together, the data indicate that DFS has anti-proliferative activity against aberrantly activated β-catenin-induced colon cancer cell and decreases the survival of SW480 and HCT116 colon cancer cells.
Figure 2. DFS suppresses colon cancer cell proliferation and induces cell death.
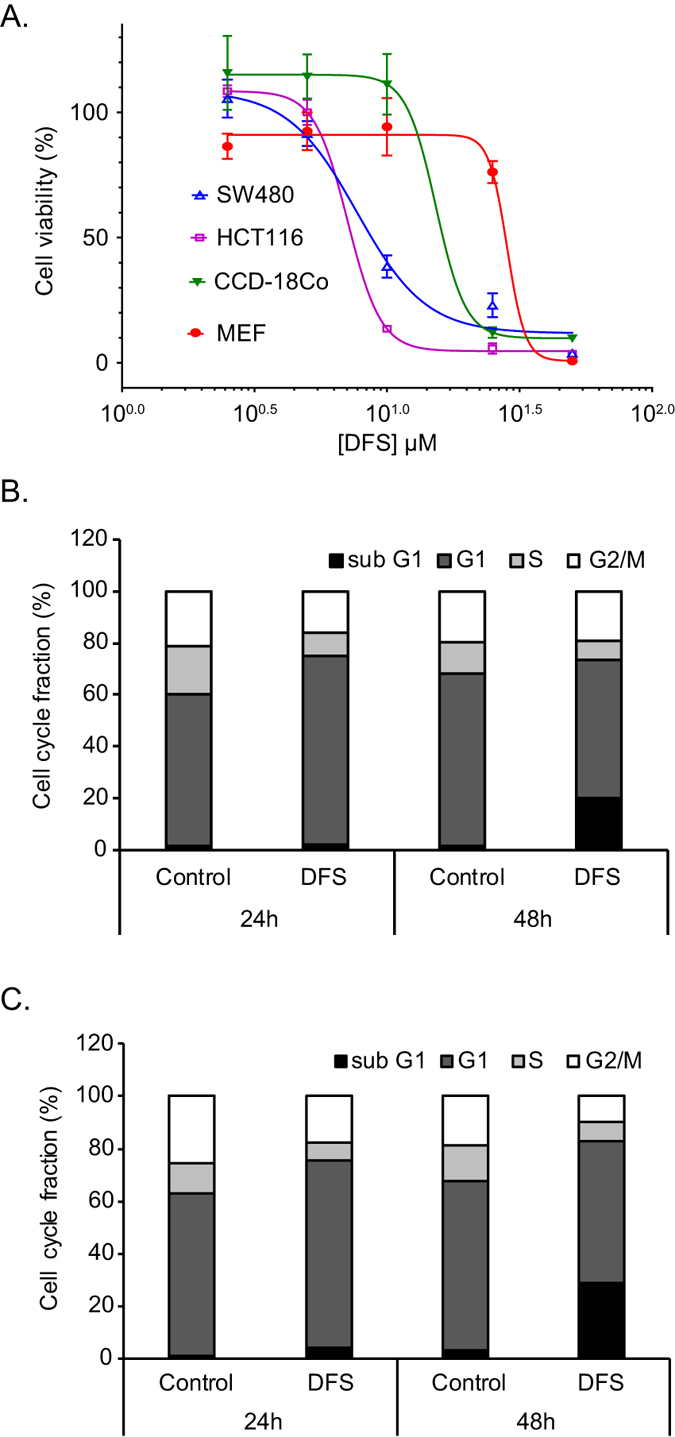
Cell viability was determined with the MTT assay after treatment with indicated concentration of DFS for 48 h (A). SW480 and HCT116 colon cancer cell cycle was analyzed by FACS after treatment with 10 μM DFS for the indicated times (B,C).
DFS suppresses nuclear location of β-catenin by decreasing the level of FoxM1 protein
To confirm the inhibitory effects of DFS on the Wnt/β-catenin pathway, β-catenin levels and levels of its target genes were determined by Western blotting. DFS siginficantly reduced the levels of β-catenin target genes, such as cyclin D1, survivin, and c-Myc in SW480 and HCT116 cells without altering the total level of β-catenin (Fig. 3A). Interestingly, the nuclear level of β-catenin was decreased by DFS in a dose-dependent manner in colon cancer cells, whereas the cytosolic level was not changed (Fig. 3B). These data indicate that DFS suppresses the Wnt/β-catenin pathway by blocking nuclear translocation of β-catenin in colon cancer cells. As FoxM1 modulates β-catenin nuclear translocation3, we tested the effect of DFS on FoxM1 levels in colon cancer cells. Knock-down of FoxM1 decreased nuclear translocation of β-catenin in SW480 cells (Fig. 3C,D). FoxM1 protein levels were reduced by DFS treatment, while the mRNA level of FoxM1 was not affected in either SW480 or HCT116 cells (Fig. 3E). These data indicate that DFS inhibits the nuclear translocation of β-catenin in colon cancer cells by modulating the level of FoxM1 protein.
Figure 3.

DFS suppresses β-catenin nuclear translocation by reducing the level of FoxM1 protein. SW480 and HCT116 colon cancer cells were treated with the indicated concentrations of DFS for 16 h and the protein levels of β-catenin and its target genes were measured (A). The cytosolic and nuclear protein levels of β-catenin were determined by Western blot (B). SW480 cells were transfected with siRNA against FoxM1 and the localization of β-catenin was determined by Western blot (C,D). SW480 and HCT116 colon cancer cells were treated with the indicated concentrations of DFS for 16 h, and the protein and mRNA levels of FoxM1 were determined by Western blot and RT-PCR (E).
DFS destabilizes FoxM1 protein in colon cancer cells
Cycloheximide was used to investigate the mechanism of action of DFS involved in suppressing FoxM1 levels. Treatment with DFS reduced FoxM1 levels in cycloheximide-treated and -untreated SW480 and HCT116 cells (Fig. 4A). Furthermore, DFS induced degradation of endogenously expressed FoxM1 in control (LacZ) adenovirus-infected HCT116 cells (Fig. 4B, left panel). DFS also induced degradation of exogenously expressed FoxM1 in cycloheximide-treaded HCT116 cells (Fig. 4B, right panel). These data indicate that DFS destabilizes the FoxM1 protein, which suppresses β-catenin nuclear translocation.
Figure 4. DFS induces lysosomal-dependent FoxM1 protein degradation.

SW480 and HCT116 colon cancer cells were treated with DFS and cycloheximide (CHX) for 14 h and the protein levels of FoxM1 were determined by Western blot (A). HCT116 colon cancer cells were infected with adeno-FoxM1 virus and treated with DFS and CHX for 14 h, then the protein levels of FoxM1 were determined by Western blot (B). SW480 and HCT116 colon cancer cells were treated with DFS and Etoposide (ET) for 16 h and the protein and mRNA levels of FoxM1 were determined by Western blot and RT-PCR (C). SW480 and HCT116 colon cancer cells were treated with DFS and MG132 for 16 h and the protein and mRNA levels of FoxM1 were determined by Western blot and RT-PCR. Then, the membrane was reprobed by stripping buffer and HIF1α protein levels were measured to assess MG132 activity (D). SW480 cells were transfected with the indicated siRNAs and related gene expressions were determined by RT-PCR and Western blot assay (E). SiRNA-transfected SW480 cells were treated with DFS for 16 h and FoxM1 protein levels were determined by Western blot (F). Black triangle denotes original FoxM1 and the brace denotes post-modified FoxM1.
To confirm the effects of DFS on FoxM1 protein degradation, we used etoposide, a clinically available anti-cancer drug that increases FoxM1 by stabilizing FoxM1 protein20,21. Etoposide increased FoxM1 levels without changing mRNA levels (Fig. 4C). However, treatment with DFS reduced protein levels of etoposide-induced FoxM1 in both SW480 and HCT116 cells (Fig. 4C). Thus, DFS diminishes the stability of FoxM1 protein, which suppresses β-catenin nuclear translocation.
DFS induces lysosomal-dependent degradation of FoxM1 protein
Protein degradation is dependent on the proteasome22. To investigate the mechanism of action of DFS for FoxM1 protein degradation, colon cancer cells were treated with the MG132 proteasome inhibitor. Treatment with MG132 reduced FoxM1 levels in both SW480 and HCT116 cells (Fig. 4D). MG132 inhibits FoxM1 transcriptional activity and FoxM1 expression in several cancer cell types23. Presently, co-treatment with DFS and MG132 further reduced FoxM1 levels than treatment with either DFS or MG132 alone (Fig. 4D). Proteasomal inhibitory activity of MG132 was confirmed by measuring the level of hypoxia inducible factor 1α (HIF1α) in SW480 cells (Fig. 4D).
To further investigate whether the proteasomal pathway is involved in DFS-induced FoxM1 degradation, siRNAs were used (Fig. 4E). Knock-down of PSMA3 (20S proteasomal protein), PSMD3 (19S proteasomal protein), and PSME1 (11S proteasomal protein) resulted in increased size and quantity of FoxM1 compared with control colon cancer cells (Fig. 4F, brace). These observations and knowledge that FoxM1 can be ubiquitinated and degraded by proteasome20,21 indicate that knock-down of proteasomal proteins suppresses FoxM1 protein degradation, and that the 20S, 19S, and 11S proteasomes are involved in FoxM1 protein degradation. However, DFS induced FoxM1 degradation in proteasomal protein knock-down cells (Fig. 4F). These data indicate that other mechanism than proteasomal degradataion might be involved in DFS-induced FoxM1 degradation.
To assess whether the lysosomal degradation pathway is involved in DFS-induced FoxM1 degradation, the lysosomal inhibitors, chloroquine and bafilomycin A1, were used. DFS-induced FoxM1 degradation was blocked by treatment with chloroquine in colon cancer cells (Fig. 5A), while the levels of Notch 3 and E-cadherin were not changed by DFS (Fig. 5B). Notch 3 and E-cadherin were also known to be targeted for lysosomal degradation24,25. These data indicate that DFS induced FoxM1 degradation in a lysosomal-dependent manner. Treatment with chloroquine slightly restored FoxM1 levels in cycloheximide-treated colon cancer cells (Fig. 5A), which means that FoxM1 degradation is partially dependent on the lysosome under normal conditions. Similar results were observed when bafilomycin A1, another lysosomal degradation inhibitor was treated SW480 colon cancer cells (Fig. S8).
Figure 5. DFS induces lysosomal-dependent FoxM1 protein degradation.

Protein levels of FoxM1 were determined by Western blot in SW480 and HCT116 cells after treatment with DFS and chloroquine (CQ) or cycloheximide (CHX) (A). Colon cancer cells were treated with DFS and the levels of E-cadherin and Notch3 were determined by Western blot assay (B).
DFS has antitumor activity in a tumor xenograft mouse model
The antitumor activity of DFS was evaluated in a nude mouse xenograft model with implanted SW480 human colon cancer cells. Mice were treated with DFS 5 days a week by intraperitoneal injection. The tumor volumes in DFS-administered groups with 30 mg/kg were significantly reduced (*p < 0.01) compared with the vehicle at the termination of the experiment on day 21 (Fig. 6A). No overt toxicity or change in body weight was shown in the treatment groups compared to the vehicle-treated control group (Fig. 6B). These results confirm that DFS effectively inhibits the tumor growth of human colorectal cancer cells in vivo.
Figure 6. DFS inhibits the growth of tumor in vivo.
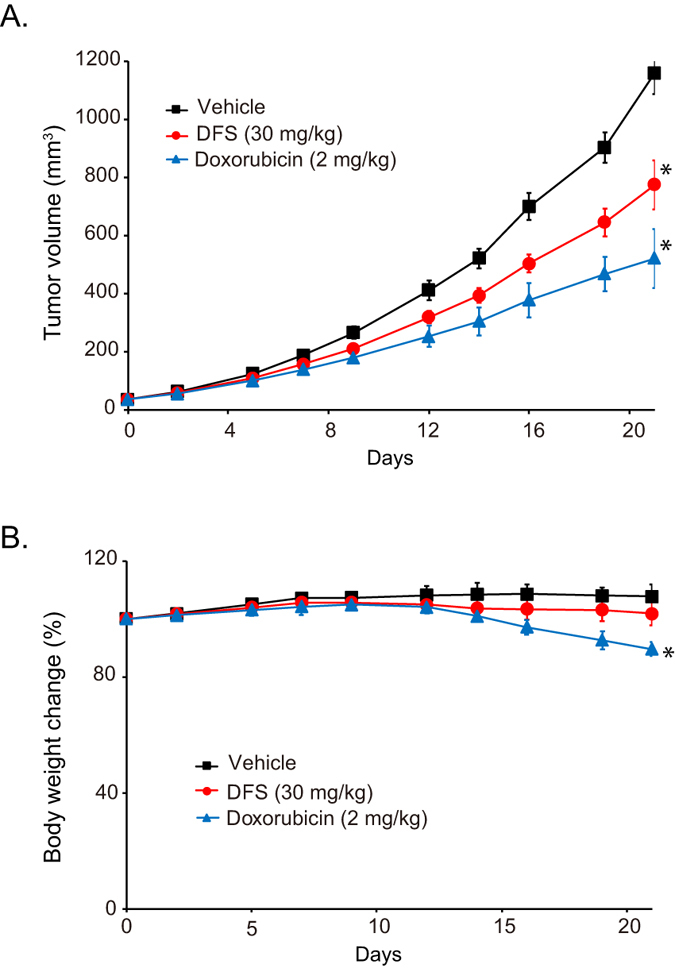
SW480 cells (9 × 106 cells/mouse) were inoculated subcutaneously into the right flank of nude mice (n = 5). Treatment was initiated when tumor volumes reached approximately ~40 mm3. DFS (30 mg/kg body weight) was administered intraperitoneally 5 days a week for 21 days. On day 21, mice were sacrificed and tumor volume was measured (A). The asterisk (*) indicated significancy compared to vehicle group (p < 0.01). Body weight was measured during the experiments as an indication of toxicity (B).
Discussion
Colorectal cancer is a common cancer that is responsible for approximately 10% of all cancer-related deaths1,26. The Wnt/β-catenin pathway is mutated in about 90% cases of sporadic and hereditary colorectal cancer1,26. Abnormal activation of the Wnt pathway promotes nuclear translocation of β-catenin, and increases initiation and maintenance of colon cancers1. Nuclear translocation of β-catenin is regulated by several oncogenes, including FoxM12,3. FoxM1 promotes the nuclear translocation of β-catenin, thereby increases the expression of β-catenin target genes3. Elevated expression of FoxM1 has been detected in most cancers from early tumor development to metastasis6,7. Elevation in FoxM1 also promotes initiation and maintenance of cancer through regulation of the cancer cell cycle, proliferation, angiogenesis, anti-apoptosis, and invasion6,7. Targeting FoxM1 is a good strategy for cancer therapeutics, because FoxM1 also regulates Wnt/β-catenin pathway.
In this study, DFS isolated from A. japonica suppressed TOPFlash activity (Fig. 1) and suppressed β-catenin nuclear translocation and expression of its target genes in both APC (SW480) or β-catenin (HCT116) mutated colon cancer cells (Fig. 3). Consequently, DFS suppressed colon cancer cell viability and cell cycle progression (Fig. 2) at 10 μM, while normal fibroblast cells (CCD-18Co and MEF cells) were quite resistant to cytotoxicity of DFS. DFS decreased the protein level of FoxM1 without changing the mRNA level in colon cancer cells (Fig. 3E). The FoxM1 destabilizing activity of DFS was also confirmed in cycloheximide-treated colon cancer cells (Fig. 4A,B). DFS decreased the etoposide stabilized FoxM1 level, which is the basis of drug resistance20,21. Thus, DFS might sensitize cancer cells to chemotherapy27. As degradation of FoxM1 depends on proteasomes22, siRNA knock-down of proteasomal proteins suppressed FoxM1 degradation (Fig. 4E,F). However, DFS reduced FoxM1 protein levels in proteasomal protein knock-down colon cancer cells (Fig. 4F). We also showed that FoxM1 protein degradation is 20S, 19S, and 11S proteasomal-dependent (Fig. 4F). These results suggest that DFS might have proteasomal independent mechanism for FoxM1 degradation.
DFS-induced FoxM1 protein degradation was blocked by the lysosomal inhibitors chloroquine and bafilomycin A1 (Fig. 5A, S8), indicating that DFS-induced FoxM1 degradation may be dependent on lysosome. This is a novel finding. The lysosomal degradation of FoxM1 might be related with autophagic regulation by DFS. This will need to be investigated in other studies. Autophagy can be tumor-promoting and tumor-suppressing depending on the stage or the type of cancers28. Presently, DFS mediated FoxM1 degradation by activating lysosomal-mediated degradation, which reduced the viability of colon cancer cells in culture (Fig. 2) and in vivo in a mouse xenograft model (Fig. 6). Although DFS might induce autophagy, and so helping promote tumorigenesis in a context dependent manner, our results suggest that DFS, at least in our colon cancer model, has anti-cancer activity. The lysosomal- dependent regulation of FoxM1 by chemicals might be a new strategy for down-regulating oncogenic transcription factor FoxM1 as a strategy to combat cancer.
Transcriptional regulation of FoxM1 was reported by the direct binding of small molecules, such as FDI-629. FoxM1 harbors an auto-regulation loop, self-binding to its own promoter induces its own expression30. This loop can be blocked by inhibiting proteasomal degradation of negative regulator of FoxM1 (NRFM). The transcriptional level of FoxM1 can be down-regulate by the antibiotics siomycin A31 and thiostrepton32 by inhibition of proteasomal degradation of NRFM33. FoxM1 knockdown sensitizes cancer cells to proteasome inhibitor-induced apoptosis but not to autophagy34.
In conclusion, DFS from A. japonica suppresses colon cancer cell proliferation by reducing β-catenin nuclear translocation. DFS induces lysosomal-dependent FoxM1 protein degradation. This is the first report on the lysosomal degradation of FoxM1 by a small molecule. DFS may be useful in treating cancers that feature the elevated expression of FoxM1.
Methods
Reagents
A. japonica was purchased from Yeongcheon, Gyungsangbuk-do, Korea in March, 2012. Cyclin D1, lamin A/C, c-Myc, and survivin antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA), β-catenin was purchased from BD Bioscience (San Jose, CA, USA), FoxM1 was purchased from Bethyl Laboratories (Montgomery, TX, USA), β-actin antibody and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Horseradish peroxidase-conjugated anti-rabbit immunoglobulin G (IgG) and anti-mouse IgG antibodies were purchased from Assay Designs (Ann Arbor, MI, USA). Lipofectamine LTX PLUS was purchased from Life Technology (Carlsbad, CA, USA). The luciferase assay system was purchased from Promega (Madison, WI, USA). Enhanced chemiluminescence agent was purchased from GE Healthcare Life Science (Uppsala, Sweden).
Extraction and isolation of DFS
A. japonica stems (5.8 kg) were extracted with methanol (MeOH) under reflux, and the concentrated extracts (294 g) were partitioned with chloroform (CHCl3) to yield a CHCl3-soluble fraction. The CHCl3 fraction (28 g) was subjected to silica gel column chromatography using hexane/ethyl acetate as an eluent to obtain three fractions. Further chromatography of fraction 2 (1.7 g) using reverse phase chromatography (gradient elution from 30% MeOH to 80% MeOH) yielded DFS (180 mg), and its purity was confirmed by high performance liquid chromatography and 1H-nuclear magnetic resonance spectrum.
Cell culture
The human colorectal adenocarcinoma SW480 cell line and normal colon fibroblast cell line CCD-18Co cells were purchased from the Korean Cell Line Bank (Seoul, Korea). Mouse embryonic fibroblast (MEF) was prepared from embryos (day 12.5) of C57BL/6 mouse as described previously35. The HCT116 human colorectal carcinoma cell line, 293 T cell line, and Wnt3a-secreting L cell line (L-Wnt3a) were purchased from the American Type Culture Collection (Manassas, VA, USA). The HEK293 reporter cell line (stably transfected with TOPFlash, a synthetic β-catenin/TCF-dependent luciferase reporter, and human Frizzled-1 expression plasmids) and a control cell line (stably transfected with FOPFlash, a negative control reporter with mutated β-catenin/TCF binding elements)36 were kindly provided by Prof. Sangtaek Oh, Kookmin University (Seoul, Korea). SW480 and CCD-18Co cells were cultured in RMPI. The other cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS), penicillin (100 U/ml), and streptomycin (10 μg/ml) at 37 °C.
Preparation of Wnt3a conditioned medium (CM)
Wnt3a CM and control CM were prepared as previously described37. L-Wnt3a cells or L-control cells (1.0 × 106 cells in a 100 mm diameter dish) were cultured in DMEM containing 10% FBS. After 4 days, the medium was collected to obtain the first batch of CM. Fresh medium was added, and the cells were cultured for another 3 days at 37 °C, at which point a second batch of CM was collected. The collected medium was passed through a sterile 0.20 μm membrane filter. Wnt3a CM and control CM were prepared by combining equal volumes of the first and second batches of media.
Cloning of FoxM1 and adenovirus preparation
FoxM1B and FoxM1C were cloned from Huh7 hepatoma cells and cloned into the pcDNA3 vector (Promega)38. FoxM1B cDNA was inserted into replication-defective E1- and E3-adenoviral vectors. FoxM1B adenovirus was then amplified in HEK293 human kidney epithelial cells, and the viral particles were purified by passage through a 0.45 μm pore size filter. Similarly, a bacterial β-galactosidase adenovirus was prepared as a control (LacZ).
Reporter gene assay
TOPFlash- or FOPFlash-transfected SW480 and HCT116 cells and TOPFlash reporter- or control FOPFlash reporter-expressing HEK293 cells were treated with the indicated concentrations of DFS for 16 h. Luciferase activity was determined with a luminometer (Promega) using a luciferase assay system (Promega). The results were normalized by the activity of FOPFlash, a negative control reporter with mutated β-catenin/Tcf binding elements.
Western blotting analysis
Cells were washed with ice-cold phosphate buffered saline and lysed with lysis buffer (1% NP-40, 0.1% sodium dodecyl sulfate, 150 mM sodium chloride, and 50 mM Tris, pH 7.4) to obtain whole-cell lysates. The cytosolic and nuclear fractions were prepared with a nuclear and cytosol extraction kit (Thermo Scientific, Rockford, IL, USA) according to the manufacturer’s instructions. Proteins in the lysates were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. The bands were detected by enhanced chemiluminescence after incubation with the indicated primary and secondary antibodies. The membranes were reprobed after removal of antibodies with stripping buffer (2% sodium dodecyl sulfate, 62.5 mM Tris-HCl, pH 6.8, 0.8% β-mercaptoethanol) for 30 min at 50 °C.
Reverse-transcription polymerase chain reaction (PCR)
Cells were treated with the indicated concentrations of DFS for 16 h. Total RNA was isolated from the cell pellets using TRIzol Reagent (Life Technology), and reverse transcribed to single-stranded cDNA using the SuperScript II first-strand cDNA synthesis system according to the manufacturer’s instructions. cDNA was amplified using a PCR thermal cycler. The PCR primer sequences were as follows: FoxM1, sense 5′-CCTTGTGTTCCCCAAGAGTAT-3′ and anti-sense 5′-CTGCAGGTCTTCCCTTCTATC-3′. Beta-actin was amplified as an internal control. The amplified DNA was separated on 2% agarose gels containing 0.01% ethidium bromide.
Cell viability and cell cycle analysis
Cell viability was determined by the MTT assay as described previously37. The cells were treated with the indicated concentrations of DFS. The cells were incubated with 50 μg/ml MTT, formazan crystals were dissolved with dimethylsulfoxide, and the absorbance was measured at 540 nm using a Versa Max microplate reader (Molecular Devices, Sunnyvale, CA, USA) at 24, 48 and 72 h.
To analyze the cell cycle, the cells were treated with 20 μM DFS for 24 and 48 h, fixed with 70% ethanol and stained with propidium iodide. The stained cells were analyzed by fluorescence-activated cell sorting (FACS, BD Biosciences, San Jose, CA, USA).
Tumor xenograft model
SW480 cells (9 × 106 cells/mouse) were suspended in Matrigel® (Corning, Tewksbury, MA, USA) and inoculated subcutaneously in the right flank of 5-week old female BALB/c nu/nu mice (Nara Biotech, Seoul, Republic of Korea). When tumor volume was ~40 mm3, mice were randomly distributed and treated with vehicle (10% dimethylacetamide, 5% Tween 80 and 85% [20% hydroxypropyl β -cyclodextrin]), 30 mg/kg of DFS or 2 mg/kg of doxorubicin by intraperitoneal injection (n = 5). Mice were treated with vehicle or DFS 5 days a week and with doxorubicin every other day for 3 weeks. Tumor volumes were estimated as length (mm) × width (mm) × height (mm)/2. On day 21, mice were sacrificed and tumor weights were measured. All animal experiment was conducted by protocols approved (Approval #: KRIBB-AEC-16001) by Institutional Animal Care and Use Committee at Korea Research Institute of Bioscience and Biotechnology (KRIBB, Daejeon, Korea), and was performed in accordance with the Guide for Care and Use of Laboratory Animals of KRIBB.
Statistical analysis
IC50 values were calculated using the GraphPad Prism computer program (GraphPad Software, Inc., La Jolla, CA, USA). Data are expressed as mean ±standard deviation. All experiments were performed at least four times. Statistical analysis was performed using the Student’s t-test. A p value < 0.01 was considered statistically significant.
Additional Information
How to cite this article: Dong, G.-Z. et al. A lignan induces lysosomal dependent degradation of FoxM1 protein to suppress β-catenin nuclear translocation. Sci. Rep. 7, 45951; doi: 10.1038/srep45951 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
This study was supported by the National Research Foundation of Korea (NRF) Grant funded by the Korean Government (MSIP) (No. 2011-0030074). We thank prof. Sung Won Kwon (S.N.U.) for measuring high resolution mass spectra.
Footnotes
The authors declare no competing financial interests.
Author Contributions G.-Z.D. designed whole experiments and performed the most of biological experiments, arranged data and prepared manuscripts and revision. J.H.J. purified DFS, measured NMR for identification of chemical structure and performed some of biological experiments for revision. Y.-i.L. performed the chemical experiments. Y.E.H. prepared DFS for animal study. J.S.S prepared DFS for animal study. Y.-J.K. synthesized DFS intermediates to confirm DFS structure. R.J. designed the organic synthesis and corrected the structure of DFS. Y.H.K. prepared FoxM1 plasmid construct and adenovirous of FoxM1 and LacZ. T.J.P. discussed biological data. K.I.K contributed key materials and critical ideas on FoxM1 degradation. J.-H.R. coordinated the overall research and experiments. All authors reviewed the manuscript.
References
- Miyaki M. et al. Characteristics of somatic mutation of the adenomatous polyposis coli gene in colorectal tumors. Cancer Res. 54, 3011–3020 (1994). [PubMed] [Google Scholar]
- Morgan R. G., Ridsdale J., Tonks A. & Darley R. L. Factors affecting the nuclear localization of beta-catenin in normal and malignant tissue. J. Cell. Biochem. 115, 1351–1361 (2014). [DOI] [PubMed] [Google Scholar]
- Zhang N. et al. FoxM1 promotes beta-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer Cell 20, 427–442 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens J. et al. Functional interaction of [beta]-catenin with the transcription factor LEF-1. Nature 382, 638–642 (1996). [DOI] [PubMed] [Google Scholar]
- Sparks A. B., Morin P. J., Vogelstein B. & Kinzler K. W. Mutational analysis of the APC/β-catenin/Tcf pathway in colorectal cancer. Cancer Res. 58, 1130–1134 (1998). [PubMed] [Google Scholar]
- Halasi M. & Gartel A. L. FOX(M1) news–it is cancer. Mol. Cancer Ther. 12, 245–254 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilarsky C., Wenzig M., Specht T., Saeger H. D. & Grutzmann R. Identification and validation of commonly overexpressed genes in solid tumors by comparison of microarray data. Neoplasia 6, 744–750 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y., Wang I. C., Yoder H. M., Davidson N. O. & Costa R. H. The forkhead box M1 transcription factor contributes to the development and growth of mouse colorectal cancer. Gastroenterology 132, 1420–1431 (2007). [DOI] [PubMed] [Google Scholar]
- Kuroyanagi M. et al. New diarylheptanoids from Alnus japonica and their antioxidative activity. Chem. Pharm. Bull. 53, 1519–1523 (2005). [DOI] [PubMed] [Google Scholar]
- Tung N. H. et al. Antioxidative and hepatoprotective diarylheptanoids from the bark of Alnus japonica. Planta Med. 76, 626–629 (2010). [DOI] [PubMed] [Google Scholar]
- Tung N. H. et al. Anti-influenza diarylheptanoids from the bark of Alnus japonica. Bioorg. Med. Chem. Lett. 20, 1000–1003 (2010). [DOI] [PubMed] [Google Scholar]
- Tung N. H. et al. An anti-influenza component of the bark of Alnus japonica. Arch. Pharm. Res. 33, 363–367 (2010). [DOI] [PubMed] [Google Scholar]
- Uto T. et al. Antiproliferative and pro-apoptotic activity of diarylheptanoids isolated from the bark of Alnus japonica in human leukemia cell lines. Am. J. Chin. Med. 43, 757–767 (2015). [DOI] [PubMed] [Google Scholar]
- Dong G.-z. et al. Diarylheptanoids from lesser galangal suppress human colon cancer cell growth through modulating Wnt/β-catenin pathway. J. Funct. Foods 18, Part A, 47–57 (2015). [Google Scholar]
- Nomura M., Tokoroyama T. & Kubota T. Further phenolic components from Alnus japonica Steud. J. Chem. Soc. Chem. Commun. 316–317 (1975). [Google Scholar]
- Moon S. S., Rahman A. A., Kim J. Y. & Kee S. H. Hanultarin, a cytotoxic lignan as an inhibitor of actin cytoskeleton polymerization from the seeds of Trichosanthes kirilowii. Bioorg. Med. Chem. 16, 7264–7269 (2008). [DOI] [PubMed] [Google Scholar]
- Wu P. L., Chuang T. H., He C. X. & Wu T. S. Cytotoxicity of phenylpropanoid esters from the stems of Hibiscus taiwanensis. Bioorg. Med. Chem. 12, 2193–2197 (2004). [DOI] [PubMed] [Google Scholar]
- Yan J. et al. Identification of two novel inhibitors of mTOR signaling pathway based on high content screening. Cancer Chemother. Pharmacol. 72, 799–808 (2013). [DOI] [PubMed] [Google Scholar]
- Bienz M. & Clevers H. Linking colorectal cancer to Wnt signaling. Cell 103, 311–320 (2000). [DOI] [PubMed] [Google Scholar]
- Tan Y., Raychaudhuri P. & Costa R. H. Chk2 mediates stabilization of the FoxM1 transcription factor to stimulate expression of DNA repair genes. Mol. Cell. Biol. 27, 1007–1016 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y. et al. Two-fold elevation of expression of FoxM1 transcription factor in mouse embryonic fibroblasts enhances cell cycle checkpoint activity by stimulating p21 and Chk1 transcription. Cell Prolif. 43, 494–504 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myatt S. S. et al. SUMOylation inhibits FOXM1 activity and delays mitotic transition. Oncogene 33, 4316–4329 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat U. G., Halasi M. & Gartel A. L. FoxM1 is a general target for proteasome inhibitors. PloS One 4, e6593 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia L., Yu G., Zhang Y. & Wang M. M. Lysosome-dependent degradation of Notch3. Int. J. Biochem. Cell Biol. 41, 2594–2598 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios F., Tushir J. S., Fujita Y. & D’Souza-Schorey C. Lysosomal targeting of e-cadherin: a unique mechanism for the down-regulation of cell-cell adhesion during epithelial to mesenchymal transitions. Mol. Cell. Biol. 25, 389–402 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A. et al. Global cancer statistics. CA Cancer J. Clin. 61, 69–90 (2011). [DOI] [PubMed] [Google Scholar]
- Halasi M. & Gartel A. L. Suppression of FOXM1 sensitizes human cancer cells to cell death induced by DNA-damage. PloS One 7, e31761 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janku F., McConkey D. J., Hong D. S. & Kurzrock R. Autophagy as a target for anticancer therapy. Nat. Rev. Clin. Oncol. 8, 528–539 (2011). [DOI] [PubMed] [Google Scholar]
- Gormally M. V. et al. Suppression of the FOXM1 transcriptional programme via novel small molecule inhibition. Nat. Commun. 5, 5165 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halasi M. & Gartel A. L. A novel mode of FoxM1 regulation: positive auto-regulatory loop. Cell Cycle 8, 1966–1967 (2009). [DOI] [PubMed] [Google Scholar]
- Radhakrishnan S. K. et al. Identification of a chemical inhibitor of the oncogenic transcription factor forkhead box M1. Cancer Res. 66, 9731–9735 (2006). [DOI] [PubMed] [Google Scholar]
- Kwok J. M. et al. Thiostrepton selectively targets breast cancer cells through inhibition of forkhead box M1 expression. Mol. Cancer Ther. 7, 2022–2032 (2008). [DOI] [PubMed] [Google Scholar]
- Gartel A. L. Thiostrepton, proteasome inhibitors and FOXM1. Cell Cycle 10, 4341–4342 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit B. & Gartel A. L. FoxM1 knockdown sensitizes human cancer cells to proteasome inhibitor-induced apoptosis but not to autophagy. Cell Cycle 10, 3269–3273 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K. I. et al. Ube1L and protein ISGylation are not essential for alpha/beta interferon signaling. Mol. Cell. Biol. 26(2), p.472–9 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. et al. Hexachlorophene inhibits Wnt/beta-catenin pathway by promoting Siah-mediated beta-catenin degradation. Mol. Pharmacol. 70, 960–966 (2006). [DOI] [PubMed] [Google Scholar]
- Dong G. z., Shim A. R., Hyeon J. S., Lee H. J. & Ryu J. H. Inhibition of Wnt/beta-catenin pathway by dehydrocostus lactone and costunolide in colon cancer cells. Phytother. Res. 29, 680–686 (2015). [DOI] [PubMed] [Google Scholar]
- Park T. J. et al. TIS21 negatively regulates hepatocarcinogenesis by disruption of cyclin B1-Forkhead box M1 regulation loop. Hepatology 47, 1533–1543 (2008). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.