

Abstract
Non-alcoholic fatty liver disease is associated with hepatocellular carcinoma. In the March 10 issue of Nature, Greten and colleagues report that this metabolic disruption affects tumor surveillance by depleting CD4+ T helper cells through lipotoxic mechanisms associated with NAFLD.
Liver cancer exacts a heavy toll on human health. It is the second most common cause of cancer deaths worldwide, and its prognosis is poor even when diagnosed early (Gelband et al., 2015). Hepatocellular carcinoma (HCC) accounts for approximately 90% of primary liver cancers (Gelband et al., 2015). HCC arises from hepatocytes, liver cells of epithelial origin. Hepatocytes perform multiple critical functions, including regulation of glucose and fatty acid metabolism, detoxification, and absorption of nutrients arriving from the gut via the portal vein. Globally, the dominant risk factor for HCC remains persistent infection with hepatitis B (HBV) or hepatitis C (HCV) viruses (Zoller and Tilg, 2016). However, the landscape is changing; many well-developed countries have seen a surge in non-alcoholic fatty liver disease (NAFLD) and associated rapidly rising rates of obesity and metabolic syndrome (Zoller and Tilg, 2016). Indeed, NAFLD could become the major risk factor for HCC in the future as effective approaches to prevent and/or treat persistent viral hepatitis become more widely adopted (Zoller and Tilg, 2016).
NAFLD is characterized by accumulation of fat in hepatocytes (Machado and Diehl, 2016). This condition, known as steatosis, is marked morphologically by the increased presence of lipid droplets containing triglycerides in hepatocytes, and it occurs when lipid uptake or synthesis outpaces export or degradation. NAFLD can be broadly grouped into two related syndromes that differ in their impact on human health. Non-alcoholic fatty liver (NAFL) is a benign and potentially reversible condition, whereas non-alcoholic steatohepatitis (NASH) is a much more serious process characterized by inflammation and death of hepatocytes in addition to steatosis (Machado and Diehl, 2016). A key difference between NAFL and NASH is the composition and toxicity of lipids that accumulate in hepatocytes. Lipid droplets in NAFL are comprised mostly of triglycerides that are not directly hepatotoxic. Lipids that accumulate in NASH include fatty acids that are mobilized from lipid droplets, in part by autophagy (“lipophagy”) and lysosomal breakdown of tryglycerides. These fatty acids undergo mitochondrial β-oxidation, generating reactive oxygen species (ROS), oxidative stress, and membrane depolarization (Wang et al., 2013). Other lipids, such as diacylglycerol, oxysterols, cholesterol, and phospholipids, can dysregulate hepatocyte signaling and metabolism, further contributing to hepatotoxicity. Hepatocyte stress caused by lipotoxicity also activates inflammatory responses and signaling pathways, such as the hedgehog pathway, that drive wound healing (Angulo et al., 2015), resulting in fibrosis, DNA damage, and mitogenesis, all of which promote development of HCC (Machado and Diehl, 2016).
A recent study by C. Ma and colleagues (Ma et al., 2016) addresses a novel hypothesis related to this all-too-common disease: are cytotoxic lipids released from steatotic hepatocytes, and are they toxic for intrahepatic T lymphocytes that provide immune surveillance against HCC in patients with NAFLD? The question is important because T cells infiltrate the liver of some patients with HCC but provide only limited clinical benefit (Prieto et al., 2015; Schmidt et al., 2012). Indeed, multiple approaches to bolstering the anti-tumor T cell response in HCC are currently being assessed; such approaches include immunization with HCC-associated tumor antigens, adoptive T cell therapy, and blockade of T cell checkpoint inhibitors such as programmed cell death 1 (PD-1) and cytotoxic-T lymphocyte-associated protein 4 (CTLA-4) (Prieto et al., 2015; Schmidt et al., 2012). The success of these approaches might depend on the fate of tumor-specific T cells in the liver and whether mechanisms that impair their activity can be identified and overcome.
Ma and colleagues employed several murine models of NAFLD and hepatocarcinogenesis to determine whether lipotoxicity impairs T cell survival and tumor surveillance that would otherwise limit the development of HCC. Diet-induced NAFLD increased susceptibility to liver tumors induced by activation of a transgenic MYC oncogene and exposure to a chemical carcinogen. Surprisingly, mice with NAFLD also displayed a significant liver-specific reduction in the number of CD4+ helper T cells but not CD8+ cytotoxic T cells. The intrahepatic CD4+ T cells displayed increased mitochondrial mass and reactive oxygen species consistent with lipid-induced oxidative stress. Death of CD4+ T cells was recapitulated by co-culture with hepatocytes from NAFLD animals. This process did not require physical contact between steatotic hepatocytes and CD4+ T cells; lipids released from the hepatocytes into the culture medium were sufficient to cause death. In a series of elegant ex vivo experiments, hepatocytes from these animals were shown to contain fatty acids associated with human NASH, including palmitic, stearic, arachidonic, and linoleic acids. The polyunsaturated fatty acid linoleic acid (C18:2) was most abundant, and when added to normal splenic CD4+ T cells it selectively altered pathways associated with oxidative phosphorylation and mitochondrial function. Linoleic acid also caused dose-dependent apoptotic death of cultured murine CD4+ T cells, a process that could be prevented by antioxidant treatment with catalase or N-acetylcysteine (NAC). Importantly, treatment of NAFLD mice with NAC also reversed CD4+ T cell loss and delayed hepatocarcinogenesis even though steatosis was not reduced. Tumor reduction in anti-oxidant-treated animals was shown to be dependent upon CD4+ T cells. These findings are summarized in Figure 1.
Figure 1. A Model of the Toxic Interaction between Hepatocytes and Intrahepatic T Lymphocytes in NAFLD.
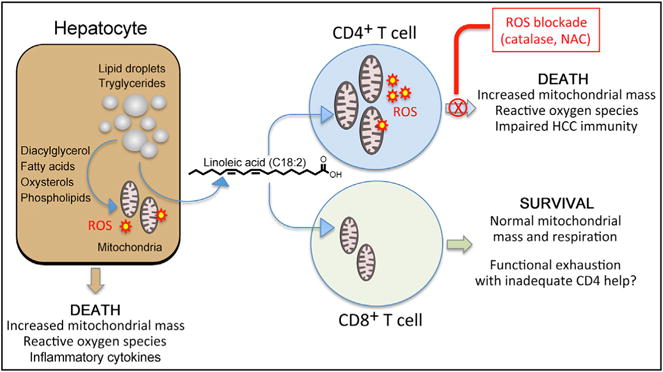
The hepatocyte. A key characteristic of NASH is accumulation of cytotoxic fatty acid metabolites in hepatocytes. Lipotoxicity results in death of hepatocytes, in part through induction of reactive oxygen species (ROS) that act on mitochondria to increase their mass and depolarize membranes.
CD4+ T cells. Linoleic acid, a polyunsaturated fatty acid consisting of an 18 carbon chain with two double bonds (C18:2), is taken up by CD4+ helper T cells after release from steatotic hepatocytes, resulting in the induction of ROS, oxidative stress, mitochondrial dysfunction, and death by apoptosis. Other hepatocyte-derived fatty acids, including palmitic (C16:0), steric (C18:0), and arachidonic (C20:4) acids, did not have a similar cytotoxic effect.
CD8+ T cells. Uptake of linoleic acid by CD8+ T cells has no impact on mitochondrial function. Why linoleic acid causes selective CD4+ T cell death is a key unresolved question. Unlike most mechanisms of T cell silencing in the liver, lipotoxicity lacks apparent antigen specificity, resulting in the potential for a much broader impact on liver immune function. Functional exhaustion of tumor-specific CD8+ T cells is common in patients with HCC. Whether apoptotic death of CD4+ T cells contributes to loss of CD8+ T cell function by restricting helper activity is as yet unknown. The possibility that ROS blockade can restore immune function in HCC, or enhance the effectiveness of T cell immunotherapy, represents an important future direction.
This study is particularly notable because CD4+ T cells targeting tumor antigens such as α-fetoprotein contribute to control of HCC in mice and possibly also in humans (Prieto et al., 2015; Schmidt et al., 2012). To establish relevance for humans, Ma and colleagues also treated peripheral blood mononuclear cells from healthy volunteers with linoleic acid, leading to enhanced death of CD4+ T cells with increased amounts of reactive oxygen species (ROS) and increased mitochondrial mass. CD8+ cytotoxic T cells were spared, thereby recapitulating findings from the murine models (Figure 1).
Could these findings be relevant to other liver diseases? Steatosis is present in most patients with persistent HCV infection and is particularly severe in those with genotype 3 virus (Goossens and Negro, 2014). Many of these patients have a clinical picture indistinguishable from NAFLD. Although prospective studies are lacking, patients with genotype 3 infection also appear to be at increased risk of HCC in comparison to those with other HCV genotypes (Goossens and Negro, 2014). A clear explanation for this is lacking, and it is intriguing to consider the possibility that lipotoxicity impairs T-cell-mediated tumor surveillance in chronic hepatitis C. Genotype 3 HCV infections are also proving particularly difficult to cure with new, direct-acting antivirals. This might reflect genotype-specific differences in antiviral EC50 concentrations, but the findings of Ma and colleagues hint at a more subtle cause that should be investigated. Further parsing of the effects of fatty acids on CD4+ T cell survival is clearly warranted in the setting of viral hepatitis.
The study is significant because it demonstrates that the toxic impact of fatty acids and their metabolites that accumulate in NASH is not restricted to hepatocytes. Linoleic acid, in particular, selectively targets CD4+ T cells that infiltrate the liver parenchyma. There is much more to learn about this process of CD4+ T cell death and its impact on cellular immunity in the liver. It seems unlikely that lipotoxicity is restricted to CD4+ T cells that target HCC tumor antigens. T cell trafficking through the liver is a key component of the tolergenic process involving dietary antigens. Moreover, the organ is targeted by mulitple hepatotropic viruses and parasites. It is conceivable that any CD4+ T cell infiltrating the liver is susceptible to lipotoxicity, regardless of antigen specificity. This possibility merits further consideration because it could indicate the potential for more generalized immune suppression in NAFLD. In addition, whereas linoleic acid was taken up equivalently by normal CD4+ and CD8+ T cells isolated from murine spleen or human peripheral blood, only the helper subset appeared to be susceptible to mitochondrial damage and apoptosis. Unraveling the selective nature of this process could provide improved targeting of therapeutics to prevent CD4+ T cell death caused by cytotoxic lipids.
Whether lipotoxic loss of CD4+ T cell helper activity more broadly impairs other arms of the cell-mediated immune response, notably CD8+ T cells, also merits further study. The most likely scenario is that the CD4+ T cells do not directly restrict the development of tumors but instead provide critical help in the generation of tumor-specific cytotoxic CD8+ T cells. Cytotoxic CD8+ T cells targeting tumor antigens are thought to be important for control of HCC but appear to lose function upon infiltration into the liver (Schmidt et al., 2012). This process, known as functional exhaustion, is also a key feature of persistent viral infections and is thought to be caused by an absence of CD4+ T cell help. Lipotoxicity directed against CD4+ T cells could therefore lead indirectly to loss of cytotoxic cell function.
It will also be important to sort out the relative contribution of lipotoxicity versus other antigen-specific mechanisms of CD4+ T cell suppression in HCC arising from NAFLD. The liver is an important immunoregulatory organ that prevents immune responses against dietary proteins from arriving from the gut (Knolle, 2016). Tolerance to these potential antigens is imposed by a variety of mechanisms, including presentation to T cells by specialized cells such as liver sinusoidal epithelial cells and even hepatocytes, resulting in T cell deletion and/or priming of suppressive regulatory cells (Knolle, 2016). That this process might be highjacked to impair responses against HCC tumor antigens that originate in the liver has been a dominant concept for many years. Getting the skinny on how lipotoxicity contributes to impairment of intrahepatic CD4+ T cells in individuals with fatty liver disease, and developing strategies to reverse the process, has the potential to substantially improve prospects for preventing and possibly treating HCC.
References
- Angulo P, Machado MV, Diehl AM. Semin Liver Dis. 2015;35:132–145. doi: 10.1055/s-0035-1550065. [DOI] [PubMed] [Google Scholar]
- Gelband H, Chen CJ, Chen W, Franceschi S, Hall SA, London WT, McGlynn KA, Wild CP. Liver Cancer. In: Gelband H, Jha P, Sankaranarayanan R, Horton S, editors. Cancer: Disease Control Priorities. Third. Vol. 3. Washington, DC: The International Bank for Reconstruction and Development; 2015. pp. 147–164. [Google Scholar]
- Goossens N, Negro F. Hepatology. 2014;59:2403–2412. doi: 10.1002/hep.26905. [DOI] [PubMed] [Google Scholar]
- Knolle PA. Curr Opin Immunol. 2016;40:36–42. doi: 10.1016/j.coi.2016.02.009. [DOI] [PubMed] [Google Scholar]
- Ma C, Kesarwala AH, Eggert T, Medina-Echeverz J, Kleiner DE, Jin P, Stroncek DF, Terabe M, Kapoor V, ElGindi M, et al. Nature. 2016;531:253–257. doi: 10.1038/nature16969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado MV, Diehl AM. Gastroenterology. 2016 Published online February 26, 2016. S0016-5085(16)00257-2. [Google Scholar]
- Prieto J, Melero I, Sangro B. Nat Rev Gastroenterol Hepatol. 2015;12:681–700. doi: 10.1038/nrgastro.2015.173. [DOI] [PubMed] [Google Scholar]
- Schmidt N, Neumann-Haefelin C, Thimme R. Dig Dis. 2012;30:483–491. doi: 10.1159/000341697. [DOI] [PubMed] [Google Scholar]
- Wang DQ, Portincasa P, Neuschwander-Tetri BA. Compr Physiol. 2013;3:1493–1532. doi: 10.1002/cphy.c130001. [DOI] [PubMed] [Google Scholar]
- Zoller H, Tilg H. Metabolism. 2016 doi: 10.1016/j.metabol.2016.01.010. S0026-0495(16)00025-1. http://dx.doi.org/10.1016/j.metabol.2016.01.010. [DOI] [PubMed]