

Abstract
Toll-like receptors (TLRs) are a major class of pattern recognition receptors, which mediate the response of innate immune cells to microbial stimuli. To systematically determine the roles of gene products in canonical TLR signaling pathways, we conducted an RNA interference (RNAi)-based screen in human and mouse macrophages. We observed a pattern of conserved signaling module dependencies across species, but found notable species-specific requirements at the level of individual gene products. Among these, we identified unexpected differences in interleukin 1 receptor-associated kinase (IRAK) protein use between the human and mouse TLR pathways. Whereas TLR signaling in mouse macrophages depended primarily on IRAK4 and IRAK2, with little or no role for IRAK1, TLR signaling and proinflammatory cytokine production in human macrophages were highly dependent on IRAK1 and were less affected by perturbations of IRAK4 or IRAK2. The differential sensitivity of human and mouse cells to the loss of IRAK4 was reflected in the inability of the IRAK4 orthologs to rescue signaling in IRAK4-deficient macrophages from the other species, and in a mouse-specific requirement for the kinase activity of IRAK4 in TLR responses in macrophages. Our study also identified a critical role for IRAK1 in TLR signaling in humans, which could potentially explain the association of IRAK1 with several autoimmune diseases. Furthermore, we demonstrated how systematic screening can be used to identify important characteristics of innate immune responses across species and more optimal therapeutic targets for regulating human TLR-dependent outputs.
INTRODUCTION
The mammalian innate immune system plays a vital role in host defense because it continually engages microbial, viral, and parasitic stimuli. This engagement is mediated through the recognition of invariant pathogen-associated molecular patterns (PAMPs) by a range of pattern recognition receptors (PRRs) expressed by the host cell (1–5). Toll-like receptors (TLRs) are a major class of PRRs, which respond to a range of PAMPs to induce a complex signaling response and the transcription of hundreds of genes required for an effective immune response (6–8). TLRs are found in multiple host cell types that encounter PAMPs, and consequently, knowledge of the canonical components that constitute TLR signaling pathways has been derived from studies encompassing different cells in different contexts (9). To date, there has been no systematic study to evaluate the contributions of the primary TLR pathway components to different ligands in a single cell type, no less in critical cells of the innate immune system, such as macrophages. RNA interference (RNAi) is a valuable research tool with which to systematically evaluate gene product contributions in a given biological system through the targeted inhibition of gene expression (10, 11). However, there are substantial challenges in the use of this technology in innate immune cells, including the efficient delivery of small RNAs into cells and the occurrence of nonspecific immune responses to double-stranded RNA (dsRNA) (12), particularly small interfering RNA (siRNA) (13, 14). The difficulty of siRNA delivery can be circumvented by the viral expression of short hairpin RNAs (shRNAs), and this approach has been used in primary dendritic cells to identify important roles for transcriptional regulators and signaling proteins whose production is induced by TLR stimulation (15, 16). However, there are very few reports of siRNA-based screens in innate immune cells, as opposed to the more easily used fibroblast or mesenchymal cell lines, which do not have the same crucial roles in host defense as those of hematopoietic cells, such as macrophages.
Beyond this lack of comprehensive analyses of the TLR pathway in a single, highly relevant cell type, a substantial proportion of our understanding of pathway architecture has come from mouse studies; knockout mice have been described for almost all of the components in the TLR pathway that were identified by in vitro studies with isolated cells (17, 18). This has shed considerable light on the similarities and differences in the response to different PAMPs engaging distinct TLRs, as well as on the broader role of TLR responses in the immune response to pathogen infection (19). Human patients deficient in several TLR pathway components have been described (20), and although the cellular response phenotypes of these patients generally support the pathway structure established from mouse studies, the broader immune phenotypes of these human patients often show considerable differences to the corresponding knockout mouse model, which suggests that there is possible divergence between the species in the regulation of TLR-dependent immune outputs at the physiological level. A direct comparison of TLR signaling responses in human and mouse innate immune cells could provide important insight into the comparative dependencies of the two species on specific pathway components. Moreover, reports have debated the question of whether mice provide a good model for human physiology or disease, especially with respect to innate immunity (21, 22). In this regard, a careful comparison of individual gene contributions at the cellular pathway level has a very important role in addressing this issue.
We previously developed a highly optimized platform for the siRNA-based screening of pathogen responses in human and mouse macrophages, with stably expressed reporters that provide dynamic readouts for activation of the transcription factor nuclear factor κB (NF-κB) and expression from the TNF-α promoter (23). In this study, we used this platform to interrogate the similarities and differences in the responses of human and mouse macrophages to different TLR ligands. We screened 126 canonical components of the human and mouse TLR pathways with six independent siRNAs per gene, and assessed signaling and inflammatory cytokine responses. We observed both conserved and distinct patterns of pathway component use for different TLR ligands within each species. Moreover, the comparison of pathway dependencies between human and mouse macrophages suggests conservation at the level of signaling modules, but substantial divergence at the level of individual signaling proteins. Through investigation of the usage patterns for different interleukin 1 receptor-associated kinase (IRAK) family members, we identified considerable differences in how human and mouse macrophages signal through IRAKs in the response to individual TLR ligands, which may have important consequences for studies of human diseases that are influenced by this protein family.
RESULTS
Targeting the canonical TLR pathway in human and mouse macrophages with a comprehensive siRNA library leads to low false-positive rates
We previously developed an assay platform for the high-throughput analysis of human and mouse macrophage activation (23). Using lentiviral constructs containing dual expression cassettes, we generated a mouse RAW264.7–derived cell line (RAW G9 cells) that had a green fluorescent protein (GFP)-tagged reporter of the nuclear translocation of the p65 subunit of NF-κB (also known as RelA) and an mCherry-based red fluorescent reporter of the activity of the mouse Tnf promoter (fig. S1A). Both of these reporters showed comparable responsiveness to multiple TLR ligands (fig. S1, B and C). Using a similar approach, we also generated a human THP1-derived cell line (THP1 B5 cells) that had a dual luciferase reporter for activation of the human TNF promoter (fig. S1D) and that was also responsive to TLR ligand activation (fig. S1E). We previously showed that these stably expressed reporter genes can be used as targets to develop highly optimized siRNA delivery protocols to achieve maximal gene silencing with minimal nonspecific activation of the dsRNA-stimulated interferon response in macrophages (23). This cell system provides a platform for systematic functional testing of putatively canonical components of the TLR signaling pathways in a relevant innate immune cell type.
As a first step towards a future full genome-wide screen of the effects of siRNA-mediated knockdown of signaling pathway components on the responses of macrophages to TLR ligands, we curated the published literature and innate immune databases to identify a comprehensive set of 126 genes whose products have reported function in TLR signaling pathways, either as direct signal propagation components or as feedback modulators (Fig. 1A). Inclusion in this list was irrespective of the cell type in which the original identification of involvement in the TLR response was made or of the species origin of the cells involved. The key goals of the present study of these putatively canonical components of TLR pathways were to determine which of these molecules were functionally important for the TLR response in a key innate immune cell type and to examine whether mouse and human macrophages required the same pathway proteins for their responses.
Fig. 1. A comprehensive siRNA screen of the TLR pathway in human and mouse macrophages.
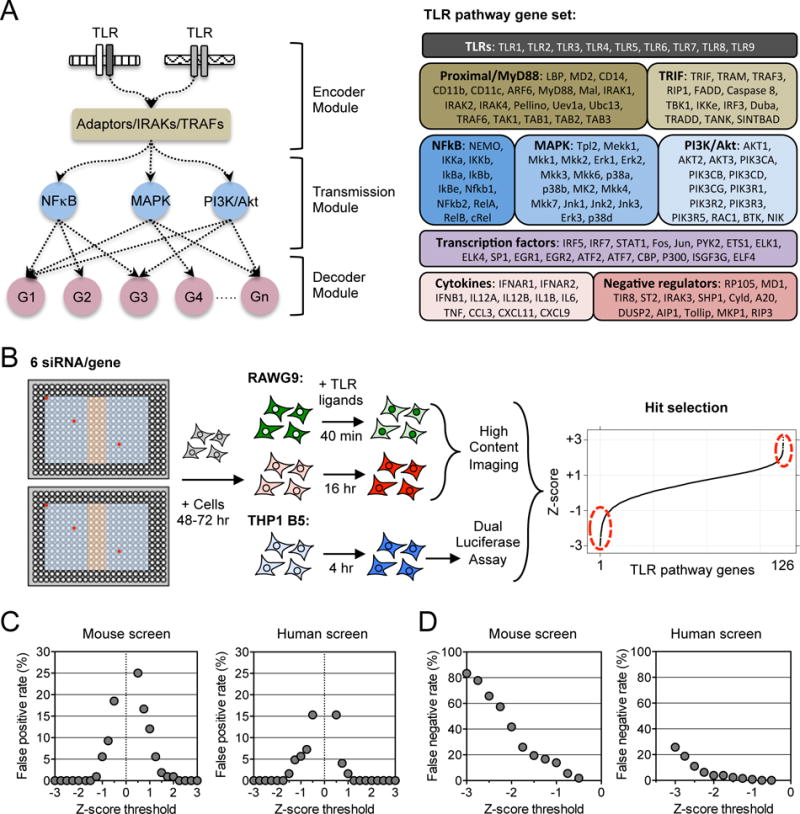
(A) Schematic of signal flow in the TLR pathway from initial signal encoding by TLR receptors, through complexes of TIR adaptor proteins, IRAKs, and TRAFs, to signal transmission through NF-κB, MAPK, and PI3K modules, and then signal decoding through gene transcription, cytokine secretion, and feedback regulation. Right: The 126 gene transcripts targeted in the screen by siRNAs. (B) Workflow for the RNAi screen using six siRNA sequences per gene distributed in separate regions of a 384-well plate. Red circles show examples of siRNA locations for a single gene. The blue region of the plate shows the locations of gene-specific siRNAs, whereas the orange region shows the positions of control siRNAs. Forty-eight to 72 hours after they were reverse-transfected with siRNAs, the mouse (RAW G9) and human (THP1 B5) reporter cell lines were assayed for their responses to simulation with different TLR ligands, and the effects of individual gene perturbations were measured (see fig. S1 and Materials and Methods for further details). (C and D) Calculation of false positive rates (C) and false negative rates (D) from negative and positive control siRNAs, respectively, in the mouse and human macrophage siRNA screens (see Materials and Methods). Data in (C) and (D) are presented as median z-scores from six siRNAs per gene. Individual siRNA scores were averaged from two (C) or three (D) independent experiments (see Materials and Methods for details).
Target gene products could be grouped into various classes and broadly classified as either initial signal encoders, mediators of signal transmission, or regulators of signal decoding at the transcriptional and feedback levels (Fig. 1A). This gene set was targeted with six independent siRNA sequences per gene, which were acquired from different commercial vendors (table S1), with previously described and optimized siRNA delivery protocols (23). Studies have shown that off-target effects in siRNA screening can be mitigated by the use of at least three individual siRNAs per gene (24, 25). Our use of six individual siRNAs in this screen thus greatly reduced the probability of off-target phenotypes and led to a more reliable evaluation of the relative contribution of each of the 126 gene products to the TLR response (Fig. 1B). To address false positive and negative rates arising from assay variability in the screens, we used data from the control siRNAs included on each screening plate (Fig. 1B and see Materials and Methods). Comparing the mouse and human assay readouts, we calculated a false positive rate in both screens of less than 5% for TLR signal propagators with z-scores of < −1.0 (Fig. 1C). This suggests that genes with median z-scores below this threshold had a high probability of being true hits (table S2). Calculation of false negative rates from the z-scores of positive control siRNAs showed that TLR pathway gene products with a median siRNA score above −0.75 had a < 2% likelihood of being false negatives in either the mouse or human screens (Fig. 1D), providing an important validation of the macrophage reporter cell lines and the RNAi screening platform.
To evaluate the relative contribution of the 126 TLR pathway gene products in both human and mouse macrophages, species-specific siRNA libraries were used. To assess the responses to different TLR ligands, the THP1 B5 and RAW G9 reporter cells were stimulated with ligands from the following group and the reporter assay readouts measured: lipopolysaccharide (LPS, for TLR4), Pam3CSK4 (P3C, for TLR1/TLR2), Pam2CSK4 (P2C, for TLR2/TLR6), peptidoglycan (PGN, for TLR2/TLR6), flagellin (FLG, for TLR5), and resiquimod 848 (R848, for TLR7 or TLR8). Data from replicate screens were averaged for each single siRNA, and z-scores were calculated from the median value of the six individual siRNAs tested for each of the 126 genes (Fig. 1B, table S2, and see Materials and Methods).
Human and mouse TLR pathways show common dependencies in modules required for signal propagation
The receptor dependency of the response to the tested TLR ligands showed the expected phenotypic patterns in the human THP1 macrophage cell line, with knockdown of TLR4, TLR2 or TLR1, TLR8, TLR2, TLR2 or TLR6, and TLR5 showing specific perturbation of TNF-α reporter activation by LPS, P3C, R848, P2C, PGN, and FLG, respectively (Fig. 2, A and B). Beyond this anticipated outcome of TLR knockdown on the TNF-α reporter response, the strongest siRNA-mediated effects resulted from the perturbation of components in receptor-proximal signaling, including NF-κB, mitogen-activated protein kinase (MAPK), and transcription factor modules (Fig. 2A and fig. S2A). As expected, many components played important roles in responses to most of the TLR ligands tested, consistent with a broadly shared pathway structure downstream of the different TLRs (6, 9). To develop a metric for identifying the most conserved pathway dependencies across the TLR ligand set in human macrophages, we ranked the genes based on their TNF-α reporter perturbation effect for each ligand, and then summed the ranks for each gene (table S3). The top four genes (lowest summed rank values) that emerged from this analysis (Fig. 2C) encode products that are all from the proximal signaling module (TRAF6, MAP3K7, IRAK1, and TAB1), followed by genes whose products are from the MAPK (MAP3K8 and MAPK14), NF-κB (NFKBIE and RELA), and transcription factor (EGR1 and ELF4) modules.
Fig. 2. Effects of siRNA gene perturbations across the human and mouse TLR pathways.
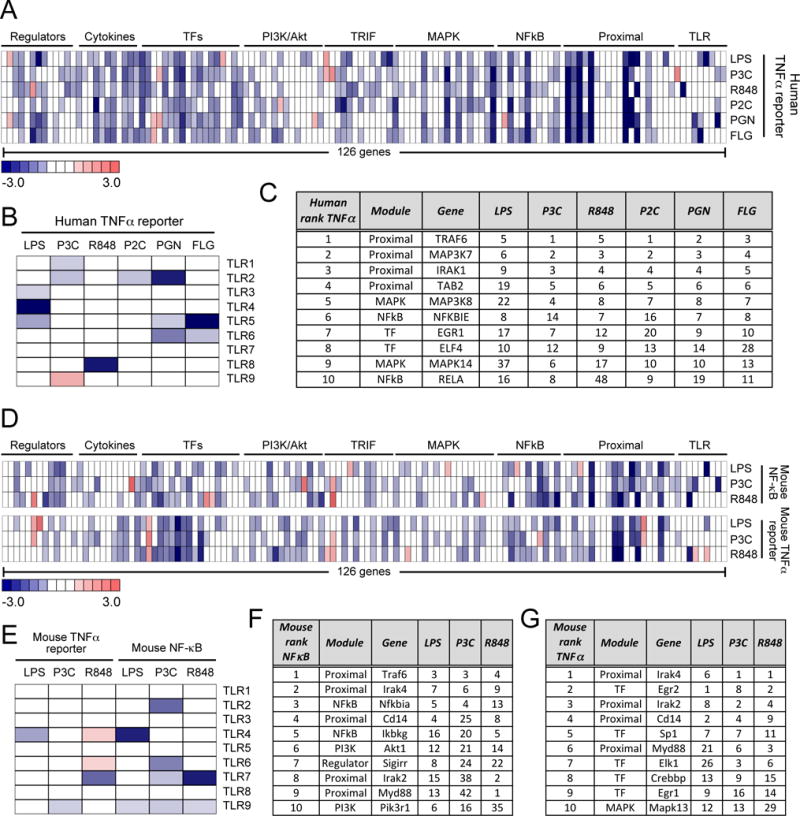
(A to G) Analysis of the effects of gene perturbations on TLR signaling. TLR pathway gene perturbation effects on (A) the TNF promoter–driven transcriptional response of the human THP1 cell line to LPS (10 ng/ml), 100 nM P3C, R848 (10 μg/ml), 1 nM P2C, 1 PGN (10 μg/ml), or FLG (10 ng/ml) and (D) on the NF-κB and Tnf promoter–dependent responses of the mouse RAW264.7 cell line to LPS (10 ng/ml), 100 nM P3C, or R848 (10 μg/ml). (B and E) Effects of the siRNA-mediated knockdown of the indicated TLRs on (B) the TNF promoter-dependent transcriptional response of the human THP1 cell line and (E) the NF-κB– and Tnf promoter–dependent responses of the mouse RAW264.7 cell line to the indicated TLR ligands. The top 10 genes with the most substantial effects on (C) the human TNF promoter–dependent response, (F) the mouse NF-κB response, and (G) the mouse Tnf promoter-driven response across all tested TLR ligands. Rankings for individual TLR ligands are shown, with overall gene order determined by the average rank calculated over all ligands (the full 126-gene ranking scores are shown in table S3). Data in (A), (B), (D), and (E) are presented as median z-scores from six siRNAs per gene. Individual siRNA scores were averaged from three (for A and B) or two (for D and E) independent experiments (see Materials and Methods).
We then assessed the effects of perturbing the same 126-gene set in mouse macrophages using the RAW G9 reporter clone. In these cells, we tested three TLR ligands, LPS, P3C, and R848, and measured two downstream readouts: the initial activation of NF-κB by measurement of the nuclear translocation of RelA and the subsequent activation of the murine Tnf promoter (Fig. 2D and fig. S1, B and C). The TLR-specificity of the responses to the different ligands in mouse macrophages showed the expected inhibition of the LPS response with Tlr4 knockdown and of the R848 response with Tlr7 knockdown (Fig. 2E). The effect of knockdown of Tlr7 was in contrast to what occurred in human macrophages, which respond to R848 through TLR8 (Fig. 2B), and is consistent with previous studies (26, 27). Knockdown of Tlr2 reduced the NF-κB response to P3C; however, unexpectedly, we did not see a strong Tlr2 dependence for the TNF-α response to P3C, despite using the same siRNA set that gave a strong NF-κB reporter effect. This is suggestive of some possible redundancy in the murine TNF-α response to this ligand or to there being different quantitative requirements for NF-κB activation vs. Tnf transcription. Among nonreceptor targets, the greatest effects on the NF-κB reporter were found upstream of NF-κB activation in the receptor-proximal signaling modules and in components of the NF-κB family itself (Fig. 2D, top, and fig. S2B). The greatest perturbations of the mouse macrophage TNF-α reporter response were observed from knockdown of receptor-proximal signaling components and transcription factors (Fig. 2D, bottom, and fig. S2C).
As described earlier for the human genes, we calculated ranks for the 126 mouse genes from the effects of their knockdown on the response to each of the three TLR ligands tested for both the the early NF-κB readout and the later TNF-α readout, and then we calculated the average rank across the TLR ligands for each output (table S3, Fig. 2, F and G). We found that the products of half of the top 10 ranked genes that controlled the mouse NF-κB response were in the receptor-proximal signaling module (Traf6, Irak4, Cd14, Irak2 and Myd88), whereas genes encoding NF-κB (Nfkbia and Ikbkg) and PI3K pathway members (Akt1 and Pik3r1) also ranked highly (Fig. 2F). The ranking of those genes with the most influence on the mouse TNF-α response across the different TLR ligands (Fig. 2G) confirmed the enrichment of genes encoding products in the proximal signaling module (Irak4, Irak2, Cd14 and Myd88) and key transcription factors for Tnf expression (Egr2, Sp1, Elk1, Crebbp and Egr1). This pattern is similar to that observed in the human macrophage reporter cells and is consistent with a pathway architecture in which single gene perturbations have the greatest effect either before the signaling system branches out through multiple modules (NF-κB, MAPK, and PI3K-Akt) or after the branches converge on the components of the Tnf transcriptional enhanceosome.
Human and mouse TLR pathways show distinct gene requirements within signaling modules
Whereas we observed similar requirements in signaling pathway modules in our analysis of human and mouse TLR pathway dependencies, we also found that the specific genes whose knockdown resulted in the strongest phenotypes were often different between macrophage cells from the two species. For example, although genes encoding proximal TLR signaling components and transcription factors were both strongly enriched among the top 10 ranked genes in human and mouse TNF-α activation by TLR ligands, only one gene, EGR1, which encodes the transcription factor EGR1, ranked highly in the screens for both species (Fig. 2, C and E). Thus, although the overall findings of the screen confirmed the expected roles of many molecules in responses to multiple TLR ligands within a species, a number of unexpected cross-species differences were discovered through this systematic analysis.
To further analyze the degree of overlap between the human and mouse macrophage responses to TLR stimulation, we compared the pathway components required for TNF-α reporter activation by the three TLR ligands that were tested in both human and mouse cells (LPS, P3C, and R848). Hierarchical clustering of the gene perturbation effects across the three TLR ligands in the two cell types showed that the gene dependencies often clustered separately for human and mouse cells (Fig. 3A and fig. S3). Whereas there were a group of shared genes required for human and mouse responses to all three TLR ligands, we identified distinct clusters of genes with stronger requirements for human or for mouse TLR signaling. Furthermore, within each species, the P3C and R848 gene dependencies were more similar, with a separate cluster of genes showing specificity for the LPS response (Fig. 3A).
Fig. 3. Human and mouse macrophages show both shared and distinct gene product dependencies in TLR signaling.
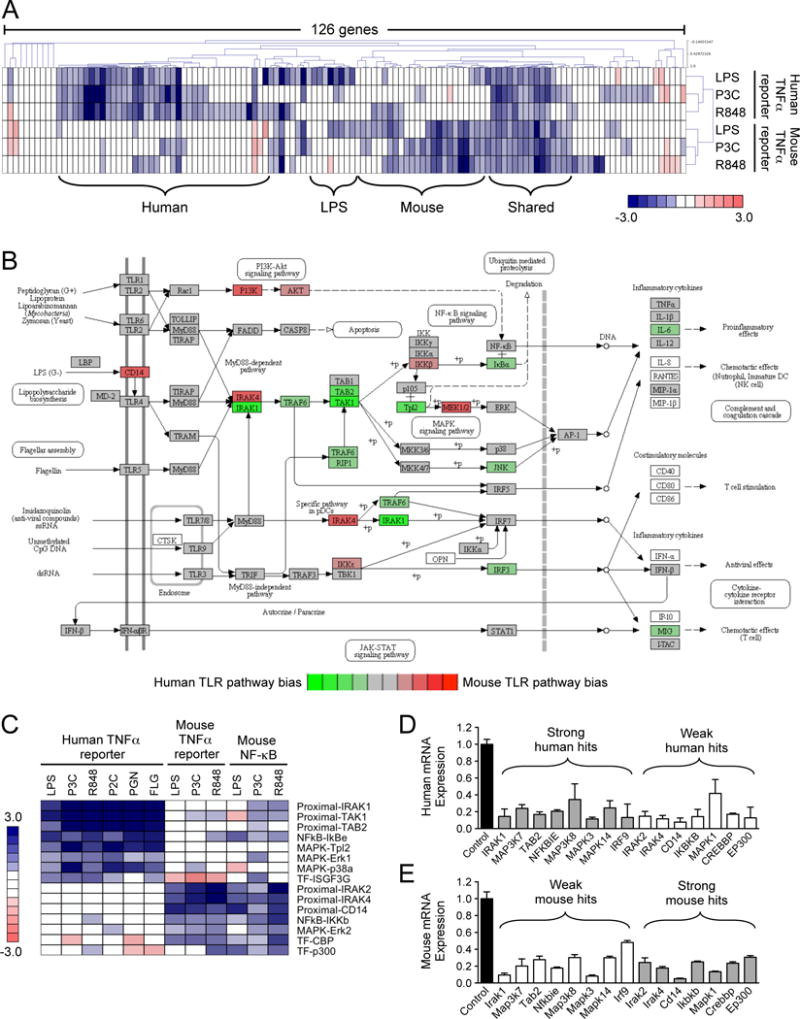
(A) Hierarchical clustering analysis (Pearson uncentered, average linkage) of the TNF/Tnf promoter–driven responses to LPS, P3C, and R848 in human THP1 cells and mouse RAW cells. (B) Average z-scores calculated from the responses to the three TLR ligands shown in (A) were compared between the human and mouse reporter cell lines (see table S4). The z-score differences between the species were overlaid onto the canonical TLR pathway from KEGG. (C) Genes whose perturbation had the most substantial effects on the human and mouse TLR pathways. (D and E) The relative abundances of the mRNAs corresponding to the genes shown in (C) in human THP1 B5 cells (D) or mouse RAW G9 cells (E) after siRNA-mediated knockdown. Data in (D) and (E) are means ± SD from two independent experiments. Data in (A) and (C) are presented as median z-scores from six siRNAs for each gene. Individual siRNA z-scores were averaged from three (for the human TNF-α readout) or two (for the mouse NF-κB and TNF-α readouts) independent experiments (see Materials and Methods).
To assess these species-specific patterns in the context of the canonical TLR pathway, we took the median value of the z-scores for each gene across the three TLR ligands, and calculated the median score difference between human and mouse cells (table S4, ‘Median hTNF-mTNF’). We overlaid these differences onto the KEGG TLR pathway to highlight pathway nodes that showed a bias towards greater signaling dependency in a particular species (Fig. 3B). This analysis highlighted several patterns. Mouse cells were more sensitive than were human cells to the knockdown of CD14, IRAK4, PI3K signaling components, IKKb, MEK1, and MEK2. Human cells were more sensitive to the knockdown of IRAK1, TRAF6, components of the TAK1/TAB complex, and Tpl2, among others. Because the KEGG pathway does not contain all of the individual genes tested in our screen, we highlighted the 15 gene products with the strongest differential dependencies in the human and mouse TLR pathways (Fig. 3C). To assess whether the observed differences could simply be a result of variation in the efficiency of knockdown, we tested the expression of each of the 15 genes in the mouse and human macrophage reporter cells after they were transfected with the appropriate siRNAs (Fig. 3, D and E). The similar degrees of knockdown observed across the 15-gene panel in both human and mouse cells suggest that differential knockdown efficiency is unlikely to be the primary cause of the phenotypic differences observed in the screen.
Of the differences between human and mouse cells that we emphasized here, we considered one of the most striking patterns to be the apparently differential usage of IRAK family proteins in the two species. Mouse cells showed a strong requirement for Irak2 and Irak4, but a weaker requirement for Irak1 (particularly in the TNF-α readout), whereas the human TNF-α readout had a very strong IRAK1 requirement, but showed very little phenotype when either IRAK2 or IRAK4 was knocked down (Fig. 3C). Given that these findings suggest a very different view of the participation of IRAKs in the TLR responses of mouse and human cells than is presently thought, we focused further efforts on this specific aspect of the overall screen findings.
IRAK4 is essential for both mouse and human TLR signaling, but the human and mouse proteins are not functionally interchangeable
Previous studies showed that upon ligand activation, TLR signaling pathways that depend on the adaptor protein myeloid differentiation primary response gene 88 (MyD88) initially recruit IRAK4, which binds to MyD88 through death domain (DD) interactions (28, 29). This complex then requires either IRAK1 or IRAK2 to signal to TRAF6 (TNF receptor–associated factor 6), because IRAK4 lacks the TRAF6-binding domain that is present in both IRAK1 and IRAK2 (30). In light of the apparently essential requirement for IRAK4 to form the initial contact with MyD88, the weak phenotype exhibited by IRAK4 knockdown cells in our human screen data was unexpected, especially considering the strong dependence on IRAK4 in the mouse dataset (Fig. 3C). We assessed whether this difference was a result of the more efficient knockdown of IRAK4 in the mouse cell line, but we found that the knockdown of IRAK4 was comparable in the THP1 and RAW264.7 reporter cell lines (Fig. 4, A and B). Furthermore, we saw a similarly weak effect of the knockdown of IRAK4 in primary human monocyte-derived macrophages (hMDMs) (Fig. 4C), despite there being a robust knockdown of IRAK mRNA and protein in these cells (Fig. 4, A and B).
Fig. 4. IRAK4 is required for human TLR responses, but the human and mouse IRAK4 proteins are not functionally interchangeable.
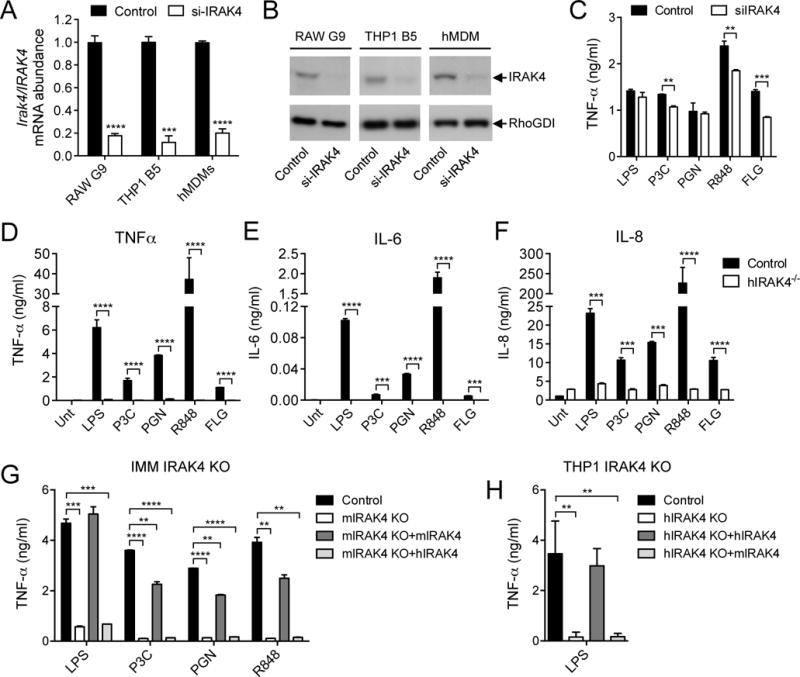
(A and B) Analysis of the relative abundances of IRAK4 mRNA (A) and protein (B) in RAW G9 cells, THP1 B5 cells, and hMDMs transfected with control or IRAK4-specific siRNAs. The relative fractions of IRAK4 protein abundance in cells treated with IRAK4-specific siRNA compared to that in control cells were as follows: 0.30 ± 0.07 for RAW G9 cells (P < 0.001); 0.28 ± 0.10 for THP1 B5 cells (P < 0.01); and 0.49 ± 0.06 for hMDMs (P < 0.01). Statistical analysis was by two-tailed t test. Western blots are representative of three independent experiments. (C) hMDMs transfected with control siRNA or IRAK4-specific siRNA were stimulated with LPS (10 ng/ml), 100 nM P3C, PGN (10 μg/ml), R848 (10 μg/ml), or FLG (10 ng/ml) for 24 hours and the amounts of TNF-α secreted by the cells were measured by ELISA. (D to F) hMDMs isolated from control and IRAK4-deficient patients were left untreated (Unt) or were stimulated with LPS (10 ng/ml), 100 nM P3C, PGN (10 μg/ml), R848 (10 μg/ml), or FLG (10 ng/ml) for 24 hours. The amounts of TNF-α (D), IL-6 (E), and IL-8 (F) secreted by the cells were measured by Bioplex assay. (G) Immortalized bone marrow–derived mouse macrophages (IMM) from IRAK4 KO mice were stably transduced with retrovirus expressing either mouse IRAK4-mCitrine or human IRAK4-mCherry. The cells were then stimulated for 24 hours with LPS (10 ng/ml), 100 nM P3C, PGN (30 μg/ml), or R848 (10 μg/ml) before the amount of TNF-α secreted by the cells was measured by ELISA. (H) IRAK4 KO THP1 cells were stably transduced with retrovirus expressing either human IRAK4-mCherry or mouse IRAK4-mCitrine. The cells were then stimulated for 24 hours with LPS (10 ng/ml) before the amount of TNF-α that they secreted was measured by ELISA. Data are means ± SD of two representative experiments for (A), (C), (G), and (H) or are means ± SD of two independent biological replicates from one set of patient blood samples for (D) to (F). **P < 0.01, ***P < 0.001, ****P < 0.0001 by two-tailed t test.
Human patients with IRAK4 deficiency have been previously described (31), and various immune cell types from these patients have defective TLR-induced cytokine responses, which are comparable to those of IRAK4-deficient mouse cells (32). We sought to determine whether hMDMs from an IRAK4-deficient patient might retain some level of TLR signaling response in light of the lack of phenotype observed in IRAK4-depleted human macrophages in our siRNA screen. However, consistent with previous studies, we found an almost complete loss of TLR-induced cytokine secretion in peripheral blood mononuclear cells (PBMCs) isolated from an IRAK4-deficient patient (fig. S4, A to C). We then tested the TLR response of MDMs from the same patient, but found a similar lack of TLR ligand–induced cytokine secretion in these cells (Fig. 4, D to F). These data suggest that human macrophages require IRAK4 to propagate a signal through the TLR pathways, but that they are less sensitive than mouse cells to IRAK4 depletion, because both THP1 cells and primary hMDMs still signaled effectively through TLR pathways when IRAK4 was depleted to <20% of its normal quantity (Fig. 4, A to C). It is possible that low amounts of human IRAK4 are simply required for the recruitment of IRAK1, which then assumes the primary function of downstream signal transduction.
To assess whether the different sensitivities of mouse and human IRAK4 to perturbation might indicate functional differences between these IRAK4 orthologs, we attempted to rescue the TLR response in IRAK4-deficient immortalized mouse macrophages (IMM) by retroviral expression of either mouse or human IRAK4. Although expression of mouse IRAK4 in these cells restored TLR-stimulated cytokine secretion to an extent similar to that of wild-type cells, human IRAK4 was completely unable to rescue the cytokine response (Fig. 4G), despite there being similar amounts of mouse Irak4 and human IRAK4 mRNA in the cells (fig. S5A). To assess whether this pattern of selective rescue of signaling was also observed in human cells, we used CRISR/Cas9-mediated genome editing to generate a THP1 cell line deficient in human IRAK4 (fig. S5, B and C), and we retrovirally expressed either human or mouse IRAK4 in this cell line (fig. S5, D and E). We found that expression of human IRAK4 fully restored TLR-stimulated TNF-α secretion, whereas the expression of mouse IRAK4 had no effect (Fig. 4H).
To further investigate the failure of the human and mouse IRAK4 protein orthologs to rescue signaling in macrophages from the other species, we immunoprecipitated endogenous Myd88 from lysates of mouse and human macrophages expressing different IRAK4 proteins and that were stimulated with LPS for various times. In mouse macrophages, mouse IRAK4 formed a sustained interaction with Myd88 for several hours (Fig. 5A), whereas human IRAK4 showed no detectable interaction with mouse Myd88 (Fig. 5B). In contrast, Myd88 immunoprecipitated from LPS-stimulated human macrophages had little detectable association with mouse IRAK4 (Fig. 5C), but clearly associated with human IRAK4 (Fig. 5D). We also observed different kinetics of Myd88 association with IRAK4 in mouse and human macrophages, with the association reaching a maximal extent at 30 min in mouse cells, but at 2 hours in human cells (Fig. 5, A and D). We then evaluated downstream signaling through the MAPK-dependent phosphorylation and nuclear accumulation of the AP1 transcription factor ATF2 in the mouse and human IRAK4 rescue cell lines. We found that in mouse macrophages, human IRAK4 was unable to restore the TLR-dependent phosphorylation of ATF2 (Fig. 5E), whereas in human macrophages, mouse IRAK4 was unable restore TLR-dependent ATF2 phosphorylation (Fig. 5F).
Fig. 5. Differential characteristics of human and mouse IRAK4 signaling properties.
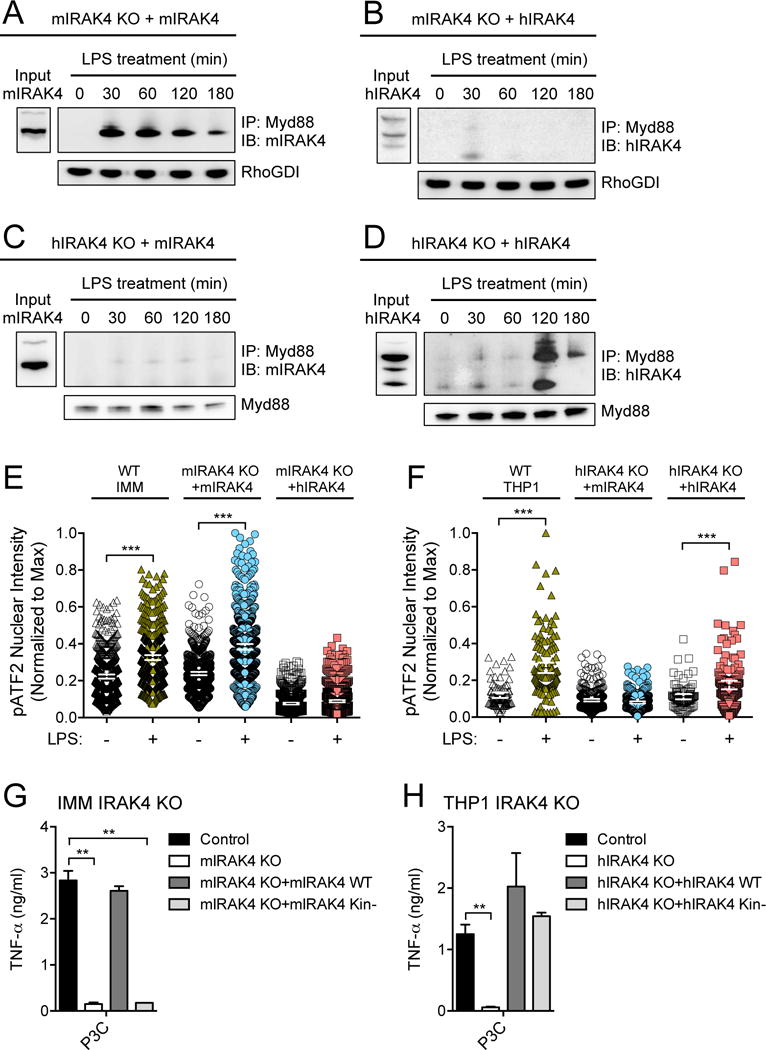
(A and B) Mouse IRAK4 KO IMMs were stably transduced with retrovirus expressing either (A) mouse IRAK4-mCitrine or (B) human IRAK4-mCherry before being treated with LPS (10 ng/ml) for the indicated times. Cell lysates were then subjected to immunoprecipitation (IP) with an antibody against Myd88 and samples were analyzed by Western blotting (IB) with antibody specific for IRAK4. RhoGDI was used as an input control for the Myd88 immunoprecipitation. (C and D) Human IRAK4 KO THP1 cells were stably transduced with retrovirus expressing either (C) mouse IRAK4-mCitrine or (D) human IRAK4-mCherry before being treated with LPS (10 ng/ml) for the indicated times. Cell lysates were then subjected to immunoprecipitation with an antibody specific for Myd88 and analyzed by Western blotting with antibody specific for IRAK4. Myd88 was used as an input control for the immunoprecipitation. (E and F) The nuclear intensity of phosphorylated ATF2 (pATF2) was measured by high-content imaging in either (E) mouse IRAK4 KO IMMs or (F) human IRAK4 KO THP1 cells stably transduced with retrovirus expressing either mouse IRAK4-mCitrine or human IRAK4-mCherry. Cells were left untreated or were stimulated for 20 (IMM) or 60 min (THP1) with 500 nM P3C. Data are shown for the central 98th percentile of cells. (G) IRAK4 KO IMMs were stably transduced with retrovirus expressing either wild-type (WT) or kinase-deficient (Kin−) mouse IRAK4. Cells were stimulated with LPS (10 ng/ml) for 24 hours and the amount of TNF-α secreted was measured by ELISA. (H) IRAK4 KO THP1 cells were stably transduced with retrovirus expressing either WT or kinase-deficient (Kin−) human IRAK4. Cells were stimulated with LPS (10 ng/ml) for 24 hours and the amount of TNF-α secreted by the cells was measured by ELISA. Western blots in (A) to (D) are representative of at least two independent experiments. Single-cell data in (E) and (F) are representative of two independent experiments (100 to 500 cells imaged per condition, error bars indicate mean ± 95% CI). Data in (G) and (H) are means ± SD of two independent experiments. **P < 0.01, ***P < 0.001 by Kolmogorov-Smirnov test (for E and F) or by two-tailed t test (for G and H).
The data described earlier suggest that the differential sensitivity to IRAK4 depletion that we observed in our screen of the responses of human and mouse cells to TLR signaling might be a result of specific properties of the IRAK4 protein that are not conserved between the species, which could underlie the failure of the human and mouse IRAK4 orthologs to rescue function in IRAK4 knockout (KO) cells from the other species. Previous studies suggested that the kinase activity of IRAK4 is required for TLR responses in mouse, but not human, fibroblasts (33, 34). To determine whether macrophages from the two species exhibited a differential dependence on the kinase activity of IRAK4, we expressed kinase-deficient mutants (Kin−) of mouse (35) and human (36) IRAK4 in their respective KO macrophage cells (fig. S6). We found that the kinase-deficient mutant mouse IRAK4 was unable to restore TLR-activated cytokine secretion in IRAK4 KO mouse macrophages (Fig. 5G), whereas the kinase-deficient human IRAK4 fully restored the cytokine response in IRAK4-deficient human macrophages (Fig. 5H).
Mouse and human primary macrophages replicate the different IRAK1 and IRAK2 dependencies identified through siRNA screening
We next sought to determine whether the different patterns of IRAK1 and IRAK2 dependency that were exhibited by the human and mouse macrophage reporter cell lines used in our screen were observed in primary cells. We first tested bone marrow–derived macrophages (BMDMs) from different IRAK KO mice (37–41). We compared the cytokine responses of BMDMs from wild-type (WT), IRAK1 KO, and IRAK2 KO mice to a range of TLR ligands. We observed an almost complete loss of TNF-α and IL-6 secretion in response to P3C, PGN, or R848 in IRAK2-deficient BMDMs, with a partial reduction in the response to LPS, consistent with the expected IRAK independence of LPS-induced TRIF (Toll-IL-1 receptor domain-containing adaptor inducing interferon-β) signaling (Fig. 6, A and B) (42–44). In contrast, we observed no substantial reduction in single TLR ligand–induced TNF-α and IL-6 secretion by IRAK1-deficient mouse BMDMs (Fig. 6, A and B). The marked effect of IRAK2 deficiency on TLR-induced cytokine secretion by mouse macrophages was demonstrated previously (39), and is partly a result of the role of IRAK2 in stabilizing pro-inflammatory cytokine transcripts (40). These data emphasize that in response to single TLR ligands, mouse macrophages show little dependence on IRAK1, as compared to IRAK2, for TNF-α and IL-6 secretion. These data mirror the effects of IRAK1 and IRAK2 depletion in the mouse macrophage reporter cell line in our siRNA screen (Fig. 3C), and validate our mouse macrophage siRNA screening platform.
Fig. 6. Differences in IRAK1 and IRAK2 protein use in mouse and human TLR responses in primary macrophages.
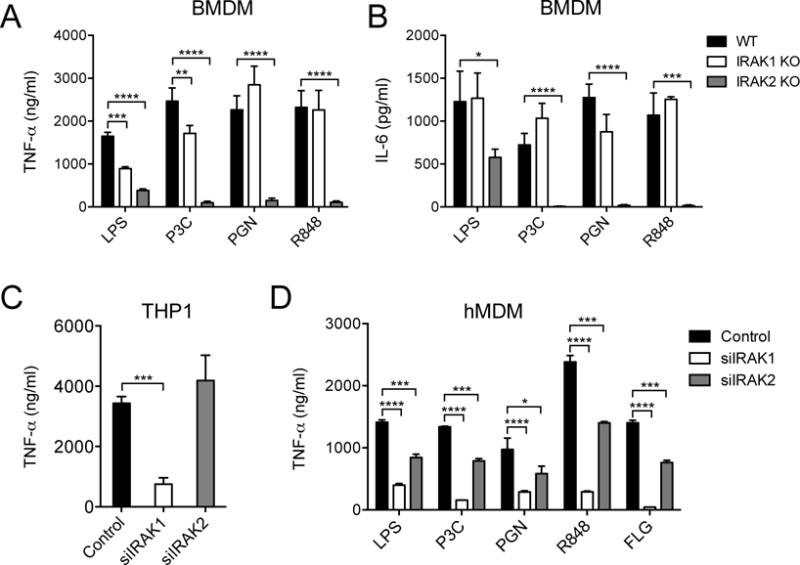
(A and B) Bone marrow-derived macrophages (BMDMs) from WT, IRAK1 KO, and IRAK2 KO mice were stimulated with LPS (10 ng/ml), 100 nM P3C, PGN (30 μg/ml), or R848 (10 μg/ml) for 24 hours before the amounts of TNF-α (A) and IL-6 (B) that were secreted by the cells were measured by ELISA. (C) THP1 cells transfected with control, IRAK1-specific, or IRAK2-specific siRNAs were stimulated with LPS (10 ng/ml) for 24 hours before the amounts of TNF-α that they secreted were measured by ELISA. (D) hMDMs transfected with control, IRAK1-specific, or IRAK2-specific siRNAs were stimulated with LPS (10 ng/ml), 100 nM P3C, PGN (10 μg/ml), R848 (10 μg/ml), or FLG (10 ng/ml) for 24 hours before the amounts of TNF-α that they secreted were measured by ELISA. Data are means ± SD of four (for A and B) or three experiments (for C and D). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by two-tailed t test.
To examine further the dependency on IRAK1 that we observed in the human macrophage reporter-based siRNA screen, we first tested the effect of depletion of IRAK1 or IRAK2 on the secretion of TNF-α by the THP1 B5 reporter cell line. TNF-α secretion induced by LPS (Fig. 6C) or P2C, P3C, or PGN (fig. S7A) was markedly reduced by IRAK1 knockdown cells, but IRAK2 depletion had little effect, which is consistent with the effects observed in the siRNA screen with the TNF-α transcriptional reporter. We tested whether differential extents of gene knockdown might contribute to the different IRAK effects observed in the human cells; however, we observed substantial and comparable reductions in the amounts of mRNAs and proteins of both IRAKs in the knockdown cells (fig. S7, B and C).
To determine whether the greater requirement for IRAK1 for TLR signaling in THP1 cells was also observed in human primary cells, we prepared hMDMs from peripheral blood and transfected them with IRAK1- or IRAK2-specific siRNAs by nucleofection. We again found that the secretion of TNF-α in response to a range of TLR ligands depended mostly on IRAK1, with much less dependence on IRAK2 (Fig. 6D). We further validated this observed pattern in hMDMs with independent siRNA sequences against IRAK1 or IRAK2 (fig. S7D) and we also confirmed that that the amount of knockdown of IRAK1 and IRAK2 mRNAs and proteins in the siRNA-treated hMDMs were similar (fig. S7, E and F). These data confirm that the pattern of IRAK1 and IRAK2 dependency that we observed in our screen was also found in primary human macrophages, and they further indicate that our analysis of the human and mouse TLR pathways identified substantial differences in how human and mouse macrophages decode bacterial PAMPs.
IRAK1 and IRAK2 have different functionalities in the human and mouse macrophage TLR signaling pathways
As described earlier, signaling through the MyD88-dependent TLR pathway requires the TRAF6-binding property of either IRAK1 or IRAK2 to propagate signals downstream to the NF-κB and MAPK signal transmission modules (Fig. 1A) (30). Previous studies in mouse macrophages suggested that IRAK1 is required for early signaling through the MyD88-dependent pathway, whereas IRAK2 is thought to be required for sustained signaling (39, 40, 45). Data from our screen suggested that the early NF-κB response to P3C and R848 at 40 min after stimulation was reduced slightly in IRAK1-depleted mouse cells, whereas the TNF-α reporter response at 16 hours was unaffected by IRAK1 perturbation (Fig. 3C). In contrast, IRAK2-depleted mouse cells had markedly reduced TNF-α reporter activity at 16 hours after stimulation, consistent with an important role for IRAK2 in sustained activation. However, we also found that knockdown of IRAK2 had a more substantial effect on LPS- and R848-induced early NF-κB activation than did knockdown of IRAK1 (Fig. 3C), suggesting a dominant role for IRAK2 in both initial and sustained responses to these TLR ligands in mouse cells.
To determine whether IRAK1 and IRAK2 contributed differently to the initial and sustained responses of human macrophages, we used CRISR/Cas9-mediated genome editing to generate THP1 cell lines deficient in human IRAK1 or IRAK2 (Fig. 7A and fig. S8). We first measured the effects of the loss of IRAK1 and IRAK2 on TLR ligand–induced TNF-α secretion by the different IRAK KO THP1 cell lines. We found that there was a marked reduction in cytokine secretion by the IRAK1 KO cells, but there was a much lesser effect of IRAK2 KO (Fig. 7B). We then tested the NF-κB and MAPK phosphoprotein responses to P3C in the IRAK KO cells by phosphoflow cytometry and saw a similarly marked reduction in signaling in the IRAK1 KO cells, but again minimal effects in the IRAK2 KO cells (Fig. 7C). This perturbation of early TLR signaling in human macrophages lacking IRAK1 was in stark contrast to the behavior of IRAK1 KO mouse macrophages, which showed relatively normal MAPK and NF-κB phosphoprotein responses to TLR stimulation (Fig. 7D).
Fig. 7. IRAK1 and IRAK2 have different functionalities in the human and mouse macrophage TLR signaling pathways.
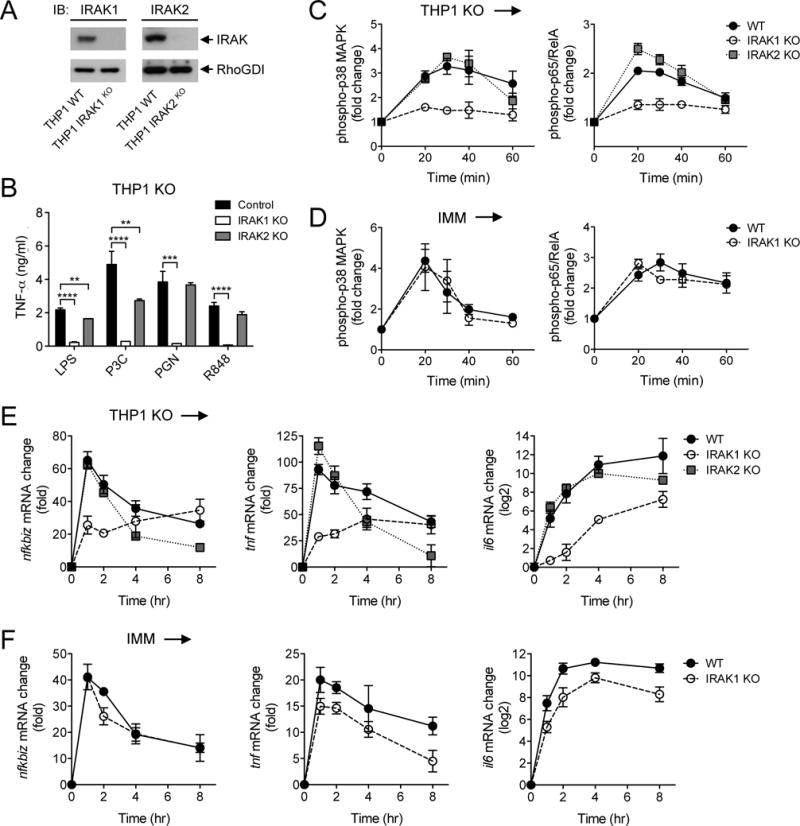
(A) The WT and indicated IRAK1 KO and IRAK2 KO THP1 cell lines were analyzed by Western blotting with antibody against IRAK proteins. RhoGDI was used as a loading control. Western blots are representative of two independent experiments. (B) The indicated control and IRAK KO THP1 cell lines were stimulated for 4 hours with LPS (10 ng/ml) before the amounts of TNF-α that they secreted were measured by ELISA. (C and D) The WT and indicated IRAK KO THP1 cell lines (C) and mouse IMMs (D) were stimulated with 500 nM P3C for the indicated times before the relative abundances of phosphorylated p38α MAPK (left) and p65 (right) were measured by phosphoflow cytometry. (E and F) The WT and indicated IRAK KO THP1 cell lines (E) and mouse IMMs (F) were stimulated with 500 nM P3C for the indicated times before the relative abundances of Nfkbiz (left), Tnf (middle), and Il6 (right) mRNAs were measured by qRT-PCR analysis. Data in (B) to (F) are means ± SD of two independent experiments. **P < 0.01, ***P < 0.001, ****P < 0.0001 by two-tailed t test.
We next determined the effect of loss of human IRAK1 and IRAK2 on the expression of genes that show different LPS response kinetics, namely early-transient (Nfkbiz), early-intermediate (Tnf), and late-sustained (Il6) expression. We found that the loss of human IRAK2 had no effect on the initial response kinetics of the expression of any of the three genes; however, IRAK2 was required to maintain mRNA amounts at times greater than 2 hours after LPS stimulation (Fig. 7E). This finding is consistent with a previously reported role for IRAK2 in maintaining the stability of cytokine mRNAs in both mouse and human macrophages (40, 46), which suggests that this is a conserved function of IRAK2 between the two species. In contrast, the loss of IRAK1 had a substantial effect on the induction of all gene classes in human macrophages (Fig. 7E), consistent with a major role for IRAK1 in the human TLR response; however, the loss of IRAK1 had little effect on the induction of the same genes in mouse macrophages (Fig. 7F). These data suggest a previously unappreciated major difference in the use of IRAK1 between human and mouse TLR signaling pathways.
To determine whether the differential effects of IRAK KO on the TLR-dependent responses of human and mouse macrophages were also observed when these cells encountered an intact pathogen, we infected cells with the gram-negative bacterium Burkholderia cenocepacia, and measured the resulting cytokine responses. We observed a similar pattern to the effects seen with individual TLR ligands, with defective cytokine secretion observed in mouse macrophages lacking IRAK2 or IRAK4 (Fig. 8A) and in human macrophages lacking IRAK1 or IRAK4 (Fig. 8B). To further assess whether the dependencies of mouse macrophages on IRAK2 and of human macrophages on IRAK1 were reflected in the signaling complexes formed after the cells were stimulated with TLR ligands, we immunoprecipitated Myd88 from both cell types after they were treated with LPS. We observed the LPS-dependent co-immunoprecipitation of IRAK2 with Myd88 in mouse IMMs throughout a 3-hour time course, with only weak and transient co-immunoprecipitation of Myd88 with IRAK1 (Fig. 8C). In contrast, we did not observe any IRAK2 in Myd88 immunoprecipitates from human THP1 cells stimulated with LPS for 3 hours, but we detected human IRAK1 in Myd88 immunoprecipitates throughout the same time course (Fig. 8D).
Fig. 8. Differential human and mouse IRAK dependencies are reflected in responses to gram-negative bacterial infection and in ligand-induced signaling complexes.
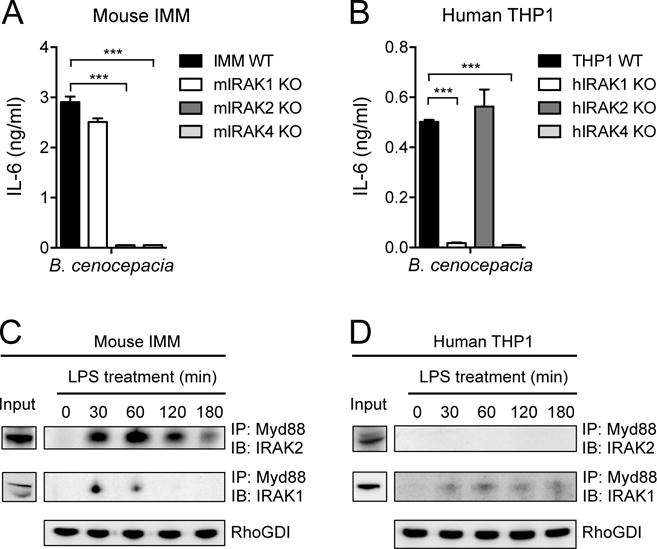
(A and B) IMMs from WT mice and the indicated IRAK KO mice (A) and the WT and indicated IRAK KO THP1 cell lines (B) were infected with B. cenocepacia at an MOI of 1. Twenty hours later, the amounts of IL-6 secreted by the cells were measured by ELISA. (C and D) IMMs from IRAK4 KO mice (C) and IRAK4 KO THP1 cells (D) were stably transduced with retrovirus expressing either mouse IRAK4-mCitrine or human IRAK4-mCherry, respectively. The cells were then treated with LPS (10 ng/ml) for the indicated times before cell lysates were subjected to immunoprecipitation (IP) with antibodies against Myd88 and analyzed by Western blotting (IB) with antibodies against either IRAK1 or IRAK2. RhoGDI is shown as an input control for the Myd88 immunoprecipitation. Western blots are representative of three independent experiments. Data in (A) and (B) are means ± SD of two independent experiments. ***P < 0.001 by two-tailed t test.
Finally, we sought to determine whether the apparent species-specific preferences in IRAK use were reflected in the extent of expression of these genes in human and mouse cells. We used the numerous expression datasets available in GEO to determine whether there were any species-specific trends in the abundances of IRAK1, IRAK2, and IRAK4 mRNAs. We used data from the Illumina platforms GPL6884 (Illumina HumanWG-6 v3.0 expression beadchip) and GPL6885 (Illumina MouseRef-8 v2.0 expression beadchip) because they used the same beadchip technology. We downloaded all available normalized gene expression data for these platforms (>6000 samples for human, >8000 samples for mouse) and calculated the expression distribution for the IRAK probes (fig. S9). We observed that IRAK1 was expressed to a greater extent than was IRAK2 in human cells, whereas in mouse cells, the trend was reversed. The expression of human IRAK1 and mouse Irak2 also shows greater variability (higher standard deviation) across the thousands of mRNA expression datasets available in GEO. This finding suggests that the expression of human IRAK1 and mouse Irak2 is specifically regulated across different cell types, which may be related to the species-specific roles in TLR signaling of their products that we have described in this study.
DISCUSSION
We previously described the development of an assay platform in human and mouse macrophages for the high-throughput, siRNA-based screening of responses to pathogenic stimuli (23). As a first step towards genome-wide screens, we described here the targeting of putatively canonical pathway components to assess TLR response requirements in a key innate immune cell type, and we also compared human and mouse pathway dependencies. Although the screen may have identified some characteristics that are distinct to the transformed macrophage cell lines used, we demonstrated validation of the findings from these cell line screens in primary human and mouse cells. Moreover, the broad use of the RAW264.7 and THP1 cell lines as model systems for the study of mouse and human macrophage biology make the presented data set a valuable resource for future studies.
At the level of signaling modules used by the TLR pathway, we observed considerable conservation between mouse and human cells in the modules containing the strongest phenotypes, namely the ligand-specific receptors, proximal signaling proteins, and the regulators of human TNF and mouse Tnf transcription. The slightly weaker phenotypes observed after branching of the pathway into the NF-κB, MAPK, and PI3K-Akt modules would suggest a degree of redundancy in the routing of TLR signals that may provide robustness to individual gene perturbations or to competition for these widely used signaling components from other pathways. One unexpected outcome in the screen was the relative lack of increased signals observed among the module of negative regulators. The assay system was capable of identifying known negative regulatory components, such as the protein TANK in the TRIF module (fig. S2); however, aside from a few cases of increased reporter output through the knockdown of the genes encoding A20, Cyld, and Mkp1 (Dusp1), we observed few strong negative regulator hits. Because many of these regulators operate through a delayed feedback mechanism that requires their transcriptional induction, it may be that our screen readouts of NF-κB activation and primary gene response were measured too early to fully capture the effect of perturbing these feedback regulators.
In comparing the hits for the different TLR ligands, we observed that the strongest hits tended to be common across all of the ligands tested, suggesting a number of nonredundant, shared TLR pathway components downstream of the different receptors; however, there were also numerous instances of gene knockdowns with ligand-selective effects. This suggests that although different TLR pathways may use a number of shared core components, there is a preferred use of certain signaling proteins in the pathways engaged by individual TLR ligands, which may lead to the activation of transcriptional programs and macrophage responses that are customized to the PAMPs presented by different pathogens. Clustering analysis showed that P3C and R848 were more similar to each other in their pathway component requirements than they were to LPS, which is likely reflective of their signaling exclusively through MyD88, whereas LPS signals through both MyD88 and TRIF. This is supported by the presence of TRIF in the gene phenotype cluster that was shared by human and mouse cell lines in response to LPS (fig. S3).
Beyond the species conservation of strongest hit enrichment among the TLR receptors, proximal signaling proteins, and human TNF/mouse Tnf transcription modules, we observed substantial cross-species differences in the individual gene requirements for TLR signal propagation in our human and mouse macrophage screens. In addition to the selectivity for the IRAK family that we have highlighted in this study, notable additional findings include a strong requirement in mouse cells for CD14, for IκB kinase β (IKKβ), and for the transcriptional regulating complex CBP/p300. Human cells appeared to be more sensitive to knockdown of TAK1, Tpl2, and ERK1, and they had an unexpected positive regulatory role for the NF-κB inhibitory protein IκBε. This latter observation may warrant further investigation, because we also found that knockdown of this protein rendered THP1 cells more sensitive to infection by gram-negative bacteria. The specific gene dependencies for human and mouse TLR pathways identified in this study may provide insight to facilitate future systematic analyses of these pathways.
Our in-depth study of the different patterns of IRAK dependency in human and mouse TLR signaling, in addition to identifying a clear difference in pathway architecture between species, also validated our screening platform for systematic analysis of macrophage responses to pathogens. We showed that the responsiveness of primary macrophages from IRAK KO mice to TLR ligands was in broad agreement with the results of our screen, and the effects of knockdown of the human IRAKs in blood-derived primary cells were also consistent with the effects that we observed in the human screen. Although we found that both primary human macrophages and THP1 cells were resistant to substantial depletion of IRAK4, human cells with a complete IRAK4 deficiency showed a loss of responsiveness to TLR ligands. This was in contrast to mouse cells in which siRNA-mediated IRAK4 knockdown led to a strong defect in responsiveness. The difference in sensitivity to IRAK4 knockdown may therefore reflect different species-specific properties of the human and mouse IRAK4 proteins, which is supported by our finding that both human and mouse IRAK4 were unable to rescue TLR responses in IRAK4-deficient cells from the other species. Previous studies demonstrated that kinase-inactive mouse IRAK4 cannot rescue IL-1 or TLR signaling in IRAK4-deficient mouse fibroblasts (34) or macrophages (47) respectively, but that kinase-inactive human IRAK4 can rescue IL-1 signaling in IRAK4-deficient human fibroblasts (33), and inhibition of the kinase activity of IRAK1 and IRAK4 has a minimal effect on TLR responses in several human immune cell types (48). We confirmed that kinase-inactive human IRAK4 rescued TLR responses in THP1 cells (Fig. 5H), which suggests that the function of IRAK4 in humans might be limited to its ability to recruit IRAK1 and that the kinase activity of IRAK1 and its TRAF6-recruitment domain may play a much more critical role in TLR signaling in human cells than in mouse cells. In light of our siRNA screen findings, the greater dependence of mouse cells on the kinase activity of IRAK4 may render them more sensitive to the depletion of this protein, whereas human cells may tolerate substantial reductions in the abundance of IRAK4 protein, because they are less reliant on this property. Furthermore, studies of mouse IRAK4 have suggested that it has important scaffolding functions for the establishment of a myddossome structure after TLR activation (28). Our data suggest the possibility that human IRAK1 compensates for a substantial depletion of IRAK4 in the human myddosome, but that mouse IRAK1 or IRAK2 are unable to similarly compensate for the depletion of mouse IRAK4.
Our data also demonstrate that the relative roles of IRAK1 and IRAK2 in the primary propagation of the TLR signal may be different between human and mouse, with there being a much stronger requirement for IRAK1 in human cells. We used the CRISPR/Cas9 genome editing technology to make human THP1 cells completely deficient in IRAK1 or IRAK2, and we showed that the initial signaling, transcriptional, and cytokine responses to TLR ligands and intact bacteria were particularly dependent on IRAK1 in those cells. Consistent with previous studies, human IRAK2 has an important role in maintaining TLR-induced gene transcription beyond 2 hours after ligand stimulation (46).
It has been known for some time that the TLR-induced cytokine response of IRAK1 KO mice is comparable to wild-type, in contrast to the dramatically reduced response observed in IRAK2 KO mice (38, 39, 41). Our cytokine data confirm that responses to individual TLR ligands and to intact bacteria in mouse IRAK1 KO cells remained relatively intact, in stark contrast to those in cells lacking IRAK2, which showed almost no cytokine secretion. Moreover, NF-κB and MAPK signaling and early transcriptional responses in TLR2-stimulated mouse macrophages were minimally affected by IRAK1 deficiency. Previous studies showed that mouse IRAK1 plays a critical role in interferon responses to endosomal TLR ligands in plasmacytoid dendritic cells (49), and mouse IRAK1 facilitates the rapid, TLR ligand–dependent priming of the NLRP3 (NLR family, pyrin domain containing 3) inflammasome (50, 51). Although these functions of IRAK1 have not been carefully compared in mouse and human cells, they suggest that mouse and human macrophages could have evolved different functions for IRAK1 that are tuned to the differential innate immune challenges faced by each organism. It is also noteworthy that among the human IRAKs, polymorphisms in IRAK1 in particular have been linked with susceptibility to a range of immune disorders (52–54), supporting an especially important role for this IRAK family member in human cells. Furthermore, in mouse models of human diseases in which IRAK1 polymorphisms are implicated, there are examples in which experimentally induced autoimmunity is defective when mouse IRAK1 is absent (55, 56); however, there are no examples in which the expression of a human IRAK1 variant that is implicated in a disease can drive disease in a mouse model. Our study establishes a critical role for IRAK1 in human TLR signal transduction that may help explain the immune consequences of genetic alterations at the IRAK1 locus. In addition, human IRAK1 is ubiquitously expressed, whereas mouse IRAK1 shows a more selective expression pattern (57, 58), again supporting a more prominent role for IRAK1 in human cells.
Other studies have compared gene expression datasets from inflammatory disease states in humans and mice and have arrived at contradictory conclusions as to how accurately mouse models can mimic human disease (21, 22). Of note, one aspect in which both of these studies agree is in the observation that genes and pathways regulating the innate immune response, such as the TLR signaling pathway, are highly enriched during responses to trauma, burns, or sepsis in both species. Despite this pathway enrichment, these studies conclude that individual gene expression changes within a pathway are often not predictive of a corresponding change in the orthologous gene in the other species, and it is proposed that the failure of numerous anti-inflammatory drugs in clinical trials can often be attributed to the targeting of an individual gene product that does not show the same behavior between human and animal models. The findings from our siRNA screens are broadly supportive of these prior studies, because we observed enrichment within the same TLR pathway modules in human and mouse cells, but relatively poor conservation in dependencies at the individual gene level. Comprehensive genetic screening of individual gene contributions between human and mouse innate immune cells thus provides important insight to this gene-level variation, and could identify more optimal anti-inflammatory drug targets in human cells.
In summary, we have performed a comprehensive siRNA-based screen of the canonical TLR pathway in human and mouse macrophages to generate a valuable resource dataset that systematically identified those proteins that are most required for the response to different TLR ligands in a key innate immune cell type. Beyond the species-conserved use of certain ligand-specific receptors and core signaling modules, we report considerable differences in human and mouse TLR pathway dependencies at the levels of individual genes and their products. In particular, we identified and experimentally validated a previously unappreciated difference in IRAK family use between mouse and human macrophages, which may have relevance for ongoing clinical efforts to target this kinase family to regulate TLR-dependent inflammatory responses, or for the development of therapeutics for autoimmune disorders that are influenced by polymorphisms in IRAK family genes.
MATERIALS AND METHODS
Cell culture and stimulation with TLR ligands
RAW264.7 G9 cells and immortalized mouse macrophages (IMMs) derived from wild-type (WT), Irak1−/−, and Irak4−/− mice were maintained in Dulbecco’s Modified Eagle medium (DMEM; 4.5 g/l glucose), 10% fetal bovine serum (FBS), 20 mM Hepes, and 2 mM glutamine. BMDMs from WT, Irak1−/−, and Irak2−/− mice were prepared by differentiation for 6 days in the same culture medium containing macrophage colony-stimulating factor (M-CSF, 60 ng/ml, R&D). THP1 B5 and the various THP1 IRAK KO cell lines were maintained in RPMI 1640, 10% FBS, and 2 mM glutamine (containing 500 μg/ml G418 for the THP1 B5 cells). After written informed consent was provided, normal donor and IRAK4-deficient patient blood was obtained at the NIH under IRB-approved protocols. The IRAK4-deficient patient has a heterozygous C to T mutation at nucleotide 877 in the IRAK4 coding sequence, which causes premature termination at Gln293. cDNA sequence analysis showed that only the mutant allele is expressed. PBMCs were prepared by a 1:1 dilution of a human blood sample with Hank’s balanced salt solution (HBSS) and layering onto the surface of lymphocyte separation medium (MP Biomedicals, LLC, 50494) at a 2:1 ratio. After centrifugation at 625 × g for 10 min, mononuclear cells were recovered from the opaque interface, erythrocytes were lysed with ACK lysing buffer (Quality Biological, 118-156-721), and the remaining cells were washed with HBSS to provide purified PBMCs. Human monocyte-derived macrophages (hMDMs) were derived from PBMCs cultured in cRPMI (RPMI-1640 medium containing 10% FBS, 10 mM Hepes, 0.1% β-mercaptoethanol and 2 mM glutamine) with granulocyte macrophage colony-stimulating factor (GM-CSF, 10 ng/ml, R&D, Cat#: 215-GM-010) for 7 days. LPS was from Alexis Biochemicals (Salmonella minnesota R595 TLRgrade, Cat. No. ALX-581-008-L002), Pam3CSK4 was from InvivoGen (P3C, Cat. No. tlrl-pms) ; peptidoglycan (PGN) was from Sigma (Staphylococcus aureus PGN, Cat. No. 77140); R848 was from InvivoGen (Cat. No. tlrl-r848) Pam2CSK4 was from EMC Microcollections (P2C, Cat. No. L2020); Flagellin was from Invivogen, (FLA-ST ultrapure, Cat. No. tlrl-epstfla).
Dual luciferase assays in THP1 B5 cells
The THP1 B5 cell clone was generated as described previously (23). Cells were differentiated into a macrophage-like state with phorbol-12-myristate-13-acetate (PMA, 5 ng/ml, Sigma, P1585) for 72 hours. The differentiated cells were treated with TLR ligands for activation or with culture medium as a control. After stimulation, firefly and renilla luciferase activities in the cell lysates were determined with the Dual-Luciferase Reporter Assay System (Promega, E1960) and the firefly:renilla luminescence ratio was calculated to reflect the response of the cells to stimulation.
High-content imaging of RAW G9 cells
The RAW G9 clone was generated as described previously (23, 59). The GFP-p65 and Tnf promoter-mCherry reporters in RAW G9 cells were imaged with a BD Pathway 855 bioimager (BD biosciences). BD AttoVision software was used to automatically identify and quantify nuclei stained with Hoechst 33342 (Invitrogen, H3570), GFP-p65, and mCherry fluorescence. GFP located within the area of nuclear staining (eroded by 2 pixels) was defined as nuclear NF-κB, whereas GFP within a 2-pixel-wide ring around the area of nuclear staining was defined as cytosolic NF-κB. To determine the extent of nuclear translocation of NF-κB after ligand stimulation, the ratio of the intensities of nuclear to cytoplasmic GFP-p65 was calculated with BD Image Data Explorer software. For mCherry expression, nuclear mCherry was quantified with the same method that was used for NF-κB. Background red fluorescence was subtracted and the average intensity was used as a measure of Tnf promoter activity.
siRNA screen
The TLR pathway 126-gene set (Fig. 1A) was targeted with six unique siRNA sequences (three each from Ambion and Qiagen) per gene (table S1), and individual siRNAs were arrayed in separate regions of 384-well plates (Fig. 1B). Three central columns in each plate contained replicate wells of negative controls [nontargeting siRNA; NTC#2, 3, and 5 (Dharmacon), lipid only and cells only] and positive controls (Mouse: Myd88, Irak4 and Ikbkg; Human: TLR4/TLR8, TIRAP and IKBKG). To control for cell passage variability, reporter cells were always thawed from a common stock 2 weeks before transfection. After the cells were transfected with siRNAs according to previously described optimized protocols (23), THP1 B5 cells were stimulated with TLR ligands for 4 hours and then were subjected to dual luciferase assays. Duplicate siRNA plates were run for RAW G9 cells to enable one plate set to be treated with TLR ligand for 40 min (for NF-κB readout) and the second plate set to be treated for 16 hours (for TNF-α reporter readout). RAW G9 cells were fixed in 4% paraformaldehyde (Polyscience, Inc), and nuclei were stained with 600 nM DAPI (Invitrogen) before high-content imaging was performed. Replicates were run for all screens with separate batches of reporter cells. Raw data from each reporter assay were normalized by the following intra-plate median/ MAD (median absolute deviation) calculation: (sample - median (all samples))/ MAD (all samples). Plate comparisons between replicates all showed Spearman correlations > 0.7. Robust z-scores relative to negative controls were calculated, and average values from replicates were calculated for each single siRNA (see table S2). The median z-score of the six individual siRNAs for each of the 126 screened genes was used as the readout for specific gene effects on the response to each TLR ligand (table S2). Estimations of false-positive and false-negative rates in the human and mouse siRNA screens were calculated with negative and positive siRNA controls that were reproducible between experiments and showed a reliable signal perturbation phenotype. Because false positive and negative rates are dependent on the selection of the screen score threshold, we estimated them over a wide range of thresholds. In this screen, we expected that the positive control wells would have high negative z-scores. Hence, the false negative rate can be calculated as the percentage of individual positive control wells with scores greater than the chosen threshold (ranging from −0.5 to −3), that is, not passing the threshold. We used negative control well scores for estimating the false positive rates. Because the screen siRNA scores were normalized to the negative controls, we expected that the individual negative control wells would have both positive and negative values (with a median value of 0), so false positive rates were estimated in both directions. The percentage of wells with scores deviating from 0 beyond a threshold (ranging from −0.5 to −3 and 0.5 to 3) were used to estimate false positive rates. The following individual human IRAK-specific siRNAs were used in post-screen experiments (all from Ambion): IRAK1 (s323 and s322), IRAK2 (s7497 and s7495), and IRAK4 (s27527 and s27528).
Quantitative RT-PCR (qRT-PCR)
Total RNA was extracted with the RNeasy Micro Kit (Qiagen, 74004) and RNeasy Mini Kit (Qiagen, 74106). Complementary DNA (cDNA) was synthesized from the extracted RNA with the iScript cDNA Synthesis Kit (Bio-rad, 170-8891). Quantitative reverse transcription polymerase chain reaction (qRT-PCR) assays were performed with Thermo Scientific Solaris qPCR Rox Master Mix (AB-4351/C) or with Power SYBR green PCR Master Mix (Applied biosystems, Cat. No. 43091655) with a Mastercycler realplex4 real-time PCR detection system (Eppendorf), according to the manufacturers’ protocols. The abundances of the mRNAs of interest in each sample were normalized to that of ACTB/Actb or HPRT1/Hprt mRNA, and fold-changes in the abundances of target mRNAs relative to their basal abundances were calculated with the 2−ΔΔCt method. The primers and probes for human and mouse IRAK1/Irak1, MAP3K7/Map3k7, TAB2/Tab2, NFKBIE/Nfkbie, MAP3K8/Map3k8, MAPK3/Mapk3, MAPK14/Mapk14, IRF9/Irf9, IRAK2/Irak2, IRAK4/Irak4, CD14/Cd14, IKBKB/Ikbkb, MAPK1/Mapk1, CREBBP/Crebbp, EP300/Ep300, TNF/Tnf, NFKBIZ/Nfkbiz, and IL6/Il6 were purchased from GE Healthcare, Dharmacon (Solaris Mouse or Human qPCR Gene Expression Assay).
Cytokine ELISA
2 × 105/ml BMDMs, IMMs, hMDMs, or differentiated THP1 cells were treated with the appropriate TLR ligands, the cell culture medium was collected, and the concentrations of TNF-α or IL-6 secreted by the cells were determined by ELISA. The mouse TNF-α ELISA kit was purchased from R&D Systems (DY410), whereas the mouse IL-6 (555240) and human TNF-α (555212) kits were purchased from BD Biosciences.
Cytokine Bioplex
PBMCs were plated into a 96-well plate at 1 × 106 cell/ml and stimulated with the appropriate TLR ligands for 16 hours before cell culture medium was collected. 1 × 107 PBMCs from the patient or a healthy control were cultured in 10 ml of cRPMI containing GM-CSF (10 ng/ml, R&D Systems, 215-GM-010) for 7 days to induce the generation of hMDMs. The hMDMs were then plated at 1.5 × 105 cell/ml and stimulated with the appropriate TLR ligands for 16 hours before the cell culture medium was collected. The amounts of secreted TNF-α, IL-6, MIP-1α, and IL-8 were determined with the Bioplex assay (Bio-rad).
Western blotting
RAW cells, THP1 cells, or hMDMs were lysed in RIPA (Radioimmunoprecipitation assay) buffer (Sigma, R0278) containing a protease inhibitor cocktail (Roche). Cell lysates were quantified by protein assay (Biorad) and equal protein amounts were resolved with a 4 to 12% Bis-tris Gel/MOPS running buffer system (Invitrogen) and transferred to nitrocellulose membranes. The membranes were analyzed by Western blotting with the following antibodies: rabbit anti-IRAK1 (Cell Signaling, 4504S), rabbit anti-IRAK4 (abcam, ab13685 or ab32511), rabbit anti-IRAK2 (abcam, ab62419, and ProSci, 3595), rabbit anti-Rho-GDI (Sigma, R3025), and horseradish peroxidase (HRP)-conjugated donkey anti-rabbit secondary antibody (GE Healthcare).
Expression of mouse or human IRAK4 fluorescent protein fusions in IRAK4 KO IMMs and THP1 cells
The pR-IRAK4-mCitrine and pR-IRAK4(Kin−)-mCitrine retroviral plasmids, which express WT or kinase-deficient (35) murine IRAK4, respectively, with an mCitrine fusion at the C terminus, were generated by amplifying the cDNA encoding murine IRAK4 from expression vectors with the following specific primers: Forward: 5′-TTTCGCGCGCGATGAACAAGCCGTTGACACCATC-3′; reverse: 5′-AAACTCGAGAGCAGACATCTCTTGTAGCAGC-3′. The resulting PCR product was digested with Mau BI and Xho I and subcloned in-frame into the corresponding sites of the pR-mCitrine retroviral vector. Entry vectors containing the human WT IRAK4 sequence (Addgene #23749: pDONR223-IRAK4) or the human IRAK4 kinase-deficient mutant (36) were recombined with pDS-FBhyg_X-mCH by Gateway cloning to generate retroviral plasmids that expressed either WT or kinase-deficient (Kin-) human IRAK4 with an mCherry fusion at the C terminus. A control retrovirus was also generated that expressed mCherry alone. Immortalized IRAK4 KO mouse macrophages or human IRAK4 KO THP1 cells expressing murine IRAK4-mCitrine (either WT and or Kin-), human IRAK4-mCherry (either WT and or Kin-), or mCherry alone were generated by retroviral transduction as previously described (60). Cells were sorted based on positive expression of mCitrine or were selected with Hygromycin B (Clontech), and then single-cell clones were expanded in 96-well tissue culture plates by limiting dilution and were functionally tested by ELISA for reconstitution of cytokine secretion in response to TLR activation. The abundances of the murine IRAK4-mCitrine and human IRAK4-mCherry fusion proteins were determined either by flow cytometric analysis or by qRT-PCR assays with the following primer pairs: Mouse Irak4 forward: TGCATGAGAAGAAAAACAGACG; mCitrine reverse: GGACACGCTGAACTTGTGG; human IRAK4 forward: GACATTAAGAAGGTTCAACAGC; mCherry reverse: GCCATGTTATCCTCCTCG.
Generation of THP1 CRISPR/Cas9 knockout lines
THP-1 cells were plated at a density of 2 × 105/ml. After 24 hours, 2.5 × 106 cells were resuspended in 250 μl of Opti-MEM, mixed with 5 μg CRISPR/Cas plasmid DNA, and electroporated in a 4-mm cuvette with an exponential pulse at 250 V and 950 mF with a Gene Pulser electroporating device (Bio-Rad Laboratories). We used a plasmid encoding a CMV-mCherry-Cas9 expression cassette and a human IRAK-specific gRNA (fig. S6) driven by the U6 promoter (61). Critical exons of the IRAK genes were targeted with the following gRNA constructs. IRAK1: 5′- GCTGAGGATGGCAACTTCCGGGG-3′; IRAK2: 5′-GCAGGGTGTGAGCATCACGCGGG-3′; IRAK4: 5′-GATGAACGACCCATTTCTGTTGG-3′. Cells were allowed to recover for 2 days in 6-well plates filled with 4 ml of medium per well before being FACS-sorted for mCherry-expression with a BD FACSAria III sorting device (BD Biosciences). For subsequent limiting dilution cloning, cells were plated at a density of 5, 10, or 20 cells per well in nine round-bottomed 96-well plates and cultured for 2 weeks. Plates were scanned for absorption at 600 nm, and growing clones were identified with customized software and were picked and duplicated by a Biomek FXp (Beckman Coulter) liquid handling system. One duplicate was used to recover gDNA and characterize the gene editing as previously described (62).
Immunoprecipitation of TLR signaling complexes
For immunoprecipitation experiments, whole-cell extracts from 1 × 107 cells per condition were prepared in 600 μl of cell lysis buffer containing 1% NP-40, 50 mM Tris-HCl (pH 7.5), and 150 mM NaCl. Lysates were centrifuged at 18,000g for 10 min at 4°C. The cleared supernatants were collected, and 50 μl of the supernatant was saved as the input sample. 1 μg of anti-MyD88 antibody (R&D; AF3109, goat) for immunoprecipitation from mouse IMMs or 10 μl of anti-MyD88 antibody (CST; 3699, Rabbit) for immunoprecipitation from human THP1 cells were added to 50 μl of Dynabeads protein G (Novex, 10003D) or Dynabeads protein A (Novex, 10001D) in 200 μl of phosphate-buffered saline (PBS) containing 0.1 % Tween-20 (PBST) for 20 min at room temperature to form bead-antibody complexes. The remaining cell lysate supernatants (around 550 μl) were added to the bead-antibody complexes and incubated with rotation overnight at 4°C. The beads were washed three times with PBST and then the immunoprecipitated complexes were eluted in 25 μl of a 1:1 dilution of Elution buffer (Invitrogen) and 2× SDS buffer (Quality biological, Cat : 351-082-661) by heating at 70°C for 10 min. Twenty microliters of eluted protein complexes were resolved with a 10% Bis-tris Gel/MOPS running buffer system (Invitrogen) and visualized by Western blotting analysis with the following antibodies. Rabbit anti-IRAK1 (Cell Signaling, 4504S), mouse anti-IRAK4 (Pierce, MA5-15883), rabbit anti-IRAK2 (abcam #ab62419 or ProSci #3595), mouse anti-MyD88 (Santa Cruz, sc-74532), rabbit anti-Rho-GDI (Sigma, R3025) and HRP-conjugated ECL anti-rabbit immunoglobulin G (IgG) or anti-mouse IgG secondary antibody (GE Healthcare).
Imaging of the TLR-activated phosphorylated ATF2 response in IMMs and THP1 cells
IMMs were seeded in 96-well plates 24 hours before they were treated with TLR ligands. THP1 cells were differentiated with PMA in 96-well plates 3 days before they were treated with ligands. After treatment with 500 nM P3C, the cells were fixed in 4% paraformaldehyde (PFA), and blocked with PBST containing 5% (w/v) bovine serum albumin (BSA). The fixed cells were treated with primary mouse antibody against pATF2 (1:200 dilution, Santa Cruz Biotechnology, sc-8398) followed by Alexa Fluor 647–conjugated goat anti-mouse IgG secondary antibody (1:1000 dilution, Life Technologies Catalog No. A-21235) or Alexa Fluor 700–conjugated goat anti-mouse IgG secondary antibody (1:1000 dilution, Life Technologies Catalog No. A-21036). The cells were imaged either on a Thermo Scientific CellInsight NXT or a Leica SP8.
Phosphoflow cytometry
THP1 cells or IMMs were treated with 0.5 mM Pam3CSK4 (InvivoGen, Cat. No. tlrl-pms) for the times indicated in the figure legends. The cells were fixed with 1% PFA, detached, and permeabilized with ice-cold methanol. The cell suspensions were incubated with purified rat anti-mouse CD16/CD32 (BD Biosciences, mouse BD Fc Block, clone 2.4G2, Cat. No. 553141) to block nonspecific binding and stained with Alexa Fluor 555–conjugated anti-phosphorylated p38 MAPK (Cell Signaling, 9836S), Alexa Fluor 647–conjugated anti-phosphorylated p65 NF-κB (Cell Signaling, 4887S), or Alexa Fluor 488–conjugated anti-phosphorylated p65 NF-κB (BD bioscience, 558421) in PBS containing 1% FCS, 2 mM EDTA.
Bacterial infection of macrophages
Infection of both THP1 cells and IMM cell lines with B. cenocepacia was performed with the infection protocol described previously (63). After incubation of cells with the bacteria, the cell culture medium was collected, and the amounts of secreted cytokines were measured by ELISA as described earlier.
Calculation of expression distributions from GEO datasets
All available normalized gene expression data for platforms GPL6884 (Illumina HumanWG-6 v3.0 expression beadchip, >6000 samples) and GPL6885 (Illumina MouseRef-8 v2.0 expression beadchip, >8000 samples) were downloaded from GEO (http://www.ncbi.nlm.nih.gov/geo/). To make the samples comparable and eliminate potential batch effects, expression data from each sample were transformed by calculating the robust z-score from the median and MAD) of individual samples. Expression z-scores for probes from all available samples corresponding to human IRAK1, IRAK2, and IRAK4 and mouse Irak1, Irak2, and Irak4 were binned into 500 equal bins between −10 and 10 to generate probability distributions.
Supplementary Material
Fig. S1. Design and TLR ligand response of mouse and human macrophage reporter cell lines for siRNA screening applications.
Fig. S2. Effects of siRNA-mediated gene perturbations across the human and mouse TLR pathways.
Fig. S3. Human and mouse macrophages show both shared and distinct gene dependencies in TLR signaling.
Fig. S4. IRAK4 is required for the responses of human PBMCs to TLR ligands.
Fig. S5. Generation of IRAK4 rescue cell lines from IRAK4 KO IMMs and THP1 cells.
Fig. S6. Expression of kinase-deficient mouse and human IRAK4 in IRAK4 KO mouse IMMs and THP1 cells.
Fig. S7. Analysis of the IRAK1-dependency of human macrophages for TLR responses.
Fig. S8. CRISPR/Cas9-mediated targeting of the human IRAK1 and IRAK2 loci in THP1 cells.
Fig. S9. Expression distributions of IRAK1/Irak1, IRAK2/Irak2, and IRAK4/Irak4 mRNAs in human and mouse GEO datasets.
Table S1. Details of the siRNAs used to target the 126 human and mouse TLR pathway genes.
Table S2. siRNA z-scores from screens of the 126 human and mouse TLR pathway genes.
Table S3. Ranking scores of human and mouse TLR pathway genes from the siRNA screens.
Table S4. Human vs mouse z-score differences across matched TLR ligands and assay readouts.
Acknowledgments
We thank R. Germain, R. Gottschalk, and colleagues in the Laboratory of Systems Biology for helpful discussions and critical reading of the manuscript; S. Holland for assistance in obtaining blood from an IRAK4-deficient patient; X. Li for provision of the human IRAK4 kinase-deficient mutant; A. Miller for assistance with the bacterial infection assay and R. Stahl for assistance in generating the mouse IRAK4-mCitrine retroviral plasmids.
Funding: This work was generously supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases (to J.S., N.L., K.O., B.D., S.J.V., B.L., J.D., N.W.L., and I.D.C.F.) and the BONFOR research commission at the University of Bonn (to D.D.). V.H. and T.S.E. are supported by grants from the German Research Foundation (SFB704 and SFB670) and the European Research Council (ERC-2009-StG 243046). E.L. is supported by grants from the National Institutes of Health (R01HL112661), the Deutsche Forschungsgemeinschaft (SFB670), and the European Research Council (ERC InflammAct). E.L. and V.H are members of the ImmunoSensation cluster of excellence. C.P. is supported by grants from NIAID (AI082265) and The Welch Foundation (I-1820). R.B. is supported by an NIH Integrative Immunology Training program grant (5T32-AI005284-35).
Footnotes
Author contributions: J.S., N.L., and I.D.C.F. designed the study; J.S. and N.L. performed the siRNA screen; B.D., J.S., N.L. and I.D.C.F. analyzed the siRNA screen data; J.S. performed all aspects of the IRAK studies; K.O. performed immunoprecipitation of TLR signaling components; S.J.V. performed high-content imaging analysis of TLR pathway components; B.L. performed qRT-PCR analysis of siRNA-mediated knockdowns; T.S.E. and V.H. generated the THP1 IRAK KO cell lines; D.D. and E.L. generated the mouse IRAK4 rescue cell lines; J.D. acquired and processed the IRAK4-deficient patient samples; R.B. and C.P. acquired and processed the IRAK1 and IRAK2 KO BMDMs; N.W.L. and B.D. performed the analysis of IRAK expression in the GEO data; J.S. and I.D.C.F. wrote the paper; and all authors commented on the manuscript.
Competing interests: The authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.Kawai T, Akira S. The roles of TLRs, RLRs and NLRs in pathogen recognition. International immunology. 2009;21:317–337. doi: 10.1093/intimm/dxp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Creagh EM, O’Neill LA. TLRs, NLRs and RLRs: a trinity of pathogen sensors that co-operate in innate immunity. Trends in immunology. 2006;27:352–357. doi: 10.1016/j.it.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 3.Barbalat R, Ewald SE, Mouchess ML, Barton GM. Nucleic acid recognition by the innate immune system. Annual review of immunology. 2011;29:185–214. doi: 10.1146/annurev-immunol-031210-101340. [DOI] [PubMed] [Google Scholar]
- 4.Wu J, Chen ZJ. Innate immune sensing and signaling of cytosolic nucleic acids. Annual review of immunology. 2014;32:461–488. doi: 10.1146/annurev-immunol-032713-120156. [DOI] [PubMed] [Google Scholar]
- 5.Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annual review of immunology. 2011;29:707–735. doi: 10.1146/annurev-immunol-031210-101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 7.Beutler BA. TLRs and innate immunity. Blood. 2009;113:1399–1407. doi: 10.1182/blood-2008-07-019307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Foster SL, Medzhitov R. Gene-specific control of the TLR-induced inflammatory response. Clinical immunology. 2009;130:7–15. doi: 10.1016/j.clim.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ostuni R, Zanoni I, Granucci F. Deciphering the complexity of Toll-like receptor signaling. Cellular and molecular life sciences : CMLS. 2010;67:4109–4134. doi: 10.1007/s00018-010-0464-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 11.Boutros M, Ahringer J. The art and design of genetic screens: RNA interference. Nat Rev Genet. 2008;9:554–566. doi: 10.1038/nrg2364. [DOI] [PubMed] [Google Scholar]
- 12.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 13.Cekaite L, Furset G, Hovig E, Sioud M. Gene expression analysis in blood cells in response to unmodified and 2′-modified siRNAs reveals TLR-dependent and independent effects. Journal of molecular biology. 2007;365:90–108. doi: 10.1016/j.jmb.2006.09.034. [DOI] [PubMed] [Google Scholar]
- 14.Judge AD, Sood V, Shaw JR, Fang D, McClintock K, MacLachlan I. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat Biotechnol. 2005;23:457–462. doi: 10.1038/nbt1081. [DOI] [PubMed] [Google Scholar]
- 15.Amit I, Garber M, Chevrier N, Leite AP, Donner Y, Eisenhaure T, Guttman M, Grenier JK, Li W, Zuk O, Schubert LA, Birditt B, Shay T, Goren A, Zhang X, Smith Z, Deering R, McDonald RC, Cabili M, Bernstein BE, Rinn JL, Meissner A, Root DE, Hacohen N, Regev A. Unbiased reconstruction of a mammalian transcriptional network mediating pathogen responses. Science. 2009;326:257–263. doi: 10.1126/science.1179050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chevrier N, Mertins P, Artyomov MN, Shalek AK, Iannacone M, Ciaccio MF, Gat-Viks I, Tonti E, DeGrace MM, Clauser KR, Garber M, Eisenhaure TM, Yosef N, Robinson J, Sutton A, Andersen MS, Root DE, von Andrian U, Jones RB, Park H, Carr SA, Regev A, Amit I, Hacohen N. Systematic discovery of TLR signaling components delineates viral-sensing circuits. Cell. 2011;147:853–867. doi: 10.1016/j.cell.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 18.Hoebe K, Beutler B. Forward genetic analysis of TLR-signaling pathways: an evaluation. Advanced drug delivery reviews. 2008;60:824–829. doi: 10.1016/j.addr.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 19.Tan Y, Kagan JC. A cross-disciplinary perspective on the innate immune responses to bacterial lipopolysaccharide. Molecular cell. 2014;54:212–223. doi: 10.1016/j.molcel.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Picard C, Casanova JL, Puel A. Infectious diseases in patients with IRAK-4, MyD88, NEMO, or IkappaBalpha deficiency. Clinical microbiology reviews. 2011;24:490–497. doi: 10.1128/CMR.00001-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takao K, Miyakawa T. Genomic responses in mouse models greatly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2014 doi: 10.1073/pnas.1401965111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, Finnerty CC, Lopez CM, Honari S, Moore EE, Minei JP, Cuschieri J, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Jeschke MG, Klein MB, Gamelli RL, Gibran NS, Brownstein BH, Miller-Graziano C, Calvano SE, Mason PH, Cobb JP, Rahme LG, Lowry SF, Maier RV, Moldawer LL, Herndon DN, Davis RW, Xiao W, Tompkins RG, Inflammation, and L. S. C. R. P. Host Response to Injury Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2013;110:3507–3512. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li N, Sun JC, Benet ZL, Wang Z, Al-Khodor S, John SP, Sung MH, Fraser ID. Development of a cell system for siRNA screening of pathogen responses in human and mouse macrophages. Scientific reports. 2015 doi: 10.1038/srep09559. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marine S, Bahl A, Ferrer M, Buehler E. Common seed analysis to identify off-target effects in siRNA screens. J Biomol Screen. 2012;17:370–378. doi: 10.1177/1087057111427348. [DOI] [PubMed] [Google Scholar]
- 25.Konig R, Chiang CY, Tu BP, Yan SF, DeJesus PD, Romero A, Bergauer T, Orth A, Krueger U, Zhou Y, Chanda SK. A probability-based approach for the analysis of large-scale RNAi screens. Nat Methods. 2007;4:847–849. doi: 10.1038/nmeth1089. [DOI] [PubMed] [Google Scholar]
- 26.Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, Horiuchi T, Tomizawa H, Takeda K, Akira S. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. 2002;3:196–200. doi: 10.1038/ni758. [DOI] [PubMed] [Google Scholar]
- 27.Jurk M, Heil F, Vollmer J, Schetter C, Krieg AM, Wagner H, Lipford G, Bauer S. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nat Immunol. 2002;3:499. doi: 10.1038/ni0602-499. [DOI] [PubMed] [Google Scholar]
- 28.Motshwene PG, Moncrieffe MC, Grossmann JG, Kao C, Ayaluru M, Sandercock AM, Robinson CV, Latz E, Gay NJ. An oligomeric signaling platform formed by the Toll-like receptor signal transducers MyD88 and IRAK-4. J Biol Chem. 2009;284:25404–25411. doi: 10.1074/jbc.M109.022392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burns K, Janssens S, Brissoni B, Olivos N, Beyaert R, Tschopp J. Inhibition of interleukin 1 receptor/Toll-like receptor signaling through the alternatively spliced, short form of MyD88 is due to its failure to recruit IRAK-4. J Exp Med. 2003;197:263–268. doi: 10.1084/jem.20021790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ye H, Arron JR, Lamothe B, Cirilli M, Kobayashi T, Shevde NK, Segal D, Dzivenu OK, Vologodskaia M, Yim M, Du K, Singh S, Pike JW, Darnay BG, Choi Y, Wu H. Distinct molecular mechanism for initiating TRAF6 signalling. Nature. 2002;418:443–447. doi: 10.1038/nature00888. [DOI] [PubMed] [Google Scholar]
- 31.Picard C, Puel A, Bonnet M, Ku CL, Bustamante J, Yang K, Soudais C, Dupuis S, Feinberg J, Fieschi C, Elbim C, Hitchcock R, Lammas D, Davies G, Al-Ghonaium A, Al-Rayes H, Al-Jumaah S, Al-Hajjar S, Al-Mohsen IZ, Frayha HH, Rucker R, Hawn TR, Aderem A, Tufenkeji H, Haraguchi S, Day NK, Good RA, Gougerot-Pocidalo MA, Ozinsky A, Casanova JL. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science. 2003;299:2076–2079. doi: 10.1126/science.1081902. [DOI] [PubMed] [Google Scholar]
- 32.Ku CL, von Bernuth H, Picard C, Zhang SY, Chang HH, Yang K, Chrabieh M, Issekutz AC, Cunningham CK, Gallin J, Holland SM, Roifman C, Ehl S, Smart J, Tang M, Barrat FJ, Levy O, McDonald D, Day-Good NK, Miller R, Takada H, Hara T, Al-Hajjar S, Al-Ghonaium A, Speert D, Sanlaville D, Li X, Geissmann F, Vivier E, Marodi L, Garty BZ, Chapel H, Rodriguez-Gallego C, Bossuyt X, Abel L, Puel A, Casanova JL. Selective predisposition to bacterial infections in IRAK-4-deficient children: IRAK-4-dependent TLRs are otherwise redundant in protective immunity. J Exp Med. 2007;204:2407–2422. doi: 10.1084/jem.20070628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qin J, Jiang Z, Qian Y, Casanova JL, Li X. IRAK4 kinase activity is redundant for interleukin-1 (IL-1) receptor-associated kinase phosphorylation and IL-1 responsiveness. J Biol Chem. 2004;279:26748–26753. doi: 10.1074/jbc.M400785200. [DOI] [PubMed] [Google Scholar]
- 34.Lye E, Mirtsos C, Suzuki N, Suzuki S, Yeh WC. The role of interleukin 1 receptor-associated kinase-4 (IRAK-4) kinase activity in IRAK-4-mediated signaling. J Biol Chem. 2004;279:40653–40658. doi: 10.1074/jbc.M402666200. [DOI] [PubMed] [Google Scholar]
- 35.De Nardo D, Nguyen T, Hamilton JA, Scholz GM. Down-regulation of IRAK-4 is a component of LPS- and CpG DNA-induced tolerance in macrophages. Cell Signal. 2009;21:246–252. doi: 10.1016/j.cellsig.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 36.Fraczek J, Kim TW, Xiao H, Yao J, Wen Q, Li Y, Casanova JL, Pryjma J, Li X. The kinase activity of IL-1 receptor-associated kinase 4 is required for interleukin-1 receptor/toll-like receptor-induced TAK1-dependent NFkappaB activation. J Biol Chem. 2008;283:31697–31705. doi: 10.1074/jbc.M804779200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kanakaraj P, Schafer PH, Cavender DE, Wu Y, Ngo K, Grealish PF, Wadsworth SA, Peterson PA, Siekierka JJ, Harris CA, Fung-Leung WP. Interleukin (IL)-1 receptor-associated kinase (IRAK) requirement for optimal induction of multiple IL-1 signaling pathways and IL-6 production. J Exp Med. 1998;187:2073–2079. doi: 10.1084/jem.187.12.2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Swantek JL, Tsen MF, Cobb MH, Thomas JA. IL-1 Receptor-Associated Kinase Modulates Host Responsiveness to Endotoxin. The Journal of Immunology. 2000;164:4301–4306. doi: 10.4049/jimmunol.164.8.4301. [DOI] [PubMed] [Google Scholar]
- 39.Kawagoe T, Sato S, Matsushita K, Kato H, Matsui K, Kumagai Y, Saitoh T, Kawai T, Takeuchi O, Akira S. Sequential control of Toll-like receptor-dependent responses by IRAK1 and IRAK2. Nat Immunol. 2008;9:684–691. doi: 10.1038/ni.1606. [DOI] [PubMed] [Google Scholar]
- 40.Wan Y, Xiao H, Affolter J, Kim TW, Bulek K, Chaudhuri S, Carlson D, Hamilton T, Mazumder B, Stark GR, Thomas J, Li X. Interleukin-1 receptor-associated kinase 2 is critical for lipopolysaccharide-mediated post-transcriptional control. J Biol Chem. 2009;284:10367–10375. doi: 10.1074/jbc.M807822200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suzuki N, Suzuki S, Duncan GS, Millar DG, Wada T, Mirtsos C, Takada H, Wakeham A, Itie A, Li S, Penninger JM, Wesche H, Ohashi PS, Mak TW, Yeh WC. Severe impairment of interleukin-1 and Toll-like receptor signalling in mice lacking IRAK-4. Nature. 2002;416:750–756. doi: 10.1038/nature736. [DOI] [PubMed] [Google Scholar]
- 42.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 43.Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, Crozat K, Sovath S, Han J, Beutler B. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. 2003;424:743–748. doi: 10.1038/nature01889. [DOI] [PubMed] [Google Scholar]
- 44.Oshiumi H, Matsumoto M, Funami K, Akazawa T, Seya T. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat Immunol. 2003;4:161–167. doi: 10.1038/ni886. [DOI] [PubMed] [Google Scholar]
- 45.Pauls E, Nanda SK, Smith H, Toth R, Arthur JS, Cohen P. Two phases of inflammatory mediator production defined by the study of IRAK2 and IRAK1 knock-in mice. J Immunol. 2013;191:2717–2730. doi: 10.4049/jimmunol.1203268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Flannery SM, Keating SE, Szymak J, Bowie AG. Human interleukin-1 receptor-associated kinase-2 is essential for Toll-like receptor-mediated transcriptional and post-transcriptional regulation of tumor necrosis factor alpha. J Biol Chem. 2011;286:23688–23697. doi: 10.1074/jbc.M111.248351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kawagoe T, Sato S, Jung A, Yamamoto M, Matsui K, Kato H, Uematsu S, Takeuchi O, Akira S. Essential role of IRAK-4 protein and its kinase activity in Toll-like receptor-mediated immune responses but not in TCR signaling. J Exp Med. 2007;204:1013–1024. doi: 10.1084/jem.20061523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chiang EY, Yu X, Grogan JL. Immune complex-mediated cell activation from systemic lupus erythematosus and rheumatoid arthritis patients elaborate different requirements for IRAK1/4 kinase activity across human cell types. J Immunol. 2011;186:1279–1288. doi: 10.4049/jimmunol.1002821. [DOI] [PubMed] [Google Scholar]
- 49.Uematsu S, Sato S, Yamamoto M, Hirotani T, Kato H, Takeshita F, Matsuda M, Coban C, Ishii KJ, Kawai T, Takeuchi O, Akira S. Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7- and TLR9-mediated interferon-{alpha} induction. J Exp Med. 2005;201:915–923. doi: 10.1084/jem.20042372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin KM, Hu W, Troutman TD, Jennings M, Brewer T, Li X, Nanda S, Cohen P, Thomas JA, Pasare C. IRAK-1 bypasses priming and directly links TLRs to rapid NLRP3 inflammasome activation. Proc Natl Acad Sci U S A. 2014;111:775–780. doi: 10.1073/pnas.1320294111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fernandes-Alnemri T, Kang S, Anderson C, Sagara J, Fitzgerald KA, Alnemri ES. Cutting edge: TLR signaling licenses IRAK1 for rapid activation of the NLRP3 inflammasome. J Immunol. 2013;191:3995–3999. doi: 10.4049/jimmunol.1301681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang H, Pu J, Wang X, Shen L, Zhao G, Zhuang C, Liu R. IRAK1 rs3027898 C/A polymorphism is associated with risk of rheumatoid arthritis. Rheumatology international. 2013;33:369–375. doi: 10.1007/s00296-012-2379-3. [DOI] [PubMed] [Google Scholar]
- 53.Arcaroli J, Silva E, Maloney JP, He Q, Svetkauskaite D, Murphy JR, Abraham E. Variant IRAK-1 haplotype is associated with increased nuclear factor-kappaB activation and worse outcomes in sepsis. American journal of respiratory and critical care medicine. 2006;173:1335–1341. doi: 10.1164/rccm.200603-341OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kaufman KM, Zhao J, Kelly JA, Hughes T, Adler A, Sanchez E, Ojwang JO, Langefeld CD, Ziegler JT, Williams AH, Comeau ME, Marion MC, Glenn SB, Cantor RM, Grossman JM, Hahn BH, Song YW, Yu CY, James JA, Guthridge JM, Brown EE, Alarcon GS, Kimberly RP, Edberg JC, Ramsey-Goldman R, Petri MA, Reveille JD, Vila LM, Anaya JM, Boackle SA, Stevens AM, Freedman BI, Criswell LA, Pons Estel BA, G. Argentine Collaborative. Lee JH, Lee JS, Chang DM, Scofield RH, Gilkeson GS, Merrill JT, Niewold TB, Vyse TJ, Bae SC, Alarcon-Riquelme ME, B. network. Jacob CO, Moser Sivils K, Gaffney PM, Harley JB, Sawalha AH, Tsao BP. Fine mapping of Xq28: both MECP2 and IRAK1 contribute to risk for systemic lupus erythematosus in multiple ancestral groups. Annals of the rheumatic diseases. 2013;72:437–444. doi: 10.1136/annrheumdis-2012-201851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deng C, Radu C, Diab A, Tsen MF, Hussain R, Cowdery JS, Racke MK, Thomas JA. IL-1 receptor-associated kinase 1 regulates susceptibility to organ-specific autoimmunity. J Immunol. 2003;170:2833–2842. doi: 10.4049/jimmunol.170.6.2833. [DOI] [PubMed] [Google Scholar]
- 56.Jacob CO, Zhu J, Armstrong DL, Yan M, Han J, Zhou XJ, Thomas JA, Reiff A, Myones BL, Ojwang JO, Kaufman KM, Klein-Gitelman M, McCurdy D, Wagner-Weiner L, Silverman E, Ziegler J, Kelly JA, Merrill JT, Harley JB, Ramsey-Goldman R, Vila LM, Bae SC, Vyse TJ, Gilkeson GS, Gaffney PM, Moser KL, Langefeld CD, Zidovetzki R, Mohan C. Identification of IRAK1 as a risk gene with critical role in the pathogenesis of systemic lupus erythematosus. Proc Natl Acad Sci U S A. 2009;106:6256–6261. doi: 10.1073/pnas.0901181106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Trofimova M, Sprenkle AB, Green M, Sturgill TW, Goebl MG, Harrington MA. Developmental and tissue-specific expression of mouse pelle-like protein kinase. J Biol Chem. 1996;271:17609–17612. doi: 10.1074/jbc.271.30.17609. [DOI] [PubMed] [Google Scholar]
- 58.Cao Z, Henzel WJ, Gao X. IRAK: a kinase associated with the interleukin-1 receptor. Science. 1996;271:1128–1131. doi: 10.1126/science.271.5252.1128. [DOI] [PubMed] [Google Scholar]
- 59.Sung MH, Li N, Lao Q, Gottschalk RA, Hager GL, Fraser ID. Switching of the relative dominance between feedback mechanisms in lipopolysaccharide-induced NF-kappaB signaling. Sci Signal. 2014;7:ra6. doi: 10.1126/scisignal.2004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wall EA, Zavzavadjian JR, Chang MS, Randhawa B, Zhu X, Hsueh RC, Liu J, Driver A, Bao XR, Sternweis PC, Simon MI, Fraser ID. Suppression of LPS-induced TNF-alpha production in macrophages by cAMP is mediated by PKA-AKAP95-p105. Sci Signal. 2009;2:ra28. doi: 10.1126/scisignal.2000202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ablasser A, Schmid-Burgk JL, Hemmerling I, Horvath GL, Schmidt T, Latz E, Hornung V. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature. 2013;503:530–534. doi: 10.1038/nature12640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schmid-Burgk JL, Schmidt T, Gaidt MM, Pelka K, Latz E, Ebert TS, Hornung V. OutKnocker: a web tool for rapid and simple genotyping of designer nuclease edited cell lines. Genome research. 2014;24:1719–1723. doi: 10.1101/gr.176701.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Al-Khodor S, Marshall-Batty K, Nair V, Ding L, Greenberg DE, Fraser ID. Burkholderia cenocepacia J2315 escapes to the cytosol and actively subverts autophagy in human macrophages. Cellular microbiology. 2014;16:378–395. doi: 10.1111/cmi.12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Design and TLR ligand response of mouse and human macrophage reporter cell lines for siRNA screening applications.
Fig. S2. Effects of siRNA-mediated gene perturbations across the human and mouse TLR pathways.
Fig. S3. Human and mouse macrophages show both shared and distinct gene dependencies in TLR signaling.
Fig. S4. IRAK4 is required for the responses of human PBMCs to TLR ligands.
Fig. S5. Generation of IRAK4 rescue cell lines from IRAK4 KO IMMs and THP1 cells.
Fig. S6. Expression of kinase-deficient mouse and human IRAK4 in IRAK4 KO mouse IMMs and THP1 cells.
Fig. S7. Analysis of the IRAK1-dependency of human macrophages for TLR responses.
Fig. S8. CRISPR/Cas9-mediated targeting of the human IRAK1 and IRAK2 loci in THP1 cells.
Fig. S9. Expression distributions of IRAK1/Irak1, IRAK2/Irak2, and IRAK4/Irak4 mRNAs in human and mouse GEO datasets.
Table S1. Details of the siRNAs used to target the 126 human and mouse TLR pathway genes.
Table S2. siRNA z-scores from screens of the 126 human and mouse TLR pathway genes.
Table S3. Ranking scores of human and mouse TLR pathway genes from the siRNA screens.
Table S4. Human vs mouse z-score differences across matched TLR ligands and assay readouts.