

Abstract

The replicative polymerases cannot accommodate distortions to the native DNA sequence such as modifications (lesions) to the native template bases from exposure to reactive metabolites and environmental mutagens. Consequently, DNA synthesis on an afflicted template abruptly stops upon encountering these lesions, but the replication fork progresses onward, exposing long stretches of the damaged template before eventually stalling. Such arrests may be overcome by translesion DNA synthesis (TLS) in which specialized TLS polymerases bind to the resident proliferating cell nuclear antigen (PCNA) and replicate the damaged DNA. Hence, a critical aspect of TLS is maintaining PCNA at or near a blocked primer/template (P/T) junction upon uncoupling of fork progression from DNA synthesis by the replicative polymerases. The single-stranded DNA (ssDNA) binding protein, replication protein A (RPA), coats the exposed template and might prohibit diffusion of PCNA along the single-stranded DNA adjacent to a blocked P/T junction. However, this idea had yet to be directly tested. We recently developed a unique Cy3-Cy5 Forster resonance energy transfer (FRET) pair that directly reports on the occupancy of DNA by PCNA. In this study, we utilized this FRET pair to directly and continuously monitor the retention of human PCNA at a blocked P/T junction. Results from extensive steady state and pre-steady state FRET assays indicate that RPA binds tightly to the ssDNA adjacent to a blocked P/T junction and restricts PCNA to the upstream duplex region by physically blocking diffusion of PCNA along ssDNA.
During S phase of the cell cycle, genomic DNA must be faithfully copied in a short time period. To achieve the high degree of processivity required for efficient DNA replication, the eukaryotic replicative DNA polymerases (pols), δ and ε, anchor to PCNA sliding clamp rings encircling the DNA. The highly conserved structure of the PCNA ring has a central cavity that is sufficiently large to encircle double-stranded DNA and slide freely along it. Thus, such an association effectively tethers these pols to DNA, substantially increasing the extent of continuous replication.1 However, the replicative pols have very stringent polymerase domains as well as 3′–5′ exonuclease (“proofreading”) domains and, thus, cannot accommodate distortions to the native DNA sequence.2−5 Prominent examples of these are modifications (lesions) to the native template bases from exposure to reactive metabolites and environmental mutagens such as ultraviolet (UV) radiation. Consequently, DNA synthesis on the afflicted template abruptly stops upon encountering these lesions. Such arrests may be overcome by translesion DNA synthesis (TLS), a DNA damage tolerance (DDT) pathway in which a specialized TLS pol binds to the resident PCNA and replicates the damaged DNA.6 Characterized by a more open polymerase active site, the lack of an associated proofreading activity, and one or more PCNA binding domains, TLS pols are able to support stable, yet potentially erroneous, nucleotide incorporation opposite damaged templates.2−5 Extensive studies of UV-irradiated cells from various eukaryotes suggest that TLS proceeds through at least two modes that are spatially and temporally distinct and each dependent on PCNA.
DNA synthesis by a replicative pol abruptly stops upon encountering a lesion it cannot accommodate, such as a cyclobutane pyrimidine dimer (CPD), the major DNA lesion resulting from exposure to UV irradiation. However, the replication fork progresses onward, exposing long stretches of the damaged template before eventually stalling. The single-stranded DNA (ssDNA) binding protein, replication protein A (RPA), coats the exposed ssDNA, protecting it from cellular nucleases and preventing formation of alternative DNA structures such as DNA hairpins.7 One or more TLS pols may bind to the resident PCNA and extend the blocked primer beyond the lesion, allowing DNA synthesis by the replicative pol to resume and, hence, progression of the replication fork to restart. In this “on the fly” mode, replication fork restart requires TLS.8−10 Alternatively, the damaged template may be reprimed, leaving behind a RPA-coated ssDNA gap containing the offending lesion. DNA synthesis by the replicative pols resumes from the nascent primer/template (P/T) junction, allowing fork restart, and the gap is “filled in” behind the restarted fork (i.e., postreplicatively). In this, “postreplicative gap filling” mode, replication fork restart requires repriming of the damaged template rather than TLS.11−18 The seminal studies on the cellular response to UV-induced lesions encountered during S phase suggested that human TLS occurs predominantly by postreplicative gap filling.15,16,19−27 Recent in vivo studies have provided staunch supporting evidence.13,14,28 In particular, continuous DNA chain elongation in human cells lacking pol η was severely blocked in response to UV irradiation; however, replication fork speed was only slightly reduced, and persistent replication fork arrest was not observed.13 Pol η is a Y-family TLS pol responsible for the error-free replication of CPDs in human cells.29 Thus, replication forks blocked by UV-induced DNA lesions are restarted predominantly by repriming of the damaged template (postreplicative gap filling) rather than TLS (on the fly TLS). Hence, TLS occurs within postreplicative gaps left behind restarted replication forks.13
Both modes of TLS described above require PCNA encircling damaged DNA. Hence, a critical aspect of postreplicative gap filling is maintaining PCNA at or near blocked P/T junctions during S phase. This may be achieved by (1) inhibiting the unloading of PCNA from DNA, (2) prohibiting diffusion of PCNA along DNA, (3) promoting the loading of PCNA onto DNA, or (4) a combination of one or more of these possibilities. Recent in vivo evidence suggests that enzyme-catalyzed unloading of PCNA from a P/T junction will not occur until the primer is completely extended and ligated to the downstream duplex region.30 A PCNA ring may rapidly diffuse along the newly synthesized dsDNA behind a progressing replication fork (D = 2.24 × 107 bp2/s).31 However, subnucleosomes are rapidly reassembled on this nascent duplex DNA within 250 bp of a progressing replication fork.32,33 Furthermore, high-affinity transcription factors essential for the establishment of chromatin structure also rebind to nascent DNA duplexes immediately after replication fork passage.34 Thus, diffusion of PCNA rings along the dsDNA adjacent to blocked P/T junctions is likely restricted during S phase by physical blocks, i.e., “protein roadblocks”. However, PCNA may diffuse along the adjacent ssDNA, which varies greatly in length and may be as long as 20 kb in human cells.11,35 Furthermore, the majority of postreplicative gaps generated at the onset of S phase in response to UV irradiation persist through S phase into G2/M phase.12,14,17,18,36 If PCNA can vacate a blocked P/T junction in this manner, new PCNA rings are not envisioned to be continuously reloaded at postreplicative gaps as PCNA is limiting37 and the clamp loader, replication factor C (RFC), responsible for loading PCNA onto DNA travels with progressing replication forks during S phase in human cells.37,38 Thus, diffusion of PCNA along ssDNA within postreplicative gaps must be prohibited to maintain PCNA at blocked P/T junctions.
Human RPA binds to ssDNA with an extremely high affinity (less than picomolar) at physiological ionic strength and might prohibit diffusion of PCNA along ssDNA by serving as a protein roadblock.39,40 However, this idea had yet to be directly tested. We recently developed a unique Cy3-Cy5 FRET pair that directly reports on the retention of human PCNA on DNA.41 This FRET pair was utilized in this study to monitor the effect of RPA on the occupancy of various P/T DNA substrates by PCNA under steady state and pre-steady state conditions. Results from these continuous assays indicate that RPA binds sufficiently tight to ssDNA adjacent to a P/T junction such that it restricts PCNA to the upstream duplex region by physically blocking diffusion of PCNA along ssDNA.
Experimental Procedures
Oligonucleotides
Oligonucleotides were synthesized by Integrated DNA Technologies (Coralville, IA) and purified on denaturing polyacrylamide gels. Concentrations were determined from the absorbance at 260 nm using the calculated extinction coefficients. For annealing the DNA substrates (Figure 1), the primer strands were mixed with a 1.1-fold excess of the corresponding complementary template strands in 1× annealing buffer [10 mM Tris-HCl (pH 8.0), 100 mM NaCl, and 1 mM EDTA], heated to 95 °C for 5 min, and allowed to cool slowly to room temperature.
Figure 1.

DNA substrates utilized in this study. The sequence of the primer (29 nucleotides) and template (62 nucleotides) strands comprising each DNA substrate are identical. When annealed, each DNA duplex mimics a blocked P/T junction.42 The size of the double-stranded P/T region (29 bp) is in agreement with the requirements for assembly of a PCNA ring onto DNA by RFC.41,45 The single-stranded DNA (ssDNA) region (33 nucleotides) adjacent to the 3′-end of the P/T junction is consistent with the footprint of RPA (22–30 nucleotides).39 When prebound to Neutravidin, the biotin attached to the 3′-end of a template strand prevents loaded PCNA from sliding off the dsDNA, i.e., duplex, end of the substrate. A Cy3 dye attached to the 5′-end of a primer strand serves as a FRET donor.
Proteins
Recombinant human proteins expressed and purified from Escherichia coli were utilized for all experiments within this study. Wild-type PCNA and a mutant PCNA that can be site-specifically labeled with a Cy5 dye were purified as described previously.41 The mutant PCNA was labeled with Cy5-maleimide and checked for activity as described previously.41 All concentrations of PCNA indicated in the text refer to the concentration of the PCNA homotrimer. Exonuclease-deficient pol δ (simply termed pol δ hereafter) was prepared as previously described.42,43 The concentrations of the four-subunit DNA pol δ complex were expressed as the concentration of the p125 subunit.42 A truncated form of RFC (hRFCp140ΔN555) described previously was used in all reported studies and is simply termed RFC hereafter.41 RPA was prepared as previously described, and the concentration was determined from the reported extinction coefficient.44
Fluorescence Microscopy
All experiments were performed at room temperature (23 ± 2 °C) in 1× replication assay buffer [25 mM TrisOAc (pH 7.7), 10 mM Mg(OAc)2, and 125 mM KOAc] supplemented with 0.1 mg/mL BSA and 1 mM DTT, and the final ionic strength was adjusted to 200 mM by the addition of appropriate amounts of KOAc. For steady state fluorescence, measurements were taken in Jobin Yvon fluoromax-4 fluorimeter. The assay solution contained Cy3-labeled P/T DNA (100 nM Cy3P/T or Cy3P/BioT), Neutravidin (400 nM), ATP (1 mM), and RPA (0–242 nM). Cy5-PCNA (110 nM homotrimer) and RFC (0–110 nM) were then sequentially added to this solution. Two minutes after the addition of RFC, the solution was excited at 514 nm and the fluorescence emission spectrum was recorded from 530 to 750 nm. The FRET signal (I665/I561) was calculated by dividing the fluorescence emission intensity at 665 nm (I665, Cy5 FRET acceptor fluorescence emission maximum) by the fluorescence emission intensity at 561 nm (I561, Cy3 FRET donor fluorescence emission maximum). Spectra were recorded every minute until the FRET signal maintained a constant value for at least 2 min. Data points within this region were averaged to obtain the final FRET value. For pre-steady state fluorescence, studies were performed on an Applied Photophysics SX20 stopped-flow machine equipped with a fluorescence detector. All experiments contained Neutravidin at a 4-fold excess over the total concentration of DNA regardless of whether the DNA was labeled with biotin. Each syringe contained 1 mM ATP such that the final concentration of ATP upon mixing was 1 mM. The final concentrations of all other reaction components are indicated in the respective figure legends. FRET was monitored by exciting the Cy3 donor at 514 nm and following the resulting FRET from the Cy5 acceptor using a 645 nm cutoff filter (Andover Corp., Salem, NH). FRET traces were recorded over 10–60 s by collecting 2000 time points over the initial 10 s and 833 time points over the remaining 50 s. All traces were analyzed using Kaleidagraph. Conditions for each experiment are detailed in the respective figure legends.
Results
RPA Stabilizes PCNA at a P/T Junction
To study the dynamics of PCNA during TLS, we utilized FRET to directly monitor the retention of PCNA at a P/T junction. A P/T DNA substrate in agreement with the requirements for assembly of a PCNA ring onto DNA by RFC was labeled with a 5′-Cy3 FRET donor and a 3′-biotin label.41,45 This substrate, herein termed Cy3P/BioT, resembles a blocked P/T junction (Figure 1). The 3′-biotin label in complex with Neutravidin serves as a protein roadblock and prevents the loaded clamp from sliding off the duplex end of the DNA; a ssDNA region directly abuts the P/T junction and is long enough (33 nucleotides) to accommodate a single RPA molecule. PCNA was site-specifically labeled with a Cy5 FRET acceptor on the “back” face of the PCNA ring, as previously described.41 The Cy5 dye label has no effect on the interaction of PCNA with RFC.41 The Cy3P/BioT DNA substrate was tested for PCNA loading by monitoring the steady state FRET signal in the presence of RPA (Figure 2A). Cy5-PCNA can be excited through FRET from Cy3P/BioT only when the two dye labels are in close proximity of each other (less than ∼10 nm). This is indicated by an increase in the fluorescence emission intensity at 665 nm (Cy5 FRET acceptor fluorescence emission maximum, I665) and a concomitant decrease in the fluorescence emission intensity at 561 nm (Cy3 FRET donor fluorescence emission maximum, I561). A FRET signal was observed only when both RFC and ATP were present (Figure 2B), indicating that RFC loads Cy5-PCNA onto the Cy3P/BioT DNA substrate in an ATP-dependent manner and Cy5-PCNA is stable on the Cy3/BioT DNA substrate at equilibrium.
Figure 2.
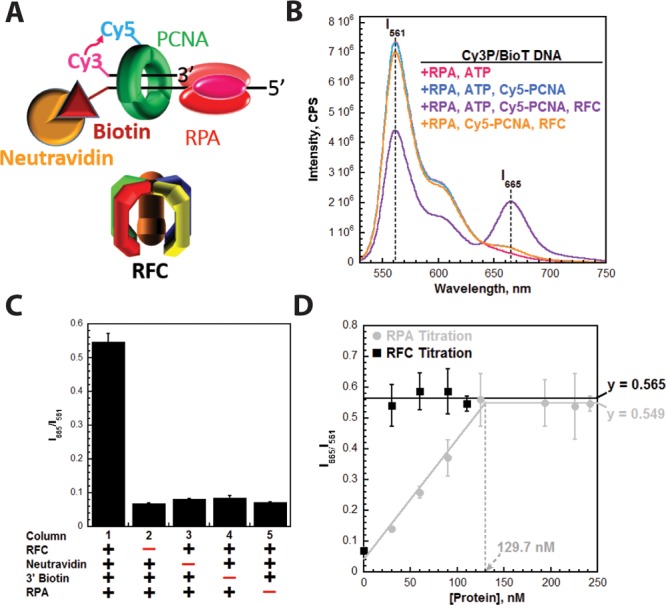
Monitoring the retention of PCNA on DNA through FRET. (A) Schematic representation of PCNA encircling a P/T junction bound by RPA. When loaded onto P/T DNA by RFC, the Cy5 FRET acceptor on PCNA faces the Cy3 FRET donor on the P/T DNA. (B) Fluorescence emission spectra in the presence of RPA. Cy3P/BioT DNA (100 nM), Neutravidin (400 nM), ATP (1 mM), and RPA (242 nM) were pre-equilibrated at 25 °C. Cy5-PCNA (110 nM homotrimer) and RFC (110 nM) were sequentially added; the solution was excited at 514 nm, and the fluorescence emission spectrum was recorded from 530 to 750 nm. The fluorescence emission intensities at 665 nm (Cy5 FRET acceptor fluorescence emission maximum, I665) and 561 nm (Cy3 FRET donor fluorescence emission maximum, I561) are indicated. Cy5-PCNA can be excited through FRET from Cy3P/BioT only when the two dyes are in close proximity of each other (less than ∼10 nm). This is indicated by an increase in I665 and a concomitant decrease in I561. (C) Characterization of the steady state FRET signal. Cy5-PCNA was assembled onto the Cy3P/BioT DNA substrate as in panel A with various components omitted, and the FRET signal (I665/I561) was measured. As a control, RFC was omitted (column 2). (D) Titrations of the steady state FRET signal. The Cy3P/BioT DNA substrate (100 nM with 400 nM Neutravidin) was either saturated with RFC (110 nM) and titrated with RPA (0–242 nM) (●) or saturated with RPA (242 nM) and titrated with RFC (30–110 nM) (■). Results are plotted vs the concentration of the respective titrant. When RPA was titrated, the FRET signal increased linearly and then plateaued. At the break point, the concentration of RPA (129.7 nM) is roughly equivalent to the concentration of RPA binding sites within the assay (120 nM). When RFC was titrated, the FRET signal remained constant at a level (0.565) equivalent to that observed at saturating concentrations of RFC and RPA (0.549).
Next, we characterized the steady state FRET signal (Figure 2C). In the presence of RFC, Neutravidin, and RPA, a robust FRET signal is observed (column 1). As a control, RFC was omitted to establish the zero FRET state (column 2). A FRET signal was not observed when Neutravidin (column 3) was omitted. Furthermore, a FRET signal was not observed in the presence of Neutravidin on the Cy3P/T DNA substrate (column 4) that lacks a 3′-biotin label (Figure 1). Together, this confirms that the Neutravidin/biotin complex alone prohibits Cy5-PCNA from diffusing off the duplex end of the Cy3P/BioT substrate and argues against RPA holding RFC at the P/T junction with PCNA.46,47 Rather, RFC releases Cy5-PCNA onto the Cy3P/BioT DNA substrate, in agreement with previous observations.41 A FRET signal was not observed in the absence of RPA (column 5), and addition of excess pol δ (200 nM) did not compensate for the lack of RPA (Figure S1). Under the conditions of the assay (200 mM ionic strength, 25 °C), pol δ binds to PCNA encircling a P/T junction with significant affinity (<10 nM) and will transiently immobilize PCNA on DNA in the absence of DNA synthesis.41,43 However, pol δ alone will not promote the retention of Cy5-PCNA on the Cy3P/BioT DNA substrate at equilibrium unless the transition state for DNA synthesis (i.e., dNTP insertion) is stabilized (Figure S1), in agreement with previous observations.48 In contrast, RPA binds to ssDNA with extremely high affinity (less than picomolar) under the same experimental conditions.39 RPA does not contain a PCNA binding domain, and a direct interaction between RPA and PCNA has yet to be reported. Altogether, this suggests that RPA is not required for RFC-catalyzed loading of Cy5-PCNA onto the Cy3P/BioT DNA substrate. Rather, tight binding of RPA to the ssDNA region of the Cy3P/BioT DNA substrate is required to stabilize Cy5-PCNA at the P/T junction, leading to a robust steady state FRET signal. To test this, we performed a titration of the steady state FRET signal (Figure 2D), as described below.
The concentrations of the Cy3P/BioT DNA substrate (100 nM), RFC (110 nM), and Cy5-PCNA (110 nM homotrimer) were held constant; increasing concentrations of RPA were added (0–242 nM), and the FRET signal (I665/I561) was monitored. RFC and PCNA were present in slight excess (10%) to ensure saturation of the DNA. At a physiological ionic strength (200 mM), RPA has an occluded site size of 24 nucleotides. Hence, a single RPA molecule may bind to the ssDNA region (33 nucleotides) within the Cy3P/BioT DNA substrate. To ensure that all of the Cy3-labeled primer is annealed as duplex DNA, a 10% excess of the biotin-labeled template was utilized during annealing of the Cy3P/BioT DNA substrate. As ssDNA, this 62-mer template strand may bind two RPA molecules. Thus, at 100 nM Cy3P/BioT DNA, the concentration of RPA binding sites is 120 nM (100 nM Cy3P/BioT + 10 nM single-stranded template × 2 = 120 nM). As observed in Figure 2D, the FRET signal increased linearly with RPA concentration up to 125 nM and plateaued thereafter. At the break point, the concentration of RPA (129.7 nM) is roughly equal to the concentration RPA binding sites in the assay (120 nM). This suggests that tight binding of a single RPA molecule to the ssDNA region of the Cy3P/BioT DNA substrate stabilizes a Cy5-PCNA ring at the P/T junction by prohibiting diffusion of Cy5-PCNA off the ssDNA end.
As mentioned above, previous indirect studies suggested that RPA holds RFC at a P/T junction with PCNA, prohibiting diffusion of PCNA in both directions.46,47 In other words, an RPA·RFC·DNA complex prevents Cy5-PCNA from sliding off the ssDNA end of the Cy3P/BioT DNA substrate. To test this, the steady state FRET signal was titrated with RFC. The concentrations of the Cy3P/BioT DNA substrate (100 nM), Cy5-PCNA (110 nM homotrimer), and RPA (242 nM) were held constant; increasing concentrations of RFC were added (0–110 nM), and the FRET signal was monitored. After loading Cy5-PCNA onto the Cy3P/BioT DNA substrate, RFC will not turn over (i.e., act catalytically) if it is held at the P/T junction by RPA. Hence, limiting concentrations of RFC will not load all Cy5-PCNA onto the Cy3P/BioT DNA substrate. On the other hand, if RFC is released into solution after loading Cy5-PCNA and RPA alone prevents Cy5-PCNA from sliding off the ssDNA end of the Cy3P/BioT DNA substrate, RFC will act catalytically. As shown in Figure 2D, the steady state FRET signal was independent of RFC concentration and remained constant at a level (0.565) equivalent to that observed at saturating concentrations of RFC and RPA (0.549), indicating that limiting concentrations of RFC can load Cy5-PCNA onto all of the Cy3P/BioT DNA substrate. Thus, RFC is released into solution after loading Cy5-PCNA, and RPA alone prevents Cy5-PCNA from sliding off the ssDNA end of the Cy3P/BioT DNA substrate. Altogether, these steady state FRET studies suggest that RPA prevents diffusion of PCNA along the ssDNA adjacent to a P/T junction.
RPA Prohibits Diffusion of PCNA along ssDNA
To monitor the dynamics of PCNA on DNA prior to equilibrium (i.e., pre-steady state), we preassembled a RFC·ATP·Cy5-PCNA complex and rapidly mixed it with ATP and a Cy3-labeled DNA substrate in a stopped-flow apparatus (Figure 3A). In the absence of RPA, a time course of the FRET traces for the Cy3P/T and Cy3P/BioT DNA substrates looked almost identical. Each displayed two distinct phases (Figure 3B). First, a very rapid, single-exponential increase in the FRET signal was observed (kinc,1), confirming that RPA is not required for RFC-catalyzed loading of Cy5-PCNA onto the Cy3-labeled DNA substrates. This was followed by a rapid, single-exponential decrease in FRET (kdec). After ∼1.0–1.5 s, the FRET signals stabilized and remained flat for up to 10 s (Figure 3C). Under the conditions of the assay, a FRET signal is not observed on either Cy3-labeled DNA substrate at equilibrium (Figure 2C). Hence, the flat regions observed in Figure 3 indicate that the reactions have reached equilibrium at the zero FRET state. Altogether, these FRET studies indicate that Cy5-PCNA initially loaded onto the Cy3-labeled DNA substrates rapidly dissociates back into solution in the absence of RPA and subsequent loadings are not observed. Thus, a Cy5-PCNA·Cy3-labeled DNA complex (i.e., loaded PCNA) is disfavored at equilibrium in the absence of RPA.
Figure 3.

RFC releases PCNA onto the duplex region of P/T DNA. Each FRET trace represents the average of at least eight shots. For comparison, all FRET traces are normalized to the same starting value (at t = 0.005 s). (A) Schematic representation of the experimental procedure for panel B. (B) Cy5-PCNA (110 nM homotrimer) was preincubated with RFC (110 nM) and ATP. This preformed RFC·Cy5-PCNA·ATP complex was mixed with Cy3-labeled DNA (100 nM Cy3P/BioT or Cy3P/T) and ATP in a stopped-flow instrument, and the FRET signal was followed. The loading traces for the Cy3P/T (gray) and Cy3P/BioT DNA (black) substrates are shown, and each was fit to a double-exponential equation. The calculated rate constants are reported for each. (C) Extended time courses (10 s) for each FRET trace from panel B. (D) Schematic representation of the experimental procedure for panel E. (E) The experiments depicted in panel B were repeated except the Cy3-labeled DNA was preincubated with excess RPA (242 nM). The loading trace for the Cy3P/T DNA substrate (gray) was fit to a double-exponential equation. The loading trace for the Cy3P/BioT DNA substrate (black) was fit to a single exponential and a linear phase. The kinetic values calculated from the fits for each trace are indicated. (F) Extended time courses (10 s) for each FRET trace from panel E.
PCNA diffuses along double-stranded DNA extremely fast (D = 2.24 × 107 bp2/s).31 Thus, Cy5-PCNA will instantly diffuse off the unblocked duplex end (29 bp) of the Cy3P/T DNA substrate once RFC releases the closed sliding clamp ring. As such, this process will be rate-limited by a preceding and obligate kinetic step along the clamp loading pathway that is much slower than PCNA diffusion. Hence, kdec measured for the Cy3P/T DNA substrate may describe ATP hydrolysis by RFC, the concomitant closure of the sliding clamp ring, or RFC release of the closed PCNA ring onto the duplex DNA region. On the Cy3P/BioT substrate, the Neutravidin/biotin complex prohibits Cy5-PCNA from diffusing off the duplex end (Figure 2C). However, the rate constant for the disappearance of the FRET signal on the Cy3P/BioT DNA substrate (kdec = 3.40 ± 0.0561 s–1) is quite similar to that observed on the Cy3P/T DNA substrate (kdec = 4.84 ± 0.379 s–1). This suggests that Cy5-PCNA initially loaded onto these Cy3-labeled P/T DNA substrates dissociates back into solution via the same pathway, diffusion of PCNA along the DNA. Next, we analyzed the effect of RPA on the pre-steady state FRET traces (Figure 3D).
For the Cy3P/T DNA substrate, the FRET trace in the presence of RPA (Figure 3E) appeared similar to that observed in the absence of RPA (Figure 3B), a very rapid, single-exponential increase in FRET (kinc,1 = 20.9 ± 0.337 s–1) followed by a rapid, single-exponential decrease in FRET (kdec = 9.87 ± 0.464 s–1) down to the zero FRET state where it remained flat for up to 10 s (Figure 3F). This confirms that RPA does not prohibit diffusion of PCNA off the duplex end of this DNA substrate and, hence, does not hold RFC at a P/T junction with PCNA.46,47 Rather, RFC releases the closed sliding clamp ring onto the Cy3P/T DNA substrate after which Cy5-PCNA immediately diffuses off the duplex end back into solution, and subsequent loadings are not observed. The FRET increase (kinc,1 = 20.9 ± 0.337 s–1) was similar to that observed in the absence of RPA [kinc,1 = 28.5 ± 0.293 s–1 (Figure 3B)]. Interestingly, the FRET decrease is ∼2-fold faster in the presence of RPA (kdec = 9.87 ± 0.464 s–1) compared to that observed in the absence of RPA [kdec = 4.84 ± 0.379 s–1 (Figure 3B)]. As discussed above, this phase represents the rate-limiting step for diffusion of Cy5-PCNA off the duplex end of the Cy3P/T DNA substrate. Thus, RPA may promote ATP hydrolysis by RFC (and concomitant closure of the sliding clamp ring) or release of the closed PCNA ring from RFC.
On the Cy3P/BioT DNA substrate, the Neutravidin/biotin complex prohibits Cy5-PCNA from diffusing off the duplex end. The FRET signal observed on this P/T DNA substrate at equilibrium requires RPA (Figure 2), suggesting that RPA prohibits diffusion of Cy5-PCNA along the ssDNA adjacent to the P/T junction. According to this model, a decrease in FRET should not be observed for the Cy3P/BioT DNA substrate when RPA is present in the pre-steady state FRET experiments depicted in Figure 3D. As shown in Figure 3E, FRET rapidly increased in the presence of RPA (kinc,1 = 26.3 ± 3.24 s–1) in a manner similar to that observed in the absence of RPA [kinc,1 = 21.2 ± 2.51 s–1 (Figure 3B)]. Such behavior agrees with that observed for the Cy3P/T DNA substrate and, hence, reaffirms that RPA is not required for RFC-catalyzed loading of Cy5-PCNA onto the Cy3-labeled DNA substrates. Interestingly, the rapid FRET increase observed for the Cy3P/BioT DNA substrate in the presence of RPA is followed by what appears to be a linear increase in FRET [slope of (0.153 ± 8.47) × 10–4 V/s] over the remainder of the time course (Figure 3F). This suggests that (1) RPA prohibits diffusion of Cy5-PCNA off the ssDNA end of the Cy3P/BioT DNA substrate and (2) upon release from RFC, Cy5-PCNA repositions to an equilibrium FRET state in which the Cy5 label on PCNA is closer to the Cy3-labeled duplex end of the Cy3P/BioT DNA substrate. However, the concentrations of Cy5-PCNA (110 nM homotrimer) and RFC (110 nM) are slightly greater than that of the Cy3P/BioT DNA substrate (100 nM). Thus, it cannot be concluded unequivocally that the linear increase in FRET observed in Figure 3F in the presence of RPA does not simply reflect the slow loading of more than one Cy5-PCNA onto the Cy3P/BioT DNA substrate. Furthermore, the absence of a FRET signal at equilibrium when both ends of the Cy3-labeled DNA substrates are not blocked (Figures 2 and 3) is quite puzzling given the rapid FRET increase (kinc,1) observed in the pre-steady state assays upon mixing (Figure 3). To clarify these ambiguities, we studied the loading of Cy5-PCNA onto the Cy3-labeled DNA substrates in greater detail, as described below.
First, the assays depicted in Figure 3D were repeated with excess Cy3-labeled DNA. RFC (100 nM) was preincubated with Cy5-PCNA (100 nM homotrimer) and ATP (Figure 4A). RFC binds tightly to PCNA (KD = 0.2 nM) in the presence of ATP and opens the PCNA ring for assembly on P/T DNA.41,49 This process requires only ATP binding, not hydrolysis.49,50 The preassembled RFC·ATP·Cy5-PCNA complex was then rapidly mixed with a 2-fold excess of Cy3-labeled DNA (200 nM Cy3P/T or Cy3P/BioT), saturating RPA (285 nM), and ATP in a stopped-flow apparatus, and the FRET was monitored over an extended time course [60 s (Figure 4B)]. Under these conditions, the appearance of a FRET signal after mixing will be rate-limited by the binding of the preassembled RFC·ATP·Cy5-PCNA complex to the Cy3-labeled DNA. Thus, kinc,1 is a second-order rate constant describing a bimolecular association and, hence, should increase when the concentration of Cy3-labeled DNA is increased. Indeed, kinc,1 was similar for the Cy3P/T (39.8 ± 8.00 s–1) and Cy3P/BioT (42.1 ± 6.45 s–1) DNA substrates, and each value was significantly elevated compared to the corresponding values observed at 100 nM Cy3-labeled DNA (Figure 3). On the Cy3P/T DNA substrate lacking a 3′-biotin label, the fast FRET increase was followed by a rapid, single-exponential decrease in FRET (kdec = 6.80 ± 0.351 s–1) down to the zero FRET state where it remained flat for up to 60 s (Figure 4B). This agrees with that observed in Figure 3E (kdec = 9.87 ± 0.464 s–1) and reaffirms that RPA does not hold RFC at the P/T junction with PCNA.46,47 Rather, RFC releases the closed sliding clamp ring onto the Cy3P/T DNA substrate after which Cy5-PCNA immediately diffuses off the duplex end back into solution, and subsequent loadings are not observed. On the Cy3P/BioT DNA substrate in which the Neutravidin/biotin complex prohibits diffusion of Cy5-PCNA off the duplex end, the fast FRET increase was followed by a slow, single-exponential increase in FRET (kinc,2 = 0.0232 ± 0.00191 s–1) over the remainder of the time course such that the total amplitude (AT) for the FRET increase was 3.40 ± 0.132. This confirms that upon release from RFC onto the Cy3P/BioT DNA substrate, Cy5-PCNA repositions to an equilibrium FRET state in which the Cy5 label on PCNA is closer to the Cy3-labeled duplex end.
Figure 4.

RPA prohibits diffusion of PCNA along ssDNA. Each FRET trace represents the average of at least seven shots. For comparison, all FRET traces are normalized to the same starting value (at t = 0.005 s). (A) Schematic representation of the experimental procedure for panel B. (B) Cy5-PCNA (100 nM homotrimer) was preincubated with RFC (100 nM) in the presence of ATP. This preformed RFC·Cy5-PCNA·ATP complex was mixed with Cy3-labeled DNA (200 nM Cy3P/BioT or Cy3P/T), RPA (285 nM), and ATP in a stopped-flow instrument, and the FRET signal was followed. The loading traces for Cy3P/T (gray) and Cy3P/BioT (black) DNA substrates are shown, and each was fit to a double-exponential equation. The calculated rate constants are reported for each. (C) Initial 3 s of the FRET traces from panel B. (D) Schematic representation of the experimental procedure for panel E. (E) Cy5-PCNA (100 nM homotrimer) was preincubated with Cy3-labeled DNA (200 nM Cy3P/BioT or Cy3P/T), RPA (285 nM), and ATP. This solution was mixed with RFC (100 nM) and ATP in a stopped-flow instrument, and the FRET signal was followed. The loading trace for the Cy3P/BioT DNA substrate (black) was fit to a double-exponential equation, and the calculated rate constants are reported. The loading trace for the Cy3P/T DNA substrate (gray) did not change over time. (F) Initial 3 s of the FRET traces from panel E.
Next, these experiments were repeated with identical concentrations except the order of addition was rearranged (Figure 4D). Cy3-labeled DNA, saturated with RPA, was preincubated with ATP and Cy5-PCNA. This solution was then rapidly mixed with RFC and ATP. Under these conditions, RFC must first bind to the Cy5-PCNA ring and open it for assembly onto the Cy3-labeled DNA substrates. This process may be decelerated by nonproductive binding of the RFC·ATP complex to the Cy3-labeled DNA or free RPA.46 On the Cy3P/BioT DNA substrate, the slow increase in FRET (kinc,2 = 0.0203 ± 0.000335 s–1) as well as the total amplitude for the FRET increase (AT = 3.37 ± 0.625) were unaffected (Figure 4E). Together, this further validates that RPA prohibits diffusion of Cy5-PCNA along the ssDNA adjacent to the P/T junctions and confirms that kinc,2 describes the repositioning of loaded Cy5-PCNA to an equilibrium FRET state after release from RFC rather than additional loading of Cy5-PCNA. Furthermore, the initial increase in FRET (kinc,1) was reduced to 0.134 ± 0.0104 s–1, more than 300-fold slower than that observed in Figure 4B (42.1 ± 6.51 s–1) when the RFC·ATP·Cy5-PCNA complex was assembled prior to mixing. This indicates that loading of free Cy5-PCNA onto the Cy3-labeled DNA substrates and, hence, the initial increase in FRET (kinc,1) are now rate-limited by a kinetic step along the clamp loading pathway that occurs prior to and much slower than binding of the RFC·ATP·Cy5-PCNA complex to the Cy3-labeled DNA substrates. If so, then a FRET signal should not be observed under these conditions for the Cy3P/T DNA substrate where dissociation of Cy5-PCNA into solution (kdec = 6.80 ± 0.351 s–1) is more than 50-fold faster than loading of Cy5-PCNA onto Cy3-labeled DNA (kinc,1 = 0.134 ± 0.0104 s–1). Indeed, a FRET signal is not observed on the Cy3P/T DNA substrate when the RFC·ATP·Cy5-PCNA complex is not preassembled (Figure 4E). This provides a likely explanation for the absence of FRET changes after the initial loading of Cy5-PCNA onto a Cy3-labeled DNA substrate in which both ends are not blocked (Figures 3 and 4). When the RFC·ATP·Cy5-PCNA complex is preassembled, the initial loading of Cy5-PCNA (FRET increase; kinc,1 = 39.8 ± 8.00 s–1) is ∼6-fold faster than dissociation of Cy5-PCNA from the Cy3P/T DNA substrate (FRET decrease; kdec = 6.80 ± 0.351 s–1). Thus, an increase in FRET is observed, and the FRET signal has a finite lifetime. In subsequent loadings (i.e., turnovers) where RFC, ATP, and Cy5-PCNA are free in solution, dissociation of Cy5-PCNA from the Cy3P/T DNA substrate (FRET decrease; kdec = 6.80 ± 0.351 s–1) is more than 50-fold faster than the loading of free Cy5-PCNA (FRET increase; kinc,1 = 0.134 ± 0.0104 s–1). Hence, Cy5-PCNA immediately dissociates from the Cy3P/T DNA substrate after loading such that changes in FRET and, hence, a FRET signal are not observed after the initial loading. To confirm this, we directly monitored RFC turnover on RPA-bound DNA as described below.
RFC Slowly Dissociates into Solution after Loading PCNA onto a RPA-coated P/T Junction
Results from the experiments described above indicate that Cy5-PCNA repositions to an equilibrium FRET state after release from RFC. Hence, this repositioning is rate-limited by a slow kinetic step (kinc,2) in the clamp loading pathway that occurs after ATP hydrolysis by RFC, concomitant closure of the PCNA ring, and RFC release of PCNA onto DNA. Given the rapid diffusion constant of human PCNA on duplex DNA (D = 2.24 × 107 bp2/s),31 repositioning (kinc,2) is not rate-limited by diffusion of Cy5-PCNA along the duplex region of the CyP/BioT DNA substrate. The human clamp loader binds directly to RPA in the absence of ATP, and such an interaction enhances the retention of RFC on RPA-bound ssDNA.46,48 Thus, kinc,2 may describe the slow dissociation of RFC (or an RFC·ADP complex) from an RPA-bound P/T junction after loading PCNA. If so, RFC turnover will be rate-limited by kinc,2. To test this, we monitored the loading of Cy5-PCNA onto the Cy3P/BioT DNA substrate under various conditions (Figure 5). First, the RFC·ATP·Cy5-PCNA complex was preassembled and rapidly mixed with Cy3P/BioT DNA, saturating RPA, and ATP (Figure 5A). Under these conditions, all Cy5-PCNA (180 nM homotrimer) will be loaded onto the Cy3P/BioT DNA substrate (200 nM) by RFC (180 nM) and stabilized by RPA (285 nM). An extended time course (Figure 5B) displayed two distinct FRET increases as expected (kinc,1 = 46.7 ± 3.30 s–1, and kinc,2 = 0.0305 ± 0.00380 s–1) and a total amplitude (AT) of 5.58 ± 0.644. Next, these experiments were repeated by adding unlabeled P/BioT DNA (200 nM) and saturating RPA (285 nM) to the RFC·ATP·Cy5-PCNA complex prior to mixing (Figure 5C). Under these conditions, all Cy5-PCNA (180 nM homotrimer) will be loaded onto the unlabeled P/BioT DNA substrate (200 nM) by RFC (180 nM) and stabilized by RPA prior to mixing. In the absence of RFC-catalyzed unloading of PCNA and diffusion of PCNA off the DNA ends, the only pathway for dissociation of Cy5-PCNA from the unlabeled P/BioT DNA substrate is through spontaneous opening of the subunit–subunit interfaces within the PCNA ring [kopen = (6.30 ± 1.70) × 10–3 s–1],41 which is approximately 5-fold slower than the purported dissociation of RFC from an RPA-bound P/T junction (kinc,2 = 0.0305 ± 0.00380 s–1). Hence, loading of Cy5-PCNA onto the Cy3P/BioT DNA substrate will be rate-limited by opening of the PCNA ring.41 Indeed, a single phase was observed for the FRET increase (Figure 5D) with a rate constant (kinc,1 = 3.71 × 10–3 ± 2.54 × 10–4 s–1) that agreed very well with spontaneous opening of the PCNA ring. This validates that RPA stabilizes all Cy5-PCNA on the Cy3P/BioT DNA substrate by prohibiting diffusion of the sliding clamp ring off the ssDNA end. Thus, the total amplitude observed above in the absence of the unlabeled P/BioT DNA [AT = 5.58 ± 0.644 (Figure 5B)] indicates the amount of Cy5-PCNA (180 nM homotrimer) loaded onto the Cy3P/BioT DNA substrate (200 nM). Finally, these experiments were repeated by adding unlabeled P/T DNA (200 nM) and saturating RPA (285 nM) to the RFC·ATP·Cy5-PCNA complex prior to mixing (Figure 5E). Under these conditions, all Cy5-PCNA (180 nM homotrimer) will be loaded onto the unlabeled P/T DNA substrate (200 nM) by RFC (180 nM) prior to mixing. In the absence of the Neutravidin/biotin complex, all loaded Cy5-PCNA instantly diffuses off the unblocked duplex end of the P/T DNA substrate and is available for reloading. Hence, loading of Cy5-PCNA onto the Cy3P/BioT DNA substrate will be rate-limited by dissociation of RFC from the RPA-bound P/T DNA substrate. Indeed, a single-exponential increase in FRET was observed (Figure 5F) with a rate constant (0.0184 ± 0.00228 s–1) that agreed with the kinc,2 measured in Figure 5B (0.0305 ± 0.00380 s–1). Furthermore, the total amplitude (AT = 2.97 ± 0.701) for the FRET increase was half (0.533) that observed in the absence of RPA-bound P/BioT DNA [AT = 5.58 ± 0.644 (Figure 5B)]. Such behavior is expected on this time scale as the unlabeled and labeled DNA substrates are identical and, hence, RFC will have an equal probability of loading Cy5-PCNA onto the unlabeled P/T DNA substrate and the Cy3P/BioT DNA substrates. Collectively, this indicates that RFC turnover is rate-limited by the slow dissociation of RFC (or RFC·ADP) from an RPA-bound P/T junction upon release of a closed PCNA ring. This process (kinc,2 = 0.0184 ± 0.00228 s–1) is >370-fold slower than the rate-limiting step for release of PCNA onto DNA (kdec). Therefore, RFC turnover and, hence, an equilibrium FRET signal are not observed when both ends of the Cy3-labeled DNA substrates are not blocked because Cy5-PCNA immediately dissociates back into solution after loading.
Figure 5.

Catalytic loading of PCNA is rate-limited by dissociation of RFC from RPA-coated ssDNA. Each FRET trace represents the average of at least eight shots. For comparison, all FRET traces are normalized to the same starting value (at t = 0.005 s). (A) Schematic representation of the experimental procedure for panel B. (B) Cy5-PCNA (180 nM homotrimer) was preincubated with RFC (180 nM) in the presence of ATP. This preformed RFC·Cy5-PCNA·ATP complex was mixed with Cy3P/BioT DNA (200 nM), RPA (285 nM), and ATP in a stopped-flow instrument, and the FRET signal was followed. The loading trace was fit to a double-exponential equation, and the kinetic values are reported. (C) Schematic representation of the experimental procedure for panel D. (D) Unlabeled P/BioT DNA (200 nM) was preincubated with RPA (285 nM), Cy5-PCNA (180 nM homotrimer), RFC (180 nM), and ATP. This solution was then mixed with Cy3P/BioT DNA (200 nM), RPA (285 nM), and ATP in a stopped-flow instrument, and the FRET signal was followed. The loading trace was fit to a single-exponential equation, and the rate constant is reported. (E) Schematic representation of the experimental procedure for panel F. (F) The experiments depicted in panels C and D were repeated except RPA (285 nM), Cy5-PCNA (180 nM homotrimer), RFC (180 nM), and ATP were preincubated with unlabeled P/T DNA (200 nM). The loading trace was fit to a single-exponential equation, and the kinetic values are reported.
Discussion
A critical aspect of human TLS is maintaining PCNA at or near blocked P/T junctions within postreplicative gaps. As discussed above, PCNA does not vacate a blocked P/T by enzymatic unloading or extensive diffusion along the nascent duplex DNA adjacent to the blocked P/T junction.30−34 However, PCNA may diffuse along the adjacent ssDNA, which may be as long as 20 kb in human cells,35 and new PCNA rings will not be continuously reloaded.37,38 Thus, diffusion of PCNA along ssDNA must be prohibited. In the study presented here, we utilized a unique Cy3-Cy5 FRET pair developed in the Benkovic lab41 to directly and continuously monitor the retention of human PCNA on a DNA substrate that mimics a blocked P/T junction (Figure 1). Results from the extensive steady state and pre-steady state FRET assays clearly indicate that RPA restricts PCNA to the upstream duplex region by directly prohibiting diffusion of PCNA along ssDNA. This may be achieved by one of three pathways: (1) significantly decreasing the diffusion coefficient of PCNA along ssDNA by increasing the frictional drag of the PCNA ring,31 (2) effectively tethering loaded PCNA to the P/T junction, or (3) binding tightly to ssDNA and physically blocking diffusion, i.e., a protein roadblock. Below, we discuss the results from this study in the context of these pathways.
RPA Is a Protein Roadblock to Diffusion of PCNA along ssDNA
Human RPA does not contain a PCNA binding domain, and a direct interaction between RPA and PCNA has yet to be reported in the literature, ruling out pathway 1. The human clamp loader, RFC, interacts directly with RPA in the absence of ATP.46 Thus, it is possible that RPA may hold RFC at a P/T junction with PCNA (pathway 2).46,47 However, upon closing a PCNA ring around a P/T junction, a process that requires ATP hydrolysis,40,50 RFC’s affinity for PCNA decreases by ∼3 orders of magnitude49,51 and PCNA is released onto the dsDNA adjacent to the P/T junction.51−55 Experiments in this study on the Cy3P/T DNA substrate (Figures 2–5) indicate that PCNA immediately diffuses off the unblocked duplex end upon release from RFC, confirming this mechanism. Furthermore, steady state titration experiments on the Cy3P/BioT DNA substrate (Figure 2) indicate that RPA does not hold RFC at a P/T junction as limiting RFC can load all Cy5-PCNA onto the Cy3P/BioT DNA substrate in the presence of RPA. Interestingly, the pre-steady state assays on the Cy3P/BioT DNA substrate depicted in Figures 4 and 5 reveal that RFC turnover after loading PCNA onto a P/T junction is rate-limited by dissociation of RFC from the adjacent RPA-coated ssDNA. This process is rather slow (t1/2 ∼ 30 s) and may explain why previous studies on biotin-labeled P/T DNA substrates observed RFC, RPA, and PCNA after isolation with biotin affinity beads.46,47 Altogether, this indicates that RPA does not hold RFC at a P/T junction with PCNA (pathway 2). Human RPA binds to ssDNA with an extremely high affinity (less than picomolar) at physiological ionic strength and might prevent diffusion of PCNA along ssDNA by acting as a protein roadblock (pathway 3).39 The steady state titration experiments on the Cy3P/BioT DNA substrate (Figure 2) indicate that a single RPA molecule stabilizes a stoichiometric amount of Cy5-PCNA on the Cy3P/BioT DNA substrate. The pre-steady state assays on the Cy3P/T and Cy3P/BioT DNA substrates (Figures 3–5) indicate that RPA alone prohibits diffusion of PCNA along the ssDNA adjacent to the P/T junction but has no effect on diffusion of PCNA along the duplex region. Altogether, results from these experiments indicate that RPA binds tightly to ssDNA adjacent to a P/T junction and directly prohibits diffusion of PCNA along the ssDNA by acting as a physical block (pathway 3).
The Ability of RPA To Prohibit Diffusion of PCNA along ssDNA Is Likely Unique
Human TLS involves a large ensemble of proteins, many of which bind directly to PCNA, DNA, or both.7 Thus, it is possible that additional proteins apart from RPA may prohibit diffusion of PCNA along ssDNA by one or more of the aforementioned pathways. In a recent biophysical study of human PCNA, the diffusion coefficient of human PCNA decreased only 2.1-fold when its molecular radius and, hence, frictional drag were increased 4-fold by the attachment of a large object (Q-Dot).31 This suggests that a large protein simply binding to PCNA encircling a P/T junction will not prohibit diffusion of PCNA along the adjacent ssDNA (pathway 1). Indeed, human pol δ will not promote the retention of Cy5-PCNA on the Cy3P/BioT DNA substrate at equilibrium (Figure S1). Under the conditions of the assay, human pol δ binds to PCNA encircling a P/T junction with significant affinity (<10 nM) but has a dramatically low affinity for P/T DNA in the absence of DNA synthesis.42,43,48,56 Perhaps a protein (or protein complex) with multiple binding domains tethers loaded PCNA to a blocked P/T junction by concurrently binding PCNA and the adjacent P/T junction (or ssDNA) with a high affinity (pathway 2). The replicative pols, δ and ε, are obvious possibilities. In agreement with that described above for Figure S1, a recent report from our lab revealed that human pol δ rapidly dissociates into solution upon stalling at a DNA lesion it cannot accommodate, leaving PCNA behind on the DNA.42 Furthermore, pol δ alone retains Cy5-PCNA on the Cy3P/BioT DNA substrate only when the transition state for DNA synthesis (i.e., dNTP insertion) is stabilized (Figure S1), in agreement with observations from similar studies.48 Analogous studies have yet to be reported for the leading strand pol, pol ε, but similar behavior is expected. First, the affinity of pol ε for PCNA encircling DNA is rather weak compared to that of pol δ.57−61 Second, on the basis of the concentration of pol ε (550 copies of the catalytic subunit per cell) and the number of replication origins active at any given time within a human cell (∼10–15% of 30000–50000 origins/cell = 3000–7500), it is likely that human pol ε travels with progressing replication forks to keep up with ongoing DNA replication.62−64 Indeed, pol ε was not identified at or near blocked P/T junctions within human cells upon inhibition of DNA synthesis.38 Collectively, this suggests that the replicative pols, δ and ε, do not remain engaged with the blocked P/T junction and the resident PCNA upon encountering a DNA lesion they cannot accommodate. Rev1 and Rad6/Rad18 are also possibilities; both are imperative for human TLS, and each contains distinct binding sites for PCNA and ssDNA.7,65 Although the possibility cannot be ruled out, it is unlikely that these proteins serve as protein bridges to prohibit diffusion of PCNA along ssDNA (pathway 2). After irradiation with UV at the G1/S border of the cell cycle, the level of Rev1 expression peaks in G2/M phase when the majority of DNA replication, i.e., S phase, has already been completed.66 Extensive studies indicate that Rad6/Rad18 is recruited to blocked P/T junctions by persistent RPA-coated ssDNA downstream of the offending damage, i.e., after the ssDNA has been generated and coated with RPA.7 Altogether, this suggests that the ability of RPA alone to prohibit diffusion of PCNA along ssDNA within postreplicative gaps may be unique among the ensemble of proteins involved in human TLS.
Acknowledgments
The authors thank Dr. Tae-Hee Lee, Ph.D. (The Pennsylvania State University), for critical reading of the manuscript.
Glossary
Abbreviations
- pol δ
DNA polymerase δ
- P/T
primer/template
- PCNA
proliferating cell nuclear antigen
- RFC
replication factor C
- RPA
replication protein A
- FRET
Forster resonance energy transfer
- ssDNA
single-stranded DNA
- dsDNA
double-stranded DNA
- TLS
translesion DNA synthesis
- DDT
DNA damage tolerance.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.6b01213.
Steady state FRET for the Cy3-P/BioT DNA substrate requires a tight binding protein to stabilize PCNA on DNA (Figure S1) (PDF)
This work was supported by National Institutes of Health Grant GM13306 (S.J.B.).
The authors declare no competing financial interest.
Supplementary Material
References
- Hedglin M.; Kumar R.; Benkovic S. J. (2013) Replication clamps and clamp loaders. Cold Spring Harbor Perspect. Biol. 5, a010165. 10.1101/cshperspect.a010165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange S. S.; Takata K.; Wood R. D. (2011) DNA polymerases and cancer. Nat. Rev. Cancer 11, 96–110. 10.1038/nrc2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sale J. E.; Lehmann A. R.; Woodgate R. (2012) Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 13, 141–152. 10.1038/nrm3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington M. T.; Carlson K. D.; Freudenthal B. D.; Pryor J. M. (2010) Variations on a theme: eukaryotic Y-family DNA polymerases. Biochim. Biophys. Acta, Proteins Proteomics 1804, 1113–1123. 10.1016/j.bbapap.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash S.; Johnson R. E.; Prakash L. (2005) Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu. Rev. Biochem. 74, 317–353. 10.1146/annurev.biochem.74.082803.133250. [DOI] [PubMed] [Google Scholar]
- Sale J. E.; Lehmann A. R.; Woodgate R. (2012) Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 13, 141–152. 10.1038/nrm3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedglin M.; Benkovic S. J. (2015) Regulation of Rad6/Rad18 Activity During DNA Damage Tolerance. Annu. Rev. Biophys. 44, 207–228. 10.1146/annurev-biophys-060414-033841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmunds C. E.; Simpson L. J.; Sale J. E. (2008) PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol. Cell 30, 519–529. 10.1016/j.molcel.2008.03.024. [DOI] [PubMed] [Google Scholar]
- Temviriyanukul P.; van Hees-Stuivenberg S.; Delbos F.; Jacobs H.; de Wind N.; Jansen J. G. (2012) Temporally distinct translesion synthesis pathways for ultraviolet light-induced photoproducts in the mammalian genome. DNA Repair 11, 550–558. 10.1016/j.dnarep.2012.03.007. [DOI] [PubMed] [Google Scholar]
- Jansen J. G.; Tsaalbi-Shtylik A.; Hendriks G.; Gali H.; Hendel A.; Johansson F.; Erixon K.; Livneh Z.; Mullenders L. H.; Haracska L.; de Wind N. (2009) Separate domains of Rev1 mediate two modes of DNA damage bypass in mammalian cells. Mol. Cell. Biol. 29, 3113–3123. 10.1128/MCB.00071-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes M.; Foiani M.; Sogo J. M. (2006) Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol. Cell 21, 15–27. 10.1016/j.molcel.2005.11.015. [DOI] [PubMed] [Google Scholar]
- Callegari A. J.; Clark E.; Pneuman A.; Kelly T. J. (2010) Postreplication gaps at UV lesions are signals for checkpoint activation. Proc. Natl. Acad. Sci. U. S. A. 107, 8219–8224. 10.1073/pnas.1003449107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elvers I.; Johansson F.; Groth P.; Erixon K.; Helleday T. (2011) UV stalled replication forks restart by re-priming in human fibroblasts. Nucleic Acids Res. 39, 7049–7057. 10.1093/nar/gkr420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamant N.; Hendel A.; Vered I.; Carell T.; Reissner T.; de Wind N.; Geacinov N.; Livneh Z. (2012) DNA damage bypass operates in the S and G2 phases of the cell cycle and exhibits differential mutagenicity. Nucleic Acids Res. 40, 170–180. 10.1093/nar/gkr596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meneghini R. (1976) Gaps in DNA synthesized by ultraviolet light-irradiated WI38 human cells. Biochim. Biophys. Acta, Nucleic Acids Protein Synth. 425, 419–427. 10.1016/0005-2787(76)90006-X. [DOI] [PubMed] [Google Scholar]
- Meneghini R.; Cordeiro-Stone M.; Schumacher R. I. (1981) Size and frequency of gaps in newly synthesized DNA of xeroderma pigmentosum human cells irradiated with ultraviolet light. Biophys. J. 33, 81–92. 10.1016/S0006-3495(81)84873-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigaku Y.; Davies A. A.; Ulrich H. D. (2010) Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature 465, 951–955. 10.1038/nature09097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karras G. I.; Jentsch S. (2010) The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell 141, 255–267. 10.1016/j.cell.2010.02.028. [DOI] [PubMed] [Google Scholar]
- Cordeiro-Stone M.; Schumacher R. I.; Meneghini R. (1979) Structure of the replication fork in ultraviolet light-irradiated human cells. Biophys. J. 27, 287–300. 10.1016/S0006-3495(79)85218-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann A. R. (1972) Postreplication repair of DNA in ultraviolet-irradiated mammalian cells. J. Mol. Biol. 66, 319–337. 10.1016/0022-2836(72)90418-4. [DOI] [PubMed] [Google Scholar]
- Lehmann A. R.; Kirk-Bell S. (1972) Post-replication repair of DNA in ultraviolet-irradiated mammalian cells. No gaps in DNA synthesized late after ultraviolet irradiation. Eur. J. Biochem. 31, 438–445. 10.1111/j.1432-1033.1972.tb02550.x. [DOI] [PubMed] [Google Scholar]
- Buhl S. N.; Stillman R. M.; Setlow R. B.; Regan J. D. (1972) DNA chain elongation and joining in normal human and xeroderma pigmentosum cells after ultraviolet irradiation. Biophys. J. 12, 1183–1191. 10.1016/S0006-3495(72)86154-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauth A. M.; Tammemagi M.; Hunter G. (1974) Nascent DNA synthesis in ultraviolet light-irradiated mouse, human and Chinese hamster cells. Biophys. J. 14, 209–220. 10.1016/S0006-3495(74)85908-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edenberg H. J. (1976) Inhibition of DNA replication by ultraviolet light. Biophys. J. 16, 849–860. 10.1016/S0006-3495(76)85735-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. D.; Cleaver J. E. (1979) Postreplication repair: questions of its definition and possible alteration in xeroderma pigmentosum cell strains. Proc. Natl. Acad. Sci. U. S. A. 76, 3927–3931. 10.1073/pnas.76.8.3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman A. R.; Kirk-Bell S.; Arlett C. F.; Paterson M. C.; Lohman P. H.; de Weerd-Kastelein E. A.; Bootsma D. (1975) Xeroderma pigmentosum cells with normal levels of excision repair have a defect in DNA synthesis after UV-irradiation. Proc. Natl. Acad. Sci. U. S. A. 72, 219–223. 10.1073/pnas.72.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer J. C.; Kaufmann W. K.; Brylawski B. P.; Cordeiro-Stone M. (1990) Defective postreplication repair in xeroderma pigmentosum variant fibroblasts. Cancer Res. 50, 2593–2598. [PubMed] [Google Scholar]
- Niimi A.; Brown S.; Sabbioneda S.; Kannouche P. L.; Scott A.; Yasui A.; Green C. M.; Lehmann A. R. (2008) Regulation of proliferating cell nuclear antigen ubiquitination in mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 105, 16125–16130. 10.1073/pnas.0802727105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J. H.; Prakash L.; Prakash S. (2009) Highly error-free role of DNA polymerase eta in the replicative bypass of UV-induced pyrimidine dimers in mouse and human cells. Proc. Natl. Acad. Sci. U. S. A. 106, 18219–18224. 10.1073/pnas.0910121106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota T.; Katou Y.; Nakato R.; Shirahige K.; Donaldson A. D. (2015) Replication-Coupled PCNA Unloading by the Elg1 Complex Occurs Genome-wide and Requires Okazaki Fragment Ligation. Cell Rep. 12, 774–787. 10.1016/j.celrep.2015.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochaniak A. B.; Habuchi S.; Loparo J. J.; Chang D. J.; Cimprich K. A.; Walter J. C.; van Oijen A. M. (2009) Proliferating cell nuclear antigen uses two distinct modes to move along DNA. J. Biol. Chem. 284, 17700–17710. 10.1074/jbc.M109.008706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibahara K.; Stillman B. (1999) Replication-dependent marking of DNA by PCNA facilitates CAF-1-coupled inheritance of chromatin. Cell 96, 575–585. 10.1016/S0092-8674(00)80661-3. [DOI] [PubMed] [Google Scholar]
- Fennessy R. T.; Owen-Hughes T. (2016) Establishment of a promoter-based chromatin architecture on recently replicated DNA can accommodate variable inter-nucleosome spacing. Nucleic Acids Res. 44, 7189–7203. 10.1093/nar/gkw331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. J.; Whitehouse I. (2012) Intrinsic coupling of lagging-strand synthesis to chromatin assembly. Nature 483, 434–438. 10.1038/nature10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonn U.; Lonn S. (1988) Extensive regions of single-stranded DNA in aphidicolin-treated melanoma cells. Biochemistry 27, 566–570. 10.1021/bi00402a010. [DOI] [PubMed] [Google Scholar]
- Callegari A. J.; Kelly T. J. (2006) UV irradiation induces a postreplication DNA damage checkpoint. Proc. Natl. Acad. Sci. U. S. A. 103, 15877–15882. 10.1073/pnas.0607343103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. F.; Mathews M. B. (1989) Regulation of proliferating cell nuclear antigen during the cell cycle. J. Biol. Chem. 264, 13856–13864. [PubMed] [Google Scholar]
- Sirbu B. M.; McDonald W. H.; Dungrawala H.; Badu-Nkansah A.; Kavanaugh G. M.; Chen Y.; Tabb D. L.; Cortez D. (2013) Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J. Biol. Chem. 288, 31458–31467. 10.1074/jbc.M113.511337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen B.; Sokoloski J.; Galletto R.; Elson E. L.; Wold M. S.; Lohman T. M. (2014) Diffusion of human replication protein A along single-stranded DNA. J. Mol. Biol. 426, 3246–3261. 10.1016/j.jmb.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinker R. L.; Kassavetis G. A.; Geiduschek E. P. (1994) Detecting the ability of viral, bacterial and eukaryotic replication proteins to track along DNA. EMBO J. 13, 5330–5337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedglin M.; Perumal S. K.; Hu Z.; Benkovic S. (2013) Stepwise assembly of the human replicative polymerase holoenzyme. eLife 2, e00278. 10.7554/eLife.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedglin M.; Pandey B.; Benkovic S. J. (2016) Stability of the human polymerase delta holoenzyme and its implications in lagging strand DNA synthesis. Proc. Natl. Acad. Sci. U. S. A. 113, E1777. 10.1073/pnas.1523653113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedglin M.; Pandey B.; Benkovic S. J. (2016) Characterization of human translesion DNA synthesis across a UV-induced DNA lesion. eLife 5, e19788. 10.7554/eLife.19788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binz S. K.; Dickson A. M.; Haring S. J.; Wold M. S. (2006) Functional assays for replication protein A (RPA). Methods Enzymol. 409, 11–38. 10.1016/S0076-6879(05)09002-6. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Baranovskiy A. G.; Tahirov T. H.; Pavlov Y. I. (2014) The C-terminal domain of the DNA polymerase catalytic subunit regulates the primase and polymerase activities of the human DNA polymerase alpha-primase complex. J. Biol. Chem. 289, 22021–22034. 10.1074/jbc.M114.570333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzhakov A.; Kelman Z.; Hurwitz J.; O’Donnell M. (1999) Multiple competition reactions for RPA order the assembly of the DNA polymerase delta holoenzyme. EMBO J. 18, 6189–6199. 10.1093/emboj/18.21.6189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waga S.; Stillman B. (1998) Cyclin-dependent kinase inhibitor p21 modulates the DNA primer-template recognition complex. Mol. Cell. Biol. 18, 4177–4187. 10.1128/MCB.18.7.4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsurimoto T.; Stillman B. (1991) Replication factors required for SV40 DNA replication in vitro. I. DNA structure-specific recognition of a primer-template junction by eukaryotic DNA polymerases and their accessory proteins. J. Biol. Chem. 266, 1950–1960. [PubMed] [Google Scholar]
- Shiomi Y.; Usukura J.; Masamura Y.; Takeyasu K.; Nakayama Y.; Obuse C.; Yoshikawa H.; Tsurimoto T. (2000) ATP-dependent structural change of the eukaryotic clamp-loader protein, replication factor C. Proc. Natl. Acad. Sci. U. S. A. 97, 14127–14132. 10.1073/pnas.97.26.14127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. H.; Hurwitz J. (1990) Mechanism of elongation of primed DNA by DNA polymerase delta, proliferating cell nuclear antigen, and activator 1. Proc. Natl. Acad. Sci. U. S. A. 87, 5672–5676. 10.1073/pnas.87.15.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G.; Gibbs E.; Kelman Z.; O’Donnell M.; Hurwitz J. (1999) Studies on the interactions between human replication factor C and human proliferating cell nuclear antigen. Proc. Natl. Acad. Sci. U. S. A. 96, 1869–1874. 10.1073/pnas.96.5.1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J.; Gibbs E.; Uhlmann F.; Phillips B.; Yao N.; O’Donnell M.; Hurwitz J. (1997) A complex consisting of human replication factor C p40, p37, and p36 subunits is a DNA-dependent ATPase and an intermediate in the assembly of the holoenzyme. J. Biol. Chem. 272, 18974–18981. 10.1074/jbc.272.30.18974. [DOI] [PubMed] [Google Scholar]
- Uhlmann F.; Cai J.; Gibbs E.; O’Donnell M.; Hurwitz J. (1997) Deletion analysis of the large subunit p140 in human replication factor C reveals regions required for complex formation and replication activities. J. Biol. Chem. 272, 10058–10064. 10.1074/jbc.272.15.10058. [DOI] [PubMed] [Google Scholar]
- Cai J.; Uhlmann F.; Gibbs E.; Flores-Rozas H.; Lee C. G.; Phillips B.; Finkelstein J.; Yao N.; O’Donnell M.; Hurwitz J. (1996) Reconstitution of human replication factor C from its five subunits in baculovirus-infected insect cells. Proc. Natl. Acad. Sci. U. S. A. 93, 12896–12901. 10.1073/pnas.93.23.12896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao N.; Turner J.; Kelman Z.; Stukenberg P. T.; Dean F.; Shechter D.; Pan Z. Q.; Hurwitz J.; O’Donnell M. (1996) Clamp loading, unloading and intrinsic stability of the PCNA, beta and gp45 sliding clamps of human, E. coli and T4 replicases. Genes Cells 1, 101–113. 10.1046/j.1365-2443.1996.07007.x. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Meng X.; Zhang S.; Lee E. Y.; Lee M. Y. (2012) Characterization of human DNA polymerase delta and its subassemblies reconstituted by expression in the MultiBac system. PLoS One 7, e39156. 10.1371/journal.pone.0039156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J.; Shi Y.; Georgescu R. E.; Yuan Z.; Chait B. T.; Li H.; O’Donnell M. E. (2015) The architecture of a eukaryotic replisome. Nat. Struct. Mol. Biol. 22, 976–982. 10.1038/nsmb.3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston L. D.; Zhang D.; Yurieva O.; Georgescu R. E.; Finkelstein J.; Yao N. Y.; Indiani C.; O’Donnell M. E. (2014) CMG helicase and DNA polymerase epsilon form a functional 15-subunit holoenzyme for eukaryotic leading-strand DNA replication. Proc. Natl. Acad. Sci. U. S. A. 111, 15390–15395. 10.1073/pnas.1418334111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgescu R. E.; Schauer G. D.; Yao N. Y.; Langston L. D.; Yurieva O.; Zhang D.; Finkelstein J.; O’Donnell M. E. (2015) Reconstitution of a eukaryotic replisome reveals suppression mechanisms that define leading/lagging strand operation. eLife 4, e04988. 10.7554/eLife.04988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgescu R. E.; Langston L.; Yao N. Y.; Yurieva O.; Zhang D.; Finkelstein J.; Agarwal T.; O’Donnell M. E. (2014) Mechanism of asymmetric polymerase assembly at the eukaryotic replication fork. Nat. Struct. Mol. Biol. 21, 664–670. 10.1038/nsmb.2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgers P. M.; Bambara R. A.; Campbell J. L.; Chang L. M.; Downey K. M.; Hubscher U.; Lee M. Y.; Linn S. M.; So A. G.; Spadari S. (1990) Revised nomenclature for eukaryotic DNA polymerases. Eur. J. Biochem. 191, 617–618. 10.1111/j.1432-1033.1990.tb19165.x. [DOI] [PubMed] [Google Scholar]
- Jackson D. A.; Pombo A. (1998) Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J. Cell Biol. 140, 1285–1295. 10.1083/jcb.140.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck M.; Schmidt A.; Malmstroem J.; Claassen M.; Ori A.; Szymborska A.; Herzog F.; Rinner O.; Ellenberg J.; Aebersold R. (2011) The quantitative proteome of a human cell line. Mol. Syst. Biol. 7, 549. 10.1038/msb.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard A. C.; Mechali M. (2013) DNA replication origins. Cold Spring Harbor Perspect. Biol. 5, a010116. 10.1101/cshperspect.a010116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda Y.; Kamiya K. (2006) Role of single-stranded DNA in targeting REV1 to primer termini. J. Biol. Chem. 281, 24314–24321. 10.1074/jbc.M602967200. [DOI] [PubMed] [Google Scholar]
- Waters L. S.; Walker G. C. (2006) The critical mutagenic translesion DNA polymerase Rev1 is highly expressed during G(2)/M phase rather than S phase. Proc. Natl. Acad. Sci. U. S. A. 103, 8971–8976. 10.1073/pnas.0510167103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.