

Abstract
Many chemicals have been used to increase the safety of consumer products by reducing their flammability and risk for ignition. Recent focus on brominated flame retardants, such as polybrominated diphenyl ethers (PBDEs) has shown them to contribute to neurobehavioral deficits in children, including learning and memory. As the manufacture and use of PBDEs has been reduced, replacement chemicals, such as hexabromocyclododecane (HBCDD) have been substituted. Our current study evaluated the neurotoxicity of HBCDD, concentrating on dopaminergic innervation to the hippocampus. Using an in vivo model, we exposed male mice to HBCDD and then assessed alterations to the dopamine synapse 6 weeks later. These exposures elicited significant reductions in presynaptic dopaminergic proteins, including TH, COMT, MAOB, DAT, VMAT2, and alpha-synuclein. In contrast, postsynaptic dopamine receptors were not impaired. These findings suggest that the mesohippocampal dopamine circuit is vulnerable to HBCDD and the dopamine terminal may be a selective target for alteration.
Keywords: Dopamine Transporter, Hexabromocyclododecane, Hippocampus, Substantia Nigra pars compacta, Tyrosine Hydroxylase, Vesicular Monoamine Transporter 2
1. Introduction
The manufacture and use of brominated flame-retardants has seen a precipitous increase in the last several decades, given their perceived ability to inhibit or reduce the flammability of consumer products, such as electronics, plastics, furniture, and other textiles (de Wit, 2002). Unfortunately, the increased demand for these products has also led to an elevated and ubiquitous presence of these compounds in our environment that is exacerbated by their ability to leach out of many of these products and bioaccumulate. Significant levels of flame retardant compounds are found throughout the environment, which may underlie an increase in body burdens of these chemicals in the human population (Covaci et al., 2011). Of particular interest are findings that demonstrate the presence of brominated flame retardants (BFRs) in human brain tissue and the association between exposure to these compounds and neurological disruption. Indeed, one of the more recently studied class of BFRs, polybrominated diphenyl ethers (PBDE) have been found to be associated with a variety of neurobehavioral impairments, including deficits in motor and sensory development, as well as several domains associated with cognitive development (Herbstman et al., 2010, Eskenazi et al., 2013, Chen et al., 2014, Cowell et al., 2015, Chevrier et al., 2016). Most concerning is the impact these compounds seem to have on aspects of learning and memory in these exposed populations. Within this context, particular interest has been focused towards the hippocampus, which plays a critical role in mediating a variety of aspects associated with memory. Multiple laboratory-based studies have found exposure to PBDE to significantly impact the function of the hippocampus, leading to deficits in memory function (Viberg et al., 2003, Ta et al., 2011, Yan et al., 2012). From a cellular and molecular perspective, further studies have shown that exposure to PBDE impairs LTP and synaptic plasticity, critical elements associated with learning and memory processes (Dingemans et al., 2007, Xing et al., 2009). Additionally, alterations in the cholinergic and glutamatergic neurotransmitter systems have been suggested to play a role in these behavioral deficits (Viberg et al., 2003, Dingemans et al., 2007, Yan et al., 2012).
As the neurotoxic properties of PBDEs became better appreciated, their manufacture and use was discontinued or limited throughout the world, allowing similar BFR products to be introduced as potential replacement compounds. One such chemical is hexabromocyclododecane (HBCDD), which appears to embody many of the same physiochemical properties as PBDEs, being lipophilic, difficult to breakdown, leading to bioaccumulation and persistence in the environment (Covaci et al., 2006). Although not as extensively studied as PBDEs, HBCDD has been shown to have neurotoxic properties similar to PBDEs. In animal studies, HBCDD exposure appears to elicit impairments in neurodevelopment and neurobehaviors. Most interestingly, the hippocampus appears to be a target of HBCDD neurotoxicity as groups have shown disruption of neuronal migration in the hippocampus as well as deficits in spatial memory following a single exposure in the developing mouse (Eriksson et al., 2006, Saegusa et al., 2012). While these findings provide valuable evidence for the neurotoxic effects of HBCDD, the underlying cellular and molecular targets that underlie these deficits in learning and memory are unclear.
Evaluation of neurotoxicological effects on the hippocampus is critical, given the importance of this structure in a variety of elements associated with learning and memory. The hippocampus is extensively interconnected with other brain regions that are known to modulate memory (Eichenbaum et al., 1996). Although not always appreciated as a substantial contributor to hippocampal function, dopaminergic projections from the midbrain, specifically the ventral tegmental area (VTA) and substantia nigra pars compacta (SNpc), have been found to have a significant impact on modulating/regulating memory processes (Frey and Morris, 1998, Jay, 2003, Lisman and Grace, 2005, Shohamy and Adcock, 2010, McNamara et al., 2014, Rosen et al., 2015). Dopaminergic projections have been found to terminate throughout the hippocampus, having more explicit localizations to cellular layers in the CA1, CA3, and dentate gyrus (Gasbarri et al., 1994, Gasbarri et al., 1997). These projections are generally found to synapse onto dendrites and cell bodies of dentate granule and pyramidal cells, where D1 and D2 receptor groups modulate long-term potentiation (LTP) through their coordinated interaction with glutamatergic receptors and intracellular cascades that underlie LTP (Boyson et al., 1986, Frey and Morris, 1998). Over the last several decades, extensive work has better delineated the role of dopamine in hippocampal function. In vitro and in vivo studies have found manipulation of dopamine signaling at the pre- and postsynaptic terminal to have significant effects on learning and memory in the hippocampus. Indeed, blockade of D1 or D2 receptors with selective antagonists manifests in memory impairments, which have been replicated in animals lacking the D1 receptor (Ortiz et al., 2010). Moreover, dopamine signaling that has been impaired following damage to dopamine terminals has also been shown to result in deficits in learning and memory. Utilization of the selective dopaminergic neurotoxins, MPTP or 6-OHDA has found reductions in dopaminergic terminals and dopamine in the hippocampus mediate impairments in LTP and memory in the hippocampus (Gasbarri et al., 1996, Zhu et al., 2011, Costa et al., 2012, Bonito-Oliva et al., 2014). These deficits were ameliorated following treatment with dopamine replacement, including L-DOPA.
In light of the importance of dopamine signaling in the hippocampus in mediating learning and memory, we sought to further characterize the potential neurotoxic effects of HBCDD on the mesohippocampal dopamine circuit. As the dopaminergic synapse appears to be uniquely vulnerable to HBCDD, we directed our focus towards the evaluation of proteins known to be critical to dopamine signaling. Indeed, following exposure to HBCDD in adult male mice, significant damage to presynaptic dopamine proteins was observed. These findings highlight the fact that the mesohippocampal dopamine circuit is vulnerable to HBCDD exposure and identifies potential cellular and molecular targets that underlie learning and memory impairments.
2. Materials and Methods
2.1. Chemicals and Reagents
Hexabromocyclododecane (HBCDD) was purchased from Sigma-Aldrich (St. Louis, MO). The BCA protein assay kit was obtained from Pierce (Rockford, IL). Monoclonal anti-rat dopamine transporter (DAT) and polyclonal rabbit anti-tyrosine hydroxylase (TH) and rabbit anti-Catechol-O-Methyltransferase (COMT) antibodies were purchased from EMD Millipore (Billerica, MA). Polyclonal rabbit anti-dopamine D2 receptor antibody was purchased from Santa Cruz Biotechnology (Dallas, TX). Monoclonal mouse anti-norepinephrine transporter (NET) was a kind gift from Craig Heilman at Emory University. Polyclonal rabbit anti-vesicular monoamine transporter 2 (VMAT2) antibodies were generated by Covance to the C-terminal sequence in mouse (CTQNNVQPYPVGDDEESESD). Monoclonal mouse anti-β-actin and anti-dopamine D1 receptor antibodies were purchased from Sigma-Aldrich (St. Louis, MO). Monoclonal mouse anti-alpha-synuclein antibody was purchased from BD Transduction (Franklin Lakes, NJ). Polyclonal rabbit anti-monoamine oxidase B antibody was purchased from Abcam (Cambridge, MA). Secondary antibodies conjugated to horseradish peroxidase or biotin were obtained from Jackson Immunoresearch Laboratories (West Grove, PA). SuperSignal West Dura Extended duration substrate and stripping buffer were obtained from Pierce. 3,3′ Diaminobenzidine (DAB) was purchased from Sigma-Aldrich (St. Louis, MO).
2.2. Animals and Treatment
Eight-week-old male C57BL/6J mice were purchased from Charles River Laboratories (Wilmington, MA). Two month old mice were orally gavaged with 25 μl of HBCDD made up to 25 mg/kg body weight (25,000 μg/kg body weight) and dissolved in corn oil vehicle. Animals were exposed daily for 6-weeks, using a protocol similar to that previously described (n=6 for control and n=6 for treated groups) (Caudle et al., 2006, Bradner et al., 2013, Genskow et al., 2015). This dosing paradigm was intended to represent the primary route of human exposure to HBCDD. Mice were sacrificed 6-weeks following the last exposure, and unilateral hippocampi were collected for subsequent analysis. While previous studies have investigated the impact of HBCDD exposure on the hippocampus (Eriksson et al., 2006, Saegusa et al., 2012), our study was the first to assess alterations to the dopamine circuit. As the focus of our study was on the dopaminergic synapse, we relied upon our previously published and ongoing studies with HBCDD to inform our dosing paradigm (Genskow et al., 2015). Standard rodent chow and tap water were available ad libitum. All procedures were conducted in accordance with the Guide for Care and Use of Laboratory Animals (National Institutes of Health) and have been approved by the Institutional Animal Care and Use Committee at Emory University.
2.3. Western Blot Analysis
Western blots were used to quantify the amount of DAT, TH, VMAT2, D1R, D2R, NET, COMT, MAO-B, and β-actin present in samples of hippocampal tissue from treated and control mice. Analysis was performed as previously described (Caudle et al., 2006, Genskow et al., 2015). Briefly, hippocampal samples were homogenized and samples subjected to polyacrylamide gel electrophoresis and electrophoretically transferred to polyvinylidene difluoride membranes. Nonspecific sites were blocked in 7.5% nonfat dry milk in Tris-buffered saline and then membranes incubated overnight in a monoclonal antibody to the N-terminus of DAT. DAT antibody binding was detected using a goat anti-rat horseradish peroxidase secondary antibody (1:10,000) and enhanced chemiluminescence. The luminescence signal was captured on an Alpha Innotech Fluorochem imaging system and stored as a digital image. Densitometric analysis was performed and calibrated to coblotted dilutional standards of pooled hippocampus from all control samples. Membranes were stripped for 15 min at room temperature with Pierce Stripping Buffer and sequentially reprobed with β-actin (1:1,000), TH (1:1,000), VMAT2 (1:10,000), D1R (1:1,000), D2R (1:500), COMT (1:1,000), MAO-B (1:1,000), NET (1:1,000) antibodies. β-Actin blots were used to ensure equal protein loading across samples.
2.4. Immunohistochemistry
Tissue staining was performed as described previously (Caudle et al., 2007). Briefly, hemisected brains were immersion fixed in 4% paraformaldehyde and serially sectioned at 40 μm. Sections were incubated overnight with polyclonal anti-TH (1:2,500) or anti-alpha-synuclein (1:500) antibody and then incubated in a biotinylated goat anti-rabbit or anti-mouse secondary antibody for 1 hr at room temperature. Visualization was performed using DAB for 1 min at room temperature. After DAB, all sections were dehydrated and coverslipped. Bright field images were captured (4–6 images from 3 control and 3 HBCDD treated animals) at 10x magnification using a Zeiss Axio Imager M2 microscope and were qualitatively assessed by a reviewer blind to the treatment groups.
2.5. Statistical analysis
All analysis was performed to compare changes in protein expression in synaptic markers in control versus HBCDD treated samples using the Student’s t-test using GraphPad analysis software (La Jolla, CA). Post hoc analysis was performed using Tukey’s post hoc test. Significance is reported at the p < 0.05 level.
3. Results
The hippocampus plays a significant role in mediating a variety of cognitive domains associated with learning and memory. While exposure to flame retardant compounds, such as HBCDD, has been shown to impair learning and memory, we sought to evaluate potential neural circuits that may be altered following exposure to HBCDD. Building upon our previous work that demonstrated alterations to the dopamine circuit in the striatum in adult mice exposed to HBCDD, we have elaborated these findings and evaluated the dopaminergic input to the hippocampus.
Adult male mice were exposed daily to 25 mg/kg body weight HBCDD or vehicle control for 6-weeks, followed by an additional 6-weeks where no exposure was given. Exposure of to HBCDD did not elicit any signs of overt toxicity, as indicated by weight loss or explicit behavioral abnormalities, including deficits in grooming, isolation, feeding, or generalized movement. Animals were monitored throughout the duration of the experiment. Our previous work has found the dopamine synapse to be especially vulnerable to neurotoxicity. To investigate the potential for HBCDD to damage the proteins critical to dopamine signaling in the hippocampus, we focused our study on three key components of the dopamine system: 1). Enzymes involved in dopamine synthesis and degradation, 2). Transporters involved in dopamine neurotransmission, 3). Receptors involved in translating the dopamine signal.
Tyrosine hydroxylase is an intracellular rate-limiting enzyme in the synthesis of dopamine. Following exposure to HBCDD, mice demonstrated a significant reduction in the expression of TH in the hippocampus (p = 0.04), as measured by immunoblotting (Figure 1A). Further investigation of these reductions using immunohistochemical techniques also demonstrated reductions in TH expression in various regions of the hippocampus (Figure 1E). Additional investigation evaluated the expression of MAO-B and COMT in our hippocampal samples. Similar to our TH results, expression of MAO-B (p = 0.02) and COMT (p < 0.01) were significantly reduced in animals treated with HBCDD (Figure 1B and C). These findings suggest that HBCDD has perturbed normal dopamine production and breakdown, which could impact dopamine signaling in the hippocampus.
Figure 1.
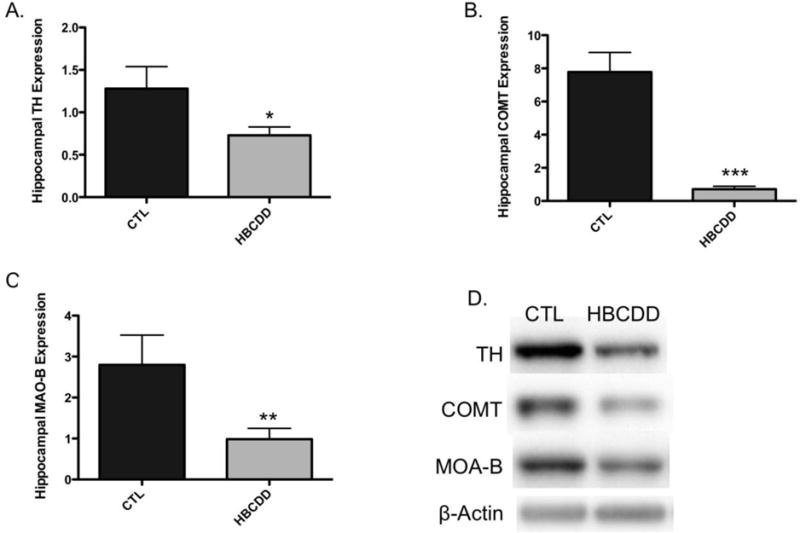
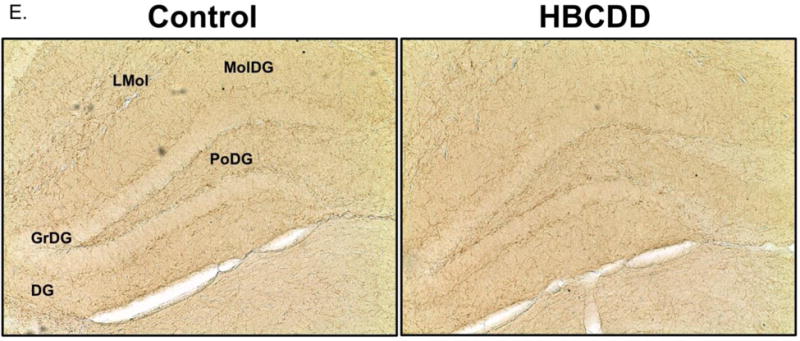
Exposure to HBCDD causes a reduction in enzymes critical to dopamine synthesis and degradation in the hippocampus. Animals received 0 or 25 mg/kg HBCDD for 6 weeks and were sacrificed 6 weeks later and evaluated for alterations in (A) TH, (B) COMT, and (C) MAO-B. (D) Representative immunoblot bands for each marker. Data represents mean ± SEM (6 animals per experimental group). (E) Representative immunohistochemical images of TH expression in the hippocampus of control and HBCDD treated animals. *Values that are significantly different from control (p<0.05). **Values that are significantly different from control (p<0.01). ***Values that are significantly different from control (p<0.001). DG: Dentate gyrus, GrDG: Granule cell layer of the dentate gyrus, LMol: Lacunosum molecular layer of CA1, MolDG: Molecular cell layer of the dentate gyrus, PoDG: Polymorph cell layer of the dentate gyrus.
Work from Genskow et al., (2015) found exposure to HBCDD to significantly alter the expression and function of transporter proteins that are critical to dopamine handling at the synapse. The VMAT2 is localized to synaptic vesicles and functions to package cytosolic dopamine into vesicles in preparation for release via neurotransmission. Working in tandem, the DAT resides on the plasma membrane and serves to sequester synaptic dopamine and transport it back into the dopamine terminal. Similar to our previous findings in the striatum, we have also demonstrated clear reductions in the expression of the DAT (p < 0.01) and VMAT2 (p = 0.02) in the hippocampus of HBCDD-exposed mice (Figure 2A and B). In addition to dopaminergic innervation, the hippocampus also received substantial noradrenergic input from the locus coeruleus. As dopamine and norepinephrine are both monoamines and utilize TH and VMAT2 for neurotransmitter synthesis and packaging, it was important to further address the specificity of our findings for the dopamine system. To this point, we used immunoblotting to evaluate the expression of NET in our hippocampal samples. In contrast to our findings with DAT and VMAT2, no change was seen in NET expression (p = 0.27), providing additional evidence that the effects of HBCDD appear to have selectivity for the dopamine circuit (Figure 2C).
Figure 2.
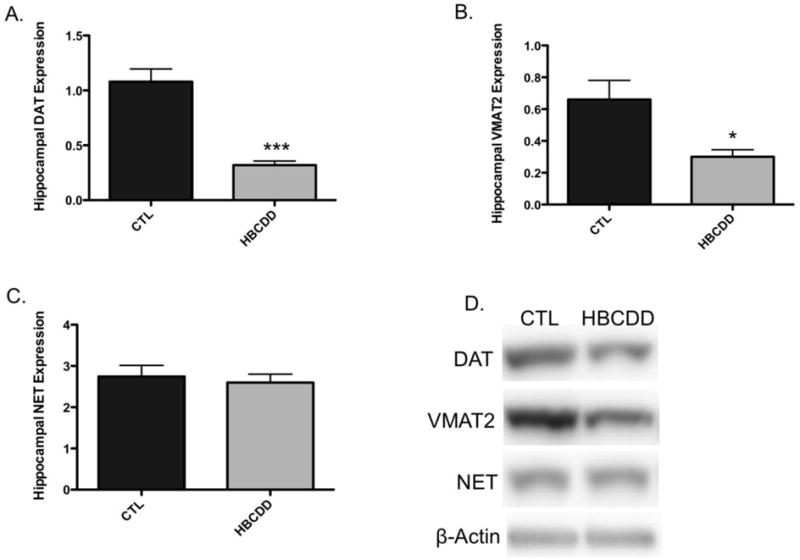
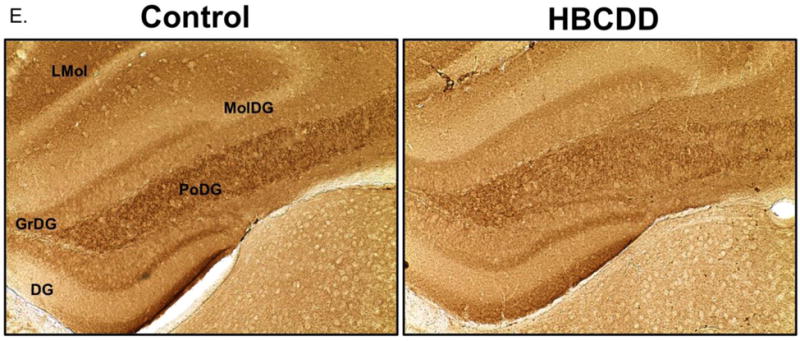
Exposure to HBCDD causes a reduction in presynaptic proteins critical to dopamine transport in the hippocampus. Animals received 0 or 25 mg/kg HBCDD for 6 weeks and were sacrificed 6 weeks later and evaluated for alterations in (A) DAT, (B) VMAT2, and (C) NET. (D) Representative immunoblot bands for each marker. Data represents mean ± SEM (6 animals per experimental group). (E) Representative immunohistochemical images of Asyn expression in the hippocampus of control and HBCDD treated animals. *Values that are significantly different from control (p<0.05). ***Values that are significantly different from control (p<0.001). DG: Dentate gyrus, GrDG: Granule cell layer of the dentate gyrus, LMol: Lacunosum molecular layer of CA1, MolDG: Molecular cell layer of the dentate gyrus, PoDG: Polymorph cell layer of the dentate gyrus.
As the presynaptic protein, alpha-synuclein (Asyn), has been shown to play a critical role in dopamine homeostasis and signaling, we also evaluated its expression in the hippocampus following HBCDD exposure. Similar to our other results, expression of Asyn demonstrated an overall reduction in expression. While these reductions appear to be ubiquitous in the hippocampus, they seem to be most pronounced within specific cell layers of the dentate gyrus and CA1 regions (Figure 2E).
Following release, dopamine traverses the synapse and interacts with dopamine receptors, which translate the chemical signal to an electrical signal at the postsynaptic neuron. Both D1 and D2 receptor families are found in the hippocampus and have been shown to be involved with mediating aspects of learning and memory. In contrast to the alterations observed in the presynaptic terminal of the dopamine projections, we did not detect a significant change to the either the D1 (p = 0.32) or D2 receptors (p = 0.63) in our samples following HBCDD exposure (Figure 3). These data demonstrate that HBCDD-mediated alterations to the hippocampal dopamine circuit may be selective for the presynaptic terminal and can elicit deficits in memory via impairment of neurotransmitter production, handling, and release.
Figure 3.
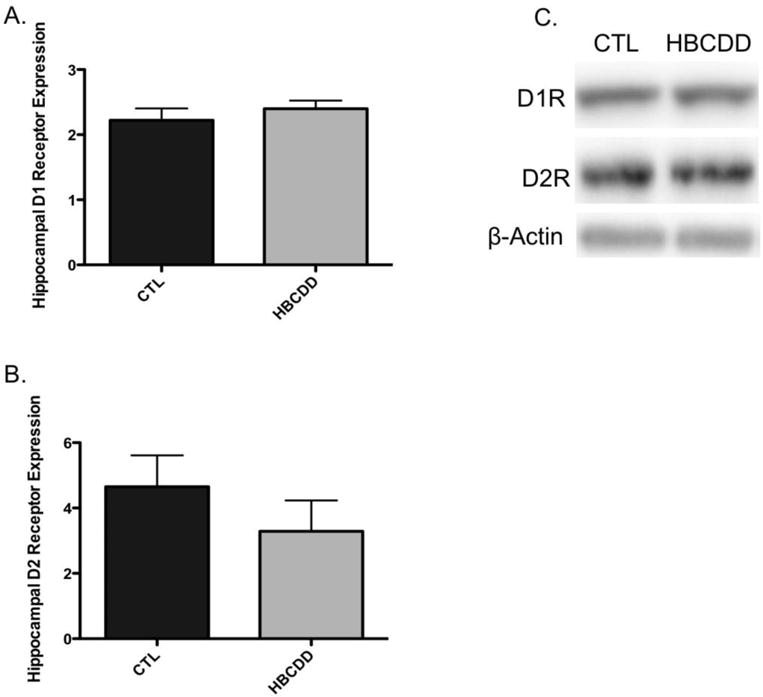
Exposure to HBCDD does not cause a reduction in postsynaptic dopamine receptors critical to dopamine signal transduction in the hippocampus. Animals received 0 or 25 mg/kg HBCDD for 6 weeks and were sacrificed 6 weeks later and evaluated for alterations in (A) D1 receptor and (B) D2 receptor. (C) Representative immunoblot bands for each marker. Data represents mean ± SEM (6 animals per experimental group).
4. Discussion
Awareness of the neurotoxic effects of brominated flame retardants has most recently been highlighted through epidemiological studies that have shown association between exposure to PBDEs and neurobehavioral deficits, particularly focused on domains of learning and memory (Herbstman et al., 2010, Eskenazi et al., 2013, Chen et al., 2014, Cowell et al., 2015, Chevrier et al., 2016). These findings have been further corroborated in animal studies, providing some insight into possible neural circuits that may underlie these deficits (Viberg et al., 2003, Dingemans et al., 2007, Xing et al., 2009, Ta et al., 2011, Yan et al., 2012). As the manufacture and use of these compounds has been or is currently being reduced the manufacture of chemically similar compounds, such as HBCDD, have been increased and have become a prominent flame retardant in many consumer products. In contrast to PBDEs, our understanding of the neurotoxicity of HBCDD is limited. However, there is some evidence that exposure to HBCDD does target the hippocampus and leads to impairments in aspects of memory (Eriksson et al., 2006, Saegusa et al., 2012). It is important to note that these studies employed significantly different exposure paradigms than currently used in our study. While Eriksson et al., (2006) administered 0.9 or 13.5 mg/kg HBCDD at a single time point (postnatal day 10), Saegusa et al., (2012) exposed pregnant rats to 800 mg/kg HBCDD from gestational day 10 through weaning at postnatal day 22, focusing their evaluation on the offspring. Recent work has also shown the dopaminergic neurotransmitter circuit to be uniquely vulnerable to damage by HBCDD (Mariussen and Fonnum, 2003, Lilienthal et al., 2009, Genskow et al., 2015). Of interest to our current study, the hippocampus receives a strong dopaminergic innervation that has been shown to play a critical role in facilitating LTP and memory formation and persistence (Gasbarri et al., 1994, Gasbarri et al., 1997, Frey and Morris, 1998, Jay, 2003, Lisman and Grace, 2005, Shohamy and Adcock, 2010, McNamara et al., 2014, Rosen et al., 2015). With these points in mind, we assessed alterations to the mesohippocampal circuit following exposure to HBCDD. Our findings identify the hippocampal dopamine pathway as a target for HBCDD damage, which could underlie or contribute to the memory impairments previously observed with HBCDD and other BFRs.
To date, evaluation of the dopaminergic input to the hippocampus has not been performed in the context of BFRs, and more specifically, HBCDD. Thus, building upon our previous findings that suggest the dopaminergic synapse to be a target for alteration by HBCDD, we focused our assessment on specific proteins known to be imperative to the maintenance and execution of normal dopamine signaling, both pre- and postsynaptically. We first directed our investigation towards enzymes that are involved in the synthesis and degradation of dopamine in the presynaptic terminal. To this end, we observed a significant decline in the expression of TH, the rate-limiting enzyme involved in the synthesis of dopamine. These reductions appear to be localized to cellular layers of the dentate gyrus and CA1 region of the hippocampus. In addition to TH, we also demonstrated a reduction in MAO-B and COMT, which are both part of the metabolic pathway that degrades dopamine to its major metabolites. Interestingly, previous studies that have evaluated the effects of BFRs on the dopamine circuit have not reported alterations in TH. Indeed, in our own work and others, expression of TH appears to be relatively stable, in contrast to alterations in other dopaminergic proteins (Richardson and Miller, 2004, Caudle et al., 2006, Bradner et al., 2013, Genskow et al., 2015). The implications of these findings can be considered from two different, yet congruent perspectives. First, HBCDD-induced reductions in TH could suggest a subsequent reduction in the synthesis of dopamine in the terminal. A reduction in dopamine would manifest in a decline in the amount of dopamine released from the terminal and an overall decrease in dopamine signaling at these synapses. In this context, as MAO-B and COMT function to degrade dopamine, a concomitant decline in their expression may indicate a compensatory response to loss of dopamine synthesis and a means to maintain basal levels of dopamine in the terminal and synapse. Such a mechanism has been suggested in mice engineered to express a reduction in TH protein. While not observed in mice that have a complete deletion of TH, animals that are heterozygous for the deletion show a 50% decline in TH expression and TH enzyme activity, yet do not show a difference in the level of dopamine in the brain (Kobayashi et al., 1995). Although not explicitly tested, it was surmised that these discrepancies in TH function and dopamine concentrations may be a result of compensatory reductions in dopamine degradation, leading to an elevation in tissue dopamine. Additional work will need to be undertaken in order to further delineate these mechanisms in our model.
Perhaps a more parsimonious explanation for our findings is that reductions in these enzymes denote explicit damage to the dopamine terminal and a loss of dopaminergic innervation to the hippocampus. Indeed, previous work from our group and others has suggested that expression of TH serves as a reliable marker of dopamine terminal integrity, in which alterations to other dopaminergic proteins occur prior to declines in TH expression and loss of dopamine terminals (Richardson and Miller, 2004, Caudle et al., 2006, Caudle et al., 2007, Caudle et al., 2008, Bradner et al., 2013, Genskow et al., 2015). This mechanism aligns more closely with our other findings related to reduction in DAT and VMAT2 in the presynaptic terminal of the hippocampus following HBCDD exposure. Given the dopamine handling functions of both DAT and VMAT2 in the dopamine terminal, it is believed that these transporters serve as critical mediators and sentinels of neurotoxicity at the dopamine synapse. The synthesis and handling of cytosolic dopamine requires a highly orchestrated series of intracellular events that aim to regulate the cytosolic concentrations of dopamine and the packaging of dopamine into synaptic vesicles by VMAT2 (Caudle et al., 2007, Caudle et al., 2008). Such a high degree of regulation seeks to prevent the generation of highly reactive oxidative species in the cytosol that can accumulate and damage intracellular targets of the dopamine terminal. Work from our group and others have shown that reductions in the expression and function of VMAT2 can facilitate the accumulation of cytosolic dopamine and the subsequent production of neurotoxic species. Indeed, utilizing mice with a 95% reduction in VMAT2 expression results in an age-dependent degeneration of the dopamine circuit that manifests as an accumulation of reactive oxygen species and a differential reduction in DAT and TH (Caudle et al., 2007, Caudle et al., 2008). While these data highlight the impact of a robust deletion of VMAT2, such alterations in VMAT2 and the dopamine terminal have also been demonstrated following exposure to specific neurotoxicants, including polychlorinated biphenyls (PCBs) and PBDEs (Mariussen and Fonnum, 2003, Richardson and Miller, 2004, Caudle et al., 2006, Bradner et al., 2013, Genskow et al., 2015). What makes these findings even more compelling is the fact that these alterations occur prior to explicit damage or loss of the dopamine terminal, as assessed by expression of the TH protein and dopamine concentrations. While our previous work with HBCDD and other similar compounds in the striatum has demonstrated a relative preservation of TH expression, in contrast to deficits in DAT and VMAT2, these findings may provide interesting insight into the temporal elements of the pathology. Indeed, unpublished data from our group suggests that alterations to the dopamine synapse, particularly the emergence of reductions in TH expression, are exacerbated when animals receiving HBCDD are evaluated at later time points, following exposure. These findings provide further support for the hypothesis that reductions in the expression and function of DAT and VMAT2 may initiate a pathological cascade that, overt time, facilitates additional damage to the dopamine terminal.
Interestingly, while impairment in the expression and function of VMAT2 offers a feasible pathological cascade in the dopamine synapse, HBCDD-induced alterations in synaptic machinery was not exhausted in our project. A variety of other intracellular proteins are involved in mediating dopaminergic neurotransmission, including voltage gated ion channels and synaptic proteins that facilitate synaptic vesicle exocytosis and endocytosis. Decrement to expression or function of any of these targets could also be involved in dysfunction of the dopamine synapse. Support for these hypotheses was demonstrated using PC12 cells and assessing the release of catecholamines following treatment with HBCDD. Interestingly, such treatments elicited a reduction in depolarization-mediated calcium influx and neurotransmitter release, suggesting a possible role for calcium handling in the HBCDD-mediated effects in the synapse (Dingemans et al., 2009).
A critical functional element of the dopamine neuron is the small intracellular protein, Asyn. Considered a neuronal protein, Asyn is enriched in the presynaptic terminal of dopamine projections (Iwai et al., 1995). Although it’s precise function in this milieu is still being determined, extensive evidence supports the role of Asyn in various aspects of dopamine synthesis, packaging, and release, through its explicit intracellular interactions with TH, VMAT2, and DAT, as well as several other proteins associated with these pathways (Lee et al., 2001, Lotharius and Brundin, 2002, Wersinger and Sidhu, 2003, Perez and Hastings, 2004). The importance of these interactions speaks to the role of Asyn as a regulator of dopamine neurotransmission. Within this context, disruption of Asyn is considered a premier pathological hallmark of Parkinson disease (PD), which is defined by dysfunction and loss of dopamine neurons in the SNpc (Braak et al., 2003, Benskey et al., 2016). Recent work has focused on the consequences to the dopamine circuit when the expression and function of Asyn has been reduced. Delivery of AAV-expressing Asyn shRNA into the SNpc results in a significant reduction in Asyn expression and degeneration of dopamine neurons (Gorbatyuk et al., 2010, Kanaan and Manfredsson, 2012, Collier et al., 2016). Based on the implicit role of Asyn in the dopamine neuron, these findings suggest that loss of Asyn culminates in a collapse of the intracellular mechanisms that underlie normal functioning of the dopamine terminal, as it affects the function of TH, VMAT2, and DAT. Unfortunately, current findings have been focused on the impact of Asyn loss in the nigrostriatal dopamine system and have not been evaluated for alterations to dopamine signaling in the hippocampus. However, as the hippocampus is innervated by dopamine projections that arise from cell bodies located in the SNpc and VTA, it is feasible to believe that similar decrements to dopamine signaling would be observed in the hippocampus using this approach (Gasbarri et al., 1994, Gasbarri et al., 1997, Benskey et al., 2016).
Given the extent of the alterations observed in presynaptic markers of dopamine terminal function and integrity, it was somewhat surprising to not see a concomitant impairment in postsynaptic markers of dopamine signaling. If we consider our findings to demonstrate explicit damage and loss of dopaminergic terminals in the hippocampus, similar to that seen in the striatum of PD patients, it could be assumed that we would observe similar alterations to postsynaptic dopaminergic elements. Indeed, imaging studies in patients with PD have shown a generalized lack of effect on D1 receptors in the striatum. However, using similar PET imaging approaches significant reduction in the D2 and D3 receptors in the striatum was found (Niccolini et al., 2014). Unfortunately, similar studies have not been initiated to evaluate the dopamine receptors in the hippocampus. The discrepancy between the striatal findings and our results could be related to the degree of terminal damage seen in PD compared to our study. A plausible explanation suggests that the magnitude of damage to the dopamine terminals by HBCDD was not robust enough or temporally extensive enough to elicit postsynaptic changes, similar to those measured in PD.
In light of these findings, a critical concern remains when translating exposure paradigms utilized in animal studies with concentration of compound in various biofluids in the human population. Studies that have evaluated relative levels of HBCDD in breast milk, plasma, and adipose tissue have found HBCDD concentrations between 1 ng/g-20 ng/g (Aylward and Hays, 2011, Kicinski et al., 2012). Data related to HBCDD concentrations was not available for our current study. However, previous work that employed a similar exposure paradigm (30 mg/kg/day for 28 days) found a concentration of 100 μg/g HBCDD isomers in liver samples (van der Ven et al., 2006). Clearly, these concentrations are well above those found in the general population. However, it can be appreciated that the discrepancy between HBCDD concentrations observed in the human population compared with animal models addresses the limitations associated with mimicking and recapitulating a lifetime of chronic human exposure to a specific chemical in an animal model. Future work delineating the HBCDD body burdens in our animal models will be extremely beneficial in informing our exposure paradigms to better align with concentrations observed in the human population.
5. Conclusions
In sum, findings from our current study suggest that the presynaptic dopamine terminal in the hippocampus is vulnerable to damage following exposure to the brominated flame retardant, HBCDD. Given the importance of dopamine signaling in the hippocampus in mediating various aspects of memory, these findings present a potential cellular target and mechanisms by which HBCDD could lead to memory impairment. Additional studies are need to further delineate the temporal aspects of this damage and correlate HBCDD-induced alterations in dopamine signaling with explicit memory deficits.
Highlights.
-Brominated flame retardants (BFR) are associated with neurobehavioral deficits
-HBCDD exposure decreased presynaptic dopaminergic proteins in the hippocampus
-Postsynaptic dopaminergic proteins in the hippocampus were not changed
-This suggests the hippocampal dopamine circuit is vulnerable to HBCDD
-Alterations to this circuit by HBCDD may underlie learning and memory deficits
Acknowledgments
This work was supported by National Institutes of Health grants R00 ES017477 (WMC) and a pilot award awarded to WMC through P30 ES019776.
Abbreviations
- Asyn
Alpha-Synuclein
- BFR
Brominated Flame Retardants
- COMT
Catechol-O-Methyltransferase
- DAB
3,3′-Diaminobenzidine
- DAT
Dopamine Transporter
- HBCDD
Hexabromocyclododecane
- MAO-B
Monoamine Oxidase-B
- NET
Norepinephrine Transporter
- PBDE
Polybrominated Diphenyl Ethers
- SNpc
Substantia Nigra pars compacta
- TH
Tyrosine Hydroxylase
- VTA
Ventral Tegmental Area
- VMAT2
Vesicular Monoamine Transporter 2
References
- Aylward LL, Hays SM. Biomonitoring-based risk assessment for hexabromocyclododecane (HBCD) International journal of hygiene and environmental health. 2011;214:179–187. doi: 10.1016/j.ijheh.2011.02.002. [DOI] [PubMed] [Google Scholar]
- Benskey MJ, Perez RG, Manfredsson FP. The contribution of alpha synuclein to neuronal survival and function - Implications for Parkinson’s disease. Journal of neurochemistry. 2016;137:331–359. doi: 10.1111/jnc.13570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonito-Oliva A, Pignatelli M, Spigolon G, Yoshitake T, Seiler S, Longo F, Piccinin S, Kehr J, Mercuri NB, Nistico R, Fisone G. Cognitive impairment and dentate gyrus synaptic dysfunction in experimental parkinsonism. Biological psychiatry. 2014;75:701–710. doi: 10.1016/j.biopsych.2013.02.015. [DOI] [PubMed] [Google Scholar]
- Boyson SJ, McGonigle P, Molinoff PB. Quantitative autoradiographic localization of the D1 and D2 subtypes of dopamine receptors in rat brain. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1986;6:3177–3188. doi: 10.1523/JNEUROSCI.06-11-03177.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiology of aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Bradner JM, Suragh TA, Wilson WW, Lazo CR, Stout KA, Kim HM, Wang MZ, Walker DI, Pennell KD, Richardson JR, Miller GW, Caudle WM. Exposure to the polybrominated diphenyl ether mixture DE-71 damages the nigrostriatal dopamine system: role of dopamine handling in neurotoxicity. Experimental neurology. 2013;241:138–147. doi: 10.1016/j.expneurol.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caudle WM, Colebrooke RE, Emson PC, Miller GW. Altered vesicular dopamine storage in Parkinson’s disease: a premature demise. Trends in neurosciences. 2008;31:303–308. doi: 10.1016/j.tins.2008.02.010. [DOI] [PubMed] [Google Scholar]
- Caudle WM, Richardson JR, Delea KC, Guillot TS, Wang M, Pennell KD, Miller GW. Polychlorinated biphenyl-induced reduction of dopamine transporter expression as a precursor to Parkinson’s disease-associated dopamine toxicity. Toxicological sciences : an official journal of the Society of Toxicology. 2006;92:490–499. doi: 10.1093/toxsci/kfl018. [DOI] [PubMed] [Google Scholar]
- Caudle WM, Richardson JR, Wang MZ, Taylor TN, Guillot TS, McCormack AL, Colebrooke RE, Di Monte DA, Emson PC, Miller GW. Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27:8138–8148. doi: 10.1523/JNEUROSCI.0319-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A, Yolton K, Rauch SA, Webster GM, Hornung R, Sjodin A, Dietrich KN, Lanphear BP. Prenatal polybrominated diphenyl ether exposures and neurodevelopment in U.S. children through 5 years of age: the HOME study. Environmental health perspectives. 2014;122:856–862. doi: 10.1289/ehp.1307562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevrier C, Warembourg C, Le Maner-Idrissi G, Lacroix A, Dardier V, Le Sourn-Bissaoui S, Rouget F, Monfort C, Gaudreau E, Mercier F, Bonvallot N, Glorennec P, Muckle G, Le Bot B, Cordier S. Childhood exposure to polybrominated diphenyl ethers and neurodevelopment at six years of age. Neurotoxicology. 2016;54:81–88. doi: 10.1016/j.neuro.2016.03.002. [DOI] [PubMed] [Google Scholar]
- Collier TJ, Redmond DE, Jr, Steece-Collier K, Lipton JW, Manfredsson FP. Is Alpha-Synuclein Loss-of-Function a Contributor to Parkinsonian Pathology? Evidence from Non-human Primates. Frontiers in neuroscience. 2016;10:12. doi: 10.3389/fnins.2016.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa C, Sgobio C, Siliquini S, Tozzi A, Tantucci M, Ghiglieri V, Di Filippo M, Pendolino V, de Iure A, Marti M, Morari M, Spillantini MG, Latagliata EC, Pascucci T, Puglisi-Allegra S, Gardoni F, Di Luca M, Picconi B, Calabresi P. Mechanisms underlying the impairment of hippocampal long-term potentiation and memory in experimental Parkinson’s disease. Brain : a journal of neurology. 2012;135:1884–1899. doi: 10.1093/brain/aws101. [DOI] [PubMed] [Google Scholar]
- Covaci A, Gerecke AC, Law RJ, Voorspoels S, Kohler M, Heeb NV, Leslie H, Allchin CR, De Boer J. Hexabromocyclododecanes (HBCDs) in the environment and humans: a review. Environmental science & technology. 2006;40:3679–3688. doi: 10.1021/es0602492. [DOI] [PubMed] [Google Scholar]
- Covaci A, Harrad S, Abdallah MA, Ali N, Law RJ, Herzke D, de Wit CA. Novel brominated flame retardants: a review of their analysis, environmental fate and behaviour. Environment international. 2011;37:532–556. doi: 10.1016/j.envint.2010.11.007. [DOI] [PubMed] [Google Scholar]
- Cowell WJ, Lederman SA, Sjodin A, Jones R, Wang S, Perera FP, Wang R, Rauh VA, Herbstman JB. Prenatal exposure to polybrominated diphenyl ethers and child attention problems at 3–7 years. Neurotoxicology and teratology. 2015;52:143–150. doi: 10.1016/j.ntt.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit CA. An overview of brominated flame retardants in the environment. Chemosphere. 2002;46:583–624. doi: 10.1016/s0045-6535(01)00225-9. [DOI] [PubMed] [Google Scholar]
- Dingemans MM, Heusinkveld HJ, de Groot A, Bergman A, van den Berg M, Westerink RH. Hexabromocyclododecane inhibits depolarization-induced increase in intracellular calcium levels and neurotransmitter release in PC12 cells. Toxicological sciences : an official journal of the Society of Toxicology. 2009;107:490–497. doi: 10.1093/toxsci/kfn249. [DOI] [PubMed] [Google Scholar]
- Dingemans MM, Ramakers GM, Gardoni F, van Kleef RG, Bergman A, Di Luca M, van den Berg M, Westerink RH, Vijverberg HP. Neonatal exposure to brominated flame retardant BDE-47 reduces long-term potentiation and postsynaptic protein levels in mouse hippocampus. Environmental health perspectives. 2007;115:865–870. doi: 10.1289/ehp.9860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichenbaum H, Schoenbaum G, Young B, Bunsey M. Functional organization of the hippocampal memory system. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:13500–13507. doi: 10.1073/pnas.93.24.13500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson P, Fischer C, Wallin M, Jakobsson E, Fredriksson A. Impaired behaviour, learning and memory, in adult mice neonatally exposed to hexabromocyclododecane (HBCDD) Environmental toxicology and pharmacology. 2006;21:317–322. doi: 10.1016/j.etap.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Eskenazi B, Chevrier J, Rauch SA, Kogut K, Harley KG, Johnson C, Trujillo C, Sjodin A, Bradman A. In utero and childhood polybrominated diphenyl ether (PBDE) exposures and neurodevelopment in the CHAMACOS study. Environmental health perspectives. 2013;121:257–262. doi: 10.1289/ehp.1205597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey U, Morris RG. Synaptic tagging: implications for late maintenance of hippocampal long-term potentiation. Trends in neurosciences. 1998;21:181–188. doi: 10.1016/s0166-2236(97)01189-2. [DOI] [PubMed] [Google Scholar]
- Gasbarri A, Packard MG, Campana E, Pacitti C. Anterograde and retrograde tracing of projections from the ventral tegmental area to the hippocampal formation in the rat. Brain research bulletin. 1994;33:445–452. doi: 10.1016/0361-9230(94)90288-7. [DOI] [PubMed] [Google Scholar]
- Gasbarri A, Sulli A, Innocenzi R, Pacitti C, Brioni JD. Spatial memory impairment induced by lesion of the mesohippocampal dopaminergic system in the rat. Neuroscience. 1996;74:1037–1044. doi: 10.1016/0306-4522(96)00202-3. [DOI] [PubMed] [Google Scholar]
- Gasbarri A, Sulli A, Packard MG. The dopaminergic mesencephalic projections to the hippocampal formation in the rat. Progress in neuro-psychopharmacology & biological psychiatry. 1997;21:1–22. doi: 10.1016/s0278-5846(96)00157-1. [DOI] [PubMed] [Google Scholar]
- Genskow KR, Bradner JM, Hossain MM, Richardson JR, Caudle WM. Selective damage to dopaminergic transporters following exposure to the brominated flame retardant, HBCDD. Neurotoxicology and teratology. 2015 doi: 10.1016/j.ntt.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbatyuk OS, Li S, Nash K, Gorbatyuk M, Lewin AS, Sullivan LF, Mandel RJ, Chen W, Meyers C, Manfredsson FP, Muzyczka N. In vivo RNAi-mediated alpha-synuclein silencing induces nigrostriatal degeneration. Molecular therapy : the journal of the American Society of Gene Therapy. 2010;18:1450–1457. doi: 10.1038/mt.2010.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbstman JB, Sjodin A, Kurzon M, Lederman SA, Jones RS, Rauh V, Needham LL, Tang D, Niedzwiecki M, Wang RY, Perera F. Prenatal exposure to PBDEs and neurodevelopment. Environmental health perspectives. 2010;118:712–719. doi: 10.1289/ehp.0901340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, Kittel A, Saitoh T. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron. 1995;14:467–475. doi: 10.1016/0896-6273(95)90302-x. [DOI] [PubMed] [Google Scholar]
- Jay TM. Dopamine: a potential substrate for synaptic plasticity and memory mechanisms. Progress in neurobiology. 2003;69:375–390. doi: 10.1016/s0301-0082(03)00085-6. [DOI] [PubMed] [Google Scholar]
- Kanaan NM, Manfredsson FP. Loss of functional alpha-synuclein: a toxic event in Parkinson’s disease? Journal of Parkinson’s disease. 2012;2:249–267. doi: 10.3233/JPD-012138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kicinski M, Viaene MK, Den Hond E, Schoeters G, Covaci A, Dirtu AC, Nelen V, Bruckers L, Croes K, Sioen I, Baeyens W, Van Larebeke N, Nawrot TS. Neurobehavioral function and low-level exposure to brominated flame retardants in adolescents: a cross-sectional study. Environmental health : a global access science source. 2012;11:86. doi: 10.1186/1476-069X-11-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Morita S, Sawada H, Mizuguchi T, Yamada K, Nagatsu I, Hata T, Watanabe Y, Fujita K, Nagatsu T. Targeted disruption of the tyrosine hydroxylase locus results in severe catecholamine depletion and perinatal lethality in mice. The Journal of biological chemistry. 1995;270:27235–27243. doi: 10.1074/jbc.270.45.27235. [DOI] [PubMed] [Google Scholar]
- Lee FJ, Liu F, Pristupa ZB, Niznik HB. Direct binding and functional coupling of alpha-synuclein to the dopamine transporters accelerate dopamine-induced apoptosis. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2001;15:916–926. doi: 10.1096/fj.00-0334com. [DOI] [PubMed] [Google Scholar]
- Lilienthal H, van der Ven LT, Piersma AH, Vos JG. Effects of the brominated flame retardant hexabromocyclododecane (HBCD) on dopamine-dependent behavior and brainstem auditory evoked potentials in a one-generation reproduction study in Wistar rats. Toxicology letters. 2009;185:63–72. doi: 10.1016/j.toxlet.2008.12.002. [DOI] [PubMed] [Google Scholar]
- Lisman JE, Grace AA. The hippocampal-VTA loop: controlling the entry of information into long-term memory. Neuron. 2005;46:703–713. doi: 10.1016/j.neuron.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Lotharius J, Brundin P. Pathogenesis of Parkinson’s disease: dopamine, vesicles and alpha-synuclein. Nature reviews Neuroscience. 2002;3:932–942. doi: 10.1038/nrn983. [DOI] [PubMed] [Google Scholar]
- Mariussen E, Fonnum F. The effect of brominated flame retardants on neurotransmitter uptake into rat brain synaptosomes and vesicles. Neurochemistry international. 2003;43:533–542. doi: 10.1016/s0197-0186(03)00044-5. [DOI] [PubMed] [Google Scholar]
- McNamara CG, Tejero-Cantero A, Trouche S, Campo-Urriza N, Dupret D. Dopaminergic neurons promote hippocampal reactivation and spatial memory persistence. Nature neuroscience. 2014;17:1658–1660. doi: 10.1038/nn.3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niccolini F, Su P, Politis M. Dopamine receptor mapping with PET imaging in Parkinson’s disease. Journal of neurology. 2014;261:2251–2263. doi: 10.1007/s00415-014-7302-2. [DOI] [PubMed] [Google Scholar]
- Ortiz O, Delgado-Garcia JM, Espadas I, Bahi A, Trullas R, Dreyer JL, Gruart A, Moratalla R. Associative learning and CA3-CA1 synaptic plasticity are impaired in D1R null, Drd1a−/− mice and in hippocampal siRNA silenced Drd1a mice. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:12288–12300. doi: 10.1523/JNEUROSCI.2655-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez RG, Hastings TG. Could a loss of alpha-synuclein function put dopaminergic neurons at risk? Journal of neurochemistry. 2004;89:1318–1324. doi: 10.1111/j.1471-4159.2004.02423.x. [DOI] [PubMed] [Google Scholar]
- Richardson JR, Miller GW. Acute exposure to aroclor 1016 or 1260 differentially affects dopamine transporter and vesicular monoamine transporter 2 levels. Toxicology letters. 2004;148:29–40. doi: 10.1016/j.toxlet.2003.12.006. [DOI] [PubMed] [Google Scholar]
- Rosen ZB, Cheung S, Siegelbaum SA. Midbrain dopamine neurons bidirectionally regulate CA3-CA1 synaptic drive. Nature neuroscience. 2015;18:1763–1771. doi: 10.1038/nn.4152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saegusa Y, Fujimoto H, Woo GH, Ohishi T, Wang L, Mitsumori K, Nishikawa A, Shibutani M. Transient aberration of neuronal development in the hippocampal dentate gyrus after developmental exposure to brominated flame retardants in rats. Archives of toxicology. 2012;86:1431–1442. doi: 10.1007/s00204-012-0824-4. [DOI] [PubMed] [Google Scholar]
- Shohamy D, Adcock RA. Dopamine and adaptive memory. Trends in cognitive sciences. 2010;14:464–472. doi: 10.1016/j.tics.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Ta TA, Koenig CM, Golub MS, Pessah IN, Qi L, Aronov PA, Berman RF. Bioaccumulation and behavioral effects of 2,2′,4,4′-tetrabromodiphenyl ether (BDE-47) in perinatally exposed mice. Neurotoxicology and teratology. 2011;33:393–404. doi: 10.1016/j.ntt.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Ven LT, Verhoef A, van de Kuil T, Slob W, Leonards PE, Visser TJ, Hamers T, Herlin M, Hakansson H, Olausson H, Piersma AH, Vos JG. A 28-day oral dose toxicity study enhanced to detect endocrine effects of hexabromocyclododecane in Wistar rats. Toxicological sciences : an official journal of the Society of Toxicology. 2006;94:281–292. doi: 10.1093/toxsci/kfl113. [DOI] [PubMed] [Google Scholar]
- Viberg H, Fredriksson A, Eriksson P. Neonatal exposure to polybrominated diphenyl ether (PBDE 153) disrupts spontaneous behaviour, impairs learning and memory, and decreases hippocampal cholinergic receptors in adult mice. Toxicology and applied pharmacology. 2003;192:95–106. doi: 10.1016/s0041-008x(03)00217-5. [DOI] [PubMed] [Google Scholar]
- Wersinger C, Sidhu A. Attenuation of dopamine transporter activity by alpha-synuclein. Neuroscience letters. 2003;340:189–192. doi: 10.1016/s0304-3940(03)00097-1. [DOI] [PubMed] [Google Scholar]
- Xing T, Chen L, Tao Y, Wang M, Chen J, Ruan DY. Effects of decabrominated diphenyl ether (PBDE 209) exposure at different developmental periods on synaptic plasticity in the dentate gyrus of adult rats In vivo. Toxicological sciences : an official journal of the Society of Toxicology. 2009;110:401–410. doi: 10.1093/toxsci/kfp114. [DOI] [PubMed] [Google Scholar]
- Yan T, Xiang L, Xuejun J, Chengzhi C, Youbin Q, Xuelan Y, Yang L, Changyan P, Hui C. Spatial learning and memory deficit of low level polybrominated diphenyl ethers-47 in male adult rat is modulated by intracellular glutamate receptors. The Journal of toxicological sciences. 2012;37:223–233. doi: 10.2131/jts.37.223. [DOI] [PubMed] [Google Scholar]
- Zhu G, Chen Y, Huang Y, Li Q, Behnisch T. MPTP-meditated hippocampal dopamine deprivation modulates synaptic transmission and activity-dependent synaptic plasticity. Toxicology and applied pharmacology. 2011;254:332–341. doi: 10.1016/j.taap.2011.05.007. [DOI] [PubMed] [Google Scholar]